

Abstract
Marburg virus causes sporadic outbreaks of severe hemorrhagic fever with high case fatality rates. Approved, effective, and safe therapeutic or prophylactic countermeasures are lacking. To address this, we used phage display to engineer a synthetic antibody, sFab H3, which binds the Marburg virus VP35 protein (mVP35). mVP35 is a critical cofactor of the viral replication complex and a viral immune antagonist. sFab H3 displayed high specificity for mVP35 and not for the closely related Ebola virus VP35. sFab H3 inhibited viral RNA synthesis in a minigenome assay, suggesting its potential use as an antiviral. We characterized sFab H3 by a combination of biophysical and biochemical methods, and a crystal structure of the complex solved to 1.7 Å resolution defined the molecular interface between sFab H3 and mVP35 interferon inhibitory domain. Our study identifies mVP35 as a therapeutic target using an approach that provides a framework for generating engineered Fabs targeting other viral proteins.
Keywords: Marburg virus, VP35, IFN antagonist, synthetic antibodies, phage display, mass spectrometry, X-ray crystallography
Graphical Abstract
Marburg and Ebola are members of the Filoviridae family of non-segmented negative strand RNA viruses (NNSVs) and are the cause of sporadic but deadly outbreaks of severe viral hemorrhagic fever (VHF)1–2. The 2005 Marburg outbreak in Angola, the 2014–2016 Ebola virus outbreak in West Africa, the 2017 Marburg virus outbreak in Uganda, and the ongoing 2018 Ebola virus outbreak in the Democratic Republic of Congo highlight the global impact on human health and underscore the critical need for prophylactic and therapeutic treatments for filoviral infections (https://www.cdc.gov/ncezid/dhcpp/vspb/outbreaks.html). Currently, there are no approved effective treatments available for filoviral infections, although there are several anti-filoviral interventions that are in development, including antibody-based therapies, siRNAs, phosphorodiamidate morpholino oligomers, small molecule antivirals, as well as immunomodulatory approaches3.
Filovirus disease is characterized by uncontrolled viral replication coupled to potent immune suppression by viral proteins. Like other NNSVs, Marburg virus (MARV) contains a single-stranded genome of ~19 kilobases, which encodes for seven proteins and is encapsidated by nucleoprotein (mNP). Genome replication is carried out by the viral RNA-dependent RNA polymerase (RdRp) complex that is comprised of Marburg viral protein 35 (mVP35), mNP, and the large protein (mL) polymerase, which is the only enzymatic subunit. Marburg VP30 (mVP30) is also essential for viral replication, but its exact role is unclear and, unlike its Ebola counterpart, may be dispensable for viral transcription4–6. mVP35 functions as a potent immune antagonist and is a cofactor required for viral RNA synthesis that bridges the interaction between mNP and mL, analogous to phosphoprotein (P) from other NNSVs6–7. mVP35 contains a coiled-coil motif at the N-terminus, which is required for oligomerization of mVP358–9, and a C-terminal interferon (IFN) inhibitory domain (mIID), which binds dsRNA and is essential for innate immune inhibition10–11. In addition, mVP35 contains an N-terminal peptide that binds mNP and maintains mNP in a non-oligomeric and RNA-free state12–15. How mVP35 simultaneously coordinates interactions between mNP and mL to facilitate viral RNA synthesis remains elusive.
Here we describe a strategy to generate molecular tools to further characterize the Marburg viral replication cycle in order to define novel therapeutic approaches. To this end, we engineered an antibody that specifically targets VP35 protein, which is important for MARV replication. Our approach utilized phage display technology to identify synthetic antibody fragments (sFabs) that bind viral replication components with high affinity and specificity. We characterized selected anti-mVP35 sFabs for their ability to differentiate between viral proteins from different species. In addition, we used structural studies of the sFab/mVP35 complex to identify the critical binding interface residues and to reveal the likely mechanism by which the sFab inhibits viral RNA synthesis. The pipeline described here for the generation and characterization of sFabs targeting mVP35 should be able to probe viral protein function generally and to validate potential targets for therapeutic development.
Results
Selection of sFabs specific for mVP35.
To isolate antibodies targeting mVP35, we utilized a previously established synthetic antibody library with chemical diversity introduced in four complementarity-determining regions (CDRs) of the light chain (CDRL3) and heavy chain (CDRH1, CDRH2, CDRH3) of a single human framework engineered for stability and minimal immunogenicity16–17. We performed four rounds of selection for binding to recombinant Marburg virus VP35 (mVP35) interferon (IFN) inhibitory domain (IID) protein and screened individual clones from the enriched phage pool for specific binding by phage ELISA. Five synthetic antigen-binding fragments (sFabs H1–5) specific for mVP35 IID were identified. Of these, sFabs H3 and H5 showed the best binding to mVP35 IID and the least cross-reactive binding to other filoviral antigens by phage ELISA (data not shown). Furthermore, sFabs H3 and H5 displayed the highest inhibition in the presence of a 50 nM competing antigen in single-point competitive phage ELISA (Figure 1A), suggesting that these two clones likely bind to the antigen with higher affinities than the others.
Figure 1. sFabs H3 and H5 bind mVP35 IID.

A. CDR sequences of anti-mVP35 antibodies. CDR numbering is according to IMGT scheme41. Residues that are identical to the template HP153 are denoted by dots. Dashes indicate gaps in the alignment. “% inhibition” indicates inhibition of Fab-phage binding to immobilized mVP35 IID by 50 nM solution-phase antigen in a competitive phage ELISA. B-C. ELISA results for immobilized B. mVP35 IID or C. eVP35 IID binding to increasing concentrations of sFab H1 (black), H2 (green), H3 (red), H4 (gold), or H5 (blue). Binding curves for sFabs H1 and H2 overlap in B.
We further screened these sFabs as purified free proteins for binding to mVP35 IID in an ELISA. sFabs were tested for specific ELISA binding on plates coated with either mVP35 IID or Ebola virus VP35 IID (eVP35 IID). Of the five sFabs that we tested, sFabs H1, H2, H3, and H5 bind to mVP35 IID (Figure 1B), with Fabs H3 and H5 displaying the highest OD450, consistent with the phage ELISA results. No appreciable binding to eVP35 IID was observed (Figure 1C).
sFabs H3 and H5 bind mVP35 IID with high specificity and affinity.
We further characterized binding of sFabs H3 and H5 to maltose-binding protein (MBP)-tagged mVP35 IID or eVP35 IID using in vitro pull-down assays. Both Fabs showed binding only to MBP-mVP35 IID (Figure 2A–B, lanes 7 and 8) and not to MBP-eVP35 IID (Figure 2C–D, lanes 7 and 8) or to the amylose resin alone (Figure 2A–D, lane 2), confirming the specificity of the interaction.
Figure 2. sFabs H3 and H5 bind mVP35 IID with high affinity.
