

Abstract
Lead halide perovskites have displayed the highest solar power conversion efficiencies of 23% but the toxicity issues of these materials need to be addressed. Lead‐free perovskites have emerged as viable candidates for potential use as light harvesters to ensure clean and green photovoltaic technology. The substitution of lead by Sn, Ge, Bi, Sb, Cu and other potential candidates have reported efficiencies of up to 9%, but there is still a dire need to enhance their efficiencies and stability within the air. A comprehensive review is given on potential substitutes for lead‐free perovskites and their characteristic features like energy bandgaps and optical absorption as well as photovoltaic parameters like open‐circuit voltage (V OC), fill factor, short‐circuit current density (J SC), and the device architecture for their efficient use. Lead‐free perovskites do possess a suitable bandgap but have low efficiency. The use of additives has a significant effect on their efficiency and stability. The incorporation of cations like diethylammonium, phenylethyl ammonium, phenylethyl ammonium iodide, etc., or mixed cations at different compositions at the A‐site is reported with engineered bandgaps having significant efficiency and stability. Recent work on the advancement of lead‐free perovskites is also reviewed.
Keywords: lead‐free perovskites, photovoltaic parameters, stability
1. Introduction
Perovskite originally referred to a mineral calcium titanium oxide, CaTiO3, discovered in 1839 in Ural Mountains of Russia by Gustav Rose, a German mineralogist and later named after a Russian mineralogist count Lev Aleksevich Perovski.1 Since then, the term perovskite has been used for any organic/inorganic compound (synthetic/natural) with the similar crystal structure and stoichiometry as of CaTiO3, that is, ABX3, where A is monovalent metallic cation, most usually from group I of the periodic table. B is divalent metallic cation, a transition metal and X is a nonmetallic anion (halide). However, for O2− anions, A and B are divalent and tetravalent cations, respectively. The size of cation A must be larger than that of cation B. Ideally, perovskite crystal structure is described as a body centered cubic structure with monovalent cation A dodecahedrally (12‐fold) coordinated by X anions as shown in Figure 1 .2 The volume occupied by A ions depends on the electronegativity and size of B and X ions, respectively. A superfluity of organic/inorganic compounds has been discovered that exists in perovskite crystal structure framework ABX3 like BaTiO3, SrTiO3, KNbO3, etc.
Figure 1.

a) Schematic view of cubic perovskite crystal structure for ABX3 compound, b) 3D crystal structure in which the A site is confined within a cage determined by the octahedral coordination of B site with X site. Reproduced with permission.1 Copyright 2019, Royal Society of Chemistry.
Generally, perovskite materials can be classified into two groups, namely, inorganic oxide perovskite and halide perovskite that further encompass alkali halide and organometal halide perovskite materials. There are some perovskite materials like MgCNi3 having neither oxygen nor halide component and hence do not belong to either of the groups.3 In alkali halide perovskites, A‐site is occupied by a monovalent organic cation such as CH3NH3 (methylammonium or MA), NH2(CH)NH2 (formamidinium or FA), or inorganic cations such as rubidium (Rb), caesium (Cs), etc., the B‐site by a divalent metal cation lead (Pb) or tin (Sn), and X‐site by a halide anion. In today's scientific world, it is the halide perovskites that have grabbed all the attention of silicon‐dominated photovoltaics industry and whole of the photovoltaics research is now focused in developing perovskite materials for solar energy conversion.
The suitability of a particular combination of cations to organize into a perovskite structure can be estimated based on two important parameters. The first one is the Goldschmidt tolerance factor (t), a dimensionless number, calculated from the ratio of ionic radii4
| (1) |
where r A and r B are the ionic radii of cations A and B, and r X is the ionic radius of anion. For a particular perovskite structure, the tolerance factor (t) can be calculated by substituting the ionic radii of cations and anions. If t = 1, it indicates the formation of an ideal cubic structure having size of cation A larger than that of B. The tolerance factor (t) must lie in the range of 0.8–1.0 for the formation of stable perovskite structures. If t < 0.8 or t > 1.0, the cation A is too small or too big to fit into BX6 octahedron, thereby resulting in the formation of alternative structures. The tolerance factor (t) leading to formation of different types of structures with examples is mentioned in Table 1 .
Table 1.
Goldschmidt tolerance factor (t) of various perovskite materials5
The second one is the octahedral factor (μ) which is the ratio between ionic radii of B and X
| (2) |
The octahedral factor (μ) must lie in the range of 0.44–0.72 for B and X in order to form a stable BX6 octahedron.2 The tolerance factor has an immense role to play in finding alternative lead halide perovskite materials as many different cations can be inserted in ABX3 structure framework leading to development of varied materials with specific engineered properties.8
The effective ionic radii of organic molecular cations and Shannon ionic radii of inorganic cations as well as the effective ionic radii of various anions are listed in Table 2 .9, 10, 11, 12, 13
Table 2.
(a) Effective ionic radii of organic molecular cations. (b) Shannon ionic radii of inorganic cations. (c) Effective ionic radii of various anions
| (a) | ||
| Cation A | Effective ionic radii (r eff) [pm] | Ref. |
| Ammonium [NH4]+ | 146 | 10 |
| Hydrazinium [NH3NH2]+ | 217 | 10 |
| Azetidinium [(CH)3NH2]+ | 250 | 10 |
| Formamidinium [CH(NH2)2]+ | 253 | 10 |
| Imidazolium [C3N2H5]+ | 258 | 10 |
| Dimethylammonium [(CH)2NH2]+ | 272 | 10 |
| Ethyl ammonium [(CH3CH2)NH3]+ | 274 | 10 |
| Guanidinium [(NH2)3C]+ | 278 | 10 |
| Tetramethylammonium [(CH3)4N]+ | 292 | 10 |
| Thiazolium [C3H4NS]+ | 320 | 11 |
| Tropylium [C7H7]+ | 333 | 11 |
| Hydroxylamine [NH3OH]+ | 216 | 10 |
| Methylammonium [CH3NH3]+ | 217 | 10 |
| Piperazinium [C4H12N2]2+ | 322 | 9 |
| Dabconium [C4H14N2]2+ | 339 | 9 |
| K+ | 164 | 12 |
| Rb+ | 172 | 12 |
| Cs+ | 188 | 12 |
| (b) | ||
| Cation B | Effective ionic radii (r eff) [pm] | Ref. |
| Be2+ | 16 | 12 |
| Mg2+ | 72 | 12 |
| Ca2+ | 100 | 12 |
| Sr2+ | 118 | 12 |
| Ba2+ | 135 | 12 |
| Mn2+ | 66 | 12 |
| Fe2+ | 78 | 12 |
| Co2+ | 58 | 12 |
| Ni2+ | 55 | 12 |
| Pd2+ | 86 | 12 |
| Pt2+ | 60 | 12 |
| Cu2+ | 73 | 12 |
| Zn2+ | 60 | 12 |
| Cd2+ | 78 | 12 |
| Hg2+ | 69 | 12 |
| Ge2+ | 73 | 12 |
| Sn2+ | 110 | 13 |
| Pb2+ | 119 | 12 |
| Eu2+ | 117 | 12 |
| Tm2+ | 103 | 12 |
| Yb2+ | 103 | 12 |
| Sn[4+] | 69 | 12 |
| Te+ | 150 | 12 |
| Au+ | 137 | 12 |
| Au3+ | 85 | 12 |
| Sb+ | 76 | 12 |
| Bi3+ | 103 | 12 |
| Te[4+] | 97 | 12 |
| La3+ | 103 | 12 |
| Ce3+ | 101 | 12 |
| Pr3+ | 99 | 12 |
| Nd3+ | 98 | 12 |
| Sm3+ | 96 | 12 |
| Eu3+ | 95 | 12 |
| Gd3+ | 94 | 12 |
| Dy3+ | 91 | 12 |
| Er3+ | 89 | 12 |
| Tm3+ | 88 | 12 |
| Lu3+ | 86 | 12 |
| Pu3+ | 100 | 12 |
| Am3+ | 98 | 12 |
| Bk3+ | 96 | 12 |
| (c) | ||
| Anion X | Effective ionic radii (r eff) [pm] | Ref. |
| Fluoride, F− | 129 | 9 |
| Chloride, Cl[−] | 181 | 9 |
| Bromide, Br[−] | 196 | 9 |
| Iodide, I[−] | 220 | 10 |
| Formate, HCOO[−] | 136 | 9 |
| BH4 [−] | 203 | 11 |
The Goldschmidt tolerance factor (t) has played a pivotal role in development of perovskites10 and is now being used to engineer/synthesize new organic–inorganic stable perovskites structures by formulating the composition of perovskite. The tolerance factor can be tuned to the stable perovskite range by mixing distinct A/B cations and X anions in a particular composition.14, 15, 16, 17
2. Perovskite Sensitized Solar Cell
Solar energy has always been sought to be converted into electrical energy through photovoltaic effect of light absorbing semiconductor in order to obtain clean and green energy. The traditional first generation crystalline silicon solar employed for this purpose enjoy a market share of more than 90% in PV market.18 The second generation solar cells consist of thin films such as cadmium telluride, copper indium gallium selenide, and amorphous silicon. The third generation has a number of thin film technologies such as dye‐sensitized solar cells (DSSCs) in development phase. The crystalline silicon solar cell has a theoretical limiting power efficiency of 33.16%19 noted as a Shockley Queisser limit in 1961. An efficiency of 25.6%20 for a silicon solar cell has been reported in 2014 that further grows to 46.1%21 in four‐junction GaInP/GaAs/GaInAsP/GaInAs solar cell reported by French‐German collaboration. The triple‐junction thin film solar cells achieved an efficiency of 13.6% in June 2015.22 The research teams at NREL, EPFL, and GSEM have reported Sun efficiencies of dual‐junction GaInP/GaAs solar cell devices up to 32.8%.23 Although, the monocrystalline silicon cells have photovoltaic conversion efficiency of more than 20%,24 they are characterized by high cost, difficult preparation conditions, and serious environmental pollution.25 Also, cadmium telluride and copper indium gallium selenium thin film solar cells' large‐scale use puts a pressure on environmental pollution. DSSCs showing an efficiency of more than 13% have low cost and easy fabrication but absorption layer in such cells is very thick26 and light dyes used in such cells suffer from phenomenon of light bleaching.
An efficient solar cell technology must ensure low raw material and finished material cost, high light absorption and solar power conversion efficiency, high abundance of raw material, low toxicity, and less environmental pollution. In order to achieve it, the organic/inorganic perovskite compounds can be used in light harvesting layer as these materials have all the requisite properties that make them suitable for use in PV27 applications. With the discovery of metal halide perovskite, especially MAPbI3, FAPbI3 as light absorbers, the use of perovskites in PV technology has been explored as they are cost effective and readily available for large‐scale use.28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38 The organic–inorganic perovskite materials have been pioneering in fabricating high solar power conversion efficiency hybrid solar cells from time to time.32, 39, 40, 41, 42, 43, 44, 45 Miyasaka and co‐workers reported the first perovskite sensitized solar cell (PSSC) in between 2006 and 2008 using CH3NH3PbI3 and CH3NH3PbBr3 absorbers and reported solar power conversion efficiency varying between 0.4 and 2% for solid‐state and liquid electrolyte cells, respectively.46, 47 A MAPbI3‐based solar cell with solar power conversion efficiency of 3.8% has been reported by Kojima et al. in 2009 and was the first peer reviewed publication on perovskite‐sensitized solar cell.28 Park and co‐workers using CH3NH3PbI3 liquid electrolyte solar cell reported an improved efficiency of 6.5%.48 In 2011, Snaith along with his co‐workers developed a solid‐state perovskite solar cell (PSC) using 2,2(7,7)‐tetrakis‐(N,N‐dimethoxyphenylamine)9,9(Spiro‐bifluorene) (Spiro‐OMeTAD) for hole transportation and produced solar power conversion efficiencies between 8 and 10%49 achieving a major breakthrough in performance efficiency of PSSC in comparison to DSSCs having only 7% efficiency.50 In 2012, Kim et al. replaced the liquid electrolyte with a solid‐state hole conducting material depositing the perovskite precursor over the mesoporous TiO2 layer achieving a solar power conversion efficiency (SPCE) of 9.7%.30 Later, increased efficiencies of 10.9% were reported by Lee et al.51 Gratzel and co‐workers reported a SPCE of 15% by using sequential deposition to produce pinhole‐free perovskite layer.52 Liu et al. Introduced Zn2SnO4 nanocrystalline thin film on PCBM buffer layer to make electron extraction process easy, thereby, increasing SPCE to 17.76.53 You et al. and Yang et al. first fabricated all metal oxide layer based perovskite solar cell reporting an efficiency of 16.1% and more stability of the material in 2016.54 Yang et al. and his team reported SPCE of 22.1%55 in defect engineered thin perovskite layers in PSSCs containing formamidinium with multiple cations and mixed halide anions in 2017. The SPCE of solid‐state PSSCs was around 10% in 2012 that later grew up to 22.1% in 2017. This has been achieved through engineering of perovskite composition and thin film deposition methods. The big issue of degradation of perovskite in polar liquid electrolyte has been solved by use of solid‐state PSSCs that have shown 500 h stability in ambient conditions without encapsulation but still the PSSCs have to prove its stability in air, on exposure to humidity,56, 57, 58 UV light,59 and high temperatures.60, 61 Research has also revealed that PSSCs also suffer from anomalous current–voltage hysteresis as reported by Snaith and co‐workers in 201462 that can have adverse effects on the stability of PSSCs.63, 64
3. Device Configuration and Working Principles of Perovskite Solar Cell
In the first perovskite solar cell fabricated in 2009, perovskite nanoparticles were used as a light absorber replacing dyes in dye‐sensitized solar cells. In the fabricated device, mesoporous TiO2 layer of several micrometer thickness acts as an anode and a platinum‐coated glass acted as a cathode in a liquid electrolyte based device.28, 29 However, the device suffered seriously from the stability issue as the perovskite light absorber layer dissolves or decomposes in the liquid electrolyte very rapidly. Hence, the liquid electrolyte was replaced by a solid‐state material to act as a hole transport material (HTM) resulting in a solid‐state mesoscopic perovskite solar cell with an improved stability. Organic Spiro‐OMeTAD was used as a hole transport material in such cells.49 The perovskite materials when used as a light absorber enhances the device stability and performance to its broad optical absorption range than the conventional dyes.65 In a mesoscopic perovskite solar cells, a compact metal oxide (TiO2) layer is deposited on a fluorine‐doped tin oxide (FTO) glass substrate by spin‐coating on which is further deposited a mesoporous TiO2 layer by spin‐coating. The perovskite light absorber layer is grown on the scaffold of mesoporous TiO2 layer which is further deposited by a HTM by spin‐coating and finally to a metal‐back electrode (Ag or Au). The device configuration of mesoporous perovskite solar cell is shown in Figure 2 a.66
Figure 2.
