

Abstract
Alzheimer’s disease (AD), the most prevalent form of dementia, is characterized by two pathological hallmarks: Tau-containing neurofibrillary tangles and amyloid-β protein (Aβ)-containing neuritic plaques. The goal of this study is to understand mild traumatic brain injury (mTBI)-related brain proteomic changes and tau-related biochemical adaptations that may contribute to AD-like neurodegeneration. We found that both phosphorylated tau (p-tau) and the ratio of p-tau/tau were significantly increased in brains of mice collected at 3 and 24 h after exposure to 82-kPa low-intensity open-field blast. Neurological deficits were observed in animals at 24 h and 7 days after the blast using Simple Neuroassessment of Asymmetric imPairment (SNAP) test, and axon/dendrite degeneration was revealed at 7 days by silver staining. Liquid chromatography-mass spectrometry (LC-MS/MS) was used to analyze brain tissue labeled with isobaric mass tags for relative protein quantification. The results from the proteomics and bioinformatic analysis illustrated the alterations of axonal and synaptic proteins in related pathways, including but not being limited to substantia nigra development, cortical cytoskeleton organization, and synaptic vesicle exocytosis, suggesting a potential axonal damage caused by blast-induced mTBI. Among altered proteins found in brains suffering blast, microtubule-associated protein 1B, stathmin, neurofilaments, actin binding proteins, myelin basic protein, calcium/calmodulin-dependent protein kinase, and synaptotagmin I were representative ones involved in altered pathways elicited by mTBI. Therefore, TBI induces elevated phospho-tau, a pathological feature found in brains of AD, and altered a number of neurophysiological processes, supporting the notion that blast-induced mTBI as a risk factor contributes to AD pathogenesis. LC/MS-based profiling has presented candidate target/pathways that could be explored for future therapeutic development.
Keywords: Alzheimer’s disease, bioinformatics, mild traumatic brain injury, open-field blast, proteomics
INTRODUCTION
Mild traumatic brain injury (mTBI) results in significant morbidity and mortality throughout the world. Overlapping neuropathological features of patients suffering Alzheimer’s disease (AD) or traumatic brain injury (TBI) suggest that pathological proteins for these two conditions may interact [1,2]. In AD, widespread neuronal loss is associated with the pathological hallmarks of neurofibrillary tangles composed of phosphorylated tau and neuritic plaques composed of amyloid-β protein (Aβ). Animal brains suffering TBI exhibit tau pathology upon following controlled cortical impact, and these transgenic mice exhibit dramatic increases in levels of tau [1]. Several studies have associated TBI with a significantly increased risk of the development of late neurodegenerative diseases, such as AD and Parkinson’s disease [3]. Soldiers exposed to even a single blast can develop behavioral and psychiatric problems within a year following injury [4]. Even a single TBI may induce long-term neuropathological changes akin to those found in neurodegenerative disease and associated with the later development of syndromes of cognitive impairment such as AD [5]. In humans, repetitive impact-induced mTBI leads to chronic traumatic encephalopathy (CTE) with increased tau pathology and other neurodegenerative changes in the brain [6]. CTE has been observed in veterans exposed to explosive blast [7]. Clinical reports describe post-concussive stress and depression symptoms along with worse cognitive performance in subjects with blast-induced mTBI, both in short- (0–7 days) and long-term (6 months to 5 years) follow-up exams after the blast [8–10]. Additional reports confirm that mTBI is positively associated with depression and increased anxiety levels [11].
Among the several mechanisms that have been proposed as possible explanations for these associations, two prime events occurring in ongoing blast injury are neuroinflammation and deposition of abnormal microtubule proteins such as tau [12]. Mutations in the tau gene cause frontotemporal dementia with Parkinsonism linked to chromosome 17 (FTDP-17) [13]. Transgenic mice expressing mutant tau show close association with tau mutation-containing, neurofibrillary tangle (NFT) formation, and neurodegeneration [14–16]. Tau protein has been used as a marker for axonal damage, and tau levels in cerebrospinal fluid (CSF) reflect these changes in the central nervous system (CNS), especially, total tau protein levels (t-tau), phosphorylated tau (p-tau), and the ratio of Aβ42/tau have been explored as potential markers for AD [17–19]. Other related proteins include neurofilament light chain (NfL), which was found to be increased in CSF and blood of patients with AD or tauopathies. In animals, blocking Aβ lesions attenuates the NfL increase [20]. These proteins may affect the formation of Aβ/tau pathologies.
Proteomic research [21, 22] using brain tissues or biofluids from either patients or animal models have made it feasible to identify a large number of proteins associated with mTBI [23,24]. MS-based proteomics possess the advantage of having no requirement of prior knowledge of the proteins being identified, allowing for unbiased, hypothesis-free biomarker discovery in complex biological samples such as plasma and brain tissue extracts. Therefore, MS-based proteomic approach can be used to unravel novel biological processes underlying TBI, neurodegeneration and repair, or to represent complex protein-protein interactions during disease progression. However, MS-based proteomic analysis has not been performed on samples derived from animals exposed to open field blast-induced mTBI.
In this study, we use proteomic profiling of mouse brains suffering blast-induced mTBI to explore contribution of mTBI toward AD pathologic process. Tau associated biological processes were found to be differentially regulated in blast-induced mTBI mouse models, and levels of phospho-tau were increased in brains of mice collected at 3 and 24 h after the exposure to blast. Behavioral and pathological alterations were observed in blast exposed mice. We employed tandem mass tag (TMT)-labelling in conjunction with liquid chromatography-tandem mass spectrometry (LC-MS/MS) approach to investigate differentially expressed proteins in mouse brain samples after exposed to a single low-intensity open-field blast for the purpose of discovering candidate proteins associated with blast-induced mTBI and AD pathology. Differentially expressed proteins were analyzed using bioinformatics tools DAVID and Metacore to determine if functional relationships were altered in mice exposed to blast. This approach allows us to evaluate effect of blast on mice by understanding the proteomic profiles of brain tissue as a way of identifying potential molecular signatures of pathological alteration that may contribute to AD pathogenesis.
MATERIALS AND METHODS
Materials and reagents
Unless indicated, all reagents used for biochemical methods and cell culture preparation were purchased from Sigma-Aldrich (St Louis, MO, USA). Reagents for BCA protein assay and sample preparation for LC-MS/MS analysis were purchased from Thermo Scientific (Rockford, IL, USA). TMT reagent six-plex and ten-plex kits were available commercially (Thermo Scientific Pierce, Rockford, IL, USA).
Open-field primary blast injury in mice
Male C57BL/6J mice (The Jackson Laboratory, Bar Harbor, ME), at 2 months old, were housed with a 12-h light/dark cycle and given unrestricted access to food and water. Open-field blast exposures were conducted at the Missouri University of Science & Technology and were recently reported [25, 26]. Briefly, for the experimental setting of open-field low-intensity blast injury with the modification as described here, mice were placed in animal holders in prone position at 2.15-meter (m) distance away from detonation of 350 g C4 explosive (both 1 m above ground) generating a magnitude of 82 kPa [e.g., 11.9 pounds per square inch (PSI)] peak overpressure and maximal impulse at 71 kPa (10.3 PSI) × ms. The Sham group underwent identical procedures as blast groups only without blast exposure. Following blast wave exposure, animals were removed from the platform and returned to the original cage. After recovery from anesthesia, mice were able to spontaneously move and continuously being monitored for at least 15–30 min. Animals were allowed access to food and water ad libitum before and after blast exposure. Mice were randomly assigned into experimental or sham control groups which were double-blinded to investigators analyzing experimental outcomes. All procedures were in accordance with the University of Missouri approved protocols for the Care and Use of Laboratory Animals and ARRIVE guidelines.
Behavioral test
The behavioral test, Simple Neuroassessment of Asymmetric imPairment (SNAP), was used after blast exposure to assess the neurological status of coordination, motor strength, proprioception, gait, and mental responses. The tests examined interaction, cage grasp, visual placing, pacing/circling, gait/posture, head tilt, visual field, and baton as we previously described [25, 27].
Brain collection and pathological examination
In total, there were thirty-seven mice in the present study, in which thirty-two mice were randomly divided into four groups at 3 h (n = 4), 24 h (n = 9), 7 days (n = 11), 30 days (n = 4), and 15 weeks (n = 4) after a single 82-kPa low-intensity blast exposure and five mice as the sham control group [25, 26, 28, 29]. Mice were euthanized using overdoing of isoflurane and cerebral cortex regions were dissected from the blast-exposed mouse brain. Silver staining and quantitative analyses were performed as previously described [25, 30]. The FD NeuroSilver Kit II was used for silver staining according to the manufacturer’s instructions. Each batch of the experiments was included the brain sections from different groups to avoid variability of the staining intensity. Tiff images of sections with the defined coordinates for stereological analysis were captured containing the regions of interests of the corpus callosum and entorhinal cortex (bregma 0.14 to 0.02 mm), cerebral peduncle (bregma −1.94 to −2.06 mm), and fornix (bregma 0.14 to 0.02 mm), based on the mouse brain atlas. These images were exported and analyzed with ImageJ software (NIH, Bethesda, MD). The background (gold color) was subtracted from each image using the color splitter and image calculator functions, so that only the gray-black silver deposits were visible. Densitometric analysis of silver staining was then performed using ImageJ. Serial sections were averaged from each animal to produce a final silver staining value. The intensity of silver staining was expressed in arbitrary units, ranging from 0 to 255.
Preparation of brain tissue for MS
Sample lysis buffer (2% SDS, 0.5M tetraethyl-ammonium bicarbonate (TEAB), protease inhibitor cocktail) were added to each brain tissue and then homogenized by TissueLyser LT (Qiagen, Valencia, CA). Tissue homogenates were centrifuged at 17,000 × g, for 20 min at 4°C. The supernatant was transferred into a new vial for protein concentration measurement by BCA assay. Preparation of tryptic peptides for TMT 10-plex labelling was carried out according to the manufacturer’s instructions. Briefly 100 μg protein from each sample was transferred into a new vial and adjusted to a final volume of 100 μL with TEAB and reduced with tris (2-carboxyethyl) phosphine (TCEP) at 55°C for 1h, and then alkylated with iodoacetamide for 30 min in the dark. Methanol-chloroform precipitation was performed prior to protease digestion. In brief, four parts methanol was added to each sample and vortexed, then one part chloroform was added to the sample and vortexed, and three parts water was added to the sample and vortexed. The sample was centrifuged at 14,000 × g for 4 min at room temperature and the aqueous phase was removed. The organic phases with protein precipitate at the surface was subsequently washed twice with four parts methanol and centrifuged with supernatant being removed subsequently. After air-drying, precipitated protein pellets were re-suspend with 100 μL of 50 mM TEAB and digested with trypsin overnight at 37°C.
TMT-labeling and sample clean up
TMT enables relative quantitation of proteins present in multiple samples by labeling peptides with isobaric stable isotope tags that fragment upon collision-induced dissociation into reporter ions used for quantitation. In this study, TMT ten tags for ten samples (blast samples and controls) were analyzed simultaneously, avoiding run-to-run variation. Tryptic digested peptides from brain samples were labeled with TMT 10-plex reagents (Thermo Fisher) according to manufacturer’s instructions. Briefly, the TMT reagents (0.8 mg) were dissolved in 41 μL of anhydrous acetonitrile. Aliquots of samples were incubated with TMT reagents for 1 h at room temperature. The reactions were quenched by 8 μL of 5% hydroxylamine solution and reacted for 15 min. The combined TMT labelled samples were dried under SpeedVac, and then reconstituted by dilute tri-fluoroacetic acid solution followed by desalting by Oasis HLB 96-well μElution plate (Waters) prior to LC-MS/MS analysis.
Phosphoprotein enrichment and analysis
For enrichment of phospho-peptides, samples were processed using the Fe-NTA phospho-peptide enrichment kit and followed by graphite spin columns according to the manufacturer’s instructions (Thermo Scientific Pierce) before LC-MS/MS analysis.
LC-MS/MS analysis
LC MS/MS was performed on a Q Exactive Orbitrap Mass Spectrometer (Thermo Fisher Scientific) coupled with a Dionex ultimate 3000 HPLC system equipped with a nano-ES ion source. The TMT labelled peptides were separated on a C18 reverse-phase capillary column (PepMap, 75 μm × 150 mm, Thermo Fisher) with linear gradients of 2–35% acetonitrile in 0.1% formic acid, at a constant flow rate of 300 nL/min for 220 min. The instrument was operated in the positive-ion mode with the ESI spray voltage set at 1.8 kV. A full scan MS spectrum (300–1800 m/z) was acquired in the Orbitrap at a mass resolution of 70,000 with an automatic gain control target (AGC) of 3e6. Fifteen peptide ions showing the most intense signal from each scan were selected for higher energy collision-induced dissociation (HCD)-MS/MS analysis (normalized collision energy 32) in the Orbitrap at a mass resolution of 35,000 and AGC value of 1e5. Maximal filling times were 100 ms in full scans and 120 ms (HCD) for the MS/MS scans. Ions with unassigned charge states and singly charged species were rejected. The dynamic exclusion was set to 50 s and a relative mass window of 10 ppm. The data were acquired using Thermo Xcalibur 3.0.63. The phospho-peptides were analyzed under the same conditions as above, other than the separation which was done with linear gradients of 1–30% acetonitrile in 0.1% formic acid, and full scan MS ranging 300–2000 m/z with NCE at 25.