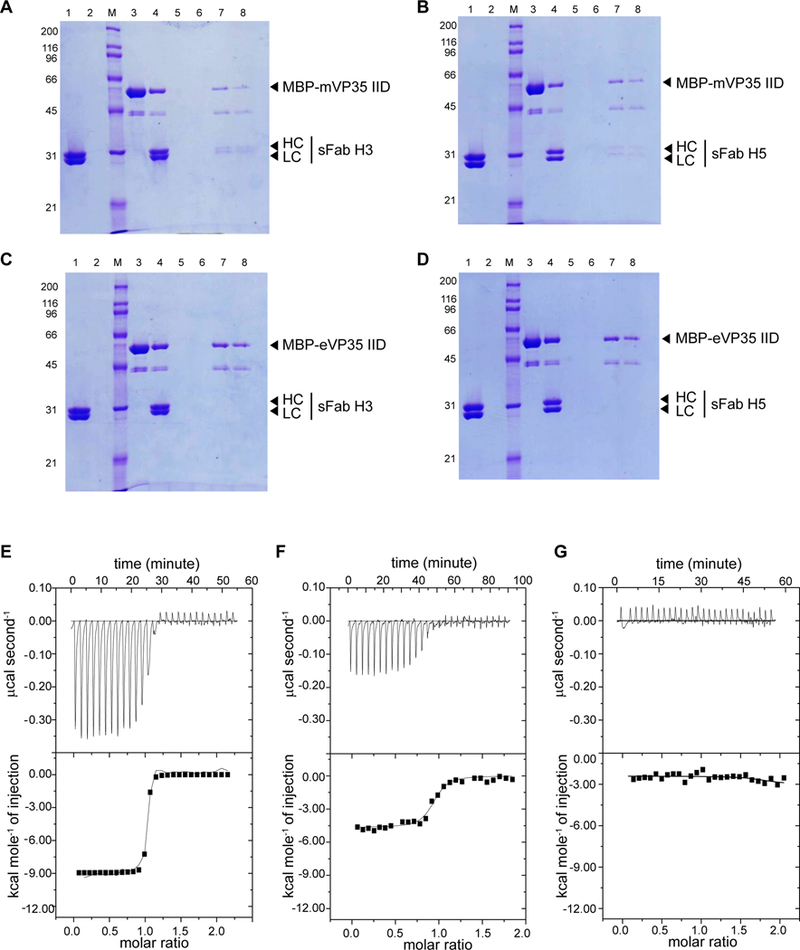
A-D. In vitro pull-down assays of A. MBP-mVP35 IID and sFab H3, B. MBP-mVP35 IID and sFab H5, C. MBP-eVP35 IID and sFab H3, and D. MBP-eVP35 IID and sFab H5. Lane 1, sFab; lane 2, sFab+ resin negative control; lane 3, immobilized MBP-fusion protein; lane 4, input; lanes 5–6, washes; lane 7, final bound resin; lane 8, eluate. Representative raw data and corresponding binding isotherms for E. mVP35 IID and sFab H3 (KD = 4.9 nM ± 1.0 nM), F. mVP35 IID and sFab H5 (KD = 80 nM ± 20 nM), and G. eVP35 IID and sFab H3 (KD not determined). Results are the average of at least two independent experiments.
We next determined dissociation constants (KD) using isothermal titration calorimetry (ITC). Analysis of the binding isotherms revealed that sFab H3 binds to mVP35 IID with high affinity (Figure 2E; KD = 4.9 ± 1 nM), whereas sFab H5 binds with moderate affinity (Figure 2F; KD = 80 ± 20 nM), and both exhibited 1:1 stoichiometry. There was no measurable enthalpy change for the interaction between sFab H3 and eVP35 IID (Figure 2G), consistent with the results from the ELISA and pull-down assays.
sFabs H3 and H5 bind the N-terminus of mVP35 IID.
We characterized sFab binding to mVP35 IID by using hydrogen-deuterium exchange coupled to mass spectrometry (HDX-MS) to identify critical binding interfaces on mVP35 IID. Peptic digestion of mVP35 IID yielded 87 peptides with 100% sequence coverage (Figure S1). Overall, both the alpha-helical subdomain and the betasheet subdomain of mVP35 IID showed increasing HDX over time (Figure 3A). The central region within the alpha helical subdomain, from H229-K237 and F248-Q254, underwent very little HDX because those backbone amides are likely participating in strong hydrogen bonding and/or are buried within the helical bundle. The loop region between alpha helices 1 and 2 (T219-T226) showed only a modest increase in HDX at longer times. In part, this attenuated HDX arises because the peptides representing this region are relatively long and serve to dilute the differences occurring over a few residues.
Figure 3. Differential deuterium uptakes clearly indicate the mVP35 IID binding epitopes with both sFab H3 and sFab H5.
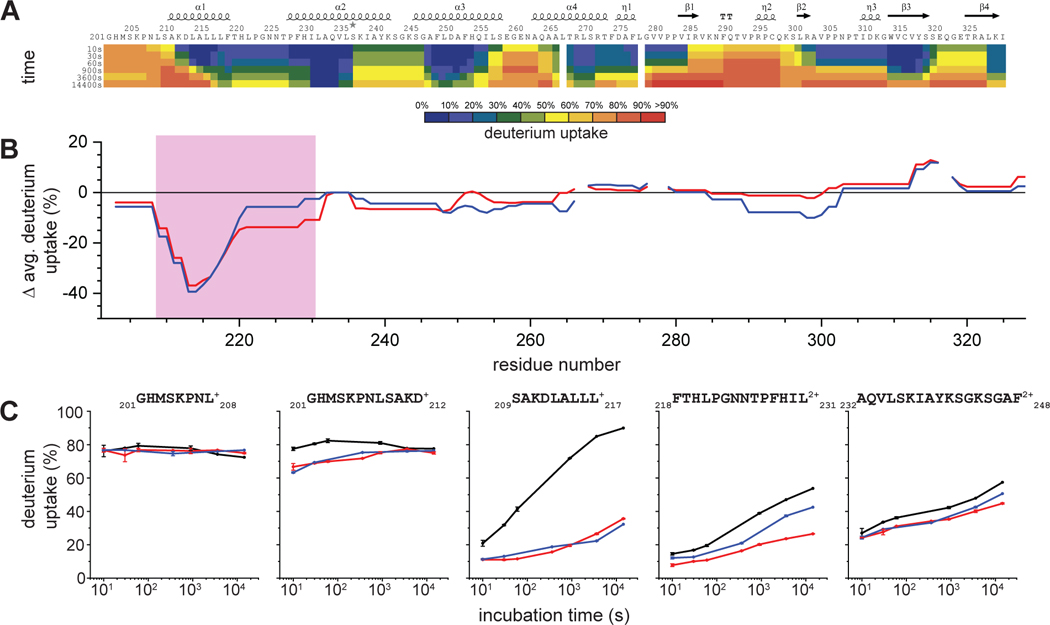
A. HDX-MS of unbound mVP35 IID.The extent of deuterium uptake is indicated by using a color gradient. B. Comparison of the per residue deuteration differences for unbound vs (red) sFabH3 bound and (blue) sFabH5 bound mVP35 IID compiled from all observed peptides and averaged across all time points. Secondary structures generated by ESPript 3.0 depicted above the heat map and difference plot. The pink box indicates a region of clearly decreased solvent exposure upon binding. C. Representative HDX kinetic curves of selected peptides in the region identified as the potential binding region in the global differential deuteration in B. See Supplementary Figure 2 for all other kinetic curves.