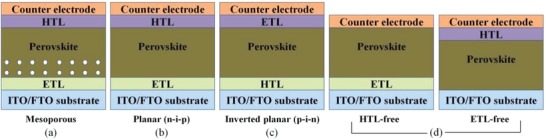
a) Device structure of mesoporous PSCs, b) planar heterojunction, c) inverted planar PCSs, and d) HTM‐free PSCs. Reproduced with permission.66 Copyright 2016, Springer Nature.
TiO2 is most commonly employed in mesoporous layer that facilitates in the formation of inner connected layer of perovskite crystals by allowing their deep penetration into the pores of mesoporous layer. Compact TiO2 layer transports electrons, blocks holes, and suppresses the recombination of electron–hole pairs. The mesoporous TiO2 layer needs a high‐temperature sintering that can consequently increase the device fabrication time. Since the perovskite materials have ambipolar nature, they have the potential of transporting electrons and holes on their own in between two electrodes so a planar structure is viable for them.67 Also, the perovskite solar cells using planar structure over time have revealed the best device performance as that of a mesoporous structure.32, 67 The device with planar configuration has reported almost 100% internal quantum efficiencies ascertaining them as an efficient device structure.43 Thus, typically there are two major device configurations for a perovskite solar cell, viz., a planar heterojunction/conventional structure (n‐i‐p) and an inverted planar structure (p‐i‐n). In a planar heterojunction structure (n‐i‐p) as shown in Figure 2b,66 a compact electron transport layer (ETL) of 30–50 nm of TiO2 (most commonly) is deposited on a transparent conducting oxide substrate that can be indium‐doped tin oxide (ITO) or FTO. The mesoporous TiO2 layer is removed and perovskite light absorber to sandwich between an ETL and a hole transport layer (HTL) by spin‐coating or by vapor deposition and vapor‐assisted solution process on a compact TiO2 32, 33 layer and finally connected to a metal electrode such as Au, Ag, or Pt. Spiro‐OMeTAD or poly‐triallylamine (PTAA) can be used in ETL. For an inverted planar structure (p‐i‐n) as shown in Figure 2c,66 a hole transport layer of poly(3,4‐ethylene dioxythiophene):poly(styrene sulfonate) (PEDOT:PSS) or NiOx is deposited on a conducting glass substrate that is most commonly ITO followed by a photoactive perovskite light absorber layer and is further covered by an electron transport layer of [6,6]‐phenyl‐C61‐butyric acid methyl ester (PC61BM) or zinc oxide (ZnO) and finally to a metal electrode of Au, Ag, or Al. The electron and holes are generated in the photoactive perovskite layer on absorption of photons of incident light and move to the opposite electrodes constituting current. HTL is used to receive the holes generated in the perovskite layer and transports them to the surface of the metal electrode whereas ETL transports electrons, block holes, and inhibits the electron–hole recombination in the FTO conductive substrate. The material used in ETL must be a n‐type semiconductor with high carrier mobilities, transparent to light, and with a suitable energy band structure matching with that of the perovskite material. ETL must have lowest unoccupied molecular orbital (LUMO) and highest occupied molecular orbital (HOMO) higher than the photoactive perovskite layer while HTL can facilitate hole motion only if the HOMO matches with the valence band of the perovskite material. The inverted planar structure has an operational edge over a conventional structure as it required a temperature of 300 °C for device fabrication in contrast to a planar heterojunction structure where a temperature up to 500 °C is required. Moreover, the hysteresis effect of perovskite solar cells is rarely observed in a planar inverted structure while this effect is most commonly observed in planar heterojunction devices.68 The highest device performance has been observed with the planar heterojunction structure using TiO2 as an electron transport layer.69 The most commonly reported structure is inverted planar device PEDOT:PSS/light absorber/PCBM as it is easily fabricated and more cost effective.68, 70 The poor SPCE of inverted planar structure may be due to a barrier at the contact interface between Fermi level of the metal electrode and lowest unoccupied molecular orbit of the ETL.126 In a planar heterojunction structure, the expensive HTL of Spiro‐OMeTAD may be removed leading to a new device framework known as planar HTM‐free architecture71, 72 as shown in Figure 2d.66
4. Why Lead‐Free?
The use of organic–inorganic lead halide perovskite such as MAPbI3 and FAPbI3 has caused an increase in solar power conversion efficiencies from 3.8% in 200928 to 22.1%34, 35, 73, 74, 75, 76 in last nine years as these materials do possess requisite optoelectronic features such as a direct bandgap, long charge carrier lifetime, diffusion length, high charge carrier mobility, and strong optical absorption coefficient.77, 78, 79, 80, 81, 82, 83, 84, 85, 86, 87, 88, 89, 90 Lead halide perovskite has high open‐circuit voltages due to photon recycling as a result of which they have long charge extraction lengths through multiple absorption–emission events within the perovskite active layer.91 The metal lead has invaluable intrinsic properties like high melting point, high density, malleability, ductility, corrosion resistance, etc. Despite having all characteristic features to be exploited in commercial PV solar market, it is the toxic nature of lead in lead halide perovskite solar cells that hinders its use in silicon‐dominated PV market. The stringent directives of European Union clearly prohibits the use of hazardous substance in electrical and electronic equipment and lead has been identified as one of the ten hazardous chemicals listed by ROHS in order to avoid its exposure to environment and people as well.92, 93 The toxicity of lead is due to its affinity for band formation with thiol and cellular phosphate groups of numerous enzymes, proteins, and cell membranes.94 Lead halide perovskite solar cells do contain a considerable portion of lead, that is, 33% by weight. Lead is carcinogenic in nature and has no safe threshold limit of exposure. It can cause serious toxicological implications on human beings leading to cardiovascular and development diseases by inflicting the functioning of liver, kidney, brain, and central nervous system. Exposure to lead can produce irreversible health damages in infants and pregnant ladies.95, 96, 97 Also, organic–inorganic lead halide perovskites are liable to degradation under moisture, rain, heat, and prolonged illumination in air.98, 99, 100 Therefore, instability is another prime issue linked with these materials that reduces their working life span which is the most important prerequisite for commercialization on large scale as PV panels are generally placed over roof tops or in open fields so their exposure to rain is inevitable.
Hailegnaw et al. have reported that in case of a catastrophic failure of a solar plant, the impact of rain of different pHs on MAPbI3 films is complete degradation of perovskite material leaving behind PbI2 in water in the order of 10−8 mol L−1 which is of course low but higher than that of CdTe, CdS, and PbS values varying from 10−27 to 10−34 so it becomes most probable that lead, being soluble in water, may leech into the underground water resources.101 Not only this, Hailegnaw et al. have analyzed the impact of leaching lead out of the damaged solar panels on the soil and reported that the leakage of lead due to broken encapsulation or sealing will induce the concentration of Pb in first cm of ground below the damaged solar panel by 70 ppm.101 Taking into consideration the repercussions of use of toxic lead halide perovskites, it becomes pertinent to investigate lead‐free perovskite materials providing better stabilities with solar power conversion efficiencies without compromising human health and environment.
5. Characteristic Features of Lead‐Free Perovskites
The perovskite based materials used in solar cells do possess such a structure that enables them to have most suitable optical bandgaps to act as a light absorber. These materials do possess a high dielectric constant, long diffusion length, and a broad optical absorption range covering the entire visible spectrum and into the infrared. Perovskite materials exhibit ambipolar properties that enable them to display both n‐type and p‐type behavior on exposure to photons of incident light. The rate of nonradiative recombination in such material is strongly suppressed that is essential for high solar power conversion efficiencies. The presence of hysteresis loss in these materials clearly indicates the presence of magnetic properties at room temperature or above. Another important characteristic of these materials is that they are deposited by low‐temperature solution methods that provide easy fabrication with low production cost. Besides they are typically flexible, light weight, and semitransparent making them more appealing for use in photovoltaic applications.
The lead‐based halide perovskites have reported a highest solar power conversion efficiency of 22% up to now within 8 years of research.102 The efficiency limit of perovskite solar cell has been envisaged to be 31% based on detailed balance calculations much closer to the Shockley–Queisser limit of 33%.103 Although the lead‐based halide perovskites have all the structural, optical, and electrical features for use in perovskite solar cell as a light absorber but due to toxicity issues of lead, it is pertinent to replace it by another suitable elements such as tin, germanium, bismuth, etc. The lead‐free perovskites have attracted the attention of the researchers at present time due to significant properties of these materials that can be engineered to make them suitable for their use as light absorbers in perovskite solar cells. Lead‐free perovskite methylammonium tin iodide MASnI3 is a direct gap semiconductor with an optical bandgap of 1.3 eV104, 105, 106 which is close to 1.5 eV of MAPbI3. It exhibits a strong photoluminescence emission corresponding to the onset at 950 nm in 700–1000 nm range of the absorption edge at room temperature. MASnI3 has an electrical conductivity of 5 × 10−2 S cm−1 at room temperature that corresponds to a Seebeck coefficient of ≈–60 µV K−1. The material exhibits a carrier concentration of the order of ≈1 × 1014 cm−3 having excellent electron mobilities of the order of ≈2000 cm2 V−1 s−1 and hole mobility of 300 cm2 V−1 s−1 in comparison to lead halide perovskites that have an electron mobility of 66 cm2 V−1 s−1 and hole mobility of 105 cm2 V−1 s−1.84 Table 3 summarizes the electron and hole mobilities of all the lead‐free perovskite materials. The Hall measurements of as‐grown crystals of MASnI3 have revealed a hole concentration of about 9 × 1017cm−3 with a hole mobility of about 200 cm2 V−1 s−1 at 250 K.107
Table 3.
Charge carrier mobility of lead‐free perovskites
| Light absorber | Architecture | Measurement method | Component | Mobilities [cm−1 V−1 s−1] | Ref. |
|---|---|---|---|---|---|
| MASnI3 | Poly c | Hall | Electron | 2320 | 105 |
| MASnI3 | Poly c | Hall | Hole | 322 | 105 |
| MASnI3 | Single c | Hall | Hole | 200 | 107 |
| MASnI3 | Poly c | Hall | Hole | 50 | 121 |
| MASnI3 | Mesostructural | THzC | Total | 1.6 | 120 |
| FASnI3 | Poly c | Hall | Electron | 103 | 105 |
| FASnI3 | Film | THzC | Total | 22 | 122 |
| CsSnI3 | Poly c | Hall | Electron | 536 | 105 |
| CsSnI3 | Poly c | Hall | Hole | 585 | 123 |
| (C6H5C2H4NH3)2SnI4 | Film | FET | Hole | 0.6 | 124 |
| PEASnI4 | Film | FET | Hole | 15 | 125 |
| (MA)3Sb2IxBr9− x | Single c | SCLC | Electron | 12.3 | 116 |
| (MA)3Sb2IxBr9− x | Single c | SCLC | Hole | 4.8 | 116 |
| Ba2BiTaO6 | Film | Hall | Hole | 30 | 126 |
| MASnI3 | Mesostructural | – | Electron | 2000 | 106 |
| MBI | Single c | Hall | Total | 1–11 | 127 |
| (MA)3Sb2I9 | Single c | SCLC | Hole | 4.8 | 116 |
| (MA)3Sb2I9 | Single c | SCLC | Electron | 12.3 | 116 |
| (MA)3Sb2I9 | Film | SCLC | Hole | 1.2 × 10−4 | 116 |
| (MA)3Sb2I9 | Film | SCLC | Electron | 1.5 × 10−4 | 116 |
| Cs2SnI6 | Poly c | Hall | Hole | 310 | 115 |
| MA3Bi2I9 | Single c | SCLC | Hole | 29.7 | 128 |
| MA3Bi2I9 | Single c | Hall | Electron | 1 | 127 |
| (MA)2SnI6 | Poly c | Hall | Electron | 3 | 129 |
Although tin halide perovskite has higher charge carrier mobilities, Sn2+ has a strong tendency to get oxidized to Sn4+ causing a p‐type self‐doping.108 The artificial hole doping of the halide‐based perovskites increases their electrical conductivity and they exhibit a metal‐like conducting behavior.107 The formamidinium tin iodide (FASnI3) has an optical bandgap of 1.41 eV that is much closer to the bandgap 1.5 eV of MAPbI3 making it a potential candidate to display an optical absorption up to 950 nm.109 The cesium tin iodides (CsSnI3) display a bandgap of ≈1.3 eV at 300 K close to the optimum value of 1.5 eV for photovoltaic performance.110 Bismuth‐based halide perovskites display lower light absorption onset at 450 nm with absorption coefficient of ≈1 × 105 cm−1 that are lower as compared to MAPbI3 that has an absorption coefficient of around 2 × 105 cm−1 at 450 nm.111
Lead‐free perovskites have high exciton binding energies that provide them stable optical properties. The exciton binding energies of bismuth‐based halide perovskites MA3Bi2I9, Cs3Bi2I9, and MA3Bi2I9Clx are of 70, 270, and 300 meV that are much higher than that of lead‐based halide perovskites (25–50 meV).111 UV–vis absorption measurements for Cs3Bi2I9 have reported a strong exciton absorption peak at room temperature. Cs3Bi2I9 exhibits an exciton absorption peak at ≈485 nm (2.56 eV) with an indirect optical bandgap of ≈2.1 eV. The films exhibited an optical absorption coefficient of ≈1 × 104 cm−1 at 450 nm. In spite of indirect bandgaps, the material is still a potential candidate for use as a light absorber due to strong exciton binding energy.112 CsSnI3 perovskite exhibits a direct bandgap of 1.32 eV with an exciton binding energy of 18 meV at room temperature. The large binding energy is on the account of exciton motion in the 2D layer of SnI4 tetragons present in the material.123