Protein identification and quantification
Raw data were processed using Proteome Discoverer (Version 2.1, Thermo Fisher Scientific). Data were searched against the mus musculus Universal Protein Resource sequence database (UniProt, August, 2013). The searches were performed with the following guidelines: Trypsin digestion with two missed cleavage allowed; fixed modification, carbamidomethyl of cysteine; variable modification, oxidation of methionine, TMT 10plex (peptide labeled) for N terminus and Lys; MS tolerance, 5 ppm; MS/MS tolerance, 0.02 Da; false discovery rate (FDR) at peptide and protein levels, <0.01; and required peptide length, ≥6 amino acids. Phosphorylation of serine, threonine, and tyrosine was defined as variable modification. Protein grouping was enabled, meaning if one protein were equal to or completely contained within the set of peptides of another protein, these two proteins were put into the same protein group. At least one unique peptide per protein group was required for identifying proteins. The relative protein abundance ratios (fold changes) between blasted and shamed groups were calculated. The changes in protein levels were considered significant if fold change was >1.2 (upregulated) or < 0.9 (down-regulated), and the p-value is less than 0.05 in two independent experiments.
Quantification of tau and phospho-tau by ELISA
Tau and p-tau peptide at Thr residue 181 were quantified by ELISA [31]. The capture antibody for tau is BT-2. To capture specific phosphorylated tau at Thr residue 181, antibody AT270 was used. The detecting antibody is the biotinylated HT7 that recognizes residue 159–163 of tau. All three tau antibodies were purchased from Thermo Scientific (Rockford, IL). ELISA was performed using Meso Scale Discovery platform (MSD, Gaithersburg, MD). The internal calibrators of p-tau and tau were purchased from Meso Scale Discovery.
Data analysis
An enrichment analysis was performed by submitting the UniProt accession numbers containing the list of differentially regulated proteins to open-source software Database for Annotation, Visualization, and Integrated Discovery (DAVID) v6.7 [32, 33]. DAVID is able to extract biological features/meanings associated with large gene lists. The gene names of the proteins were entered to analyze GO biological process (GOBP), GO molecular function (GOMF), and GO cellular components (GOCC) and KEGG pathways. Statistically significant differences (p-value < 0.05) were identified using an EASE score (a modification of Fisher’s exact test), which was provided by the DAVID gene bioinformatics online resource. The cellular and molecular process networks of the dysregulated proteins were also analyzed using Metacore. Metacore uses these proteins and their identifiers to navigate the curated literature database and extract the biologically relevant information among the candidate proteins. Associated biofunctions were generated, along with a score representing the log probability of a particular process network.
RESULTS
Increase of phospho-tau in brains of mice at 3 and 24 h after the blast
In this study, we analyzed the mouse brain samples at 3 h, 24 h, 30 days, and 15 weeks after the blast. We applied ELISA to quantify levels of tau and phospho-tau in the same brain tissue later used for LC-MS/MS analysis. We found a significant increase of phospho-tau in mouse brains collected at 3 and 24 h after the blast, while a reduction of tau was also observed in the same samples (Fig. 1). We also calculated the ratio of p-tau/tau and found that both levels of p-tau and the ratio of p-tau/tau were increased compared to those from brains of mice without exposure to blast (Sham). The impact of blast to mouse brains was manifested as early as 3 h, and remained after 24 h, when significant reduction of tau and increase of p-tau were observed. Total tau level was decreased ~20% compared to that of sham mouse brains, and the p-tau level was increased 1.2 folds, and these changes were found statistically significant. Thirty days after the blast, tau levels bounced back and remained at similar levels to those of sham mice. Similarly, p-tau dropped a statistically significant 20% after 30 days and then remained at similar levels to those of sham mice after 15 weeks (Fig. 1). The ratios of p-tau/tau reflected similar changes to the levels of p-tau in mouse brains harvested at 30 days and 15 weeks after the blast.
Fig. 1.
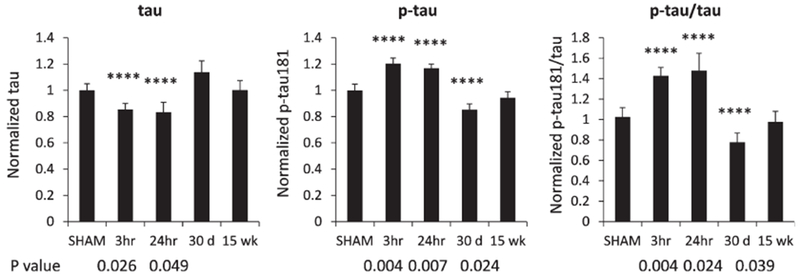
Quantification of total tau and phosphorylated tau from mouse brain at different time points after the blast. Brain lysates were subjected to ELISA using Meso Scale Discovery platform, and same samples were quantified for the levels of tau and p-tau. The ratio of p-tau to tau (p-tau/tau) was calculated for each sample, and the levels of tau, p-tau, p-tau/tau in brains of control mice were normalized to 1. Relative levels of these analytes in brains of mice harvested at 3 h, 24 h, 30 days, and 15 weeks after the blast were illustrated. The bar represents standard error of means (SEM). Asterisks represent statistically significant differences.
Behavioral and pathological alteration in mice after the blast
We performed an explorative behavioral SNAP test on the mice exposed to 82-kPa low intensity blast (LIB). SNAP provides a sensitive, reliable, time-efficient, and cost-effective means of assessing neurological deficits in mice. Each individual test had a scoring range of 0–5 based on the assessment guidelines (Fig. 2). Using the current settings and 350 g of high-energy explosive C4, SNAP assessments revealed a significant difference in the blast group compared to sham controls at both 24 h and 7 days post injury (DPI) (Fig. 2), indicating that neurological deficits affecting coordination, motor strength, proprioception, gait, and mental status occurred after LIB exposure.
Fig. 2.
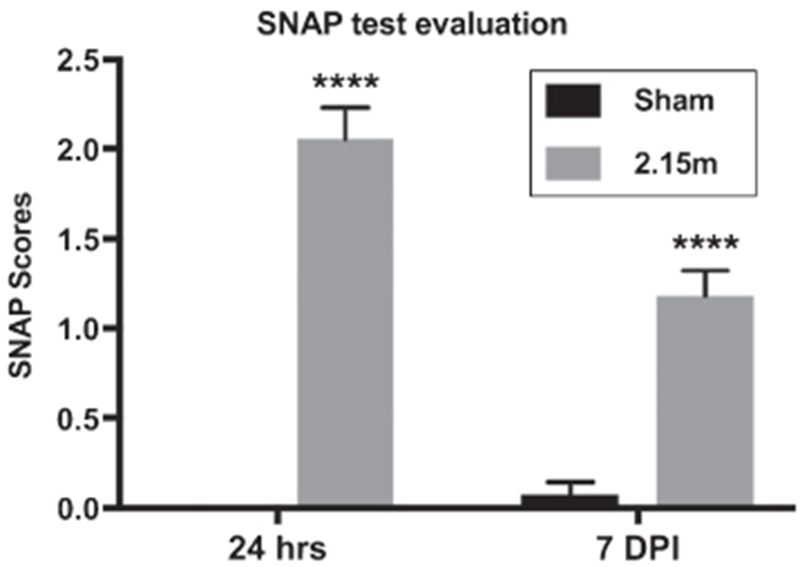
Neurological deficits after low intensity blast exposure assessed by SNAP test. Neurological status was evaluated by SNAP tests at 24 h and 7 days post injury (DPI). All comparisons were performed between blast (2.15 m, distance between the animals and the blast) and sham group on the same DPI; n = 5 for sham, n = 9 at 24 h after blast, n = 11 at 7 DPI. Differences were considered significant at p <0.05 for all analyses, with asterisks representing p < 0.0001.
Axonal injury in LIB-exposed animals occurred mainly at 7 DPI, as we previously described [25]. Degenerating axon-terminals and dendrites, with high affinity for silver (argyrophilia) were visualized on silver-stained tissue sections (Fig. 3A–C). Densitometric analyses by NIH ImageJ were examined on various white matter regions rich for axons, including corpus callosum and entorhinal cortex, cerebral peduncle, and fornix. The silver stain intensities in blast group were significantly higher than in sham controls at 7 DPI for all subregions (Fig. 3D–F). We have previously seen that the abnormalities at 7 DPI appeared to return to comparable levels of sham controls at 30 DPI (data now shown).
Fig. 3.
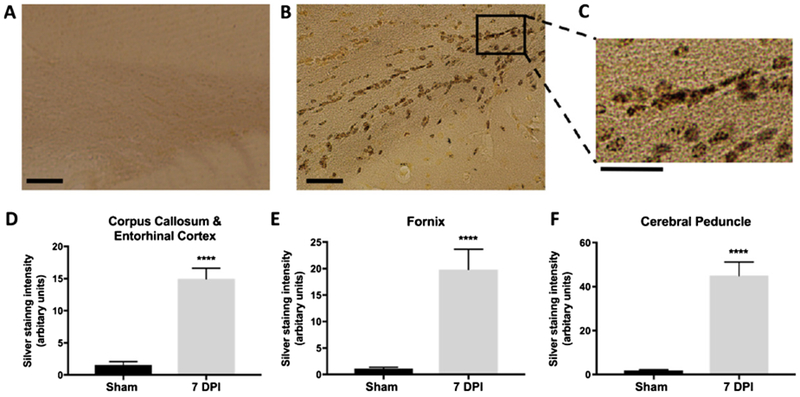
Axonal injury detected and quantified after LIB exposure by silver staining. A) Representative images from the sham group showed no axonal injury (scale bar = 50 μm). B, C) Representative images from the blast group showed axonal injury at corpus callosum and entorhinal cortex (scale bar = 50 μm). Inset box represent the higher magnification of the indicated area (scale bar = 25 μm). D-F) Intensity of silver staining in corpus callosum and entorhinal cortex, fornix, and cerebral peduncle regions in blast group at 7 DPI were significantly higher than that in the sham group. All comparisons were performed between blast and sham groups with t-test; n = 5 for sham controls, and n = 4 for blast at 7 DPI. Differences were considered significant at p < 0.05 for all analyses, with asterisks representing p < 0.0001.
Global protein alteration in brains exposed to blast
To understand impact of blast to brain proteins that drive these phenotypes, analysis of both global proteomics and phospho-proteomics were conducted using mouse brains collected at 3 h, 24 h, 30 days, and 15 weeks after the blast. We specifically searched for significant changes in proteins from mouse brains harvested at the time points (3 h and 24 h) where we also observed significant changes in tau and p-tau proteins. A total of 1,692 proteins and 296 phosphoproteins were identified at global level and analyzed in this study. By comparing to the sham mouse brain, the fold of changes (ratio ≥1.2 or ≤0.9) of proteins observed in brains of mice at 3 h and 24 h after the blast was used for functional analysis by DAVID. At the global level, relative to the proteins identified from the sham controls, only a few proteins were downregulated after the blast, including Hemoglobin subunit α(Hba) and Hemoglobin subunit β-1(Hbb-b1). They are enriched in the cellular location of hemoglobin complex. Their primary molecular functions are oxygen transporter and oxygen/heme binding. On the other hand, 130 proteins were upregulated with the fold changes ≥1.2 after the blast. DAVID functional analysis in three categories, including cellular components (CC), biological process (BP), and molecular function (MF), of upregulated proteins at 3 h after the blast were conducted (Fig. 4A–C). The top two enriched CC are cytoplasm and neuronal cell body. Among CC, axon was also significantly enriched (p = 1.2E-4), followed by other neuron related components, such as postsynaptic dendritic spine and microtubule (Fig. 4A). Besides general BP such as transport and translation, most altered proteins were enriched in cell-cell adhesion and vesicle mediated transport. In addition, proteins enriched in axon guidance, axon extension and microtubule-based process were also illustrated (Fig. 4B). The top enriched MF (Fig. 4C) include cadherin binding/cell-cell adhesion, hydrolase activity, nucleotide binding, microtubule binding, filamin binding, and β-tubulin binding activities (p ≤ 3.0E-2).
Fig. 4.