Upon sFab H3 binding, we found protection (diminished HDX) of mVP35 IID in the region of S209-L231 (approximately 20–50% when averaged across all time points) (Figure 3B). Specifically, peptides covering the alpha helical region from 209–220 showed the largest difference in HDX upon binding, diverging early and not converging in the kinetic plot even after 4 hours, whereas peptides covering residues 221–231 showed less difference in deuterium uptake (Figure 3C and Figure S2). The attenuated difference suggests a more dynamic binding interface or may be a consequence of the long peptide lengths that include nearby alpha helices, which dilute the effect of binding. Similarly, the same regions on mVP35 IID were protected from HDX upon binding to sFab H5, but residues 221–231 were less protected from exchange than when bound to sFab H3. All other regions of mVP35 IID bound to sFab H3 or H5 showed little difference in HDX. Taken together, these results suggest that the N-terminal helix-loop-helix in mVP35 IID is involved in binding to the sFabs.
Extensive interactions between mVP35 IID and the CDRs of sFab H3.
Comparison of sFabs H3 and H5 revealed identical CDRL1 and CDRL2 sequences, as these CDRs were not diversified in the library (Figure 1B). Among the diversified CDRs, CDRH2 sequences are very similar, whereas there are major differences in the sequences of CDRL3, CDRH1 and CDRH3 (Figure 1B). To assess those CDRs of sFab H3 critical for mediating high affinity interactions with mVP35 IID, we also used HDX-MS to characterize the changes in solvent accessibility/H-bonding of sFab H3 alone and bound with mVP35 IID. Peptic digestion of sFab H3 yielded 200 peptides with 100% sequence coverage of the light chain and 219 peptides with 99.6% sequence coverage of the heavy chain (Figure S3A–B). Upon mVP35 IID binding, all CDRs, except CDRL1, underwent some protection according to HDX (approximately 20–50% protection on average) but exhibited different deuteration kinetics (Figure 4).
Figure 4. The CDRs L2, L3, H1, H2, and H3 of sFabH3 show differential deuterium uptake rates when bound to mVP35IID.

Comparison of HDX kinetic curves of selected peptides containing, or in the region of, the predicted CDRs of the A. light chain (1–3) CDRs L1, L2, and L3, respectively, and B. heavy chain (1–3) CDRs H1, H2, and H3, respectively, of FabH3 when (black) unbound and (red) bound to mVP35 IID. All other kinetic curves are in Supplementary Figures 4 and 5 for the light chain and heavy chain, respectively. Global representation C. of the deuteration difference in the average across all time points for unbound and mVP35 IID bound sFabH3 clearly indicates those regions of sFabH3 (1) light chain and (2) heavy chain involved in binding. Green and blue boxes indicate the predicted CDR for the light chain and heavy chain, respectively.
HDX kinetics curves can also illuminate the dynamics of the binding event18. The deuterium uptake kinetic curves of peptide 47–542+, covering a portion of CDRL2, are consistent with a more dynamic binding event relative to peptides derived from the surrounding region of the protein, as evidenced by differences in HDX extent that converge at longer times (Figure 4A–L2 and Figure S4). In contrast, peptides observed in the region of CDRH3 exhibited parallel but regularly separated HDX kinetic curves, suggesting a strong and stable binding interface (Figure 4B–H3 and Figure S5). The HDX kinetic curves for peptides in the region of CDRL3 showed evidence for strong, non-dynamic binding interactions, as seen for peptide 87–93+, as well as dynamic binding interactions, as seen for peptide 93–98+ (Figure 4A–L3). Although strong and stable binding occurs with most of the CDRs, our data indicate that the binding of some residues in the CDR is more dynamic.
Peptides from regions of CDRH1 and CDRH2 exhibited HDX kinetic curves that overlapped at earlier time points but diverged later (Figure 4B–H1 and 4B–H2), indicating that only some residues in the peptide bound mVP35 IID. The HDX of Y33 in CDRH1 cannot be characterized because of the high propensity for pepsin to digest at aromatic residues (the CDR contains three tyrosines in sequence). Note that peptide 32–46+ showed no difference in deuterium uptake despite binding (Figure 4B–H1), likely because the first two residues of any peptide undergo ready back-exchange.
Altogether, HDX-MS showed that there are many interfacial interactions between sFab H3 and mVP35 IID (5 of the 6 CDRs), many of which are not dynamic, consistent with the measured high binding affinity (Figure 4C).
X-ray crystal structure of the mVP35 IID-sFab H3 complex.
We solved the X-ray crystal structure of the mVP35 IID-Fab H3 complex to 1.7 Å resolution by molecular replacement using mVP35 IID (PDB 4GHL11) and a synthetic anti-VEGF Fab (PDB 1TZI19) as search models. The crystallographic asymmetric unit contained one mVP35 IID molecule (mol A) and one sFab H3 molecule (mol B) (Figure 5A and Table S1). The overall structure of mVP35 IID in our crystal is similar to that observed previously 11, with an N-terminal alpha-helical subdomain linked to the C-terminal beta-sheet subdomain important for dsRNA binding, consistent with the observation of a backbone RMSD of 0.5 Å over 120 residues. The sFab framework, reported previously (PDB 3PNW, PDB 4K94), is similar to those in sFab structures generated from this antibody library17, 20. Examination of the interface between mVP35 IID mol A and sFab H3 mol B within the asymmetric unit reveals hydrogen bonds between the third and fourth alpha helices of mVP35 IID in the alpha-helical subdomain (involving residues N254, S257, E258, and R268) and residues in CDRL2 and CDRH3 of sFab H3, with a calculated buried surface area (BSA) of 1006.4 Å2 (Figure 5B–D). However, extension of the analysis to the symmetry mates revealed extensive hydrogen bonding and hydrophobic interactions across a larger interface between mVP35 IID mol A and sFab H3 mol B’, with a larger calculated BSA of 1796.3 Å2 (Figure 5A), suggesting that this is likely the biologically relevant complex (Figure 5E). Close examination of this interaction, based on Ligplot2+ analysis21, revealed that binding between mVP35 and sFab H3 likely consists of the following: three hydrogen bonds between mVP35 IID (involving residues N224 and T219) and CDRL1 and CDRL3 of sFab H3 (Figure 5F), two hydrogen bonds between mVP35 IID (involving residues N225 and K211) and CDRH3 of sFab H3 (Figure 5G), and numerous hydrophobic contacts across the interface. HDX-MS results also validate the assignment of the interface between mVP35 IID mol A and sFab mol B’ as the relevant complex as increased protection from deuterium uptake was observed for mVP35 residues 211–229 in the presence of sFab H3 (Figure 3 and Figure S3). Furthermore, we find that sFab H3 binding to the alpha-helical subdomain of mVP35 IID does not perturb the ability of the beta sheet subdomain of mVP35 IID to bind dsRNA (Figure S6).