Tin‐based perovskites are prepared by using solution methods and crystallizes at room temperature whereas lead‐based halide perovskites crystallize by heating. The variation in composition of halide anion in lead‐free perovskites has a significant effect on the absorption coefficient of these materials thus paving the way for engineering the bandgaps and optical absorption spectrum of these materials. The tin‐based hybrid halide perovskites MASnI3− xBrx (x = 0, 1, 2, 3) synthesized in an inert atmosphere in the nitrogen glove box exhibit an optical absorption onset that can be blueshifted from 954 to 577 nm by varying the composition of halide anion, that is, for x = 0 and x = 3 whereas for x = 1 and 2, optical absorption onset at 795 and 708 nm has been reported. Also ultraviolet photoelectron spectroscopy (UPS) measurements of valence band energy E VB of MASnI3− xBrx under high vacuum have revealed that the bandgaps can be engineered from 1.30 eV for MASnI3 to 2.15 eV for MASnBr3.106 Not only this, the color of the tin‐based hybrid halide perovskite MASnI3− xBrx shows a variation with increased bromine content from black (x = 0) to dark brown (x = 1) and yellow (x = 3); thus, colorful solar devices can be designed by using bandgap engineering. Thus, the composition of tin‐based mixed halide perovskite can be tailored to emit between 954 and 574 nm in contrast to lead‐based counterparts that display photovoltaic emission in between 700 and 800 nm. The emitted wavelengths are in agreement with the values of bandgaps obtained through experiments clearly indicating the presence of direct optical bandgaps in MASnI3.106
The investigation of Ge mixed halide perovskites MAGeI3− xClx [x = 0, 1, 1.5, 2, 3] by using first principle calculations has reported a bandgap of 1.8 eV for MAGeI3 (x = 0) whereas MAGeCl3 (x = 3) has a much wider bandgap of 3.8 eV clearly demonstrating the effect of doped chlorine in MAGeI3 perovskites. The absorption coefficients also display an increasing trend when the proportion of x decreases from 3 to 0 attributed to the redshift of the optical bandgap caused due to change in chemical composition of the material.113 In case of antimony‐based mixed halide perovskites MA3Sb2IxBr9− x, the optical bandgap onset for perovskite films shows a decreasing trend, that is, the optical absorption onset is blueshifted from 558 to 453 nm as x changes from 9 to 0. The hole and electron mobilities of MA3Sb2I9 single crystals have been calculated by using space charge limited current methods114, 115 and are shown in Table 3. The MA3Sb2I9 single crystals have high absorption coefficient greater than 105 cm−1 at absorption peak wavelengths. The absorption onset for [x = 0, 3, 6] in MA3Sb2IxBr9− x films are 453, 486, and 516 nm with a direct bandgap of 2.78, 2.66, and 2.49 eV, respectively.116 Lead‐free perovskites do possess suitable carrier diffusion lengths and minority charge carrier lifetimes exhibiting photovoltaic performance. The long carrier diffusion lengths of electrons and holes in MASnI3 are 279 ± 88 and 193 ± 46 nm, respectively, obtained by broadband transient absorption and time‐resolved fluorescence spectroscopy. Addition of SnF2 in MASnI3 films results in not only increase in diffusion lengths to more than 500 µm but also enhances the fluorescence lifetime up to ten times.117 The background concentration of doped holes has an effect on the diffusion lengths of MASnI3 perovskite. As the background doping level in MASnI3 decreases, there is a corresponding increase in diffusion length. For a doping concentration below 1015 cm−3, the diffusion length can be engineered to increase above 1 µm in length that is close to the value shown by lead‐based halides.120 Lead‐free CsSnI3 perovskite films synthesized by the solution method have carrier lifetime of ≈54 ps, minority carrier diffusion length of ≈1.6 nm, and a doping concentration of more than 9.2 × 1018 cm−3 obtained as a consequence of better quality of crystalline films whereas single crystals of CsSnI3 have a long minority carrier diffusion length of more than 930 nm which is comparable to that of the lead‐based perovskites110 having diffusion lengths exceeding 1 µm.74
6. Hole Transport Material and Electron Transport Material in Lead‐Free Perovskite Solar Cells
Lead‐free perovskites have been prepared by using mesoporous perovskite solar cells in planar heterojunction and inverted planar structures. Spiro‐OMeTAD is most commonly used hole transport material in lead‐free perovskites as it has the ability to penetrate deep into the pores of the perovskite layer but it has a low hole mobility and complicated device processing. Also it deteriorates the stability of the fabricated device.130, 131 Therefore, dopants are added into it in order to enhance its conductivity. The first lead‐free perovskite device was prepared by solvent engineering method by employing MASnI3 as a light absorber. Spiro‐OMeTAD has been used as a HTM on the top of the perovskite layer in a device architecture of FTO/compact TiO2/mesoporous TiO2 layer/MASnI3 light absorber/Spiro‐OMeTAD/Au.120 An additive doping of hydrogen bis(trifluoromethane sulfonyl)imide (H‐TFSI) and tert‐butyl pyridine is done into Spiro‐OMeTAD to enhance the rate of hole extraction and transport.132 The additive doping of lithium bis(trifluoro methyl sulfonyl)imide salt (Li‐TFSI) and 4‐tert‐butyl pyridine (TBP) deteriorates the stability of MASnI3 perovskite device than H‐TFSI.120 In another approach, also Spiro‐OMeTAD used as a HTM is doped with lithium bis(trifluoro methyl sulfonyl)imide and 2,6‐lutidine in order to enhance its hole mobility.49 The solar cell capacitance simulator and analytical calculations (SCAPS) have reported an efficiency of above 15% in lead‐free tin‐based MASnI3 perovskites employing Spiro‐OMeTAD as a hole transport material.133 Chlorobenzene (CB), Li‐TFSI, and TBP have been used as an additive in Spiro‐OMeTAD in lead‐free Ma3Bi2I9 perovskites.111 Oxygen‐doped Spiro‐OMeTAD employed as a HTM in Cs3Bi2I9 perovskite solar device has yielded the maximum of the reported solar power conversion efficiencies.134 The Spiro‐OMeTAD as a HTM has been used in lead‐free tin, germanium, antimony, bismuth, and copper‐based perovskite devices. Figure 3 shows the scanning electron microscopy (SEM) image of a MASnI3 perovskite device with Spiro‐OMeTAD as a HTM.106
Figure 3.

SEM image of a photovoltaic device using CH3NH3SnI3 perovskite material. Reproduced with permission.106 Copyright 2014, Springer Nature.
Cu‐based lead‐free perovskites reported so far have a planar heterojunction (n‐i‐p) structure employing Spiro‐OMeTAD as a HTM with a highest reported efficiency of 2.41%. The low efficiency is attributed to the mismatch in the energy levels between the (MA)2CuClxBr4− x and Spiro‐OMeTAD as a HTM leading to a poor hole extraction in the device.135, 136, 137 Another polymeric organic HTM PTAA has been employed in planar heterojunction n‐i‐p perovskite devices.138, 139 Owing to its large hole mobility the use of PTAA as a HTM in Cs3Sb2I9 perovskite solar cells has reported a V OC of 250–300 meV and an extremely low solar power conversion efficiency.140 The doping of bismuth‐based perovskite Cs3Bi2I9 films with N,N‐dimethyl formamide/hydroiodide (HI) solution featured a pure crystalline film with an excellent thermal stability.141 PTAA employed as a HTM in ethylene diammonium and methylammonium tin iodide en[MASnI3] has reported a SPCE of 6.63% with a very high current density of 24.3 mA cm−2 in a device architecture of FTO/C‐TiO2/mp‐TiO2/en[MASnI3]/PTAA/Au.142 Many research groups have synthesized lead‐free MASnI3, FASnI3, and CsSnI3 perovskite solar devices by using PTAA as a HTM. The presence of another polymeric organic poly(3‐hexyl thiophene) (P3HT) as a HTM in lead‐free perovskites can enhance the SPCE as well as stability of the fabricated device due to its potential to decrease the resistance from the hole transfer impedance. P3HT has been employed as a HTM instead of Spiro‐OMeTAD for Cs3Bi2I9 perovskite solar devices.111 In another research, P3HT is used as a HTM in thin films of CsBi3I10 perovskite deposited by solution processing in device architecture of glass/FTO/compact TiO2/mesoporous TiO2/CsBi3I10 light absorber/P3HT/Ag. The addition of dopant 4‐tert‐butyl pyridine in Spiro‐OMeTAD employed as a HTM in CsBi3I10 perovskite dissolves the light absorbing perovskite layer143 and suffers from stability and degradation issues. Unlike Spiro‐OMeTAD, P3HT enhances the SPCE of the fabricated perovskite device as in CsSnBr3 films, an efficiency of 0.11% was enhanced to 3.2% by replacing Spiro‐OMeTAD HTM by P3HT.144 Also, P3HT employed as a HTM in MA3Bi2I9 films enhance the overall performance of the fabricated device.127 P3HT has been employed as a HTM in MASnBr3, FASnI3, CsSnI3, en[MASnI3], Cs2SnI6, MA3Bi2I9, Cs3Bi2I9, CsBi3I10, AgBi2I7, and AgBiI5 perovskite solar devices.
In an inverted planar (p‐i‐n) perovskite device, the hole transport layer is kept under the perovskite light absorber layer that alleviates the stringent requirement of efficient conductivity of hole transport material. Polymeric organic HTM PEDOT:PSS is used in such devices. PEDOT:PSS is used as a HTM in FASnI3 perovskite solar cells in an inverted planar (p‐i‐n) architecture and at present has reported a maximum SPCE of 8.12% in FA0.75MA0.25SnI3 in lead‐free tin‐based perovskite with a V OC of 0.61 V.145 The PEDOT:PSS with intercalated polyethylene glycol (PEG) used as a HTM in FASnI3 perovskite solar cell alleviates the energy level mismatch between the perovskite light absorber and PEDOT:PSS as HTM. As a consequence, the SPCE increased from 2.01 to 5.12% in the forward scan.146 The inverted planar (p‐i‐n) device structure employed for antimony‐based MA3Sb2I9 perovskite films has reported a SPCE of 0.5% with a V OC of 0.89 V.147 Additives are added into PEDOT:PSS in order to enhance the conductivity and morphology of PEDOT:PSS films. Polyorganic solvents such as dimethyl sulfoxide (DMSO), dimethylformamide (DMF), and ethylene glycol have been used as additives in lead‐free perovskite device fabrication.148 The addition of additive in PEDOT:PSS films leads to enhancement in efficient hole extraction and collection rate attributed to the strong dipole–dipole or dipole–charge interactions between the polar additive and PEDOT:PSS used as a HTM in fabricated perovskite device.149 PEDOT:PSS as a HTM has been employed in lead‐free MASnI3, FASnI3, FASnI3Br, FA1− xMAxSnI3, MA3Bi2I9, MA3Sb2I9, and Cs3Sb2I9 reported perovskite solar devices. Figure 4 shows the device structure and energy band diagram of (FA)x(MA)1− xSnI3 perovskite solar cell.150 The use of Spiro‐OMeTAD as a HTM damages the perovskite film. In order to overcome this issue, inverted planar (p‐i‐n) perovskite solar cells of MASnI3 have been prepared by using PEDOT:PSS doped with poly‐TPD as a HTM as shown in Figure 4.150 Figure 4 shows the (a) schematic device structure of (FA)x(MA)1− xSnI3 perovskite solar cell, (b) band alignment diagram, and (c) cross‐sectional SEM image of a completed device (scale bar: 500 nm).[150] Addition of poly‐TPD layer into PEDOT:PSS resulted into suppressed charge recombination and better efficiencies.151
Figure 4.
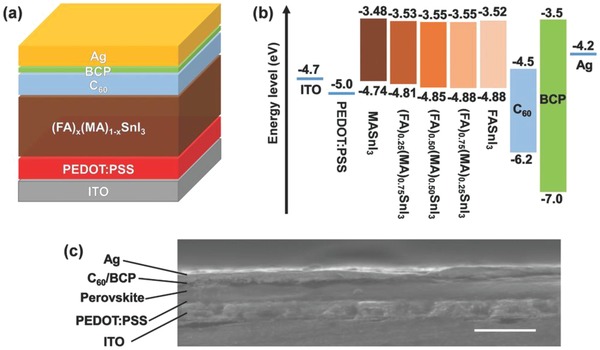
a) Schematic device structure of (FA)x(MA)1− xSnI3 perovskite solar cell. b) Band alignment diagram. c) Cross‐sectional scanning electron microscope (SEM) image of a completed device (scale bar: 500 nm). Reproduced with permission.150 Copyright 2017, Wiley‐VCH.
Besides organic HTMs, inorganic NiO (nickel oxide) and CuI (copper iodide) have also been used as a HTM in inverted planar (p‐i‐n) structures. NiO has a large work function of 15.2 eV in comparison to 5.2 eV for PEDOT:PSS that makes it viable for use as a HTM in perovskite solar cells reporting a higher V OC.152, 153, 154, 155 The thickness and morphology of NiO as a HTM has a direct impact on charge collection and recombination in perovskite solar devices. The use of NiO as a HTM leads to more air stability in FASnI3 perovskite inverted planar (p‐i‐n) solar cell using (PEA)2FA8Sn9I28 as a light absorber reporting a SPCE of 5.94%.156 NiOx has been employed as a HTM instead of Spiro‐OMeTAD in inverted planar structured B‐ϒ‐CsSnI3 PSCs in order to overcome the low conductivity of the undoped Spiro‐OMeTAD. The perovskite device exhibited an enhanced SPCE of 2.61% higher than that of Spiro‐OMeTAD used as a HTM.157 Figure 5 shows the device structure and corresponding energy diagram employing NiOx as a HTM in B‐ϒ‐CsSnI3 perovskite solar cells.157
Figure 5.
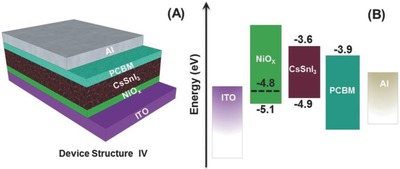
A) Scheme of the “inverted” structure planar B‐γ‐CsSnI3 PSC device employing NiOx as HTL and PCBM as ETL, and B) corresponding energy level diagram (the dashed line indicates NiOx work function). Reproduced with permission.157 Copyright 2016, Wiley‐VCH.
CuI has a hole conductivity greater than that of Spiro‐OMeTAD that enables CuI to improve the fill factor (FF) of the perovskite device employing it as a HTM.158 CuI has been used as a HTM in fabrication of CsSnI3 perovskite solar cells reporting a V OC of 0.55 V and enhanced air stability.159 Figure 6 shows the device architecture of CsSnI3 using CuI as a HTM and SEM image of CsSnI3 films.159
Figure 6.