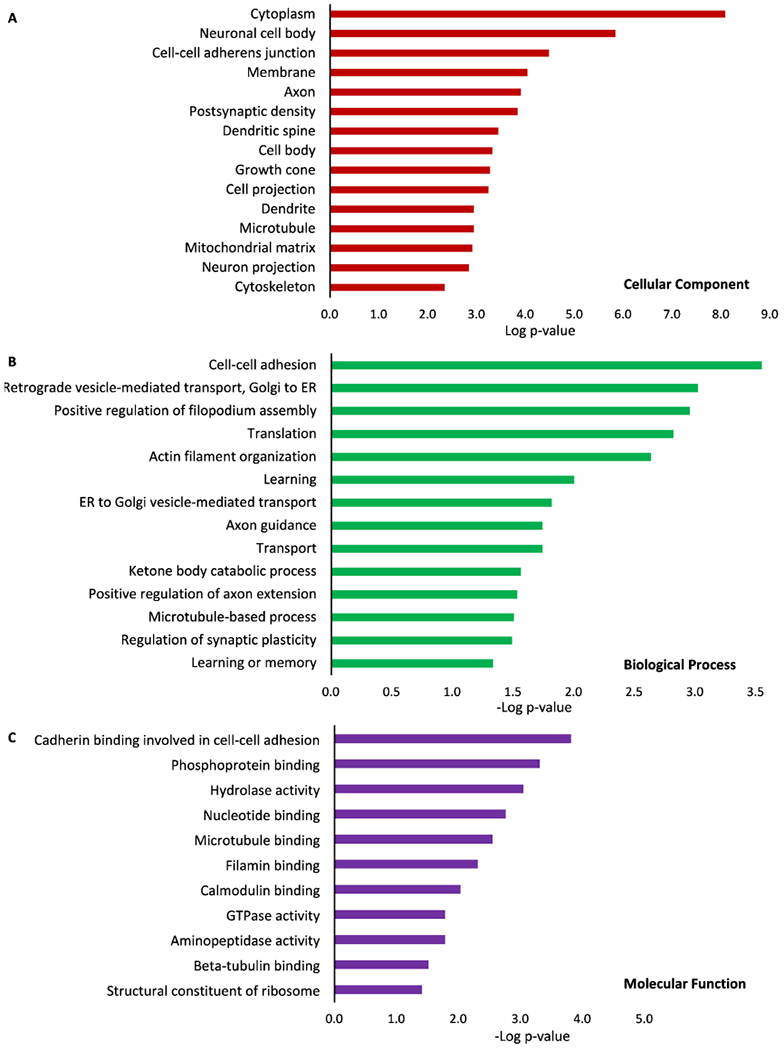
GO ontology analysis of altered proteins at global level. Proteins from mouse brains harvested at 3 h after the blast were analyzed, and each of the three categories, cellular components (A), biological process (B) and molecular functions (C) were listed. Proteins from blast-exposed animals were compared to those from control/sham animals, and –Log of p values was plotted.
We have examined proteins involved in each of the three top enriched categories BP, CC, and MF that are related to neuronal functions and listed upregulated proteins from animals harvested at 3 h post the blast (Table 1). The BP included positive regulation of axon extension and axon-dendritic transport. The upregulated proteins highly enriched in postsynaptic density and axon included huntingtin (Htt), which may play a role in microtubule-mediated transport or vesicle function. Proteins involved in microtubule-based process include Tubulin α-1A chain (Tubα1a) and Dynein light chain 1, cytoplasmic (Dynll1). GO enrichment analysis in each of the three categories (BP, CC, MF) revealed proteins related to those microtubule associated proteins, and network analysis of upregulated proteins at 3 h were illustrated by STRING (Fig. 5A). Proteins related to microtubule, such as Actr1b, Tubα1a, Tubβ2a, Dunll1, are among the proteins with the extensive network connections. These proteins are also observed in mouse brains harvested at 24 h (Fig. 5B), and less in mouse brains harvested at 30 days (Fig. 5C) and 15 weeks (Fig. 5D) after the blast exposure.
Table 1.
DAVID functional analysis of upregulated proteins at global level at 3 h after the blast and proteins enriched in each category
| Categories | GO Terms | ACCESSION | GENE NAME |
|---|---|---|---|
| BP | Positive regulation of filopodium assembly | ||
| Q99JY9 | ARP3 actin-related protein 3(Actr3) | ||
| Q62188 | Dihydropyrimidinase-like 3(Dpysl3) | ||
| Q61553 | Fascin actin-bundling protein 1(Fscn1) | ||
| P35802 | Glycoprotein m6a(Gpm6a) | ||
| Actin filament organization | |||
| Q99JY9 | ARP3 actin-related protein 3(Actr3) | ||
| Q641P0 | ARP3 actin-related protein 3B(Actr3b) | ||
| Q9QXS6 | Drebrin 1(Dbn1) | ||
| Q61553 | Fascin actin-bundling protein 1(Fscn1) | ||
| Q60875 | Rho/rac guanine nucleotide exchange factor (GEF) 2(Arhgef2) | ||
| Learning | |||
| O88587 | Catechol-O-methyltransferase(Comt) | ||
| O35927 | Catenin (cadherin associated protein), delta 2(Ctnnd2) | ||
| P42859 | Huntingtin(Htt) | ||
| P31324 | Protein kinase, cAMP dependent regulatory, type II beta(Prkar2b) | ||
| Axon guidance | |||
| P97427 | Collapsin response mediator protein 1(Crmp1) | ||
| Q9EQF6 | Dihydropyrimidinase-like 5(Dpys15) | ||
| O08917 | Flotillin 1(Flot1) | ||
| P06837 | Growth associated protein 43(Gap43) | ||
| P13595 | Neural cell adhesion molecule 1(Ncam1) | ||
| Positive regulation of axon extension | |||
| Q9QXS6 | Drebrin 1(Dbn1) | ||
| P10637 | Microtubule-associated protein tau(Mapt) | ||
| Q9D8E6 | Ribosomal protein L4(Rpl4) | ||
| Microtubule-based process | |||
| P63168 | Dynein light chain LC8-type 1(Dynll1) | ||
| P68369 | Tubulin, alpha 1A(Tuba1a) | ||
| Q7TMM9 | Tubulin, beta 2A class IIA(Tubb2a) | ||
| Regulation of synaptic plasticity | |||
| O35927 | Catenin (cadherin associated protein), delta 2(Ctnnd2) | ||
| P42859 | Huntingtin(Htt) | ||
| P13595 | Neural cell adhesion molecule 1(Ncam1) | ||
| CC | Axon | ||
| P63054 | Purkinje cell protein 4(Pcp4) | ||
| O88587 | Catechol-O-methyltransferase(Comt) | ||
| P48320 | Glutamic acid decarboxylase 2(Gad2) | ||
| P06837 | Growth associated protein 43(Gap43) | ||
| P42859 | Huntingtin(Htt) | ||
| P33173 | Kinesin family member 1A(Kif1a) | ||
| Q9CQV6 | Microtubule-associated protein 1 light chain 3 beta(Map1lc3b) | ||
| P10637 | Microtubule-associated protein tau(Mapt) | ||
| Q9JM52 | Misshapen-like kinase 1 (zebrafish)(Mink1) | ||
| P13595 | Neural cell adhesion molecule 1(Ncam1) | ||
| O35136 | Neural cell adhesion molecule 2(Ncam2) | ||
| Postsynaptic density | |||
| O35927 | Catenin (cadherin associated protein), delta 2(Ctnnd2) | ||
| Q9JLM8 | Doublecortin-like kinase 1(Dclk1) | ||
| Q9QXS6 | Drebrin 1(Dbn1) | ||
| P06837 | Growth associated protein 43(Gap43) | ||
| P42859 | Huntingtin(Htt) | ||
| P10637 | Microtubule-associated protein tau(Mapt) | ||
| Q9JM52 | Misshapen-like kinase 1 (zebrafish)(Mink1) | ||
| Q60875 | Rho/rac guanine nucleotide exchange factor (GEF) 2(Arhgef2) | ||
| Dendritic spine | |||
| Q5SSM3 | Rho GTPase activating protein 44(Arhgap44) | ||
| P47757 | Capping protein (actin filament) muscle Z-line, beta(Capzb) | ||
| O88587 | Catechol-O-methyltransferase(Comt) | ||
| Q9QXS6 | Ddrebrin 1(Dbn1) | ||
| P35802 | Glycoprotein m6a(Gpm6a) | ||
| P31324 | Protein kinase, cAMP dependent regulatory, type II beta(Prkar2b) | ||
| P62137 | Protein phosphatase 1, catalytic subunit, alpha isoform(Ppp1ca) | ||
| Growth cone | |||
| Q3UNH4 | G protein-regulated inducer of neurite outgrowth 1(Gprin1) | ||
| P97427 | Collapsin response mediator protein 1(Crmp1) | ||
| Q62188 | Dihydropyrimidinase-like 3(Dpysl3) | ||
| Q9QXS6 | Drebrin 1(Dbn1) | ||
| Q61553 | Fascin actin-bundling protein 1(Fscn1) | ||
| P10637 | Microtubule-associated protein tau(Mapt) | ||
| P13595 | Neural cell adhesion molecule 1(Ncam1) | ||
| Microtubule | |||
| Q9Z0H8 | CAP-GLY domain containing linker protein 2(Clip2) | ||
| P63168 | Dynein light chain LC8-type 1(Dynll1) | ||
| P33173 | Kinesin family member 1A(Kif1a) | ||
| P28738 | Kinesin family member 5 C(Kif5c) | ||
| Q9CQV6 | Microtubule-associated protein 1 light chain 3 beta(Map1lc3b) | ||
| P10637 | Microtubule-associated protein tau(Mapt) | ||
| Q60875 | Rho/rac guanine nucleotide exchange factor (GEF) 2(Arhgef2) | ||
| P68369 | Tubulin, alpha 1A(Tuba1a) | ||
| Q7TMM9 | Tubulin, beta 2A class IIA(Tubb2a) | ||
| MF | Microtubule binding | ||
| Q9Z0H8 | CAP-GLY domain containing linker protein 2(Clip2) | ||
| Q9EQF6 | Dihydropyrimidinase-like 5(Dpysl5) | ||
| P33173 | Kinesin family member 1A(Kif1a) | ||
| Q9CQV6 | Microtubule-associated protein 1 light chain 3 beta(Map1lc3b) | ||
| P10637 | Microtubule-associated protein tau(Mapt) | ||
| Q60875 | Rho/rac guanine nucleotide exchange factor (GEF) 2(Arhgef2) | ||
| Q9QY76 | Vesicle-associated membrane protein, associated protein B and C(Vapb) | ||
| Filamin binding | |||
| P97427 | Collapsin response mediator protein 1(Crmp1) | ||
| Q62188 | Dihydropyrimidinase-like 3(Dpysl3) | ||
| O35098 | Dihydropyrimidinase-like 4(Dpysl4) | ||
| Calmodulin binding | |||
| Q61656 | DEAD (Asp-Glu-Ala-Asp) box polypeptide 5(Ddx5) | ||
| Q61545 | Ewing sarcoma breakpoint region 1(Ewsr1) | ||
| P63054 | Purkinje cell protein 4(Pcp4) | ||
| P08414 | Calcium/calmodulin-dependent protein kinase IV(Camk4) | ||
| P06837 | Growth associated protein 43(Gap43) | ||
| Q61481 | Phosphodiesterase 1A, calmodulin-dependent(Pde1a) | ||
| Beta-tubulin binding | |||
| P47757 | Capping protein (actin filament) muscle Z-line, beta(Capzb) | ||
| P42859 | Huntingtin(Htt) | ||
| Q9QY76 | Vesicle-associated membrane protein, associated protein B and C(Vapb) |
BP, biological process; CC, cellular components; MF, molecular functions.
Fig. 5.

Network analysis of proteins was illustrated using the web-based tool STRING 10.0. Proteins from mouse brains at 3 h (A), 24 h (B), 7 days (C), and 15 weeks (D) after the blast were compared to those from the control group using LC-MS/MS.
KEGG pathway analysis of proteins shows that synthesis and degradation of ketone bodies (p = 4.1E-3), dopaminergic synapse (p = 6.8E-3), and ribosome (p = 4.0E-2) were enriched. Those proteins involved in the dopaminergic synapse pathway include catechol-O-methyltransferase (Comt), guanine nucleotide binding protein (G protein) β4 (Gnb4), guanine nucleotide binding protein, αq polypeptide (Gnaq), guanine nucleotide binding protein, αstimulating, olfactory type (Gnal), kinesin family member 5 C (Kif5c) and protein phosphatase 1, catalytic subunit, α isoform (Ppp1ca). How these proteins connect to each other and respond to blast-induced protein alteration remain to be investigated.
Alteration of phospho-proteomics in brains of mice exposed to blast
We compared phosphoproteins from mouse brains and captured 296 phosphorylated proteins for quantification. Among the list of altered phosphoproteins at 3 h and 24 h after the blast (Table 2), 29 phosphoproteins were downregulated and only one was upregulated at 3 h, whereas four were downregulated and 17 proteins were upregulated at 24 h. Several clusters were enriched in these dysregulated proteins, such as synapse, cytoskeleton/actin binding, dendritic spine, microtubule, growth cone, and neural projection. Based on DAVID analysis, the top enriched protein domains at 24 h include Stathmin family, C2 calcium-dependent membrane targeting protein and Synaptotagmin. The phosphoproteins with largest increase include Add1, Camk2b, Syt1, and Stmn1. Ap3b2, Sgip1, Basp1, and Rph3a were among those significantly downregulated phosphoproteins (Table 2).
Table 2.