Figure 5. Structure of mVP35 IID/sFab H3 complex.

A. Cartoon representation of the mVP35 IID/sFab H3 complex. The crystallographic asymmetric unit contains one molecule of mVP35 IID (mol A, magenta) and one molecule of sFab H3 (mol B, gray). The complex formed between mol A and the sFab H3 symmetry mate (mol B’; heavy chain, blue; light chain, green) is used for subsequent analysis. B. Enlarged view of the interface between mVP35 IID mol A and sFab H3 mol B. The complex is rotated 90° along the x-axis from the view in A. Ligplot+ analyses of the interactions between C. mVP35 IID mol A and sFab H3 mol B light chain and D. mVP35 IID mol A and sFab H3 mol B heavy chain. Amino acid residues of CDRs involved in contacts are labeled. Hydrogen bonding is indicated by dashed lines and hydrophobic contacts represented by spoked arcs. E. Enlarged view of the interface between mVP35 IID mol A and sFab H3 mol B’. Ligplot+ analyses of the interactions between F. mVP35 IID mol A’ and sFab H3 mol B’ light chain and G. mVP35 IID mol A and sFab H3 B’ heavy chain.
Species specific determinants of mVP35 IID binding to sFab H3.
Structural alignment of the structures of mVP35 IID (PDB 4GHL) and of eVP35 IID (PDB 3FKE) reveal near identical structures with a backbone RMSD of 1.03 Å over 120 residues11, 22. To define the molecular determinants for the specificity of sFab H3 for mVP35 IID over eVP35 IID, we observed in the crystal structure that three of the four mVP35 residues involved in hydrogen bonding with sFab H3, namely T219, N224, and N225, are located in the linker between alpha-helices 1 and 2 and are not conserved between mVP35 and eVP35 (Figure 6A). Mutation of these mVP35 IID residues to the corresponding eVP35 IID residues resulted in the complete loss of mVP35 IID T219D/N224F/N225G binding to sFab H3, as determined by using the in vitro pull-down assay (Figure 6B). Furthermore, mutation of eVP35 IID residues to the corresponding mVP35 IID residues produced eVP35 IID D230T/F235N/G236N, which can now bind to sFab H3 (Figure 6C). Similar binding behavior was observed with sFab H5 (Figure S7). Taken together, these results provide a molecular basis to explain the differences between sFabs H3 and H5 in terms of quantitative binding data. These data also support the ability to discriminate between VP35 proteins from Marburg virus and Ebola virus, despite the high degree of structural similarity among VP35 orthologs.
Figure 6. sFab H3 binds with high specificity to mVP35 IID.

A. Sequence alignment of mVP35 IID and eVP35 IID (Accession # YP0015311154.1 and AAD14582, respectively). Secondary structural elements are drawn above the sequence. Residues that are identical between the two sequences are highlighted in red. Residues that are involved in interfacial contacts between mVP35 IID and Fab H3 and are not conserved are highlighted in gray. In vitro pull-down assays of B. MBP-tagged mVP35 IID T219D/N224F/N225G and C. MBP-tagged eVP35 IID D230T/F235N/G236N with sFab H3. Lane 1, sFab; lane 2, sFab + resin negative control; lane 3, immobilized MBP-fusion protein; lane 4, input; lanes 5–6, washes; lane 7, final bound resin; lane 8, eluate. D. Marburg mini-genome assay and E. Ebola mini-genome assay with increasing concentrations of scFv H3 vector (6.25, 12.5, 25, 62.5 ng). Corresponding Western blots of β-tubulin and scFv H3 are shown below. The error bars indicate standard deviation of three independent replicates. **p = 0.002 and ****p < 0.0001, determined by one-way ANOVA followed by Tukey’s multiple comparison test.
scFv H3 inhibits viral RNA synthesis.
Marburg viral RNA synthesis depends on formation of the viral RdRp complex that is minimally comprised of mVP35, mNP and mL. To test if sFab H3 binding interferes with mVP35 function in viral replication, we generated a mammalian expression plasmid encoding a single chain variable fragment (scFv), containing just the VH and VL of H3 connected by a flexible linker. In this assay, the scFv fragment was expressed in the cytosol without fusion of a secretion or nuclear localization signal peptide, and thus is expected to fold in the cytoplasm where it would be able to inhibit VP35 activity, as observed. scFv H3 was coexpressed with the components of the viral polymerase complex, and viral RNA synthesis was determined by measuring expression of a Renilla luciferase reporter gene expressed from a non-infectious, minigenome RNA. We found that scFv H3 expression inhibited the Marburg polymerase complex in a dose-dependent manner according to the minigenome reporter gene expression (Figure 6D) but did not inhibit Ebola minigenome activity (Figure 6E). These results suggest that scFv H3 either locks mVP35 IID in a conformation that is incompatible with or sterically hinders a critical interaction interface required for viral RNA synthesis. These results are also consistent with our findings that sFab H3 binds only to mVP35 IID and with our previous findings that the alpha-helical subdomain of mVP35 IID contains interaction surfaces that are critical for viral replication11.
Discussion
Filoviral RNA synthesis is incompletely understood, and thus new tools are required both to improve understanding and to target Marburg virus replication with antivirals. Importantly, differences in host and viral structural requirements for viral RNA replication among species have only been minimally defined. In the case of Ebola virus, viral replication requires a complex that includes eVP35, eNP and eL along with eVP30 for transcription. For Marburg virus, despite its significant homology to eVP30, mVP30 is not a required component for viral transcription according to a minigenome assay. However, mVP30 enhances transcription and is essential for viral growth, suggesting that mVP30 may have distinct roles that modulate the RdRp replication/transcription complex4–6. We are only just beginning to decipher this process at the molecular level23–24.
Although these previous studies shed some light on the Marburg replication/transcription process, there are many important details that remain incompletely defined. To better understand these roles and to define critical interfaces in the viral RdRp complex, we implemented a pipeline to select and characterize antibody reagents that can perturb viral RNA synthesis and provide insight into this process. We used phage display to screen a synthetic antibody library and identified a series of sFabs that bind to mVP35. Using HDX-MS and X-ray crystallography, we evaluated the binding interface and defined the paratope on sFab H3 and the epitope on mVP35. The two structural characterization techniques used in this pipeline provide a wealth of complementary information that can be used in multiple ways. For example, sFab H3 binds mVP35 tightly, but nonetheless, knowledge of the sFab CDR residues involved in antigen recognition can be used to further optimize the paratope for improved affinity and broadened specificity. We found that although the HDX-MS results validated the assignment of the biologically relevant complex of mVP35 with the symmetry related sFab H3 molecule, HDX-MS also revealed a more dynamic paratope interaction where CDRs L1 and L2 are likely involved in binding mVP35. Conversely, the defined structural epitope provides a potential target site that appears to be critical for viral RNA synthesis and could help to elucidate the role of the protein during viral replication.