a) Schematic of the device architecture used in this work; b) SEM image of a CsSnI3 film prepared with a 10 mol% excess SnI2 and spin cast at 4000 rpm from 8 wt% solution onto an ITO glass substrate coated with a 100 nm layer of CuI. Reproduced with permission.159 Copyright 2015, Royal Society of Chemistry.
Perovskite solar cells without a HTM layer have the advantage of having simple structures, easy fabrication process, and higher stability if the work function of the metal electrode used in perovskite solar cells is close to the maximum valence band of perovskite light absorber, then absence of hole transport layer has no impact on the built‐in electric field.160 The perovskite material in HTL‐free devices works as a light absorber and a hole transport layer in such cells.161 A HTL‐free solar cell of MASnI3 has reported an efficiency of 3.15%, J SC of 21.4 mA cm−2 that has been prepared through a solvent engineering method having a device architecture of the form FTO/c‐TiO2/mp‐TiO2/MASnI3 light absorber/Au.162 HTL‐free CsSnI3 PSC has stability ten times greater than the devices using same device architecture using MAPbI3 as a light absorber.163 Inorganic metal oxides like TiO2, ZnO, SnO2 and organic fullerene derivatives like phenyl‐C60‐butynic and methyl ester (PC60BM) or PC70BM have been employed as an ETM for perovskite solar cells. The efficient ETM should have the capability to engineer the optical bandgap for maximum absorption of incident light by the perovskite light absorber layer and must have a better electron extraction and hole blocking property in order to suppress the electron–hole recombination at the interface of the device. TiO2 as an ETM has been employed in device fabrication of most of the reported lead‐free perovskite solar cells. On the top of a mesoporous TiO2 layer, the perovskite film of MASnI3 is crystallized upon spin‐coating and it penetrates into the pores of ETM. The MASnI3 films fabricated on the top of 400 nm thick mp‐TiO2 layer are better than that of prepared on an 80 nm thick mp‐TiO2 layer. The mesoporous MAPbI3− xClx perovskite films have a better film morphology than that of MASnI3 films fabricated in a similar way and architecture as shown in Figure 7 .120 Figure 8 shows the schematic energy‐level diagram of CH3NH3SnI3− xBrx compounds.106
Figure 7.

SEM images. a) Top view of a film of CH3NH3SnI3 spin‐coated onto mesoporous TiO2 (80 nm thickness). b) Top view of a spin‐coated film of CH3NH3PbI3− xClx on mesoporous TiO2 (400 nm thickness). c) Top view of a spin‐coated film of CH3NH3SnI3 on mesoporous TiO2 (400 nm thickness). d) Cross‐sectional view of a complete device active layer composed of FTO glass/compact TiO2 (50 nm)/mesoporous TiO2 infiltrated with CH3NH3SnI3 (400 nm)/Spiro‐OMeTAD (600 nm). Reproduced with permission.120 Copyright 2014, Royal Society of Chemistry.
Figure 8.
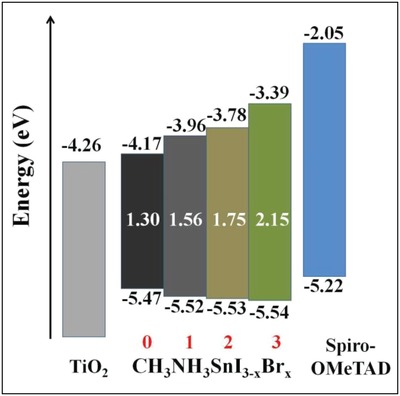
Schematic energy‐level diagram of CH3NH3SnI3− xBrx compounds. Reproduced with permission.106 Copyright 2014, Springer Nature.
By controlled crystallization, it is possible to enhance the quality of film formation.120 In another approach, solvent engineering method was employed to prepare thin films of MASnI3. A 30 nm thick TiO2 compact layer as an ETM is deposited on the substrate by atomic layer deposition system. The perovskite light absorber crystals infiltrate into the pores of mp‐TiO2 layer and remaining pores of mesoporous TiO2 layer are filled up by the HTM forming a 200 nm thick capping layer on the top of the composite structure.106
In a planar heterojunction (n‐i‐p) structure, a compact TiO2 layer is deposited on a glass that is further covered by a mesoporous TiO2 layer in order to enhance the electron collection and to avoid hysteresis loss during V–I measurements.164 By employing mp‐TiO2 as an ETM, the homogenous MASnI3 films prepared by vapor‐assisted solution process165, 166 have reported a J SC of 17.4 mA cm−2 when used as a light absorber in perovskite solar cells. The SPCE of pristine FASnI3 films was 0.003% by using mesoporous TiO2 layer as an ETM. The low value of SPCE is attributed to high background carrier density of 1019 cm−3 that leads to a metal like conductivity and device short circuiting.156 However, the addition of Br2 into FASnI3 films lowers the background carrier density of the perovskite. As a consequence of reduction in tin vacancies, the leakage current of the device is reduced that further increases the recombination lifetime and finally V OC and FF of the fabricated device and SPCE up to 5.5%.167 TiO2 as an ETM has an intrinsic low mobility and this has been a generation of deep traps by UV light that results in charge accumulation, recombination classes, and severe V–I hysteresis.41, 59, 62, 168, 169 The evaporation‐assisted method combining thermal evaporation with solution method has been employed to obtain uniform, full coverage, dense, and pinhole‐free CsSnI3 films eliminating the direct contact between HTM and ETM and reduces the consequent recombination. Evaporation‐assisted solution method makes it feasible for convenient tuning of SnF2 addition as a solvent. The conventional mesoporous n‐i‐p structure PSCs with an architecture FTO/bl‐TiO2/mp‐TiO2/CsSnI3/OMeTAD/Au has reported an efficiency of 2.2% in the device based on a 66 nm thickness of CsI.170 In CsSnBr3, the best reported SPCE so far is 2.1% that is due to the significant role of SnF2 addition as a solvent that alleviates the serious mismatch of band energy levels between the perovskite light absorber and TiO2 layer as an ETM.102
Germanium‐based perovskites have been fabricated by employing compact and mesoporous TiO2 layer as ETM and Spiro‐OMeTAD as a HTM. The fabrication films of CsGeI3 and MAGeI3 displayed a smooth morphology with a SPCE of 0.11 and 0.20% whereas that of FAGeI3 exhibited a poor morphology leading to no photovoltaic behavior. The poor performance of the device is attributed to oxidation of Ge2+ into Ge4+ during fabrication process.144 However, TiO2 requires a high‐temperature sintering and exhibits degradation in SPCE on exposure to UV light. TiO2 requires high‐temperature annealing but the substrate cannot withstand such a high temperature. The mesoporous TiO2 (n‐i‐p) devices have exhibited better efficiencies whereas inverted planar (p‐i‐n) devices suffer from hysteresis losses. Tin‐based lead‐free perovskites are considered unsuitable for planar heterojunction solar cells due to their short diffusion lengths, a SPCE of 1.72% is shown by FASnI2Br films as a light absorber with C60 as ETM suggesting the significance of perovskite film morphology on the device performance. FASnI2Br films with an architecture ITO/PEDOT:PSS/FASnI2Br/ C60/Ca/Al reported a J SC of 6.82 mA cm−2 and V OC of 0.46 mV.171 Figure 9 shows the structure of (a) FASnI2Br (SEM image)171 and (b) FASnI3 (SEM image) and energy band diagram.172
Figure 9.
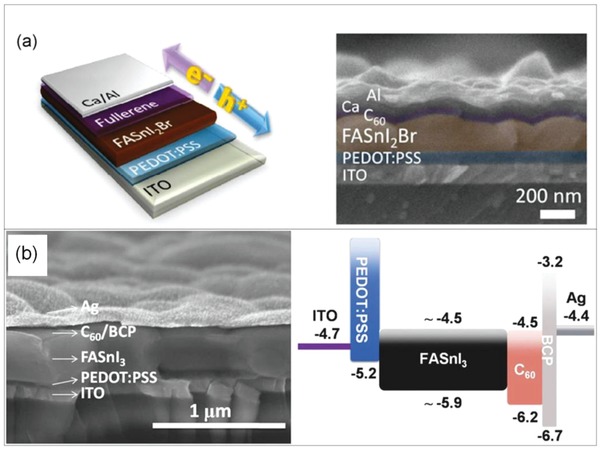
a) Configuration of the FASnI2Br‐based p‐i‐n heterojunction solar cells and its cross‐sectional SEM image of a typical device. Reproduced with permission.171 Copyright 2016, Springer Nature. b) Cross‐sectional SEM image of the entire device with 10 mol% SnF2 additives, in which each layer is labeled, and schematic of energy level diagram of our FASnI3 perovskite solar cells. Reproduced with permission.172 Copyright 2016, Wiley‐VCH.
Anatase, rutile, and brookite are three stable phases in TiO2 when used as an ETM. For anatase TiO2‐based perovskite solar cells, the electron diffusion constant was ten times higher but the time constant for recombination was ten times lower than for rutile TiO2‐based one. Fast charge recombination in anatase TiO2‐based device is the result of poor charge separation in TiO2/perovskite interface.173 Figure 10 shows the MBI perovskite layer deposited on a compact, mesoporous, and brookite TiO2.174 Fullerene C60 and its derivatives such as PC60BM or PC70BM have been employed as an interfacial material at the interface between TiO2 and perovskite layer because of its high electron mobility. A self‐assembled C60 monolayer was introduced on TiO2 surface that enhances the charge separation, reduces the capacitance of TiO2 and V–I hysteresis.175 Organic ETM of PC60BM or PC70BM is more efficient to collect electrons in comparison to mp‐TiO2 in inverted planar p‐i‐n devices as they can passivate the charge traps of metal oxide176, 177, 178 and hence can reduce the nonradiative recombination channels at the surface leading to an improved SPCE with a very low hysteresis.179, 180 The perovskite layer of Cs3Sb2I9 was prepared through a single‐step spin‐coating process for an inverted planar p‐i‐n structure using architecture glass/ITO/PEDOT:PSS/Cs3Sb2I9/PC71BM/C60/bathocuproine (BCP)/Al. PC71BM/C60 is a double fullerene layer employed as an ETM to minimize the trap densities.181 Also, the perovskite solar cells with a p‐i‐n structure of ITO/PEDOT:PSS/(NH4)3Sb2I9/PC61BM/Al were synthesized to study the photovoltaic performance of (NH4)3Sb2I9 reporting a SPCE of 0.51%.147
Figure 10.

Top and cross‐sectional SEM view of MBI perovskite layer deposited on a,b) TiO2 compact layer and c,d) brookite mesoporous layer. Reproduced with permission.174 Copyright 2016, American Chemical Society.
The selection of charge extraction layers by modulating a desirable energy band alignment between the conduction band edge of CsSnI3 and LUMO of ETL is another feasible strategy. A V OC of 0.55 V was reported for CsSnI3 perovskite solar device using p‐i‐n structure ITO/CuI/CsSnI3/indane‐C60‐bisadduct ICBA/BCP/Al architecture. Here, ICBA acted as an ETM.159 BCP is used as an interfacial material in between C60 derivatives and the metal electrodes. The FF was significantly improved by using electrode interfacial layer. In an inverted planar (p‐i‐n) FASnI3 perovskite device, C60 has been employed as an ETM for efficient electron extraction.172 A solution gel derived ZnO used as an ETL bilayer fabricated at <110 °C facilitates the improved energy level alignment and enhanced charge carrier extraction and a PCBM layer is used to reduce the hysteresis and enhance the perovskite thermal stability.
ZnO can be a potential candidate to replace TiO2 as an ETM layer without causing a marked effect on the performance of PSCs.182, 183 The doping of pure ZnO nanorods with Au/Al results in high electron mobility and high electron density.184 From SCAPS‐1D, the use of ZnO nanorods as an ETM and Cu2O as a HTM for MASnI3 perovskite devices has displayed the best performance among all the PSCs. Cu2O is a suitable HTM layer in PSCs due to its high hole mobility and low electron affinity. The device displayed a maximum SPCE for ZnO nanorods/MASnI3/Cu2O structure exhibiting a J SC of 32.26 mA cm−2, V OC of 0.85 V, FF of 0.74, and SPCE of 20.23%.185 The ETM used in a perovskite solar cell has a significant impact on the SPCE of the device when bl‐TiO2 layer in Ag2Bi3I11 is replaced by bl‐SnO2, there is a significant increase in current density from 1.33 mA cm−2 (TiO2) to 2.31 mA cm−2 (SnO2) attributed to better electron extraction by SnO2 ETM.186
7. Lead‐Free Perovskites
7.1. Tin‐Based Perovskites
Tin is the most suitable candidate for substitution of lead for lead‐free perovskite solar cell because of its similar valence electronic configuration as that of lead and approximate same ionic radius of Sn2+ (115 pm) as that of Pb2+ (119 pm). It has lower value of electronegativity Sn2+ (1.96) than that of Pb2+ (2.33).237 Tin‐based perovskites have optical bandgap of 1.2–1.6 eV most suitable for their use as light absorbers, large carrier mobilities, and low exciton binding energies of 18 meV.105, 119, 187 Tin‐based perovskites are represented by the general formula ASnX3 where A can be MA+, Fa+ or Cs+ cation, and X is a halogen anion.