Altered phosphoproteins in mouse brain at 3 h and 24 h after the blast
| Time | Accession | Description | Score | Coverage | Ratio | MW [kDa] |
|---|---|---|---|---|---|---|
| 3 h | Q9QYC0 | Alpha-adducin (Add1) | 114 | 17 | 1.96 | 80.6 |
| Q8BTI8 | Serine/arginine repetitive matrix protein 2 (Srrm2) | 54 | 6 | 0.77 | 294.7 | |
| Q3UHJ0 | AP2-associated protein kinase 1 (Aak1) | 65 | 14 | 0.74 | 103.3 | |
| Q61120 | SHC-transforming protein 3 (Shc3) | 12 | 5 | 0.71 | 52.1 | |
| Q9QYG0 | Protein NDRG2 (Ndrg2) | 45 | 9 | 0.67 | 40.8 | |
| Q8BGT8 | Phytanoyl-CoA hydroxylase-interacting protein-like (Phyhipl) | 14 | 7 | 0.67 | 42.3 | |
| O55131 | Septin-7 (Sept7) | 29 | 12 | 0.66 | 50.5 | |
| P04370 | Myelin basic protein (Mbp) | 24 | 23 | 0.65 | 27.2 | |
| Q80XU3 | Nuclear ubiquitous casein and cyclin-dependent kinase substrate (Nucks1) | 19 | 22 | 0.64 | 26.3 | |
| Q9WV18 | Gamma-aminobutyric acid type B receptor subunit 1 (Gabbr1) | 16 | 6 | 0.64 | 108.1 | |
| Q8C437 | PEX5-related protein (Pex51) | 15 | 5 | 0.63 | 63.1 | |
| Q8BPN8 | DmX-like protein 2 (Dmxl2) | 104 | 5 | 0.61 | 338.0 | |
| Q9JIX8 | Apoptotic chromatin condensation inducer in the nucleus (Acin1) | 21 | 6 | 0.61 | 150.6 | |
| Q9QXS1 | Plectin (Plec) | 20 | 1 | 0.60 | 533.9 | |
| Q8VDN2 | Sodium/potassium-transporting ATPase subunit alpha-1 (Atp1a1) | 150 | 21 | 0.60 | 112.9 | |
| P08553 | Neurofilament medium polypeptide(Nefm) | 61 | 18 | 0.59 | 95.9 | |
| Q9CYT6 | Adenylyl cyclase-associated protein 2 (Cap2) | 21 | 11 | 0.59 | 52.8 | |
| Q8C419 | Probable G-protein coupled receptor 158(Gpr158) | 24 | 6 | 0.59 | 134.3 | |
| A2AN08 | E3 ubiquitin-protein ligase UBR4 (Ubr4) | 13 | 1 | 0.58 | 571.9 | |
| P14873 | Microtubule-associated protein 1B (Map1b) | 376 | 22 | 0.58 | 270.1 | |
| Q9WV92 | Band 4.1-like protein 3 (Epb4113) | 89 | 19 | 0.57 | 103.3 | |
| Q80TE7 | Leucine-rich repeat-containing protein 7 (Lrrc7) | 44 | 5 | 0.57 | 166.8 | |
| Q811P8 | Rho GTPase-activating protein 32 (Arhgap32) | 32 | 6 | 0.55 | 229.6 | |
| Q6I6G8 | E3 ubiquitin-protein ligase HECW2(Hecw2) | 10 | 2 | 0.55 | 176.1 | |
| Q9WV69 | Dematin (Dmtn) | 43 | 21 | 0.55 | 45.4 | |
| Q9JIS5 | Synaptic vesicle glycoprotein 2A (Sv2a) | 8 | 4 | 0.54 | 82.6 | |
| Q9JME5 | AP-3 complex subunit beta-2 (Ap3b2) | 17 | 4 | 0.52 | 119.1 | |
| P54285 | Voltage-dependent L-type calcium channel subunit beta-3 (Cacnb3) | 7 | 6 | 0.52 | 54.5 | |
| Q8VD37 | SH3-containing GRB2-like protein 3-interacting protein 1 (Sgip1) | 93 | 20 | 0.46 | 86.0 | |
| Q91XV3 | Brain acid soluble protein 1 (Basp1) | 98 | 63 | 0.43 | 22.1 | |
| 24 h | Q9QYC0 | Alpha-adducin (Add1) | 114 | 17 | 2.83 | 80.6 |
| P28652 | Calcium/calmodulin-dependent protein kinase type II subunit beta (Camk2b) | 71 | 20 | 2.28 | 60.4 | |
| P48962 | ADP/ATP translocase 1 (Slc25a4) | 11 | 13 | 2.06 | 32.9 | |
| Q04735 | Cyclin-dependent kinase 16 (Cdk16) | 6 | 4 | 1.98 | 55.9 | |
| P46096 | Synaptotagmin-1 (Syt1) | 23 | 11 | 1.95 | 47.4 | |
| Q5SNZ0 | Girdin (Ccdc88a) | 10 | 2 | 1.81 | 215.8 | |
| O70166 | Stathmin-3 (Stmn3) | 11 | 18 | 1.74 | 20.9 | |
| P54227 | Stathmin (Stmn1) | 153 | 42 | 1.73 | 17.3 | |
| Q6NS60 | F-box only protein 41 (Fbxo41) | 8 | 4 | 1.72 | 94.3 | |
| Q3USB7 | Inactive phospholipase C-like protein 1 (Plcl1) | 17 | 4 | 1.71 | 122.6 | |
| Q8C9H6 | Striatin-interacting proteins 2 (Strip2) | 11 | 4 | 1.64 | 96.2 | |
| Q91XM9 | Disks large homolog 2 (Dlg2) | 37 | 9 | 1.62 | 94.8 | |
| O70439 | Syntaxin-7 (Stx7) | 15 | 11 | 1.61 | 29.8 | |
| G5E829 | Plasma membrane calcium-transporting ATPase 1 (Atp2b1) | 46 | 13 | 1.55 | 134.7 | |
| P18760 | Cofilin-1 (Cfl1) | 9 | 23 | 1.53 | 18.5 | |
| P97427 | Dihydropyrimidinase-related protein 1 (Crmp1) | 40 | 14 | 1.51 | 62.1 | |
| Q8BGU5 | Cyclin-Y (Ccny) | 12 | 13 | 1.41 | 39.4 | |
| B1AZP2 | Disks large-associated protein 4 (Dlgap4) | 29 | 9 | 0.84 | 108 | |
| P51954 | Serine/threonine-protein kinase Nek1 (Nek1) | 7 | 4 | 0.82 | 136.6 | |
| Q8K4G5 | Actin-binding LIM protein 1 (Ablim1) | 22 | 9 | 0.78 | 96.7 | |
| P47708 | Rabphilin-3A (Rph3a) | 4 | 4 | 0.48 | 75.4 |
GO ontology analysis of CC of dysregulated phosphoproteins revealed alteration in synaptic proteins (Fig. 6A). At both time points, the downregulated phosphoproteins are highly enriched in postsynaptic density, synapse, cell projection, and cytoskeleton (p ≤ 4.40E-3). Those dysregulated phosphoproteins were enriched in brains harvested at 3 h and 24 h (Table 2) after the blast, respectively. At 3 h post blast, nearly 50% of the downregulated phosphoproteins were enriched in cellular components related to synapse, postsynaptic density, myelin sheath and microtubule (Table 3). At 24 h post blast, 33% of the proteins are enriched at synapse, followed by cytoskeleton (33%), neuron projection (24%), dendrite (24%), and synaptic vesicle (14%) (Table 4).
Fig. 6.
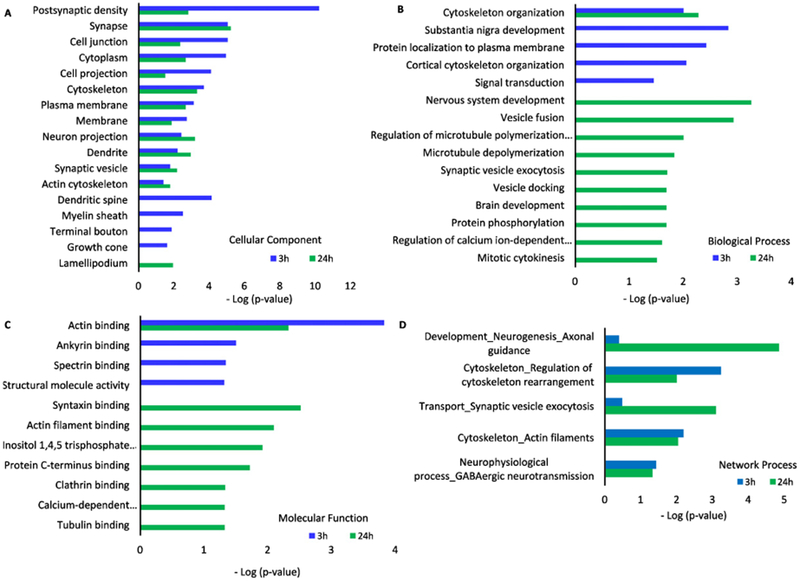
GO ontology analysis of altered brain phospho-proteomics at 3 h and 24 h post impact. Cellular components (A), biological process (B), molecular functions (C), and network process (D) of phosphoproteins from mouse brains tissue at 3 h and 24 h after the blast were compared to those from control/sham group of animals.
Table 3.
The list of altered phosphoproteins (3 h post blast) enriched in DAVID functional analysis
| Categories | GO Terms | ACCESSION | GENE NAME |
|---|---|---|---|
| BP | Substantia nigra development | Q9QYG0 | N-myc downstream regulated gene 2(Ndrg2) |
| Q91XV3 | Brain abundant, membrane attached signal protein 1(Basp1) | ||
| P04370 | Myelin basic protein(Mbp) | ||
| Cortical cytoskeleton organization | Q9WV92 | Erythrocyte membrane protein band 4.1 like 3(Epb41l3) | |
| O55131 | Septin 7(Sept7) | ||
| Signal transduction | Q9CYT6 | CAP, adenylate cyclase-associated protein, 2 (yeast)(Cap2) | |
| Q8C419 | G protein-coupled receptor 158(Gpr158) | ||
| Q9QYG0 | N-myc downstream regulated gene 2(Ndrg2) | ||
| Q811P8 | Rho GTPase activating protein 32(Arhgap32) | ||
| Q9WV18 | Gamma-aminobutyric acid (GABA) B receptor, 1(Gabbr1) | ||
| Q61120 | Src homology 2 domain-containing transforming protein C3(Shc3) | ||
| CC | Postsynaptic density | Q8VDN2 | ATPase, Na+/K+ transporting, alpha 1 polypeptide(Atp1a1) |
| Q9CYT6 | CAP, adenylate cyclase-associated protein, 2 (yeast)(Cap2) | ||
| Q811P8 | Rho GTPase activating protein 32(Arhgap32) | ||
| Q9QYC0 | Adducin 1 (alpha)(Add1) | ||
| Q9WV69 | Dematin actin binding protein(Dmtn) | ||
| Q9WV92 | Erythrocyte membrane protein band 4.1 like 3(Epb41l3) | ||
| Q80TE7 | Leucine rich repeat containing 7(Lrrc7) | ||
| P14873 | Microtubule-associated protein 1B(Map1b) | ||
| P08553 | Neurofilament, medium polypeptide(Nefm) | ||
| Dendritic spine | Q811P8 | Rho GTPase activating protein 32(Arhgap32) | |
| Q9QYC0 | Adducin 1 (alpha)(Add1) | ||
| Q9WV18 | Gamma-aminobutyric acid (GABA) B receptor, 1(Gabbr1) | ||
| Q80TE7 | Leucine rich repeat containing 7(Lrrc7) | ||
| P14873 | Microtubule-associated protein 1B(Map1b) | ||
| Cytoskeleton | Q9QYC0 | Adducin 1 (alpha)(Add1) | |
| P28652 | Calcium/calmodulin-dependent protein kinase II, beta(Camk2b) | ||
| Q9WV69 | Dematin actin binding protein(Dmtn) | ||
| Q9WV92 | Erythrocyte membrane protein band 4.1 like 3(Epb41l3) | ||
| P14873 | Microtubule-associated protein 1B(Map1b) | ||
| P08553 | Neurofilament, medium polypeptide(Nefm) | ||
| Q9QXS1 | Plectin(Plec) | ||
| O55131 | Septin 7(Sept7) | ||
| A2AN08 | Ubiquitin protein ligase E3 component n-recognin 4(Ubr4) | ||
| Myelin sheath | Q8VDN2 | ATPase, Na+/K+ transporting, alpha 1 polypeptide(Atp1a1) | |
| P04370 | Myelin basic protein(Mbp) | ||
| P08553 | Neurofilament, medium polypeptide(Nefm) | ||
| O55131 | Septin 7(Sept7) | ||
| MF | Actin binding | Q9CYT6 | CAP, adenylate cyclase-associated protein, 2 (yeast)(Cap2) |
| Q9QYC0 | Adducin 1 (alpha)(Add1) | ||
| Q9WV69 | Dematin actin binding protein(Dmtn) | ||
| Q9WV92 | Erythrocyte membrane protein band 4.1 like 3(Epb41l3) | ||
| P14873 | Microtubule-associated protein 1B(Map1b) | ||
| Q9QXS1 | Plectin(Plec) | ||
| Ankyrin binding | Q8VDN2 | ATPase, Na+/K+ transporting, alpha 1 polypeptide(Atp1a1) | |
| Q9QXS1 | Plectin(Plec) | ||
| Spectrin binding | Q9QYC0 | Adducin 1 (alpha)(Add1) | |
| Q9WV69 | Dematin actin binding protein(Dmtn) | ||
| Structural molecule activity | Q9QYC0 | Adducin 1 (alpha)(Add1) | |
| Q9WV92 | Erythrocyte membrane protein band 4.1 like 3(Epb41l3) | ||
| P08553 | Neurofilament, medium polypeptide(Nefm) |
Table 4.