Our work provides a potential target for anti-viral therapeutics, as mVP35 appears to be critical for viral RNA synthesis. When we tested scFv H3 in minigenome assays, we found that expression of scFv H3 inhibited Marburg but not Ebola viral RNA synthesis. This suggests that the region between the first two helices of the Marburg VP35 IID protein is important for this process or that scFv H3 sterically hinders formation of a functional replication complex. Even though the structures of Marburg and Ebola VP35 IID are nearly identical, our sFabs discriminate between different species of viral proteins, likely through sequence specific interactions and subtle conformational changes. These reagents can now facilitate studies that identify functionally relevant regions that are unique to each member of the filovirus family and provide suitable targets for development of diagnostics and therapeutics.
Many challenges remain in targeting protein-protein interactions in the cell. Advances in engineering alternative protein scaffolds have overcome many hurdles, including immunogenicity, delivery and toxicity, to produce highly functional reagents25–29. The general inability of large proteins to cross the plasma membranes currently precludes the use of antibodies for targeting intracellular proteins. Nonetheless, our in vitro cell-based studies provide strong support that inhibition of VP35, through the target defined by the sFab/mIID complex, is a previously unrecognized opportunity to inhibit viral replication. Furthermore, our structural studies reveal the molecule details of an epitope that may be targeted with conventional small molecule screening approaches. Thus, our work describes a viable candidate target for anti-viral therapies and represent a step forward in developing a diagnostic or therapeutic for a group of viral pathogens that have resisted traditional approaches.
Methods
Synthetic Fab library design and construction.
A highly functional synthetic antigen-binding fragment (sFab) library, Lib F, was utilized to identify antibodies targeting viral protein mVP35 IID. Full details for Lib F construction and characterization were published previously17. Briefly, Lib F was designed based on an understanding of natural antibody repertoires and on our previous study of a series of synthetic antibody libraries with minimalist design30. It was constructed on a single human Fab framework with variable domains from subgroups VH3 and Vk1. Chemical diversity was introduced in four of the six CDRs. Binomial Tyr/Ser diversity was introduced at solvent accessible paratope positions of CDR-H1 and CDR-H2. In CDR-L3 and CDR-H3, more extensive chemical diversity, totaling nine different amino acids with a bias for Tyr, Ser, Gly and Ala, was used to mimic the observed amino acid distribution in CDRs of natural antibody repertoires31. In addition, further diversity was introduced by varying the lengths of CDR-L3 and CDR-H3.
Phage display selections, direct phage ELISAs, and competitive phage ELISAs were performed as described32. Briefly, to identify antibodies bound to mVP35 IID, phage particles from Lib F were cycled through four rounds of binding selections with the antigen directly immobilized on 96-well Maxisorp immunoplates (Fisher Scientific, Nepean, ON, Canada). Phage particles were produced from individual clones of the enriched phage pool grown in a 96-well format and the culture supernatants were used in phage ELISAs to detect specific binding clones. Clones that bound specifically to mVP35 IID were subjected to DNA sequencing for decoding the sequences of the displayed Fabs. To estimate antigen-binding affinities of these antibodies, a competitive phage ELISA was used, whereby binding of Fab-phage to immobilized mVP35 IID was assessed in the presence of solution-phase mVP35 IID, as described.
Cloning, expression and protein purification.
The mVP35 IID was expressed and purified as previously described11. Fabs were expressed and purified as described previously33. Briefly, vectors encoding for Fabs were transformed into E. coli, and a single colony was inoculated into a 50 mL starter culture containing 2YT medium and ampicillin at 37 °C for 16–18 h at 230 rpm. The starter culture was used to inoculate 2 L of TB ampicillin broth and incubated at 37 °C for 3 to 3.5 h at 230 rpm until reaching an OD600 of 0.8. Fab expression was induced by adding 1 mM IPTG for 3 h at 37 °C. Cells were harvested and resuspended in PBS pH 7.4 with protease inhibitors. Cells were lysed using an EmulsiFlex-C5 Homogenizer (Avestin) and clarified at 47000 × g for 40 min at 4 °C. The supernatant was applied onto a 2 mL Protein A Sepharose column, washed with PBS pH 7.4, and eluted with buffer containing 50 mM sodium phosphate monobasic pH 3.0, 50 mM sodium citrate, and 300 mM NaCl. The eluted antibody was neutralized with 5 mL of 0.5 M sodium phosphate dibasic to a pH of 6.0, and the salt concentration diluted to 50 mM with 25 mM sodium phosphate buffer pH 6.0. The Fab was further purified using a 2 mL SOURCE 15S column prior to application onto a size exclusion column by using buffer containing 10 mM Hepes pH 7.0, 150 mM NaCl, and 200 mM arginine pH 7.0. The purity of samples at each purification step was analyzed on SDS-PAGE gels.
In vitro pull-down assay.
Purified Fabs were applied to amylose resin pre-bound to MBP-tagged mVP35 IID WT or mutant. The resin was washed five times with PBS pH 7.4 prior to elution with 1 % maltose. Samples taken at each step of the assay were visualized by Coomassie staining of SDS-PAGE.
ELISA.
High binding Immulon 2HB 96 well plates were coated with 100 μL of 1 μg/mL antigen and incubated overnight at 4 °C. The plate was blocked with 1 % bovine serum albumin for 1 h at room temperature and washed with PBS containing 0.1 % Tween-20 (PBST). Different concentrations of Fab, starting from 10 μg/mL were added into the wells and incubated for 2 h at 25 °C. The plate was washed 10 times with PBST. Afterwards, 100 μL/well of anti-FLAG conjugated to HRP was added (1:20,000 dilution in PBST) and incubated for 30 min at 25 °C. The plate was again washed 10 times with PBST. The reaction was developed by adding 50 μL/well of TMB substrate and quenched by adding 50 μL/well of 2 M sulphuric acid after 2 to 3 minutes of incubation at room temperature. The plate was read using a plate reader with a 450 nm filter.
ITC.
Protein samples were dialyzed against buffer containing 10 mM HEPES (pH 7.0) and 150 mM NaCl overnight in cold room. Titrations were set up using a VP-ITC microcalorimeter (Malvern) with 100 μM antigen in the syringe, 10 μM sFab in the cell, and a reference power of 4μcals−1. Raw ITC data were processed using Origin 7.0 software, and data were fit by nonlinear least-squares analysis to yield Kd and n. Results are an average of at least two independent experiments.
HDX-MS experiments.
All chemical reagents used in HDX-MS experiments were purchased from Sigma-Aldrich (St. Louis, MO) unless otherwise noted. All mass spectral analyses (LC/MS and LC/MS/MS) were performed by using a custom-built liquid chromatography (LC) assembly incorporating online digestion using an in-house packed pepsin column (2 mm × 20 mm), trapping and desalting on a ZORBAX Eclipse XDB C8 column (2.1 mm × 15 mm, Agilent, Santa Clara, CA), and incorporating analytical separation with a Hypersil Gold C18 column (2.1 mm × 50 mm, Thermo Fisher, Waltham, MA). All valves, tubes, and columns (except for the pepsin column, which suffered a loss in activity at low temperature) were submerged in an ice-water bath to minimize back-exchange. The peptides were then introduced via electrospray ionization into an LTQ-FTICR mass spectrometer (Thermo Fisher, Waltham, MA). All experiments were conducted in duplicate unless otherwise indicated. Data analysis of continuous HDX was carried out with HDExaminer (version 1.1.0, Sierra Analytics, Inc., Modesto, CA).