Methylammonium tin halides MASnX3 have a direct bandgap of 1.20–1.35 eV, electron mobility of 2320 cm2 V−1 s−1, hole carrier mobility of 322 cm2 V−1 s−1,105, 188 and long charge carrier diffusion length of more than 500 nm.117 The first completely lead‐free Sn‐based perovskite MASnI3 was processed on a mesoporous TiO2 scaffold that achieved SPCE of 8.4% under 1 sun illumination in a highly inert atmosphere in a glove box with V OC of 0.88 V, J SC of 16.8 mA cm−2, and FF of 0.42 obtained from material having optical bandgap of 1.23 eV.120 A Sn‐based perovskite model with the novel architecture of glass/ZnO:Al/TiO2/Ch3NH3Sncl3/CuI/Au, devised by Mandadapu et al.,189 has been analyzed by using the solar cell capacitance simulator (SCAPS‐ID), with the predicted parameters such as thickness 0–6 µm, defect density of 1014 cm−3 of light absorber layer, and bandgap 1.3 eV. The model achieved a SPCE of 24.82%, V OC of 1.04 V, J SC of 3.50 mA cm−2, and FF of 0.78. The excellent results of this model clearly signify the enormous potential of Sn‐based perovskites for their efficient use in solar cells. Since then an extensive work has been carried out on preparation and characterization of Sn‐based perovskites material to examine their structural, optical, and charge transport abilities for efficient use as light absorber in perovskite solar cells.188 The perovskite solar cells with CH3NH3SnBr3 as light absorber reported a SPCE of 0.35% for coevaporation and 0.12% for sequential deposition method.190 The composition of a halide anion in mixed halide tin‐based perovskites has an influence on the photovoltaic performance exhibited by them. The mixed halide tin‐based perovskite MASnI3− xBrx was investigated by altering the Br−/I− ratio, it was reported that MASnBr3 as a light absorber displays more V OC (0.88 V) and less J SC (8.26 mA cm−2) in comparison to MASnIBr2 having V OC (0.82 V) and J SC (12.33 mA cm−2). Among all MASnI3− xBrx perovskites, MASnIBr2 has the highest reported SPCE of 5.73% under stimulated full sunlight.98 Also, the position of band edge of mixed halides perovskites, MASnI3− xBrx can be tuned from 954 nm (MASnI3) to 577 nm (MASnBr3) thus displaying a remarkable tunability of color. Also the mixed halides tin‐based perovskites MASnIBr2− xClx has been fabricated for carbon‐based mesoscopic cells devoid of ETM and HTM layers by varying the composition of SnCl2/SnBr2. The solar device with MASnIBr1.8Cl0.2 achieved the best photovoltaic performance of 3.11% with a long‐term stability in air. The device exhibited excellent charge recombination and dielectric relaxation properties.191 However, tin‐based perovskites have low values of SPCE due to fast oxidation of divalent Sn2+ into a more stable state Sn4+ and easy formation of Sn vacancies due to small value of formation energy. As a consequence of it, there is a large charge carrier recombination and high levels of self p‐doping in Sn‐based perovskites films. Thus, a lot of research has been carried out to suppress oxidation of divalent Sn2+. SnF2 has been added to such films to inhibit the oxidation process so as to reduce the background carrier hole density by filling Sn vacancies. The entire fabrication process is carried out in an inert atmosphere in the glove box encapsulated by hot melt polymer film, a glass cover slide with sealed edges so as to avoid the oxidation of perovskite film on exposition to ambient air that could cause its fast degradation. Addition of 5‐ammonium valeric acid iodide to MASnI3 suppressed oxidation of Sn2+ for better stability of the perovskite device.192 Hypophosphorous acid was also used for reducing the oxidation of divalent Sn2+ thereby reducing the number of Sn vacancies and charge carrier density. As a consequence of it, there is enhancement in charge recombination lifetime by fourfold than that of the control device.193
A SPCE of 2.14% was reported for a perovskite solar cell having MASnI3 with SnF2 additive as a light absorber. The fabricated device achieved V OC of 0.45 V, J SC of 11.48 mA cm−2, and FF of 0.48 and has long lifetimes of 200 h under 1 sun degradation conditions.194 However, an excess of SnF2 deteriorates the perovskite film morphology and device performance indicating that SnF2 concentration must be kept very low; as a result, the background charge carrier density remains too large to achieve high efficiency, thus it becomes mandatory to explore new and more efficient ways to alleviate the background charge carrier density for better performance of the perovskite solar cell. It was also proposed that the fabrication of perovskite film must be carried out under a reducing vapor atmosphere to reduce the hole density in MASnI3 films by inhibiting the oxidation of Sn2+ during the fabrication process. The excess use of SnF2 induces the phase separation in perovskite films. As a result of exposure to excess SnF2, plate like aggregates are formed in the film, thus it was resolved to use nonsolvent dripping process along with SnF2 via the formation of SnF2–pyrazine complex. Pyrazine has a strong binding affinity to SnF2 thereby suppressing the phase separation induced by the excess use of SnF2.195 Table 4 shows some photovoltaic parameters of methylammonium tin halides.
Table 4.
Photovoltaic parameters of methylammonium tin halides (MASnX3)
| Light absorber | E g | V OC | J SC | FF | SPCE | Architecture | Ref. |
|---|---|---|---|---|---|---|---|
| MASnI3 | 1.23 | 0.88 | 16.8 | 0.42 | 6.4 | FTO/c‐TiO2/mp‐TiO2/absorber/Spiro‐OMeTAD/Au | 120 |
| MASnI3 | 1.3 | 0.716 | 15.18 | 0.50 | 5.44 | FTO/c‐TiO2/absorber/Spiro‐OMeTAD/Au | 196 |
| MASnI3+SnF2 | 1.3 | 0.32 | 21.4 | 0.46 | 3.15 | FTO/c‐TiO2/mp‐TiO2/absorber/Au | 162 |
| MASnI3 | 1.3 | 0.38 | 12.1 | 0.36 | 1.7 | ITO/PEDOT:PSS/poly‐TPD/absorber/C60/BCP/Ag | 151 |
| MASnI3 | 1.26 | 0.27 | 17.4 | 0.39 | 1.86 | FTO/c‐TiO2/mp‐TiO2/absorber/PTAA/Au | 197 |
| MASnIBr2 | 1.75 | 0.82 | 12.33 | 0.57 | 5.73 | FTO/c‐TiO2/mp‐TiO2/absorber/Spiro‐OMeTAD/Au | 106 |
| MASnBr3 | 2.15 | 0.88 | 8.26 | 0.59 | 4.27 | FTO/c‐TiO2/mp‐TiO2/absorber/Spiro‐OMeTAD/Au | 106 |
| MASnBr3 | 2.2 | 0.50 | 4.27 | 0.49 | 1.12 | FTO/c‐TiO2/mp‐TiO2/absorber/P3HT/Au | 190 |
| MASnBr3 | 1.41 | 0.20 | 4.5 | 0.36 | 0.3 | ITO/PEDOT:PSS/absorber/PCBM/Bis‐C60/Ag | 198 |
| MASnI3+hydrazine vapor | 1.3 | 0.38 | 19.9 | 0.51 | 3.80 | FTO/c‐TiO2/mp‐TiO2/absorber/PTAA/Au | 199 |
| MASnI3+SnF2 | – | 0.45 | 11.8 | 0.40 | 2.14 | FTO/PEDOT:PSS/absorber/C60/BCP/Ag | 194 |
| en[MASnI3]+SnF2 | 1.4 | 0.43 | 24.3 | 0.63 | 6.63 | FTO/c‐TiO2/mp‐TiO2/absorber/PTAA/Au | 142 |
| MASnI3+SnF2 | 0.46 | 21.4 | 0.42 | 4.29 | ITO/PEDOT:PSS/absorber/C60/BCP/Ag | 150 | |
| (FA)0.75(MA)0.25SnI3 | 1.33 | 0.61 | 21.2 | 0.62 | 8.12 | ITO/PEDOT:PSS/absorber/C60/BCP/Ag | 150 |
| (FA)0.75(MA)0.5SnI3 | 1.33 | 0.53 | 21.3 | 0.52 | 5.92 | ITO/PEDOT:PSS/absorber/C60/BCP/Ag | 150 |
| FASnI3 | 1.36 | 0.48 | 21.3 | 0.64 | 6.60 | ITO/PEDOT:PSS/absorber/C60/BCP/Ag | 150 |
| MASnIBr2− xClx +0%SnCl2+100% SnBr2 | 1.81 | 0.31 | 13.37 | 0.52 | 2.18 | Glass/FTO/TiO2/absorber/carbon | 191 |
| 10% SnCl2+90% SnBr2 | 1.87 | 0.38 | 13.99 | 0.57 | 3.11 | Glass/FTO/TiO2/absorber/carbon | 191 |
| 25% SnCl2+75% SnBr2 | 1.97 | 0.35 | 11.06 | 0.47 | 1.87 | Glass/FTO/TiO2/absorber/carbon | 191 |
| 50% SnCl2+50% SnBr2 | 1.49 | 0.24 | 9.33 | 0.47 | 1.07 | Glass/FTO/TiO2/absorber/carbon | 191 |
| 75% SnCl2+25% SnBr2 | 1.36 | 0.19 | 13.34 | 0.32 | 0.81 | Glass/FTO/TiO2/absorber/carbon | 191 |
| 100% SnCl2+0% SnBr2 | 1.25 | 0.12 | 19.12 | 0.30 | 0.74 | Glass/FTO/TiO2/absorber/carbon | 191 |
Formamidinium tin iodide FASnI3 has a direct bandgap of 1.41 eV closer to the requisite bandgap value for use in perovskite solar cells and do possess a single stable phase over a broad temperature range up to 200 °C. Sn‐based perovskite FASnI3 is more stable than MASnI3 due to suppression of oxidation of Sn2+ by FA+.188, 200 FASnI3 is first used as light absorber in perovskite solar cell by Koh et al.109 The fabricated films displayed a SPCE of 2.1%, J SC of 24.5 mA cm−2, V OC of 0.2 V, and FF of 0.36. Additive SnF2 is incorporated into FASnI3 to suppress the oxidation of Sn2+ for better film morphology. A SPCE of 4.8% has been achieved by incorporating SnF2 in FASnI3 to form a complex with SnF2 thereby improving the morphology of the perovskite film and slowing down the rate of crystallization of perovskite thin film. Antisolvent process can play a very significant role in preparing the uniform and pinhole‐free compact thin film with the use of diethyl ether as an antisolvent. A significant SPCE of 6.22% has been achieved in FASnI3 by using antisolvent process under forward scan with a small photocurrent hysteresis and a highly reproducibility.172
SnF2–pyrazine complex has been used to further enhance the morphology of FASnI3 perovskite that achieved a SPCE of 4.8%, V OC of 0.32 V, J SC of 23.7 mA cm−2, and improved stability. The encapsulated FASnI3 films displayed a stable performance for over 100 d maintaining 98% of their initial efficiency.195Chlorobenzene is also used as an antisolvent for FASnI3 films. The A‐site cation in Sn‐based perovskite has a significant effect on photovoltaic performance. The use of diethylammonium (en) and FA+ at A‐site of tin‐based perovskite results in a wider bandgap and an improved stability of photovoltaic performance. The complex en [FASnI3] displayed a SPCE of 7.1% and retained 96% of its initial efficiency over 1000 h without encapsulation. Also, the addition of en at A‐site cation along with (FA/MA/Cs) SnI3 cannot reduce dimensionality of the perovskite to 2D.192
The first mixed design composition in tin‐based perovskite was reported on FA1− xMAxSnBr3 with a cubic structure. The bandgap of the perovskite film was varied from 2.4 eV (x = 0) to 1.92 eV (x = 0.82) but the device displayed no photovoltaic performance.201
Another mixed A‐site cation perovskite (FA)x(MA)1− xSnI3 has been investigated for its use as a light absorber in a perovskite solar cell with an invested structure. By tuning the ratio of FA and MA yields the different values of SPCE. A SPCE of 8.12% is achieved for (FA)0.75(MA)0.25SnI3 with V OC of 0.61 V and bandgap of 1.33 eV. The high SPCE is attributed to the improved perovskite film morphology and reduced recombination process.150
Phenylethylammonium (PEA) is substituted at A‐site in tin‐based perovskite and pure 3D, 2D‐3D mixture and pure 2D perovskite are fabricated by tuning the ratio of PEA from 0 to 100% such that inside every two metal halide octahedral layers, there is bilayer of PEA. The resulting mixed cation perovskite is (PEA)2(FA)n −1SnnI3 n +1 where n is the number of tin iodide layers in the structure unit (n ≥ 1). For n = 3, 4, optical bandgaps of 1.5 and 1.42 eV are achieved. The 2D tin perovskite displayed better moisture stability than their 3D counterparts. Incorporation of 20% PEA into FA leads to low‐dimensional mixed perovskite (PEA)2FA8Sn9I28. The as‐fabricated solar cells achieved a SPCE of 5.9% with V OC of 0.55 V and J SC of 14.4 mA cm−2.156
The incorporation of phenylethylammonium iodide (PEAI) obtained by evaporating it at the bilateral interface of a FASnI3 film enhances the V OC and FF of the mixed perovskite solar cell due to improvement in service coverage and formation of 2D‐3D bulk heterojunction structure whereas the presence of LiF at A‐site in tin‐based perovskite by evaporating it at the bilateral interface of FASnI3 film reduces the work function of PEDOT:PSS and aids in hole extracting at the ITO/PEDOT:PSS interfacial layer. A SPCE of 6.98% is achieved for LiF thickness of 5 nm in perovskite films with a V OC of 0.47 V and FF of 0.74, respectively.202 The incorporation of BAI as an additive to the tin‐based perovskite film as a light absorber changes the orientation of the crystal growth thereby enhancing the connectivity of the crystal grains. The fabricated FASnI3 devices doped with 15% BAI exhibited a SPCE of 5.5% in contrast to the pristine FASnI3 films having 4% efficiency.203 Similarly, the doping of EDAI2 into FASnI3 films results in the removal of pinholes in the perovskite films, inhibition of oxidation of Sn2+ into Sn4+, and passivation of surface defect states. The 1% EDAI2‐doped FASnI3 perovskite films displayed an efficiency of 8.9% with a stability of over 1400 h with only slight degradation for more than 200 h in contrast to pristine FASnI3 films with a SPCE of 7.4% only. The high efficiency is attributed to improved perovskite film morphology and passivation of surface defects that enable better separation of charge carriers and suppression of oxidation of Sn2+ to Sn4+.203 Figure 11 a shows the schematic representations of perovskite crystals in the presence of BAI and EDAI2 additives; top‐view SEM images of (b) pristine FASnI3, (c) FASnI3‐BAI 15%, and (d) FASnI3‐EDAI2 1%; (e) current–voltage curves, (f) corresponding IPCE spectra with integrated current densities, (g) histograms of 30 fresh cells fabricated under the same experimental conditions, (h) Mott–Schottky plots, (i) Nyquist plots obtained from electrochemical impedance spectra (EIS), and (j) stabilized power‐conversion efficiencies and photocurrent densities of the FASnI3‐BAI 15% and FASnI3‐EDAI2 1% devices for 240 s.203
Figure 11.