The list of altered phosphoproteins (24 h post blast) enriched in DAVID functional analysis
| Category | GO Terms | ACCESSION | GENE NAME |
|---|---|---|---|
| BP | Nervous system development | P28652 | Calcium/calmodulin-dependent protein kinase II, beta(Camk2b) |
| Q5SNZ0 | Coiled coil domain containing 88A(Ccdc88a) | ||
| P97427 | Collapsin response mediator protein 1(Crmp1) | ||
| Q91XM9 | Discs, large homolog 2 (Drosophila)(Dlg2) | ||
| P54227 | Stathmin 1(Stmn1) | ||
| Cytoskeleton organization | Q8K4G5 | Actin-binding LIM protein 1(Ablim1) | |
| P18760 | Cofilin 1, non-muscle(Cfl1) | ||
| Q8C9H6 | Striatin interacting protein 2(Strip2) | ||
| Microtubule depolymerization | P54227 | Stathmin 1(Stmn1) | |
| O70166 | Stathmin-like 3(Stmn3) | ||
| Synaptic vesicle exocytosis | P47708 | Rabphilin 3A(Rph3a) | |
| P46096 | Synaptotagmin I(Syt1) | ||
| CC | Synapse | G5E829 | ATPase, Ca++ transporting, plasma membrane 1(Atp2b1) |
| Q9QYC0 | Adducin 1 (alpha)(Add1) | ||
| Q04735 | Cyclin-dependent kinase 16(Cdk16) | ||
| Q91XM9 | Discs, large homolog 2 (Drosophila)(Dlg2) | ||
| B1AZP2 | Discs, large homolog-associated protein 4 (Drosophila)(Dlgap4) | ||
| P47708 | Rabphilin 3A(Rph3a) | ||
| P46096 | Synaptotagmin I(Syt1) | ||
| Cytoskeleton | P51954 | NIMA (never in mitosis gene a)-related expressed kinase 1(Nek1) | |
| Q8K4G5 | Actin-binding LIM protein 1(Ablim1) | ||
| Q9QYC0 | Adducin 1 (alpha)(Add1) | ||
| P28652 | Calcium/calmodulin-dependent protein kinase II, beta(Camk2b) | ||
| P18760 | Cofilin 1, non-muscle(Cfl1) | ||
| P97427 | Collapsin response mediator protein 1(Crmp1) | ||
| P54227 | Stathmin 1(Stmn1) | ||
| Neuron projection | Q04735 | Cyclin-dependent kinase 16(Cdk16) | |
| P47708 | Rabphilin 3A(Rph3a) | ||
| P54227 | Stathmin 1(Stmn1) | ||
| O70166 | Stathmin-like 3(Stmn3) | ||
| P46096 | Synaptotagmin I(Syt1) | ||
| Postsynaptic density | Q8K4G5 | Actin-binding LIM protein 1(Ablim1) | |
| Q9QYC0 | Adducin 1 (alpha)(Add1) | ||
| P28652 | Calcium/calmodulin-dependent protein kinase II, beta(Camk2b) | ||
| Q91XM9 | Discs, large homolog 2 (Drosophila)(Dlg2) | ||
| MF | Calmodulin binding | G5E829 | ATPase, Ca++ transporting, plasma membrane 1(Atp2b1) |
| Q9QYC0 | Adducin 1 (alpha)(Add1) | ||
| P28652 | Calcium/calmodulin-dependent protein kinase II, beta(Camk2b) | ||
| P46096 | Synaptotagmin I(Syt1) | ||
| Syntaxin binding | P47708 | Rabphilin 3A(Rph3a) | |
| P46096 | Synaptotagmin I(Syt1) | ||
| O70439 | Syntaxin 7(Stx7) | ||
| Actin binding | Q8K4G5 | Actin-binding LIM protein 1(Ablim1) | |
| Q9QYC0 | Adducin 1 (alpha)(Add1) | ||
| P18760 | Cofilin 1, non-muscle(Cfl1) | ||
| Q5SNZ0 | Coiled coil domain containing 88A(Ccdc88a) | ||
| Tubulin binding | P54227 | Stathmin 1(Stmn1) | |
| O70166 | Stathmin-like 3(Stmn3) |
The BP of dysregulated phosphoproteins was examined using GO ontology analysis (Fig. 6B). The only common enriched biological process at both time points is cytoskeleton organization. In addition, substantia nigra development and signal transduction at 3 h time point were also enriched. The top five enriched biological processes at 24 h are nervous system development, vesicle fusion, and cytoskeleton organization, microtubule depolymerization and synaptic vesicle exocytosis. MF of altered phosphoproteins from mouse brains at 3 h and 24 h after the blast were analyzed (Fig. 6C). Actin binding is the only common MF among the dysregulated phosphoproteins at both time points. Other cytoskeleton related MF, such as ankyrin binding and spectrin binding were also enriched at 3 h (Table 3). At 24 h, in additional to structural related MF, such as syntaxin binding and tubulin binding, calcium-dependent phospholipid is also enriched (Table 4).
The top three GO process network analysis of up- and downregulated proteins were revealed using Metacore (Fig. 6D). The networks related to cytoskeleton, such as Plectin 1, actin-binding Lim protein 1 (Ablim1) and neurofilament medium polypeptide (Nefm), were downregulated at both time points. Proteins related to the networks of synaptic contact and neurogenesis, such as Syt1, Stx7, Camk2b, and Dlg2, were upregulated at 24 h (Fig. 6D). Metacore analysis illustrated Cofilin as the top network for upregulated phosphoproteins at 24 h after the blast.
When we analyzed mouse brains harvested at 30 days and 15 weeks after the blast exposure, we obtained similar results. Analysis of cellular components of phosphoproteins illustrated changes in synaptic proteins (Fig. 7A). Those phosphoproteins were enriched in brains harvested at 30 days (Table 5) and 15 weeks (Table 6) after the blast, respectively. The BP of dysregulated phosphoproteins was examined using GO ontology analysis (Fig. 7B). Similar to what we have observed in tissue collected at earlier time points, one enriched process at all four time points is cytoskeleton organization. Actin binding is the common MF among the phosphoproteins at all time points (Fig. 7C). The networks process of up- and downregulated proteins includes cytoskeleton and actin filament and development of neurogenesis and synaptogenesis (Fig. 7D), similar to those identified at earlier time points (Fig. 6D).
Fig. 7.
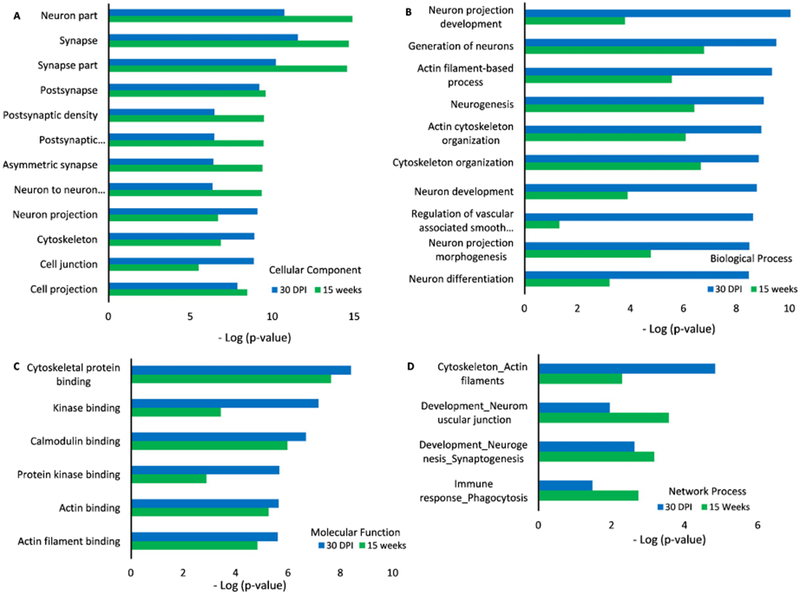
GO ontology analysis of altered brain phospho-proteomics at 30 days and 15 weeks post impact. Cellular components (A), biological process (B), molecular functions (C), and network process (D) of phosphoproteins from mouse brains tissue at 30 days and 15 weeks after the blast were compared to those from control/sham group of animals.
Table 5.
The list of altered phosphoproteins (30 days post blast) enriched in DAVID functional analysis
| Category | GO Terms | ACCESSION | GENE NAME |
|---|---|---|---|
| BP | Neurotransmitter secretion | Q64332 | synapsin II(Syn2) |
| Q8JZP2 | synapsin III(Syn3) | ||
| P60879 | synaptosomal-associated protein 25(Snap25) | ||
| Actin cytoskeleton organization | Q3UHD9 | ArfGAP with GTPase domain, ankyrin repeat and PH domain 2(Agap2) | |
| Q8R0S2 | IQ motif and Sec7 domain 1(Iqsec1) | ||
| O88643 | p21 protein (Cdc42/Rac)-activated kinase 1(Pak1) | ||
| Q4JIM5 | v-abl Abelson murine leukemia viral oncogene 2 (arg, Abelson-related gene)(Abl2) | ||
| Neuron projection morphogenesis | Q62418 | drebrin-like(Dbnl) | |
| P35802 | glycoprotein m6a(Gpm6a) | ||
| O88643 | p21 protein (Cdc42/Rac)-activated kinase 1(Pak1) | ||
| Synaptic vesicle priming | Q80TJ1 | Ca2+-dependent secretion activator(Cadps) | |
| P60879 | synaptosomal-associated protein 25(Snap25) | ||
| CC | Cytoskeleton | Q9Z0H8 | CAP-GLY domain containing linker protein 2(Clip2) |
| A2AJI0 | MAP7 domain containing 1(Map7d1) | ||
| P62484 | abl-interactor 2(Abi2) | ||
| Q8K4G5 | actin-binding LIM protein 1(Ablim1) | ||
| Q80VC9 | calmodulin regulated spectrin-associated protein family, member 3(Camsap3) | ||
| Q62418 | drebrin-like(Dbn1) | ||
| Q8VDD5 | myosin, heavy polypeptide 9, non-muscle(Myh9) | ||
| P26645 | myristoylated alanine rich protein kinase C substrate(Marcks) | ||
| Q62417 | sorbin and SH3 domain containing 1(Sorbs1) | ||
| Q4JIM5 | v-abl Abelson murine leukemia viral oncogene 2 (arg, Abelson-related gene)(Abl2) | ||
| Q8BG89 | zinc finger protein 365(Zfp365) | ||
| Lamellipodium | P62484 | ab1-interactor 2(Abi2) | |
| Q8K4G5 | actin-binding LIM protein 1(Ablim1) | ||
| Q62418 | drebrin-like(Dbnl) | ||
| O88643 | p21 protein (Cdc42/Rac)-activated kinase 1(Pak1) | ||
| P60879 | synaptosomal-associated protein 25(Snap25) | ||
| Q4JIM5 | v-abl Abelson murine leukemia viral oncogene 2 (arg, Abelson-related gene)(Abl2) | ||
| Synapse | Q80TJ1 | Ca2+-dependent secretion activator(Cadps) | |
| Q61361 | brevican(Bcan) | ||
| Q04735 | cyclin-dependent kinase 16(Cdk16) | ||
| Q62418 | drebrin-like(Dbnl) | ||
| Q64332 | synapsin II(Syn2) | ||
| Q8JZP2 | synapsin III(Syn3) | ||
| P60879 | synaptosomal-associated protein 25(Snap25) | ||
| P63044 | vesicle-associated membrane protein 2(Vamp2) | ||
| Postsynaptic density | UNIPROT_ACCESSION | GENE NAME | |
| D3YVF0 | A kinase (PRKA) anchor protein 5(Akap5) | ||
| Q8K4G5 | actin-binding LIM protein 1(Ablim1) | ||
| Q62418 | drebrin-like(Dbnl) | ||
| O88643 | p21 protein (Cdc42/Rac)-activated kinase 1(Pak1) | ||
| Q64332 | synapsin II(Syn2) | ||
| Q8JZP2 | synapsin III(Syn3) | ||
| MF | Calmodulin binding | D3YVF0 | A kinase (PRKA) anchor protein 5(Akap5) |
| Q80VC9 | calmodulin regulated spectrin-associated protein family, member 3(Camsap3) | ||
| Q8VDD5 | myosin, heavy polypeptide 9, non-muscle(Myh9) | ||
| P26645 | myristoylated alanine rich protein kinase C substrate(Marcks) | ||
| P63044 | vesicle-associated membrane protein 2(Vamp2) | ||
| Protein kinase binding | D3YVF0 | A kinase (PRKA) anchor protein 5(Akap5) | |
| Q3UHD9 | ArfGAP with GTPase domain, ankyrin repeat and PH domain 2(Agap2) | ||
| Q80TJ1 | Ca2+-dependent secretion activator(Cadps) | ||
| Q9Z2V6 | histone deacetylase 5(Hdac5) | ||
| O88643 | p21 protein (Cdc42/Rac)-activated kinase 1(Pak1) | ||
| Q62417 | sorbin and SH3 domain containing 1(Sorbs1) | ||
| Actin binding | D3YVF0 | A kinase (PRKA) anchor protein 5(Akap5) | |
| Q8K4G5 | actin-binding LIM protein 1(Ablim1) | ||
| Q62418 | drebrin-like(Dbn1) | ||
| Q8VDD5 | myosin, heavy polypeptide 9, non-muscle(Myh9) | ||
| P26645 | myristoylated alanine rich protein kinase C substrate(Marcks) | ||
| Calcium-dependent protein binding | Q64332 | synapsin II(Syn2) | |
| P60879 | synaptosomal-associated protein 25(Snap25) | ||
| P63044 | vesicle-associated membrane protein 2(Vamp2) |
Table 6.