HDX-MS of the mVP35 IID binding epitopes.
HDX analyses of the binding epitope were carried out on stock solutions of unbound mVP35 IID (30 μM protein in PBS, pH 7.4) and sFab H3 and sFab H5 bound mVP35 IID (1:1.2 molar ratio mVP35IID:Fab H3 or Fab H5 in PBS, pH 7.4). Samples were equilibrated at 25 °C for 2 h before HDX analysis. Continuous deuterium labeling was triggered by diluting 2 μL of the stock solution 10-fold with PBS buffer in D2O. The HDX samples were incubated for various time intervals (10 s, 30 s, 60 s, 360 s, 900 s, 3600 s, and 14400 s) at 25 °C, and the reactions quenched using 30 μL of ice-cold 3 M urea and 1% (v/v) trifluoroacetic acid (TFA). The resulting sample was immediately injected into the aforementioned LC/MS system, for 3 min of digestion and desalting at a flow rate of 200 μL/min 0.1% TFA, followed by LC separation of the peptic peptides using a 5.5 minute linear gradient of 4% to 40% acetonitrile with 0.1% formic acid.
HDX-MS of the Fab H3 paratope.
HDX analyses were carried out on stock solutions of unbound sFab H3 (40 μM in PBS, pH 7.4) and mVP35 IID bound sFab H3 (1:2.2 molar ratio sFab H3:mVP35IID in PBS, pH 7.4). Stock samples were equilibrated at 25 °C for 2 h before HDX analysis. Continuous deuterium labeling was triggered by diluting 2 μL of the stock solution 10fold with PBS buffer in D2O. The HDX samples were incubated for various time intervals (10 s, 30 s, 60 s, 360 s, 900 s, 3600 s, and 14400 s) on ice. The HDX reaction was then quenched using a solution of 500 mM tris(2-carboxyethyl)phosphine hydrochloride (TCEP) and 4 M guanidine hydrochloride in PBS, pH 2.4, and the disulfide bonds within sFab H3 were reduced upon incubation for 3 min at 25 °C. The samples were then flash frozen in liquid nitrogen for later injection onto the LC/MS platform for 3 min of digestion and desalting at a flow rate of 200 μL/min 0.1 % TFA, followed by LC separation using a 10.5 min linear gradient of 4 % to 40 % acetonitrile with 0.1 % formic acid.
HDX-MS mapping analysis.
To identify the peptides produced in digestion, separate experiments without deuterium labeling were performed, thereby generating a list of peptides that could be followed during HDX-MS data acquisition and analysis. Mass spectra of product-ions (MS/MS) were collected in a data-dependent mode, in which the six most abundant ions from each scan were selected for MS/MS analysis. For the mapping of mVP35 IID, the MS/MS files from three parallel runs were converted to mzXML files by using MM File Conversion and submitted to MassMatrix for peptide identification34. For the mapping of sFab H3, the MS/MS files from two parallel runs were analyzed using Byonic™ and Byologic™ (Protein Metrics, San Carlos, CA). Both searches were similarly carried out against a reverse sequence to discard ambiguous identifications.
X-ray crystallography.
Crystallization trials of the mVP35 IID/sFab H3 complex were initiated by using commercially available screens (Hampton Research). Initial mVP35 IID/sFab H3 complex crystals were subsequently optimized by using in-house reagents. The crystal of mVP35 IID/sFab H3, from which synchrotron data were collected, grew in 0.01 M Bis Tris (pH 6.5), 20% PEG MME 5000, and a mVP35 IID:sFab H3 ratio of 1:1.25 mg/ml. The structure of the complex was solved by molecular replacement using PDB 4GHL and 1TZI as the search models in Phaser. The final model was built following iterative rounds of BUCCANEER35, manual model building in Coot36, and refinement with REFMAC537. Final structures figures were generated by using PyMOL38–39. Structure quality was assessed with MolProbity40. Interface analysis were performed using LigPlot+21, 41.
Minigenome assay.
The Ebola and Marburg minigenomes are previously described42–43. The gene encoding scFv H3 was cloned into pcDNA3.1/nFLAG-Dest vector (a modified version of pcDNA3.1/nV5-Dest (Invitrogen) where a V5 tag is replaced with a FLAG tag) for intracellular expression of N-FLAG tagged scFv H3 fragments. Briefly, increasing amounts of scFv H3 plasmid (50, 100, 250, 500 ng) were co-transfected with the plasmids required to reconstitute the EBOV RNA polymerase complex (L (125 ng), NP (62.5 ng), VP35 (31.25 ng), VP30 (25 ng)) or MARV RNA polymerase complex (L (125 ng), NP (62.5 ng), VP35 (31.25ng), VP30 (25 ng)) along with minigenome plasmid encoding Renilla luciferase (50 ng) and firefly plasmid (1 ng) as a control for transfection efficiency and a T7 (50 ng) expression plasmid in HEK293T cells. The plasmid amounts are indicated for one 96 well plate. The luciferase activity is determined 48 hours post transfection using Dual Glo reagent (Promega). Relative minigenome activity is determined by normalizing Renilla luciferase activity to firefly luciferase activity, setting the activity of the “no scFv H3” (empty vector only) samples as 100 percent. The error bars indicate standard deviation of three independent replicates. **p = 0.002 and ****p < 0.0001 determined by one-way ANOVA followed by Tukey’s multiple comparison test.
Corresponding western blots show expression of β-tubulin and scFv H3, which was detected by anti-FLAG antibody.
Fluorescence polarization assay (FPA).
FPAs were performed on a Cytation 5 plate reader using Gen5 software (BioTek), as described previously44. Briefly, excitation and emission wavelengths were set to 485 and 529 nm, respectively, with a bandpass width of 20 nm and a read height of 8.5 mm. FP signals were measured for samples containing 1 nM FITC-labeled 19 bp dsRNA (Genscript) alone and in the presence of 100 nM mVP35 IID, 50 nM sFab H3, or 100 nM MBP. Data were plotted using Origin software.
Protein Structure Accession Numbers.
Coordinates and structure factor files are deposited in the Protein Data Bank under identifier 6OTC.
Supplementary Material
Acknowledgements
We would like to thank members of the D.W.L lab for assistance and support. We also acknowledge the support of National Institutes of Health (P01AI120943 to D.W.L., C.F.B., D.M.B, S.S., and M.L.G; R01AI140758 to D.W.L.; P41GM103422 to M.L.G.). Results shown are partly derived from work performed at Argonne National Laboratory, Structural Biology Center (SBC) Beamline 19-ID at the Advanced Photon Source. SBC-CAT is operated by U Chicago Argonne, LLC, for the U.S. Department of Energy, Office of Biological and Environmental Research under contract DE-AC02–06CH11357. C.F.B. is a Georgia Research Alliance Eminent Scholar in Microbial Pathogenesis.