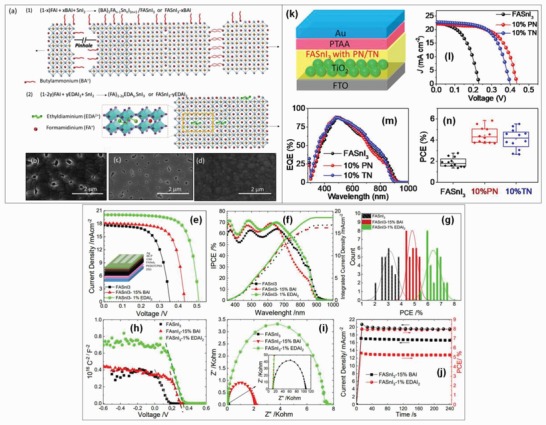
a) Schematic representations of perovskite crystals in the presence of BAI and EDAI2 additives; top‐view SEM images of b) pristine FASnI3, c) FASnI3‐BAI 15%, and d) FASnI3‐EDAI2 1%; e) current–voltage curves, f) corresponding IPCE spectra with integrated current densities, g) histograms of 30 fresh cells fabricated under the same experimental conditions, h) Mott–Schottky plots, i) Nyquist plots obtained from electrochemical impedance spectra (EIS), and j) stabilized power‐conversion efficiencies and photocurrent densities of the FASnI3‐BAI 15%and FASnI3‐EDAI2 1% devices for 240 s; Reproduced with permission.203 Copyright 2018, Royal Society of Chemistry. k) Device structure, l) J–V curves, m) EQE curves, and n) PCE statistics of the FASnI3 solar cells with and without 10% PN and 10% TN; Reproduced with permission.206 Copyright 2018, American Chemical Society.
Crystalline FASnI3 with the orthorhombic a‐axis out‐of‐plane direction was fabricated by mixing (0.08 m) 2D tin perovskite with (0.92 m) 3D tin perovskite in a planar p‐i‐n structure and achieved a SPCE of 9% with a V OC of 0.25 V with more efficient collection of charge and stability in ambient air.145 The introduction of a low‐dimensional interlayer to the interface in a FASnI3 perovskite solar cell can improve the morphology of the film reducing the number of trap sites. As a result, the charge accumulation and recombination in the device is suppressed leading to a high SPCE of 7.05%.204
The bifunctional ammonium cations, 2‐hydroxyethyl ammonium OH(CH2)NH3 +(HEA+), are incorporated into FASnI3 resulting in a mixed tin‐based perovskite HEAxFA1− xSnI3 where x = 0–1 and can act as a light absorber in carbon‐based mesoscopic solar cells. As a consequence of incorporation of HEA+, the crystal lattice changed from orthorhombic to rhombohedra (x = 0.2–0.4). For x ≥ 0.6, a 3D vacant perovskite (HEA)xFA1− xSn0.67I2.33 with a tetragonal structure is formed. The light absorbers in this series are synthesized by employing mesoporous solar cells using one‐step drop‐cast (DC), two‐step solvent–solvent extraction (SE), and a solvent extraction by using ethylenediammonium diiodide (EDAI2) as an additive. The fabricated solar device HEA0.4FA0.6SnI3 displayed the photovoltaic parameters with V OC of 0.371 V, J SC of 18.52 mA cm−2, FF of 0.562, and a stable SPCE of 3.9% for a period of 340 h.205 The FASnI3 perovskite light absorbers incorporated with a diammonium cation such as propylenediammonium (PW) and trimethylenediammonium (TN) display better efficiency than the pristine FASnI3 solar cell. The FASnI3 light absorbers mixed with 10% TN and 10% PN displayed a SPCE of 5.53 and 5.58% with a better film morphology along with retaining their 3D perovskite structure.206 Figure 11 shows the (k) device structure, (l) J–V curves, (m) EQE curves, and (n) PCE statistics of the FASnI3 solar cells with and without 10% PN and 10% TN.206 The addition of bromide into FASnI3 crystal lattice reduces the p‐doping in the perovskite film by reducing the Sn vacancies thereby lowering the current density of the light absorbers. As a result, there is an enhancement in charge recombination lifetime that increases V OC and FF of the devices having M‐TiO2 as electron transport layer. The fabricated devices achieved SPCE of 5.5% with high stability of encapsulated devices over 1000 h under continuous illumination including UV region.167 Table 5 shows some photovoltaic parameters of formamidinium tin halides.
Table 5.
Photovoltaic parameters of formamidinium tin halides (FASnX3)
| Light absorber | E g | V OC | J SC | FF | SPCE | Architecture | Ref. |
|---|---|---|---|---|---|---|---|
| FASnI3+SnF2 | 1.41 | 0.24 | 24.5 | 0.36 | 2.1 | FTO/c‐TiO2/mp‐TiO2/absorber/Spiro‐OMeTAD/Au | 195 |
| FASnI3+SnF2 pyrazine | 1.4 | 0.32 | 23.7 | 0.63 | 4.8 | FTO/c‐TiO2/mp‐TiO2/absorber/Spiro‐OMeTAD/Au | 109 |
| FASnI3+diethyl ether | 1.36 | 0.47 | 22.1 | 0.60 | 6.22 | ITO/PEDOT:PSS/absorber/C60/BCP/Ag | 172 |
| FASnI3+SnF2 | 1.4 | 0.48 | 21.3 | 0.64 | 6.6 | ITO/PEDOT:PSS/absorber/C60/BCP/Ag | 150 |
| FASnI3+SnF2 | 1.4 | 0.38 | 23.1 | 0.60 | 5.27 | FTO/c‐TiO2/mpTiO2/ZnS/absorber/PTAA/Au | 207 |
| en[FASnI3] | 1.5 | 0.48 | 22.5 | 0.66 | 7.14 | FTO/c‐TiO2/mp‐TiO2/absorber/PTAA/Au | 208 |
| FA0.25MA0.75SnI3+SnF2 | 1.28 | 0.48 | 20.7 | 0.45 | 4.49 | ITO/PEDOT:PSS/absorber/C60/BCP/Ag | 150 |
| FA0.50MA0.50SnI3+SnF2 | 1.33 | 0.53 | 21.3 | 0.52 | 5.92 | ITO/PEDOT:PSS/absorber/C60/BCP/Ag | 150 |
| FA0.75MA0.25SnI3+SnF2 | 1.33 | 0.61 | 21.2 | 0.62 | 8.12 | ITO/PEDOT:PSS/absorber/C60/BCP/Ag | 150 |
| FA0.8Cs0.2SnI3 | 1.41 | 0.24 | 16.05 | 0.35 | 1.4 | ITO/PEDOT:PSS/absorber/PCBM/Bis‐C60/Ag | 198 |
| FASnI3 | 1.37 | 0.31 | 18.36 | 0.67 | 3.85 | ITO/PEDOT:PSS/absorber/C60/BCP/Ag | 202 |
| FASnI3+50% PEAI | 1.38 | 0.38 | 19.96 | 0.69 | 5.28 | ITO/PEDOT:PSS/absorber/C60/BCP/Ag | 202 |
| (PEA)2(FA)2Sn2I28+SnF2 | 1.789 | 0.59 | 14.4 | 0.69 | 5.94 | ITO/NiOx/absorber/PCBM/Al | 156 |
| (BA)2(MA)3Sn4I13+SnF2 | 1.42 | 0.229 | 24.1 | 0.45 | 2.53 | FTO/c‐TiO2/mp‐TiO2/absorber/PTAA/Au | 209 |
| FASnI3+LiF (0 nm) | – | 0.38 | 19.65 | 0.71 | 5.04 | ITO/PEDOT:PSS/absorber/C60/BCP/Ag | 202 |
| FASnI3+LiF (3 nm) | – | 0.42 | 19.83 | 0.73 | 5.96 | ITO/PEDOT:PSS/absorber/C60/BCP/Ag | 202 |
| FASnI3+LiF (5 nm) | – | 0.47 | 20.00 | 0.73 | 6.77 | ITO/PEDOT:PSS/absorber/C60/BCP/Ag | 202 |
| FASnI3+LiF (8 nm) | – | 0.45 | 16.66 | 0.65 | 4.71 | ITO/PEDOT:PSS/absorber/C60/BCP/Ag | 202 |
| FASnI3 | – | 0.36 | 17.6 | 0.62 | 4.0 | ITO/PEDOT:PSS/absorber/C60/BCP/Ag | 203 |
| FASnI3+15% (BAI) | 1.40 | 0.44 | 18.0 | 0.69 | 5.5 | ITO/PEDOT:PSS/absorber/C60/BCP/Ag | 203 |
| FASnI3+1% EDAI2 | 1.43 | 0.51 | 20.0 | 0.71 | 7.4 | ITO/PEDOT:PSS/absorber/C60/BCP/Ag | 203 |
| FASnI3+2% EDAI2 | 1.44 | 0.58 | 21.3 | 0.71 | 9.6 | ITO/PEDOT:PSS/absorber/C60/BCP/Ag | 203 |
| FASnI3 | – | 0.525 | 24.1 | 0.71 | 9.0 | ITO/PEDOT:PSS/absorber/C60/BCP/Al | 145 |
| HEA0.4FA0.6SnI3 +3%EDAI2 | – | 0.371 | 18.52 | 0.562 | 3.9 | – | 205 |
| FASnI3+10%TN | – | 0.398 | 22.72 | 0.610 | 5.53 | FTO/mp‐TiO2/absorber/PTAA/Au | 206 |
| FASnI3+10%PN | – | 0.435 | 22.15 | 0.606 | 5.85 | FTO/mp‐TiO2/absorber/PTAA/Au | 206 |
Cesium tin iodide perovskite CsSnI3 possess a direct bandgap of 1.30 eV, a melting point of 435 °C indicating its better thermal stability and a 3D orthorhombic structure110, 210, 211 whereas cesium tin bromide perovskite CsSnBr3 has a bandgap of 1.7 eV.102 Cesium‐based tin perovskite has a high hole mobility of 585 cm−1 V−1 s−1, low exciton binding energy (180 meV) than MAPbI3.119, 187 The melt‐synthesized CsSnI3 ingots containing high‐quality large single crystal grains have been reported to have bulk carrier lifetime more than 6.6 ns, doping concentration of about 4.5 × 1017 cm−3, and minority carrier diffusion lengths approaching to 1 µm.118 A SPCE of 23% was predicted for optimized single crystal solar cells CsSnI3 highlighting their great potential for use in perovskite solar cell. The CsSnI3 was first used in a Schottky‐type perovskite solar cell consisting of simple layer architecture of ITO/CsSnI3/Au/Ti on a glass substrate that achieved an efficiency of 0.9%. A HTM‐free CsSnI3 perovskite solar cell with SnI2 as an additive displayed an efficiency of up to 2.76% with a V OC of 0.43 V and FF of 0.39.212 The use of excess SnI2 as an additive in CsSnI3 not only suppress Sn2+ vacancies but also reduces p‐type conductivity thereby producing a SPCE of 4.8% in CsSnI3 perovskite solar cells.213 The thin films of CsSnIBr3 were fabricated with the addition of hypophosphorous acid (HPA) with thermal stability up to 473 K achieving a SPCE of 3% that last for over 77 d.193 The results of a computational study on mixed cesium perovskite RbyCs1− ySn (BrxI3− x)3 as a light absorber have revealed that the substitution of Rb+ for Cs+ enhanced the quality of perovskite film and its practical applicability in perovskite solar cells.214 Another study on CsSnI3 and CsSnI3− xBrx as light absorbers in n‐i‐p devices structure reported an efficiency of 2%. CsSnI3 has a small bandgap of 1.27 eV to a near‐infrared absorption onset to 950 nm and exhibited a high charge carrier density up to27.67 mA cm−2.215
An excess of SnCl2 and SnI2 to CsSnI3 perovskite films can have masked influence on both stability and SPCE of the corresponding cells reported to be of 3%. An extensive monitoring of oxidation of CsSnI3 in the air by using additives SnCl2, SnBr2, and SnI2 has been carried out to measure electronic, optical absorption spectrum with time and reported that it exhibits the highest stability by inhibiting the crystallization/decomposition.163, 192 The addition of SnF2 lowers the background charge carrier density by neutralizing traps.214, 216 The mesostructured CsSnI3 displayed a SPCE of 2.02% with the addition of 20% SnF2 as an additive. Also a spectral response of 950 nm is demonstrated with SnF2 addition. As a result, the concentration of the defect is reduced that further suppressed the background charge carrier density.215 The anionic substitution of Br− in CsSnI3− xBrx (0 ≤x ≤ 3) results in change in crystal structure from orthorhombic to cubic framework for CsSnBr3 enhancing the V OC and J SC as a result of decrease in tin vacancies and low charge carrier densities of 1015 cm−3.217 The carrier lifetime gets enhanced and the PL line width has reduced when the temperature decreases below 110 K due to the phase transition from orthorhombic to tetragonal phase in CsSnX3 that improved the solar cell performance.218
The evaporation method comprising of thermal evaporation with solution method has been used to produce smooth uniform dense pinhole‐free CsSnI3 films that achieved a SPCE of 1.86% with V OC of 0.265 V, J SC of 15.25 mA cm−2, and FF of 0.46, by using the architecture (FTO/bl‐TiO2/mp‐TiO2/absorber/Spiro‐OMeTAD).102The undesirable p‐doping of CsSnI3 perovskite films can be reduced by the addition of piperazine that can improve the morphology of the film as well as can alleviate the crystallization of excess SnI2 at the same time. With the use of piperazine as an additive, CsSnI3 perovskite devices displayed a SPCE of 3.83%.219 Table 6 shows some photovoltaic parameters of cesium‐based perovskites. Figure 12 shows schematic diagram along with the performance of CsSnI3‐based perovskite solar cell.170
Table 6.