The list of altered phosphoproteins (15 weeks post blast) enriched in DAVID functional analysis
| Category | GO Terms | ACCESSION | GENE NAME |
|---|---|---|---|
| BP | Negative regulation of phosphatase activity | F8VPU2 | FERM, RhoGEF (Arhgef) and pleckstrin domain protein 1 (chondrocyte-derived)(Farp1) |
| Q91XM9 | discs, large homolog 2 (Drosophila)(Dlg2) | ||
| Q5XJV6 | lemur tyrosine kinase 3(Lmtk3) | ||
| Axonogenesis | Q5RJI5 | BR serine/threonine kinase 1(Brsk1) | |
| Q69Z98 | BR serine/threonine kinase 2(Brsk2) | ||
| P54227 | stathmin 1(Stmn1) | ||
| Regulation of synaptic transmission, GABAergic | Q04690 | neurofibromatosis 1(Nf1) | |
| Q3USB7 | phospholipase C-like 1(Plcl1) | ||
| Regulation of cell shape | P05064 | aldolase A, fructose-bisphosphate(Aldoa) | |
| Q9WV69 | dematin actin binding protein(Dmtn) | ||
| Q6URW6 | myosin, heavy polypeptide 14(Myh14) | ||
| CC | Synapse | Q5RJI5 | BR serine/threonine kinase 1(Brsk1) |
| Q80TJ1 | Ca2+-dependent secretion activator(Cadps) | ||
| F8VPU2 | FERM, RhoGEF (Arhgef) and pleckstrin domain protein 1 (chondrocyte-derived)(Farp1) | ||
| Q811P8 | Rho GTPase activating protein 32(Arhgap32) | ||
| Q9QYC0 | adducin 1 (alpha)(Add1) | ||
| O70174 | cholinergic receptor, nicotinic, alpha polypeptide 4(Chrna4) | ||
| Q91XM9 | discs, large homolog 2 (Drosophila)(Dlg2) | ||
| Q80TE7 | leucine rich repeat containing 7(Lrrc7) | ||
| Q8BSS9 | protein tyrosine phosphatase, receptor type, f polypeptide (PTPRF), interacting protein (liprin), alpha 2(Ppfia2) | ||
| Q9EQZ7 | regulating synaptic membrane exocytosis 2(Rims2) | ||
| Q8BJI1 | solute carrier family 6 (neurotransmitter transporter), member 17(Slc6a17) | ||
| Q71LX4 | talin 2(Tln2) | ||
| Postsynaptic density | Q9QYE3 | B cell CLL/lymphoma 11A (zinc finger protein)(Bcl11a) | |
| Q811P8 | Rho GTPase activating protein 32(Arhgap32) | ||
| Q8K4G5 | actin-binding LIM protein 1(Ablim1) | ||
| Q9QYC0 | adducin 1 (alpha)(Add1) | ||
| Q80YN3 | breast carcinoma amplified sequence 1(Bcas1) | ||
| Q9WV69 | dematin actin binding protein(Dmtn) | ||
| Q91XM9 | discs, large homolog 2 (Drosophila)(Dlg2) | ||
| Q80TE7 | leucine rich repeat containing 7(Lrrc7) | ||
| Q04690 | neurofibromatosis 1(Nf1) | ||
| Myelin sheath | P05064 | aldolase A, fructose-bisphosphate(Aldoa) | |
| P07901 | heat shock protein 90, alpha (cytosolic), class A member 1(Hsp90aa1) | ||
| P04370 | myelin basic protein(Mbp) | ||
| Q6URW6 | myosin, heavy polypeptide 14(Myh14) | ||
| Q9Z1B3 | phospholipase C, beta 1(Plcb1) | ||
| Q60932 | voltage-dependent anion channel 1(Vdac1) | ||
| MF | Calmodulin binding | P28667 | MARCKS-like 1(Marcksl1) |
| Q9QYC0 | adducin 1 (alpha)(Add1) | ||
| Q8C078 | calcium/calmodulin-dependent protein kinase kinase 2, beta(Camkk2) | ||
| Q6URW6 | myosin, heavy polypeptide 14(Myh14) | ||
| Q9Z1B3 | phospholipase C, beta 1(Plcb1) | ||
| Actin binding | P28667 | MARCKS-like 1(Marcksl1) | |
| Q8K4G5 | actin-binding LIM protein 1(Ablim1) | ||
| Q9QYC0 | adducin 1 (alpha)(Add1) | ||
| Q9WV69 | dematin actin binding protein(Dmtn) | ||
| Q6URW6 | myosin, heavy polypeptide 14(Myh14) | ||
| Q71LX4 | talin 2(Tln2) | ||
| Tau-protein kinase activity | Q5RJI5 | BR serine/threonine kinase 1(Brsk1) | |
| Q69Z98 | BR serine/threonine kinase 2(Brsk2) |
Network analysis revealed up- and downregulated phosphoproteins at 3 h (Fig. 8A), 24 h (Fig. 8B), 30 days (Fig. 8C), and 15 weeks (Fig. 8D) using STRING. At the early time point (3 h), ATPase, Ca2+ transporting, plasma membrane 1 (ATP2b1), calcium/calmodulin-dependent protein kinase II beta (Camk2b) and solute carrier family 25 (mitochondrial carrier, adenine nucleotide translocator), member 4 (Slc25a4) are all changed. KEGG pathway analysis shows that calcium signaling pathway is enriched among these altered proteins. At later time points, Cdk16 was a common protein identified by network analysis (Fig. 8B–D).
Fig. 8.
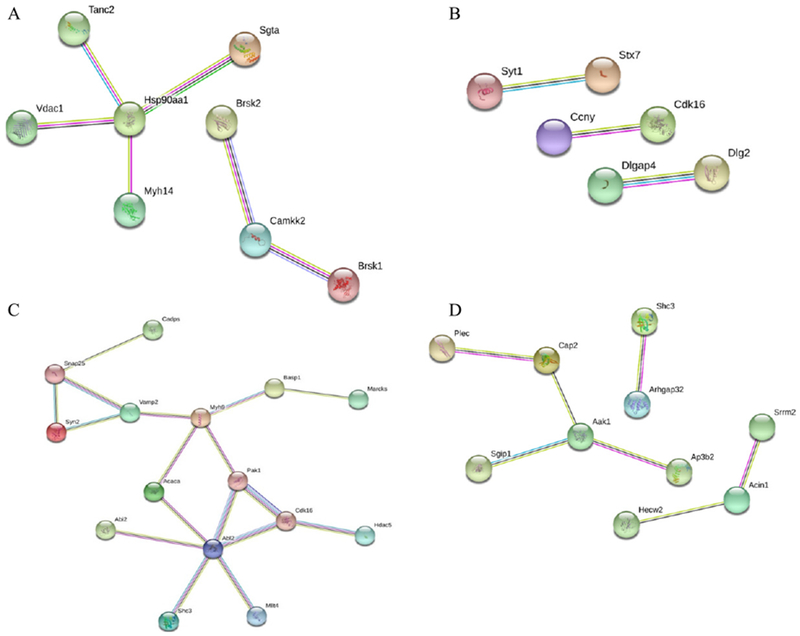
Network analysis of all captured phosphoproteins was illustrated using the web-based tool STRING. Proteins from mouse brains at 3 h (A), 24 h (B), 30 days (C), and 15 weeks (D) after the blast were compared to those from the control group using LC-MS/MS, and the enriched cellular components and altered proteins were analyzed by DAVID.
DISCUSSION
The significance of this study is highlighted by an urgent need to identify critical players involved in neuropathological development in people who suffer mTBI and to investigate how these factors contribute to neuronal damage and neurodegenerative diseases like AD. We have used an animal model exposed to open field blast to model mTBI [25, 26]. This unique platform allows us to compare mice responses to brain injury at the molecular level of in-depth proteomics and biochemistry of brain tissue. Currently we lack mechanistic understanding of neuronal damages caused by mTBI and we do not have a target for therapeutic intervention for those suffering mTBI or in people with a history of mTBI. Based on the proteomic profiles of mouse brain exposed to blast, we are exploring novel targets and candidate proteins linked to axonal injury, microtubule cytoskeleton organization, and tau pathology.
Proteomic analysis of the moue brain tissues revealed significant changes in protein expression representing axonal injury and microtubule cytoskeleton organization in response to injury
This study utilized TMT which offers the ability to simultaneously detect ten samples under a single condition. DAVID analysis of altered proteins show that several functions significantly changed after the blast. Specifically, we found negative effect of blast on a number of cellular processes.
Diffuse axonal injury is a hallmark pathological consequence of non-penetrative mTBI [34]. In neuronal culture model, very mild stretch injury to a localized region of the cortical axon is able to trigger a degenerative response characterized by growth cone collapse and significant abnormal cytoskeleton rearrangement, resulting in the formation of smaller growth cones at the tips of axons and a significantly higher number of collapsed structures at both 24 h and 72 h post injury [34]. Axonal degeneration following stretch injury involves destabilization of the microtubule cytoskeleton. Microtubules are the stiffest structural components of axons and thus may be at risk of damage during mTBI. Loss of axonal microtubule was found after stretch injury and progressive disassembly of microtubules along the breakage points [35]. Stabilization of microtubule resulted in a significant reduction in the number of fragmented axons following injury [34]. Consistent with previous reports [36], our studies also revealed depolymerization of microtubules.
We identified several proteins, such as Map1B, Stmn1, Shc3, Sept7, Nefm, Ap3b2, and Mapt. They play important roles in maintaining microtubule stability and axonal integrity. Map1B belongs to a group of microtubule-associated proteins that support microtubule-stabilization, tubulin assembly/disassembly, and physical interactions between microtubules and components of the cytoskeleton. Map1B and its phosphorylated isoform are present in growing axons and concentrated in distal end near the growth cone. Map1B plays a crucial role in the stability of the cytoskeleton, and its function is modulated by the state of phosphorylation. Immuno-histochemical staining showed that phosphorylated Map1B was abundant in developing axons, suggesting its essential role in axonal elongation [37]. Map1B is upregulated after ischemia in aged rats [38] but downregulated in optic nerve after mTBI [36]. In this study, downregulation of phosphorylated Map1B was indicative of axonal injury and cytoskeletal damage following TBI. It is possible that the structural reorganization at the cellular level and shifts in cytoskeletal dynamic associate with injury. This observation is consistent with our previous finding of downregulated phosphorylated Map1B from the inferior prefrontal cortex region of postmortem brain tissues from AD patients [39], clearly linking the TBI-induced cytoskeletal damage to pathological changes in AD brain.
Stathmin (Stmn) plays an essential role in the maintenance of axonal integrity. It is involved in the regulation of the microtubule filament system by destabilizing microtubules, and phosphorylation of Stmn regulates microtubule disassembly. In our study, phosphorylated Stmn1and Stmn3 were found upregulated at 24 h after the blast, and the level of Stmn1 was found 2-fold of that of the control group at 15 weeks after the blast (data not shown). A recent study demonstrated that Stmn-dependent changes in microtubule stability are crucial for synaptic function and memory formation [40]. Stmn acts on microtubule disruption and/or tubulin sequestration, and elevation of Stmn1 after blasts was found in rat model of mTBI [41]. High Stmn expression induced microtubule structures disruption and enhanced tau hyperphosphorylation [42]. Similarly, brain injury causes reduced polymerization of Gephyrin and subsequent synapse destabilization [43], and progressive disassembly and loss of axonal microtubule along the breakage points [35].
A number of proteins altered in our mice exposed to open field blast were also reported from previous proteomic analysis of optic nerves from mice suffering repetitive mTBI (rmTBI) [36]. Various negative effects of rmTBI on cellular processes were reported, including depolymerization of microtubules, disassembly of filaments and loss of neurons, evidenced by a reduction of several proteins, including neurofilaments, tubulin (Tubb2A, Tuba4A), and microtubule-associated proteins (Map1A, Map1B) [36]. In our study, phosphorylated neurofilament medium polypeptide (Nefm) was downregulated. Nefm belongs to the group of neurofilament triplet proteins, the major cytoskeletal components of neurons in the CNS. They interact with microtubules and microfilaments to form and maintain the axonal structural integrity and neuronal shape, and also participate to the axonal mechanism of transport. Significant decrease in neurofilament levels in optic nerve of rat retina after 21 days mTBI was reported [36]. Furthermore, increased level of Nefm was observed in CSF and serum from patients with a history of mTBI, but the effect could come from peripheral nerve injury [44].