Footnotes
Supporting Information. Data collection and refinement statistics (Table S1), peptide coverage maps for mVP35 IID and sFab H3 (Figure S1), HDX-MS heatmaps for mVP35 IID and sFab H3 (Figure S2), HDX kinetics curves of mVP35 IID peptides (Figure S3), HDX kinetics curves of sFab H3 light chain peptides (Figure S4), HDX kinetics curves of sFab H3 heavy chain peptides (Figure S5), fluorescence polarization assay for mVP35 IID binding to sFab H3 with dsRNA (Figure S6), and pull-down assay of eVP35 IID mutant binding to sFab H5 (Figure S7).
References
- 1.Marburg-and ebolaviruses. Springer Berlin Heidelberg: New York, NY, 2017; p pages cm. [Google Scholar]
- 2.Messaoudi I; Amarasinghe GK; Basler CF, Filovirus pathogenesis and immune evasion: insights from Ebola virus and Marburg virus. Nat Rev Microbiol 2015, 13 (11), 663–76 DOI: 10.1038/nrmicro3524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cross RW; Mire CE; Feldmann H; Geisbert TW, Post-exposure treatments for Ebola and Marburg virus infections. Nat Rev Drug Discov 2018, 17 (6), 413–434 DOI: 10.1038/nrd.2017.251. [DOI] [PubMed] [Google Scholar]
- 4.Enterlein S; Volchkov V; Weik M; Kolesnikova L; Volchkova V; Klenk HD; Muhlberger E, Rescue of recombinant Marburg virus from cDNA is dependent on nucleocapsid protein VP30. J Virol 2006, 80 (2), 1038–43 DOI: 10.1128/JVI.80.2.1038-1043.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Muhlberger E; Lotfering B; Klenk HD; Becker S, Three of the four nucleocapsid proteins of Marburg virus, NP, VP35, and L, are sufficient to mediate replication and transcription of Marburg virus-specific monocistronic minigenomes. J Virol 1998, 72 (11), 8756–64 DOI. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Muhlberger E; Weik M; Volchkov VE; Klenk HD; Becker S, Comparison of the transcription and replication strategies of marburg virus and Ebola virus by using artificial replication systems. J Virol 1999, 73 (3), 2333–42 DOI. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schmidt KM; Muhlberger E, Marburg Virus Reverse Genetics Systems. Viruses 2016, 8 (6), DOI: 10.3390/v8060178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bruhn JF; Kirchdoerfer RN; Urata SM; Li S; Tickle IJ; Bricogne G; Saphire EO, Crystal Structure of the Marburg Virus VP35 Oligomerization Domain . J Virol 2017, 91 (2), DOI: 10.1128/JVI.01085-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Reid SP; Cardenas WB; Basler CF, Homo-oligomerization facilitates the interferonantagonist activity of the ebolavirus VP35 protein. Virology 2005, 341 (2), 179–89 DOI: 10.1016/j.virol.2005.06.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bale S; Julien JP; Bornholdt ZA; Kimberlin CR; Halfmann P; Zandonatti MA; Kunert J; Kroon GJ; Kawaoka Y; MacRae IJ; Wilson IA; Saphire EO, Marburg virus VP35 can both fully coat the backbone and cap the ends of dsRNA for interferon antagonism. PLoS Pathog 2012, 8 (9), e1002916 DOI: 10.1371/journal.ppat.1002916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ramanan P; Edwards MR; Shabman RS; Leung DW; Endlich-Frazier AC; Borek DM; Otwinowski Z; Liu G; Huh J; Basler CF; Amarasinghe GK, Structural basis for Marburg virus VP35-mediated immune evasion mechanisms. Proc Natl Acad Sci USA 2012, 109 (50), 20661–6 DOI: 10.1073/pnas.1213559109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Leung DW; Borek D; Luthra P; Binning JM; Anantpadma M; Liu G; Harvey IB; Su Z; Endlich-Frazier A; Pan J; Shabman RS; Chiu W; Davey RA; Otwinowski Z; Basler CF; Amarasinghe GK, An Intrinsically Disordered Peptide from Ebola Virus VP35 Controls Viral RNA Synthesis by Modulating Nucleoprotein-RNA Interactions. Cell Rep 2015, 11 (3), 376–89 DOI: 10.1016/j.celrep.2015.03.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu B; Dong S; Li G; Wang W; Liu X; Wang Y; Yang C; Rao Z; Guo Y, Structural Insight into Nucleoprotein Conformation Change Chaperoned by VP35 Peptide in Marburg Virus. J Virol 2017, 91 (16), DOI: 10.1128/JVI.00825-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhu T; Song H; Peng R; Shi Y; Qi J; Gao GF, Crystal Structure of the Marburg Virus Nucleoprotein Core Domain Chaperoned by a VP35 Peptide Reveals a Conserved Drug Target for Filovirus. J Virol 2017, 91 (18), DOI: 10.1128/JVI.00996-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kirchdoerfer RN; Abelson DM; Li S; Wood MR; Saphire EO, Assembly of the Ebola Virus Nucleoprotein from a Chaperoned VP35 Complex. Cell Rep 2015, 12 (1), 140–149 DOI: 10.1016/j.celrep.2015.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen G; Sidhu SS, Design and generation of synthetic antibody libraries for phage display. Methods Mol Biol 2014, 1131, 113–31 DOI: 10.1007/978-1-62703-992-5_8. [DOI] [PubMed] [Google Scholar]
- 17.Persson H; Ye W; Wernimont A; Adams JJ; Koide A; Koide S; Lam R; Sidhu SS, CDR-H3 diversity is not required for antigen recognition by synthetic antibodies. J Mol Biol 2013, 425 (4), 803–11 DOI: 10.1016/j.jmb.2012.11.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wei H; Mo J; Tao L; Russell RJ; Tymiak AA; Chen G; Iacob RE; Engen JR, Hydrogen/deuterium exchange mass spectrometry for probing higher order structure of protein therapeutics: methodology and applications. Drug Discov Today 2014, 19 (1), 95–102 DOI: 10.1016/j.drudis.2013.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fellouse FA; Wiesmann C; Sidhu SS, Synthetic antibodies from a four-amino-acid code: a dominant role for tyrosine in antigen recognition. Proc Natl Acad Sci U S A 2004, 101 (34), 12467–72 DOI: 10.1073/pnas.0401786101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Reshetnyak AV; Nelson B; Shi X; Boggon TJ; Pavlenco A; Mandel-Bausch EM; Tome F; Suzuki Y; Sidhu SS; Lax I; Schlessinger J, Structural basis for KIT receptor tyrosine kinase inhibition by antibodies targeting the D4 membrane-proximal region. Proc Natl Acad Sci U S A 2013, 110 (44), 178327 DOI: 10.1073/pnas.1317118110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Laskowski RA; Swindells MB, LigPlot+: multiple ligand-protein interaction diagrams for drug discovery. J Chem Inf Model 2011, 51 (10), 2778–86 DOI: 10.1021/ci200227u. [DOI] [PubMed] [Google Scholar]