Photovoltaic parameters of cesium‐based perovskites
| Light absorber | E g | V OC | J SC | FF | SPCE | Architecture | Ref. |
|---|---|---|---|---|---|---|---|
| CsSnI3+SnI2 | 1.3 | 0.38 | 25.71 | 0.49 | 4.81 | FTO/c‐TiO2/mp‐TiO2/absorber/PTAA/Au | 213 |
| CsSnI3+SnI2 | 1.3 | 0.43 | 12.3 | 0.39 | 2.76 | ITO/CuI/absorber/ICBA/BCP/Al | 159 |
| CsSnI3 | 1.3 | 0.52 | 10.2 | 0.62 | 3.31 | ITO/NiOx/absorber/PCBM/Al | 157 |
| CsSnI3+SnCl2 | 1.3 | 0.50 | 9.89 | 0.68 | 3.56 | ITO/absorber/PC61 BM/BCP/Al | 163 |
| CsSnI3+SnF2 | 1.27 | 0.20 | 22.7 | 0.29 | 1.66 | FTO/TiO2/mp‐TiO2/absorber/Spiro‐OMeTAD/Au | 217 |
| CsSnI3+SnF2 | 1.25 | 0.17 | 30.8 | 0.34 | 1.83 | FTO/c‐TiO2/mp‐TiO2/absorber/PTAA/Au | 199 |
| CsSnI3+SnF2 | 1.3 | 0.24 | 27.7 | 0.37 | 2.0 | FTO/c‐TiO2/mp‐TiO2/absorber/m‐MTDATA/Au | 215 |
| CsSnI3 | 1.3 | 0.42 | 4.8 | 0.22 | 0.88 | ITO/absorber/Au/Ti | 212 |
| CsSnBr3+SnF2 | 1.8 | 0.45 | 2.4 | 0.55 | 0.55 | ITO/MoO3/absorber/C60/BCP/Ag | 220 |
| CsSnBr3+SnF2 | 1.75 | 0.41 | 3.99 | 0.58 | 0.95 | ITO/mp‐TiO2/absorber/Spiro‐OMeTAD/Au | 217 |
| CsSnIBr2+SnF2 | 1.65 | 0.31 | 11.6 | 0.43 | 1.56 | ITO/mp‐TiO2/absorber/Spiro‐OMeTAD/Au | 217 |
| CsSnIBr2+HPA‐SnF2 | 1.63 | 0.31 | 17.4 | 0.57 | 3.2 | FTO/c‐TiO2/Al2O3/absorber/C | 193 |
| CsSnBr3+SnF2 | 1.79 | 0.37 | 14.0 | 0.59 | 3.04 | FTO/c‐TiO2/mp‐TiO2/absorber/PTAA/Au | 199 |
| CsSnBr3+SnF2 | 1.75 | 0.42 | 9.1 | 0.58 | 2.1 | FTO/c‐TiO2/mp‐TiO2/absorber/Spiro‐OMETAD/Au | 102 |
| CsSnI2.9Br0.1+SnF2 | – | 0.22 | 24.16 | 0.33 | 1.76 | FTO/c‐TiO2/mp‐TiO2/absorber/Spiro‐OMETAD/Au | 217 |
| CsSnI3 | – | 0.265 | 15.25 | 0.46 | 1.86 | FTO/bl‐TiO2/mp‐TiO2/absorber/Spiro‐OMETAD/Au | 170 |
Figure 12.

a) Schematic diagram for development of evaporation‐assisted solution (EAS) method using CsSnI3. b) J–V curves of the device by EAS method in both forward and reverse directions, c) IPCE spectrum of the optimized device (V oc = 0.265 V, J sc = 15.25 mA cm−2, FF = 46.05%, and PCE = 1.86%), d) steady‐state current density of champion device at a bias of 0.18 V, and d) PCE histogram of 25 tested devices. Reproduced with permission.170 Copyright 2018, Wiley‐VCH.
Tin in +4 oxidation state shows more air and moisture stability with enhanced photovoltaic properties. To combat the challenge of oxidation of Sn2+ to Sn4+, tin‐based perovskite structures like A2SnX6 are investigated for their use in a perovskite solar cell.105, 221, 222 Tin‐based Cs2SnI6 as a light absorber has reported a SPCE of almost 1%. These perovskites have been investigated for their use as hole transport material in solar cells.223 Cs2SnI3Br3 222 and Cs2SnI6 224 as light absorbers in solid‐state dye‐sensitized solar cells have displayed an efficiency of 7.8%224 by using classical dyes as a light absorber. Cs2SnI6 used as a hole transport material in solid‐state DSSCs reported an efficiency close to 8.6% in air.224 Cs2SnI6 has a direct bandgap of 1.3–1.6 eV, high absorption coefficient, high electron carrier concentration of the order of 1 × 1014 cm−3, electron mobility of 310 cm2 V−1 s−1, and better stability in air with moisture than that of CsSnI3 as Sn4+ is chemically more stable than Sn2+.105, 216, 225 The cesium‐based perovskite Cs2SnI6 as a light absorber was first studied in 2016 that reported an efficiency of 1%.216, 223 Cs2SnI6 do possess defects of iodide vacancies and interstitial Sn atoms that give rise to the intrinsic n‐type behavior completely opposite to p‐type behavior in CsSnI3. Optimization of thickness of perovskite light absorption layers leads to spontaneous oxidation conversion of unstable B‐ϒCsSnI3 to air stable Cs2SnI6 that has bandgap of 1.48 eV and a high absorption coefficient of 105cm−1.216 The bandgap of A2SnX6 perovskite depends upon the composition of halide anion. With increase in bromide composition CsSnI6− xBrx, the bandgap can be tuned from 1.3 to 2.9 eV and the color of the film changes from dark brown to brown red then to yellow. Cs2SnI4Br2 reported an efficiency of 2.03% highest among all the fabricated compositions. The fabrication of all the reported composition was done in ambient air without the use of any additive and the perovskite film exhibited thermal stability.226 The polycrystalline films of (MA)2SnI6 have been proposed by using thermal evaporation method having a direct bandgap of 1.81 eV with a strong absorption coefficient of 7 × 104 cm−1, carrier concentration of 2 × 1015 cm−3, and electron mobility of ≈3 cm2 V−1 s−1.129 Table 7 shows some photovoltaic parameters of cesium‐based perovskites.
Table 7.
Photovoltaic parameters of tin‐based perovskites A2SnX6
| Light absorber | E g | J SC | V OC | FF | SPCE | Architecture | Ref. |
|---|---|---|---|---|---|---|---|
| Cs2SnI6 | 1.48 | 5.41 | 0.51 | 0.35 | 0.96 | FTO/TiO2/absorber/P3HT/Ag | 216 |
| Cs2SnI6 | 1.30 | 6.75 | 0.37 | 0.59 | 1.47 | FTO/bl‐TiO2/2% Sn‐TiO2/absorber/Cs2SnI6HTM/LPAH/FTO | 226 |
| Cs2SnI5Br | 1.38 | 6.58 | 0.44 | 0.55 | 1.60 | FTO/bl‐TiO2/2% Sn‐TiO2/absorber/Cs2SnI6HTM/LPAH/FTO | 226 |
| Cs2SnI4Br2 | 1.40 | 6.23 | 0.56 | 0.57 | 2.03 | FTO/bl‐TiO2/2% Sn‐TiO2/absorber/Cs2SnI6HTM/LPAH/FTO | 226 |
| Cs2SnI2Br4 | 1.63 | 3.41 | 0.58 | 0.54 | 1.08 | FTO/bl‐TiO2/2% Sn‐TiO2/absorber/Cs2SnI6HTM/LPAH/FTO | 226 |
| Cs2SnIBr5 | 2.36 | 0.01 | 0.57 | 0.37 | 0.002 | FTO/bl‐TiO2/2% Sn‐TiO2/absorber/Cs2SnI6HTM/LPAH/FTO | 226 |
| Cs2SnBr6 | 2.85 | – | – | – | – | FTO/bl‐TiO2/2% Sn‐TiO2/absorber/Cs2SnI6HTM/LPAH/FTO | 226 |
7.2. Germanium‐Based Perovskites
Germanium is another candidate for substitution of lead for lead‐free perovskite solar cells because of its valence electronic configuration as that of Pb2+. Ge2+ has a small ionic radius (73 pm) as compared to that of divalent metal cation Pb2+ (119 pm) and Sn (110 pm). Ge2+ is low in toxicity than Pb2+.227 However, germanium is prone to oxidation than tin. It has a value of electronegativity (2.1) as compared to Pb (3.2) and Sn (1.96). Methyl ammonium germanium halides MAGeX3 are the most potential candidate for perovskites solar cells as Goldschmidt tolerance factor for MAGeX3[X‐Cl, Br, I] has value of 1.005,0.988, and 0.965, respectively, that is close to the optimum range 0.99 < t < 1.03 for a material to form a stable 3D perovskite structure. MAGeI3 has an optical bandgap 1.63 eV which is greater in magnitude than that of MAPbI3(1.55 eV) and MASnI3(1.30), excellent hole and electron conducting behavior and better stability in air as compared to MaPbI3.217 However, Ge2+ cation being smaller in size (73 pm) deviates from its regular [GeI6] octahedral center as it replaces cation of much larger ionic radius as that of Pb2+ (119 pm) and Sn2+ (110 pm).228 As a consequence, it forms three short Ge—I bonds (2.73–2.77 Å)228 and three long in Ge—I bonds (3.26–3.58 Å). The Ge‐based perovskites have been extensively studied by carrying computational work based on density functional theory (DFT).229, 230 The size of constituent halide ion has a remarkable effect on the bandgap of Ge‐based perovskite. The DFT calculations of bandgap values of CsGeX3[X‐Cl, Br, I] showed the decreasing trend of 3.67, 2.32, and 1.53 eV, respectively.231 Similar trend is noticed in MAGeI3[X‐Cl, Br, I] whose DFT calculations reveal that with the increase in size of halide anion, the bandgaps have decreasing values of 3.7, 2.81, and 1.61 eV.230 The cation at A‐site also plays a pivotal role for the size of bandgap of AGeI3.144, 230 Bandgaps show an increasing trend when small Cs+ cation(1.6 eV) is replaced by a larger counterpart such as CH3NH3 + (1.9 eV) and CH(NH2)+ (2.2 eV), acetamidinium (2.5 eV), trimethylammonium (2.8 eV), guanidinium (2.7 eV), and isopropyl ammonium (2.7 eV).229
A study of Ge‐based perovskite AGeX3 (A‐Cs+, CH3NH3 +, HC(NH2)2 +) reported the estimated values of optical bandgap derived from tauc plot for CsGeI3 (1.63 eV), MAGeI3 (2.0 eV), and FAGeI3 (2.3 eV).144 The replacement of Cs with MA and FA decreases the valence band level as evident from measured value of valence band of CsGeI3, MAGeI3, and FAGeI3 that has the value of −5.10, −5.2, and −5.5 eV, respectively, by photoemission spectroscopy in air.144 CsGeI3 displays a higher stability up to 850 °C in contrast to up to ≈250 °C stability shown by MAGeI3 and FAGeI3. Ge‐based perovskite solar cells have two values of V OC due to its oxidation into Ge4+ during the fabrication process. The poor quality of FAGeI3 films results in loss of photoconductivity in them.144 The small A‐site cations like Cs+, CH3NH3 +, and HC(NH2)2 + in AGeX3 lead to 3D structure framework based on corner‐sharing octahedral and the perovskite materials do exhibit direct bandgaps whereas large A‐size cations lead to distortion of the crystal structure. As a result, 1D chain like perovskite structures are formed having indirect bandgaps.144, 229 The introduction of bromide ions into MAGeI3 perovskites enhances not only photovoltaic performance but also stability to a slight extent.232 The substitution of 10% of the iodide content by bromide results in MAGeI2.7Br0.3 perovskite that reported a SPCE of 0.57% as a light absorber in solar cells fabricated with planar p‐i‐n architecture having PEDOTS:PSS as HTM and PC70BM as ETM.232
The mixed Ge‐based perovskite RbSn0.5Ge0.5I3 displays a direct optical bandgap in the range of 0.9–1.6 eV with sufficient optical absorption spectrum comparable to MAPX3 perovskites. The material exhibited favorable effective masses for higher carrier mobility and good stability in water.233 A 2D perovskite (C6H5(CH2)2NH3)2GeI4 [(PEA)2 GeI4] consisting of inorganic germanium iodide planes separated by organic PEAI layers has a direct bandgap of 2.12 eV that is very close to the value 2.17 eV obtained through DFT calculations. The perovskite material exhibits luminescence at room temperature with a medium lifetime and is a potential candidate for PV applications. The 2D (PEA)2GeI4 shows more stability in air than 3D MAGeI3 that is attributed to the presence of a hydrophobic organic long chain.234 On the basis of DFT calculations, one more 2D Ruddlesden–Popper hybrid organic–inorganic perovskite BA2MAn −1MnI3 n +1 [M = Sn or Ge, n = 2–4] has been reported that has suitable excitonic and optical light absorbing properties for application in lead‐free perovskites. Moreover, 2D Ge‐based perovskites have enhanced thermodynamic stability in comparison to their 3D counterparts that enables 2D Ge‐based perovskites with a thickness of a few tens of unit cells to be used as light absorbers in perovskite solar cell.235 Table 8 shows some photovoltaic parameters of Ge‐based perovskites. Figure 13 shows the crystal structure, band diagram, and the I–V characteristics of Ge‐based perovskites in a solar cell (a) CsGeI3 and (b) MAGeI3,229 (c) optical absorption spectrum of CsGeI3, MAGeI3, and FAGeI3, in comparison with CsSnI3, and (d) calculated band structure and projected density of states of CsGeI3. The energy of the highest occupied state is set to 0 eV. (e) Photoelectron spectroscopy in air (PESA) of powder samples and (f) schematic energy level diagram of CsGeI3, MAGeI3, and FAGeI.144
Table 8.
Ge‐based perovskites and their photovoltaic parameters
Figure 13.
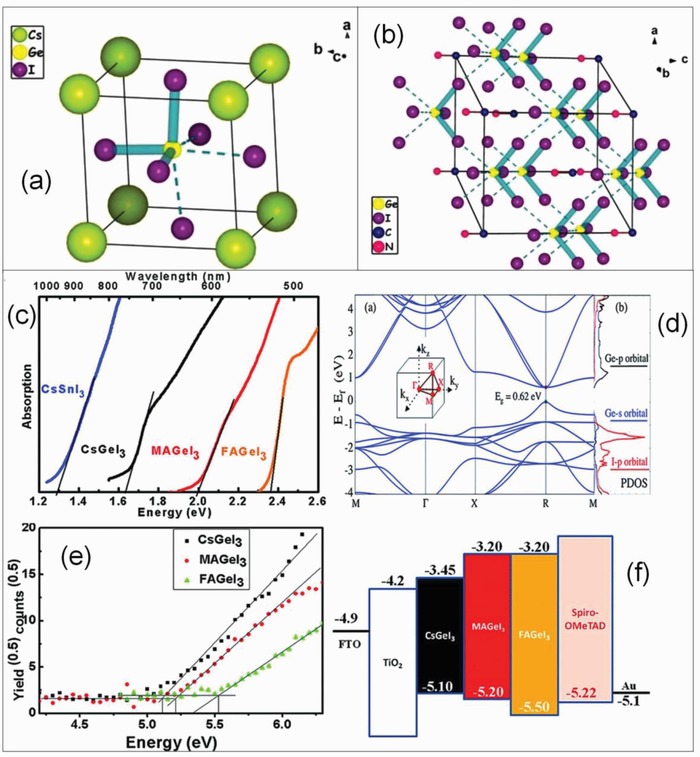
Schematic diagram for the unit cell of a) CsGeI3 and b) MAGeI3; Reproduced with permission.229 Copyright 2015, American Chemical Society. c) Optical absorption spectrum of CsGeI3, MAGeI3, and FAGeI3, in comparison with CsSnI3. d) Calculated band structure and projected density of states of CsGeI3. The energy of the highest occupied state is set to 0 eV. e) Photoelectron spectroscopy in air (PESA) of powder samples and f) schematic energy level diagram of CsGeI3, MAGeI3, and FAGeI3; Reproduced with permission.144 Copyright 2012, Royal Society of Chemistry.