We also observed several altered dynein proteins in addition to those proteins related to microtubule and tubulin binding. Axonal transport is related to microtubule stability and essential for transducing extracellular signals and recycling misfolded proteins from the nerve terminals to cell soma, thus avoiding the build-up of toxic aggregates. Axonal transport requires the cytoskeletal microtubule scaffold to serve as tracks, and the motor proteins kinesin and dynein that exert mechanical force to move cargoes.
In this study, we found changes in a large proportion of proteins located in dendritic spines, those highly dynamic membranous protrusions on post-synaptic dendrites. Structural changes in dendritic spines form the basis for learning and memory formation, making them central hubs for the processing and storage of information in the brain [45]. Study in Map1B-deficient mice showed that Map1B was required for dendritic spine development and synaptic maturation [46]. Study of mouse hippocampal neuron cultures showed inhibition of microtubule dynamics alters dendritic spine morphology through the actin cytoskeleton, suggesting that the microtubule and actin cytoskeleton act together in dendritic spines to regulate synaptic plasticity [47]. We have observed changes in several proteins with actin binding activities. Three phosphorylated actin binding proteins, Cofilin, Add1, and Ccdc88a, were found upregulated 24 h after the blast in our study. Cofilin is a major component of intranuclear and cytoplasmic actin rods, and Ccdc88a is required for formation of actin stress fibers and lamellipodia.
Consistent with previous study [36], phosphomyelin basic protein (Mbp) was found reduced in mice exposed to blast in our study. Mbp is a major constituent of CNS myelin and mature oligodendrocytes, and the decrease in Mbp levels could be a direct reflection of an ongoing demyelination. We observed continuous decrease of phospho-Mbp at 15 weeks (data not shown), suggesting that demyelination was a long-lasting outcome of TBI. In Schwann cells responding to 2 h of sustained shear stress, Mbp appeared to be downregulated, suggesting that shear stress alone is sufficient to induce pathological changes [48]. In rats, extensive degradation of Mbp isoforms was observed after TBI [49], suggesting that TBI-mediated axonal injury causes secondary structural damage to the adjacent myelin membrane and instigate Mbp degradation. This could initiate myelin sheath instability and demyelination, which might further promote axonal vulnerability [49].
Upregulated calcium signaling after TBI
In our study, several phosphorylated proteins involved in calcium signaling pathway were upregulated. There is strong evidence in support of the concept that abnormal neurotransmission and Ca2+-mediated signal transduction pathways could be responsible for neuronal dysfunction following trauma. Calcium is crucial for several aspects of plasticity at glutamatergic synapses. Changes in the levels of intracellular Ca2+ or alterations of Ca2+ signaling could contribute to a variety of pathologies. One of the major causes of secondary injury following TBI is related to an excess influx of Ca2+ into cells in the brain [50].
Calcium/calmodulin-dependent protein kinase type II subunit beta (Camk2b) is essential for the integrity of the actin cytoskeleton and for cell migration. Camk2b is expressed in motor neurons during axon outgrowth and is part of slow axonal transport [51]. Consistent with previous findings [52, 53], we found increased phosphorylation of CamkIIb at 24 h after the TBI. Atp2b1, which mediates calcium efflux, was upregulated in our blast-induced mouse brains. Slc25a4, which catalyzes the exchange of ADP and ATP across the mitochondrial inner membrane, was also upregulated. Since the response to CNS injury includes an apparent increase in energy mobilization capacity, elevated intracellular Ca2+ after TBI may be related to reduced ATP levels, as previously reported [50]. Another key protein, phosphorylated Synaptotagmin I (Syt1), was upregulated at 24 h after the blast. Synaptotagmins are integral membrane proteins of synaptic vesicles thought to serve as Ca2+ sensors in the process of vesicular trafficking and exocytosis. Calcium binding Syt-1 participates in triggering neurotransmitter release at the synapse [53].
Increases of p-tau, behavioral and pathological changes in mice exposed to blast link mTBI to AD
Our study revealed pathways related to p-tau (Fig. 1), which is the component of neurofibrillary tangles found in brains of AD patients. In addition, our exploratory SNAP tests revealed a significant difference in behavioral outcomes in the blast group compared to sham controls (Fig. 2). Degenerating axon-terminals and dendrites were visualized on silver-stained tissue sections from mouse brains exposed to blast (Fig. 3), supporting a contributing role of mTBI toward neurodegenerative process commonly found in brains of AD patients.
Tau as axonal injury marker has been reported in previous studies following mTBI [54, 55]. Traumatic brain injury can lead to diffuse traumatic axonal injury and blood–brain barrier disruption. Shearing of axons results in the disruption of tau binding to tubulin [56]. Subsequent hyperphosphorylation of tau leads to formation of tau oligomers in the neuronal soma. When tau becomes hyperphosphorylated, it binds to other normal tau proteins, which leads to aggregation. Accumulation of insoluble tau within neurons contributes to the development of tau oligomers [56]. Tau oligomers are granular intracellular buildups of mutated tau, which precede the development of NFTs. Tau hyperphosphorylation is common after mTBI and other brain injuries [57]. Previous study in patients with mTBI showed the level of phosphorylated tau was significantly higher and correlated with the Glasgow Coma Scale (GCS) score [58]. We found a similar increase of p-tau in brains of mice collected at 3 and 24 h after open field blast exposure, which is consistent with previous findings in humans and animals [58–60]. While increase in tau immunostaining in brain tissue was not observed in mini-pig from 2 weeks to 8 months after blast-induced TBI [61], our ELISA-based quantitation of tau and phospho-tau in mice exposed to blast illustrate a significant difference at 3 and 24 h. Levels of tau and p-tau were significantly changed within one day after the blast and returned to pre-blast stage at 15 weeks after blast (Fig. 1). It seems that blast-exposed animals were capable of recovering from impact, and the amount of ptau gradually reduced to the pre-blast levels. Several protein kinases (e.g., GSK3β) phosphorylate tau at multiple sites, and activities of these kinases might be enhanced upon low impact blast, leading to an increase in phosphorylated tau levels [7]. Once the animals were recovered from the blast, kinase activities might return to pre-blast levels, and diminish transient increases of ptau. This is consistent with previous findings in mini-pigs [61] and mice [62]. In transgenic mice expressing human tau (hTau), only transient increases in p-tau was observed after single or repetitive mild TBI, and no tau pathology was found at one year after TBI [62].
Tau-related pathological change is a common feature linking mTBI to AD. Whether mTBI-induced phospho-tau increase triggers AD pathogenesis needs to be investigated. Elucidation of tau-related molecular changes in people with a history of mTBI will open a new window to determine whether personnel who have suffered mTBI have asymptomatic AD. This is essential for designing future preventive measures for AD. Future effort to suppress p-tau-related neuropathological changes will potentially reduce the burden on affected individuals, especially those vulnerable to developing AD.
ACKNOWLEDGMENTS
We thank Dr. Guy Surpris and Benjamin Morris-Eppolito for critical discussions. This study was supported by the award I01BX003527 and I21 BX003807 from the Biomedical Laboratory Research and Development Service of the Veterans Affairs Office of Research and Development (WX) and the Cure Alzheimer’s Fund (WX). This publication was also made possible by funding from the DoD Congressionally Directed Medical Research Programs (CDMRP) for the Peer Reviewed Alzheimer’s Research Program Convergence Science Research Award (PRARP-CSRA; AZ140109) and the research funds of the University of Missouri (ZG). The views expressed in this article are those of the authors and do not represent the views of the US Department of Veterans Affairs or the US Government.
Footnotes
Authors’ disclosures available online (https://www.j-alz.com/manuscript-disclosures/18-0726r1).
REFERENCES
- [1].Tran HT, LaFerla FM, Holtzman DM, Brody DL (2011) Controlled cortical impact traumatic brain injury in 3xTg-AD mice causes acute intra-axonal amyloid-beta accumulation and independently accelerates the development of tau abnormalities. J Neurosci 31, 9513–9525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].DeKosky ST, Abrahamson EE, Ciallella JR, Paljug WR, Wisniewski SR, Clark RS, Ikonomovic MD (2007) Association of increased cortical soluble abeta42 levels with diffuse plaques after severe brain injury in humans. Arch Neurol 64, 541–544. [DOI] [PubMed] [Google Scholar]
- [3].Ikonomovic MD, Uryu K, Abrahamson EE, Ciallella JR, Trojanowski JQ, Lee VM, Clark RS, Marion DW, Wisniewski SR, DeKosky ST (2004) Alzheimer’s pathology in human temporal cortex surgically excised after severe brain injury. Exp Neurol 190, 192–203. [DOI] [PubMed] [Google Scholar]
- [4].Bailes JE, Petraglia AL, Omalu BI, Nauman E, Talavage T (2013) Role of subconcussion in repetitive mild traumatic brain injury. J Neurosurg 119, 1235–1245. [DOI] [PubMed] [Google Scholar]
- [5].Johnson VE, Stewart W, Smith DH (2012) Widespread tau and amyloid-Beta pathology many years after a single traumatic brain injury in humans. Brain Pathol 22, 142–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Gavett BE, Cantu RC, Shenton M, Lin AP, Nowinski CJ, McKee AC, Stern RA (2011) Clinical appraisal of chronic traumatic encephalopathy: Current perspectives and future directions. Curr Opin Neurol 24, 525–531. [DOI] [PubMed] [Google Scholar]
- [7].Goldstein LE, Fisher AM, Tagge CA, Zhang XL, Velisek L, Sullivan JA, Upreti C, Kracht JM, Ericsson M, Wojnarowicz MW, Goletiani CJ, Maglakelidze GM, Casey N, Moncaster JA, Minaeva O, Moir RD, Nowinski CJ, Stern RA, Cantu RC, Geiling J, Blusztajn JK, Wolozin BL, Ikezu T, Stein TD, Budson AE, Kowall NW, Chargin D, Sharon A, Saman S, Hall GF, Moss WC, Cleveland RO, Tanzi RE, Stanton PK, McKee AC (2012) Chronic traumatic encephalopathy in blast-exposed military veterans and a blast neurotrauma mouse model. Sci Transl Med 4, 134ra160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Terrio H, Brenner LA, Ivins BJ, Cho JM, Helmick K, Schwab K, Scally K, Bretthauer R, Warden D (2009) Traumatic brain injury screening: Preliminary findings in a US Army Brigade Combat Team. J Head Trauma Rehabil 24, 14–23. [DOI] [PubMed] [Google Scholar]
- [9].Mac Donald CL, Adam OR, Johnson AM, Nelson EC, Werner NJ, Rivet DJ, Brody DL (2015) Acute post-traumatic stress symptoms and age predict outcome in military blast concussion. Brain 138, 1314–1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Mac Donald CL, Barber J, Jordan M, Johnson AM, Dikmen S, Fann JR, Temkin N (2017) Early clinical predictors of 5-year outcome after concussive blast traumatic brain injury. JAMA Neurol 74, 821–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Vanderploeg RD, Belanger HG, Horner RD, Spehar AM, Powell-Cope G, Luther SL, Scott SG (2012) Health outcomes associated with military deployment: Mild traumatic brain injury, blast, trauma, and combat associations in the Florida National Guard. Arch Phys Med Rehabil 93, 1887–1895. [DOI] [PubMed] [Google Scholar]
- [12].Menon DK, Maas AI (2015) Traumatic brain injury in 2014. Progress, failures and new approaches for TBI research. Nat Rev Neurol 11, 71–72. [DOI] [PubMed] [Google Scholar]
- [13].Hutton M (2000) Molecular genetics of chromosome 17 tauopathies. Ann N Y Acad Sci 920, 63–73. [DOI] [PubMed] [Google Scholar]
- [14].Lewis J, McGowan E, Rockwood J, Melrose H, Nacharaju P, Van Slegtenhorst M, Gwinn-Hardy K, Paul Murphy M, Baker M, Yu X, Duff K, Hardy J, Corral A, Lin WL, Yen SH, Dickson DW, Davies P, Hutton M (2000) Neurofibrillary tangles, amyotrophy and progressive motor disturbance in mice expressing mutant (P301L) tau protein. Nat Genet 25, 402–405. [DOI] [PubMed] [Google Scholar]
- [15].Gotz J, Chen F, van Dorpe J, Nitsch RM (2001) Formation of neurofibrillary tangles in P301l tau transgenic mice induced by Abeta 42 fibrils. Science 293, 1491–1495. [DOI] [PubMed] [Google Scholar]