- 22.Leung DW; Ginder ND; Fulton DB; Nix J; Basler CF; Honzatko RB; Amarasinghe GK, Structure of the Ebola VP35 interferon inhibitory domain. Proc Natl Acad Sci U S A 2009, 106 (2), 411–6 DOI: 10.1073/pnas.0807854106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kirchdoerfer RN; Moyer CL; Abelson DM; Saphire EO, The Ebola Virus VP30-NP Interaction Is a Regulator of Viral RNA Synthesis. PLoS Pathog 2016, 12 (10), e1005937 DOI: 10.1371/journal.ppat.1005937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xu W; Luthra P; Wu C; Batra J; Leung DW; Basler CF; Amarasinghe GK, Ebola virus VP30 and nucleoprotein interactions modulate viral RNA synthesis. Nat Commun 2017, 8, 15576 DOI: 10.1038/ncomms15576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Miersch S; Sidhu SS, Intracellular targeting with engineered proteins. F1000Res 2016, 5, DOI: 10.12688/f1000research.8915.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hanke L; Schmidt FI; Knockenhauer KE; Morin B; Whelan SP; Schwartz TU; Ploegh HL, Vesicular stomatitis virus N protein-specific single-domain antibody fragments inhibit replication. EMBO Rep 2017, 18 (6), 1027–1037 DOI: 10.15252/embr.201643764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Radwanska MJ; Jaskolowski M; Davydova E; Derewenda U; Miyake T; Engel DA; Kossiakoff AA; Derewenda ZS, The structure of the C-terminal domain of the nucleoprotein from the Bundibugyo strain of the Ebola virus in complex with a pan-specific synthetic Fab. Acta Crystallogr D Struct Biol 2018, 74 (Pt 7), 681–689 DOI: 10.1107/S2059798318007878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Seesuay W; Jittavisutthikul S; Sae-Lim N; Sookrung N; Sakolvaree Y; Chaicumpa W, Human transbodies that interfere with the functions of Ebola virus VP35 protein in genome replication and transcription and innate immune antagonism. Emerg Microbes Infect 2018, 7 (1), 41 DOI: 10.1038/s41426-018-0031-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen G; Koellhoffer JF; Zak SE; Frei JC; Liu N; Long H; Ye W; Nagar K; Pan G; Chandran K; Dye JM; Sidhu SS; Lai JR, Synthetic antibodies with a human framework that protect mice from lethal Sudan ebolavirus challenge. ACS Chem Biol 2014, 9 (10), 2263–73 DOI: 10.1021/cb5006454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fellouse FA; Esaki K; Birtalan S; Raptis D; Cancasci VJ; Koide A; Jhurani P; Vasser M; Wiesmann C; Kossiakoff AA; Koide S; Sidhu SS, High-throughput generation of synthetic antibodies from highly functional minimalist phage-displayed libraries. J Mol Biol 2007, 373 (4), 924–40 DOI: 10.1016/j.jmb.2007.08.005. [DOI] [PubMed] [Google Scholar]
- 31.Zemlin M; Klinger M; Link J; Zemlin C; Bauer K; Engler JA; Schroeder HW Jr.; Kirkham PM, Expressed murine and human CDR-H3 intervals of equal length exhibit distinct repertoires that differ in their amino acid composition and predicted range of structures. J Mol Biol 2003, 334 (4), 733–49 DOI. [DOI] [PubMed] [Google Scholar]
- 32.Sidhu SS; Fellouse FA, Synthetic therapeutic antibodies. Nat Chem Biol 2006, 2 (12), 682–688 DOI: 10.1038/nchembio843. [DOI] [PubMed] [Google Scholar]
- 33.Studier FW, Protein production by auto-induction in high density shaking cultures. Protein Expr Purif 2005, 41 (1), 207–34 DOI. [DOI] [PubMed] [Google Scholar]
- 34.Xu H; Freitas MA, MassMatrix: a database search program for rapid characterization of proteins and peptides from tandem mass spectrometry data. Proteomics 2009, 9 (6), 1548–55 DOI: 10.1002/pmic.200700322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cowtan K, The Buccaneer software for automated model building. 1. Tracing protein chains. Acta Crystallogr D Biol Crystallogr 2006, 62 (Pt 9), 1002–11 DOI: 10.1107/S0907444906022116. [DOI] [PubMed] [Google Scholar]
- 36.Emsley P; Cowtan K, Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 2004, 60 (Pt 12 Pt 1), 2126–32 DOI: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 37.Collaborative Computational Project N, The CCP4 suite: programs for protein crystallography. Acta Crystallogr D Biol Crystallogr 1994, 50 (Pt 5), 760–3 DOI: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 38.DeLano WL, Use of PYMOL as a communications tool for molecular science. Abstr Pap Am Chem S 2004, 228, U313–U314 DOI. [Google Scholar]
- 39.DeLano WL, PyMOL molecular viewer: Updates and refinements. Abstr Pap Am Chem S 2009, 238, DOI. [Google Scholar]
- 40.Davis IW; Murray LW; Richardson JS; Richardson DC, MOLPROBITY: structure validation and all-atom contact analysis for nucleic acids and their complexes. Nucleic Acids Res 2004, 32 (Web Server issue), W615–9 DOI: 10.1093/nar/gkh398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wallace AC; Laskowski RA; Thornton JM, LIGPLOT: a program to generate schematic diagrams of protein-ligand interactions. Protein Eng 1995, 8 (2), 127–34 DOI. [DOI] [PubMed] [Google Scholar]
- 42.Luthra P; Liang J; Pietzsch CA; Khadka S; Edwards MR; Wei S; De S; Posner B; Bukreyev A; Ready JM; Basler CF, A high throughput screen identifies benzoquinoline compounds as inhibitors of Ebola virus replication. Antiviral Res 2018, 150, 193–201 DOI: 10.1016/j.antiviral.2017.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Luthra P; Naidoo J; Pietzsch CA; De S; Khadka S; Anantpadma M; Williams CG; Edwards MR; Davey RA; Bukreyev A; Ready JM; Basler CF, Inhibiting pyrimidine biosynthesis impairs Ebola virus replication through depletion of nucleoside pools and activation of innate immune responses. Antiviral Res 2018, 158, 288–302 DOI: 10.1016/j.antiviral.2018.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liu G; Nash PJ; Johnson B; Pietzsch C; Ilagan MX; Bukreyev A; Basler CF; Bowlin TL; Moir DT; Leung DW; Amarasinghe GK, A Sensitive in Vitro High-Throughput Screen To Identify Pan-filoviral Replication Inhibitors Targeting the VP35-NP Interface. ACS Infect Dis 2017, 3 (3), 190–198 DOI: 10.1021/acsinfecdis.6b00209. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.