- [16].Santacruz K, Lewis J, Spires T, Paulson J, Kotilinek L, Ingelsson M, Guimaraes A, DeTure M, Ramsden M, McGowan E, Forster C, Yue M, Orne J, Janus C, Mariash A, Kuskowski M, Hyman B, Hutton M, Ashe KH (2005) Tau suppression in a neurodegenerative mouse model improves memory function. Science 309, 476–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Fagan AM, Roe CM, Xiong C, Mintun MA, Morris JC, Holtzman DM (2007) Cerebrospinal fluid tau/beta-amyloid(42) ratio as a prediction of cognitive decline in nondemented older adults. Arch Neurol 64, 343–349. [DOI] [PubMed] [Google Scholar]
- [18].Hansson O, Zetterberg H, Buchhave P, Londos E, Blennow K, Minthon L (2006) Association between CSF biomarkers and incipient Alzheimer’s disease in patients with mild cognitive impairment: A follow-up study. Lancet Neurol 5, 228–234. [DOI] [PubMed] [Google Scholar]
- [19].Shaw LM, Vanderstichele H, Knapik-Czajka M, Figurski M, Coart E, Blennow K, Soares H, Simon AJ, Lewczuk P, Dean RA, Siemers E, Potter W, Lee VM, Trojanowski JQ (2011) Qualification of the analytical and clinical performance of CSF biomarker analyses in ADNI. Acta Neuropathol 121, 597–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Bacioglu M, Maia LF, Preische O, Schelle J, Apel A, Kaeser SA, Schweighauser M, Eninger T, Lambert M, Pilotto A, Shimshek DR, Neumann U, Kahle PJ, Staufenbiel M, Neumann M, Maetzler W, Kuhle J, Jucker M (2016) Neurofilament light chain in blood and CSF as marker of disease progression in mouse models and in neurodegenerative diseases. Neuron 91, 56–66. [DOI] [PubMed] [Google Scholar]
- [21].Sarkis GA, Mangaonkar MD, Moghieb A, Lelling B, Guertin M, Yadikar H, Yang Z, Kobeissy F, Wang KK (2017) The application of proteomics to traumatic brain and spinal cord injuries. Curr Neurol Neurosci Rep 17, 23. [DOI] [PubMed] [Google Scholar]
- [22].Boutte AM, Yao C, Kobeissy F, May Lu XC, Zhang Z, Wang KK, Schmid K, Tortella FC, Dave JR (2012) Proteomic analysis and brain-specific systems biology in a rodent model ofpenetrating ballistic-like brain injury. Electrophoresis 33, 3693–3704. [DOI] [PubMed] [Google Scholar]
- [23].Song H, Fang S, Gao J, Wang J, Cao Z, Guo Z, Huang Q, Qu Y, Zhou H, Yu J (2018) Quantitative proteomic study reveals up-regulation of cAMP signaling pathway-related proteins in mild traumatic brain injury. J Proteome Res 17, 858–869. [DOI] [PubMed] [Google Scholar]
- [24].Zupanc GK (2007) Proteomics of traumatic brain injury and regeneration. Proteomics Clin Appl 1, 1362–1372. [DOI] [PubMed] [Google Scholar]
- [25].Song H, Konan LM, Cui J, Johnson CE, Langenderfer M, Grant D, Ndam T, Simonyi A, White T, Demirci U, Mott DR, Schwer D, Hubler GK, Cernak I, DePalma RG, Gu Z (2018) Ultrastructural brain abnormalities and associated behavioral changes in mice after low-intensity blast exposure. Behav Brain Res 347, 148–157. [DOI] [PubMed] [Google Scholar]
- [26].Song H (2018) Ultrastructural brain abnormalities and associated behavioral changes in mice after low-intensity blast exposure. Behav Brain Res 347, 148–157. [DOI] [PubMed] [Google Scholar]
- [27].Shelton SB, Pettigrew DB, Hermann AD, Zhou W, Sullivan PM, Crutcher KA, Strauss KI (2008) A simple, efficient tool for assessment of mice after unilateral cortex injury. J Neurosci Methods 168, 431–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Song H, Cui J, Simonyi A, Johnson CE, Hubler GK, DePalma RG, Gu Z (2018) Linking blast physics to biological outcomes in mild traumatic brain injury: Narrative review and preliminary report of an open-field blast model. Behav Brain Res 340, 147–158. [DOI] [PubMed] [Google Scholar]
- [29].Johnson MC, Song H, Cui J, Mossine VV, Gu Z, Greenlief CM (2016) Development of a method and validation for the quantitation ofFruArg in mice plasma and brain tissue using UPLC-MS/MS. ACS Omega 1, 663–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Bennett RE, Brody DL (2014) Acute reduction of microglia does not alter axonal injury in a mouse model of repetitive concussive traumatic brain injury. J Neurotrauma 31, 1647–1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Lee HK, Sanchez C, Chen M, Morin P, Wells J, Hanlon E, Xia W (2016) Three dimensional human neuro-spheroid model of Alzheimer’s disease based on differentiated induced pluripotent stem cells. PLoS One 11, e0163072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Huang DW, Sherman BT, Lempicki RA (2009) Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4, 44–57. [DOI] [PubMed] [Google Scholar]
- [33].Huang DW, Sherman BT, Tan Q, Collins JR, Alvord WG, Roayaei J, Stephens R, Baseler MW, Lane HC, Lempicki RA (2007) The DAVID Gene Functional Classification Tool: A novel biological module-centric algorithm to functionally analyze large gene lists. Genome Biol 8, R183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Yap YC, King AE, Guijt RM, Jiang T, Blizzard CA, Breadmore MC, Dickson TC (2017) Mild andrepetitive very mild axonal stretch injury triggers cystoskeletal mislocalization and growth cone collapse. PLoS One 12, e0176997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Tang-Schomer MD, Patel AR, Baas PW, Smith DH (2010) Mechanical breaking of microtubules in axons during dynamic stretch injury underlies delayed elasticity, microtubule disassembly, and axon degeneration. FASEB J 24, 1401–1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Tzekov R, Dawson C, Orlando M, Mouzon B, Reed J, Evans J, Crynen G, Mullan M, Crawford F (2016) Sub-chronic neuropathological and biochemical changes in mouse visual system after repetitive mild traumatic brain injury. PLoS One 11, e0153608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Sato-Yoshitake R, Shiomura Y, Miyasaka H, Hirokawa N (1989) Microtubule-associated protein 1B: Molecular structure, localization, and phosphorylation-dependent expression in developing neurons. Neuron 3, 229–238. [DOI] [PubMed] [Google Scholar]
- [38].Dieterich DC, Trivedi N, Engelmann R, Gundelfinger ED, Gordon-Weeks PR, Kreutz MR (2002) Partial regeneration and long-term survival of rat retinal ganglion cells after optic nerve crush is accompanied by altered expression, phosphorylation and distribution of cytoskeletal proteins. Eur J Neurosci 15, 1433–1443. [DOI] [PubMed] [Google Scholar]
- [39].Chen M, Lee HK, Moo L, Hanlon E, Stein T, Xia W (2018) Common proteomic profiles of induced pluripotent stem cell-derived three-dimensional neurons and brain tissue from Alzheimer patients. J Proteomics 182, 21–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Uchida S, Martel G, Pavlowsky A, Takizawa S, Hevi C, Watanabe Y, Kandel ER, Alarcon JM, Shumyatsky GP (2014) Learning-induced and stathmin-dependent changes in microtubule stability are critical for memory and disrupted in ageing. Nat Commun 5, 4389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Elder GA, Dorr NP, De Gasperi R, GamaSosa MA, Shaughness MC, Maudlin-Jeronimo E, Hall AA, McCarron RM, Ahlers ST (2012) Blast exposure induces post-traumatic stress disorder-related traits in a rat model of mild traumatic brain injury. J Neurotrauma 29, 2564–2575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Miyasaka T, Sato S, Tatebayashi Y, Takashima A (2010) Microtubule destruction induces tau liberation and its subsequent phosphorylation. FEBS Lett 584, 3227–3232. [DOI] [PubMed] [Google Scholar]
- [43].Bonislawski DP, Schwarzbach EP, Cohen AS (2007) Brain injury impairs dentate gyrus inhibitory efficacy. Neurobiol Dis 25, 163–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Martinez-Morillo E, Childs C, Garcia BP, Alvarez Menendez FV, Romaschin AD, Cervellin G, Lippi G, Diamandis EP (2015) Neurofilament medium polypeptide (NFM) protein concentration is increased in CSF and serum samples from patients with brain injury. Clin Chem Lab Med 53, 1575–1584. [DOI] [PubMed] [Google Scholar]
- [45].Bourne JN, Harris KM (2008) Balancing structure and function at hippocampal dendritic spines. Annu Rev Neurosci 31, 47–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Tortosa E, Montenegro-Venegas C, Benoist M, Hartel S, Gonzalez-Billault C, Esteban JA, Avila J (2011) Microtubule-associated protein 1B (MAP1B) is required for dendritic spine development and synaptic maturation. J Biol Chem 286, 40638–40648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Merriam EB, Millette M, Lumbard DC, Saengsawang W, Fothergill T, Hu X, Ferhat L, Dent EW (2013) Synaptic regulation of microtubule dynamics in dendritic spines by calcium, F-actin, and drebrin. J Neurosci 33, 16471–16482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Gupta R, Truong L, Bear D, Chafik D, Modafferi E, Hung CT (2005) Shear stress alters the expression of myelin-associated glycoprotein (MAG) and myelin basic protein (MBP) in Schwann cells. J Orthop Res 23, 1232–1239. [DOI] [PubMed] [Google Scholar]
- [49].Liu MC, Akle V, Zheng W, Kitlen J, O’Steen B, Larner SF, Dave JR, Tortella FC, Hayes RL, Wang KK (2006) Extensive degradation of myelin basic protein isoforms by calpain following traumatic braininjury. JNeurochem 98, 700–712. [DOI] [PubMed] [Google Scholar]
- [50].Weber JT (2012) Altered calcium signaling following traumatic brain injury. Front Pharmacol 3, 60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Lund LM, McQuarrie IG (2002) Calcium/calmodulin-dependent protein kinase Ilbeta isoform is expressed in motor neurons during axon outgrowth and is part of slow axonal transport. J Neurosci Res 67, 720–728. [DOI] [PubMed] [Google Scholar]
- [52].Folkerts MM, Parks EA, Dedman JR, Kaetzel MA, Lyeth BG, Berman RF (2007) Phosphorylation of calcium calmodulin-dependent protein kinase II following lateral fluid percussion brain injury in rats. J Neurotrauma 24, 638–650. [DOI] [PubMed] [Google Scholar]
- [53].Zhang M, Shan H, Gu Z, Wang D, Wang T, Wang Z, Tao L (2012) Increased expression of calcium/calmodulin-dependent protein kinase type II subunit delta after rat traumatic brain injury. J Mol Neurosci 46, 631–643. [DOI] [PubMed] [Google Scholar]
- [54].Li J, Li XY, Feng DF, Pan DC (2010) Biomarkers associated with diffuse traumatic axonal injury: Exploring pathogenesis, early diagnosis, and prognosis. J Trauma 69,1610–1618. [DOI] [PubMed] [Google Scholar]
- [55].Evans TM, Van Remmen H, Purkar A, Mahesula S, Gelfond JA, Sabia M, Qi W, Lin AL, Jaramillo CA, Haskins WE (2014) Microwave & Magnetic (M(2)) proteomics of a mouse model of mild traumatic brain injury. Transl Proteom 3, 10–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Lucke-Wold BP, Turner RC, Logsdon AF, Bailes JE, Huber JD, Rosen CL (2014) Linking traumatic brain injury to chronic traumatic encephalopathy: Identification of potential mechanisms leading to neurofibrillary tangle development. J Neurotrauma 31, 1129–1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Mannix R, Meehan WP, Mandeville J, Grant PE, Gray T, Berglass J, Zhang J, Bryant J, Rezaie S, Chung JY, Peters NV, Lee C, Tien LW, Kaplan DL, Feany M, Whalen M (2013) Clinical correlates in an experimental model of repetitive mild brain injury. Ann Neurol 74, 65–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Yang WJ, Chen W, Chen L, Guo YJ, Zeng JS, Li GY, Tong WS (2017) Involvement of tau phosphorylation in traumatic brain injury patients. Acta Neurol Scand 135, 622–627. [DOI] [PubMed] [Google Scholar]
- [59].Albayram O, Kondo A, Mannix R, Smith C, Tsai CY, Li C, Herbert MK, Qiu J, Monuteaux M, Driver J, Yan S, Gormley W, Puccio AM, Okonkwo DO, Lucke-Wold B, Bailes J, Meehan W, Zeidel M, Lu KP, Zhou XZ (2017) Cis P-tau is induced in clinical and preclinical brain injury and contributes to post-injury sequelae. Nat Commun 8, 1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Rubenstein R, Chang B, Yue JK, Chiu A, Winkler EA, Puccio AM, Diaz-Arrastia R, Yuh EL, Mukherjee P, Valadka AB, Gordon WA, Okonkwo DO, Davies P, Agarwal S, Lin F, Sarkis G, Yadikar H, Yang Z, Manley GT, Wang KKW, Cooper SR, Dams-O’Connor K, Borrasso AJ, Inoue T, Maas AIR, Menon DK, Schnyer DM, Vassar MJ (2017) Comparing Plasma phospho tau, total tau, and phospho tau-total tau ratio as acute and chronic traumatic brain injury biomarkers. JAMA Neurol 74, 1063–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Goodrich JA, Kim JH, Situ R, Taylor W, Westmoreland T, Du F, Parks S, Ling G, Hwang JY, Rapuano A, Bandak FA, de Lanerolle NC (2016) Neuronal and glial changes in the brain resulting from explosive blast in an experimental model. Acta Neuropathol Commun 4, 124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Mouzon B, Bachmeier C, Ojo J, Acker C, Ferguson S, Crynen G, Davies P, Mullan M, Stewart W, Crawford FC (2018) Chronic white matter degeneration, but no tau pathology at 1-year post-repetitive mild traumatic brain injury in tau transgenic model. J Neurotrauma, doi: 10.1089/neu.2018.5720. [DOI] [PMC free article] [PubMed] [Google Scholar]