7.3. Bismuth‐Based Perovskites
Bismuth can form +3 ions with similar valence electronic configuration as that of Pb2+, having ionic radius (103 pm) in comparison to divalent Pb2+ (119 pm) and Sn2+ (110 pm). The value of electronegativity of bismuth is 2.02 in comparison to that of Pb (2.33) and Sn (1.96).237 Bismuth‐based perovskites are represented with a general formula A3Bi2X9, where A can be MA, Cs, NH3, Ag. These materials have attracted large attention due to their low toxic nature.238 They can have 0D dimer, 1D chain like, 2D layered, or 3D double perovskite elpasolite frameworks,238 containing A‐site cations such as MA+, Cs+, Rb+, K+, guanidinium, cyclohexylammonium, imidazolium to form a 0D dimer perovskite structure.143 Bismuth‐based methylammonium single crystal MABi2I9 (MBI) shows a regular hexagon shape with a diameter ranging from 100 to 200 nm. The MBI crystals exhibit a dark red color with an optical bandgap of ≈2.11 eV. The 0D MBI consists of face‐sharing bi‐octahedral [Bi2I9] clusters surrounded by MA+ cations. The fabricated solar cells using MBI as light absorbing layer reported an efficiency of 0.08% with J SC (≈0.36 mA cm−2), V OC of 0.51 V, and FF of 0.44. The photovoltaic performance was enhanced by using thick mesoporous TiO2 layer (1.8 µm) to V OC of 0.51 V, J SC of 1.16 mA cm−2, FF of 0.46, and SPCE of 0.19%.127
The positive Hall coefficient of MBI film reveals p‐type charge carrier with carrier concentration of 1015–1016 cm−3 for solution‐processed MBI films. MBI films have got excellent stability against exposure to humidity level of 50% and ambient air at room temperature for 40 d.127 The first study on bismuth‐based perovskite (MA)3Bi2I9 as a light absorber was reported by preparing simple (MA)3Bi2I9 perovskite and mixed (MA)3Bi2I9− xClx perovskite thin films with a hexagonal crystalline phase. The mesostructured solar cells displayed a better SPCE of 0.12%, V OC of 0.68 V, J SC of 0.52 mA cm−2, and FF of 0.33 as compared to (MA)3Bi2I9− xClx displaying SPCE of 0.003, V OC of 0.04 V, J SC of 0.18 mA cm−3, and FF of 0.38. Also the substitution of iodine with chloride in (MA)3Bi2I9− xClx shifted the bandgap from 2.1 to 2.4 eV.111 An efficiency of 0.42% is achieved by using a mesoporous TiO2 substrate for fabricating a (MA)3Bi2I9 perovskite film with V OC of 0.67 V, J SC of 1.0 mA cm−3, and FF of 0.62.240 MA3Bi2I9 films fabricated by evaporation‐spin‐coating process produced better quality films which produced SPCE of 0.39% in an inverted planar device with a V OC of 0.83 V, J SC of 1.39 mA cm−2, and FF of 0.34.241 The gas‐assisted deposition method enhances the morphology of active light absorber layer. The fabricated (MA)3Bi2I9 light absorber layer by gas‐assisted deposition process reported an enhanced value of SPCE of 0.08% and V OC of 0.686 V.242 The solvent annealing in (MA)3Bi2I9 films enhances its electrical conductivity. The DMF‐induced solvent annealing impacts the charge transport through the films.243 The morphology of (MA)3Bi2I9 perovskite film can also be enhanced by incorporating a small amount of N‐methyl‐2 pyrrolidone (NMP) into the MBI‐DMF solution. The addition of various concentration of NMP into the precursor solution not only controls the rate of crystallization but also enhanced SPCE to a value of 0.31% and stability for 30 d in a relative humidity of 50–60%.244 The optical measurement of solution‐processed perovskite film (MA)3Bi2I9 fabricated by spin‐coating process showed a strong absorption band around 500 nm on further heating. The devices made on anatase TiO2 mesoporous layer exhibited a current density of 0.8 mA cm−2 whereas those fabricated by using brookite TiO2 layer do not display any current density.174 There is considerable effect of solvent treatment and substrate temperature on the morphology and structure of bismuth‐based perovskite films of MA3Bi2I9. The electron transport layer of fluorinated perylene diimide (FPD) treated by solvent vapor annealing with chloroform reported an efficiency of 0.06% for substrate temperature at 75 °C. The perovskite solar cell MA3Bi2I9 exhibited a small degradation after 17 d storage in ambient air conditions.245
The concentration of perovskite solution also impacts the morphology and photovoltaic performance of spin‐coated MA3Bi2I9 solar cells. The fabricated cells displayed an efficiency of V OC (0.73 V) and efficiency of 0.17% after 48 h in air. The solar cells exhibited 56% of peak efficiency and 84% of open‐circuit voltage even after 300 h exposure in ambient air.246 The vapor‐assisted solution process (VASP) applied to BiI3 films by exposing them to CH3NH3I vapors results in enhancement of film morphology, efficiency, and stability in ambient air. The solar cell fabricated using pure BiI3 films and CH3NH3 vapors on mesoporous TiO2 substrate displayed high SPCE up to 3.17% attributed to better morphology, improved device composition, reduced metallic content, and suitable optoelectronic properties of the fabricated material that maintained a stability for 60 d with only 0.1% drop in efficiency.247 The wide bandgap of lead‐free perovskite devices (E g > 1.9 eV) can be engineered to a narrow bandgap by incorporating triiodide into (4‐methyl piperidinium)3 Bi2I9 (MP‐Bi2I9) that resulted in 0D perovskite (MP‐T‐BiI6) (4‐methyl piperidinium)4I3BiI6. MP‐T‐BiI6 displayed a narrow bandgap of 1.58 eV comparable to 1.5 eV of MAPbI3, hole mobility (≈12.8 cm2 V−1 s−1), and charge trap density (≈1.13 × 1010 cm−3). The narrow bandgap signifies its potential to be used as an effective light absorber in perovskite solar cells.248 The solvent engineering method can be applied at bismuth‐based perovskite to produce pinhole‐free films of MA3Bi2I9, Cs3BiI9, or (MA)3Bi2I9. The fabricated MA3Bi2I9 films are most suitable for efficient and stable perovskite solar cells than the pristine MA3Bi2I9 films with pinholes.249 An enhanced open‐circuit voltage of 0.84 V is obtained in (MA)3Bi2I9 perovskite by using ethanol as solvent.250 The film quality of (MA)3Bi2I9 can be enhanced by high‐vacuum BiI3 deposition and low‐vacuum transformation of BiI3 to (MA)3Bi2I9. The fabricated perovskite solar cells exhibited a SPCE of 1.64%, J SC of 2.95 mA cm−2, V OC of 0.81 V, FF of 0.69, and long stability.251 0D Cs3Bi2I9 perovskite films in mesostructured perovskite solar cells exhibited a SPCE of 1.09% with a V OC of 0.81 V, J SC of 2.95 mA cm−2, and FF of 0.69111 with a bandgap of 2.2 eV.
1D iodobismuthates consisting of 1D chain like BiI4 − anions with edge‐sharing BiI6 octahedra have been prepared from aqueous solutions. The four reported compounds LiBiI4·5H2O, MgBi2I8·8H2O, MnBi2I8·8H2O, and KBiI4·H20 have direct bandgaps of 1.70–1.76 eV and can be used as potential light absorber.252 The 1,6 hexadiammonium bismuth halide perovskite (HDABiI5) showing 1D chain like structure is prepared by solution method. The (HDABiI5) m‐str. perovskite displayed a SPCE of 0.027%, V OC of 0.40 V, J SC of 0.12 mA cm−2, and FF of 0.43 with an optical bandgap of 2.05 eV.253 K3Bi2I9 and Rb3Bi2I9 are 2D layered defect perovskites prepared by solution method or solid‐state reactions. K3Bi2I9 and Rb3Bi2I9 have a direct bandgap of 2.1 eV.239 The perovskite film CsBi3I10 has a layered 2D structure as evident from X‐ray diffraction (XRD) pattern with a bandgap of 1.77 eV which is smaller than the bandgap of Cs3Bi2I9 (2.03 eV), absorption coefficient 1.4 × 105 cm−1. The perovskite solar cell with CsBi3I10 achieved a photocurrent up to 700 nm leading to better scope for use in solar cells.143 The Cs3Bi2I9 films have a better film morphology and pinhole‐free layers. The CsBi3I10 films as a light absorber in mesostructured solar cells displayed a SPCE of 0.4% whereas Cs3Bi2I9 solar cells have displayed a SPCE of 0.02% only in same device architecture.143 Another 2D layered perovskite is MA3Bi2I9 which is prepared from solution and has a bandgap of 2.04 eV which is smaller than that of k3Bi2I9 and Rb3Bi2I9 (2.1 eV).254, 255 Bismuth‐based 3D double perovskite has been proposed with a chemical formula A2 IBIBiIIIX6 to maintain a charge neutrality of the perovskite material. The double perovskites like Cs2AgBiX6 (Br, Cl)254, 255, 256, 257 and (MA)2KBiCl6 258 have been synthesized by using a solution method. It has been reported that Cs2AgBiBr6 254 and Cs2AgBiCl6 have an indirect bandgap of 2.19 and 2.77 eV. (MA)2KBiCl6 has too large bandgap of 3.04 eV to be suitable for use in perovskite solar cells.258 The DFT calculations have revealed that double perovskite (MA)2TlBiI6 has a bandgap of 2.00 V potential to be used as a lead‐free perovskite material due to similar property as that of MAPbI3 but Tl is toxic in nature.259 The bimetal iodide thin films AgBi2I7 show a SPCE of 1.22%, V OC of 0.56 V, J SC of 3.30 mA cm−2, and FF of 1.87 with a better stability under ambient conditions.260 Using first principle calculations, a 3D double perovskite family has been revealed with optical bandgaps in the visible range and low carrier effective masses. The members of this family Cs2CuBiX6, Cs2AgBiX6, and CsAuBiX6 have optical bandgaps in the range of 1.3–2.0, 1.6–2.7, and 0.5–1.6 eV, respectively.261
The bismuth‐based perovskite solar cells Cs3Bi2I9 fabricated by glass/FTO/TiO2/Cs3Bi2I9/PTAA/Au architecture displayed an efficiency of 8%.The perovskite film of Cs3Bi2I9 exhibited a pure crystalline phase and excellent thermal stability. The encapsulated perovskite cell displayed constant efficiency for more than 500 h as light absorber at 65 °C with humidity at 60–70% level. The stability is attributed to the large size of bismuth‐based perovskite structure than lead‐based perovskite structure.141 The lattice compression of 0D perovskite Cs3Bi2I9 results in change in their structural, optical, and electrical properties. It is a result of lattice compression that there is an increase in exciton binding energy leading to an enhancement in emission under mild pressure. Bi—I bond contraction causes bandgap narrowing and an increase in metal halide orbital overlapping resulting from decrease in bridging Bi—I—Bi angle. These changes are reversible on decomposition. There is a semiconductor to conductor transition at ≈28 GPa due to decrease in resistance thus leading to metallization of Cs3Bi2I9.262 The high‐quality polycrystalline films of Cs3Bi2I9, Rb3Bi2I9, and AgBi2I7 can be fabricated by two‐step Co‐evaporation process involving two square evaporation of CsI, RbI or AgI and BiI3 and further annealing under BiI3 vapors producing films with better pinhole‐free morphology of films with average grain size >200 nm.263 Bismuth‐based perovskite films can be further engineered to produce pinhole‐free films of MA3Bi2I9 and Cs3Bi2I9. The MA3Bi3I9 films are more suitable for perovskite solar cells than the pristine.14 Table 9 shows some photovoltaic parameters of bismuth‐based perovskites. Figure 14 shows the SEM images of (MA)3Bi2I9 without and with different concentration of NMP additives.244
Table 9.
PV parameters of bismuth‐based perovskites
| Light absorber | E g | V OC | J SC | FF | SPCE | Architecture | Ref. |
|---|---|---|---|---|---|---|---|
| MA3Bi2I9 | 2.11 | 0.35 | 1.16 | 0.46 | 0.19 | FTO/TiO2/mp‐TiO2/absorber/P3HT/Au | 127 |
| MA3Bi2I9 | 2.1 | 0.68 | 0.52 | 0.33 | 0.12 | FTO/TiO2/mp‐TiO2/absorber/Spiro‐OMeTAD/Ag | 111 |
| MA3Bi2I9 | 2.26 | 0.72 | 0.49 | 0.31 | 0.11 | FTO/TiO2/absorber/Spiro‐OMeTAD/Au | 128 |
| MA3Bi2I9 | 2.1 | 0.68 | 0.37 | 0.32 | 0.08 | FTO/TiO2/absorber/Spiro‐OMeTAD/Au | 242 |
| MA3Bi2I9 | – | 0.56 | 0.83 | 0.49 | 0.26 | FTO/TiO2/mp‐TiO2/absorber/Spiro‐OMeTAD/Au | 173 |
| MA3Bi2I9 | – | 0.51 | 0.94 | 0.61 | 0.31 | FTO/TiO2/mp‐TiO2/absorber/Spiro‐OMeTAD/Au | 244 |
| MA3Bi2I9 | 2.1 | 0.65 | 1.10 | 0.50 | 0.36 | FTO/TiO2/mp‐TiO2/absorber/Spiro‐OMeTAD/Au | 264 |
| MA3Bi2I9 | 2.22 | 0.83 | 1.39 | 0.34 | 0.39 | ITO/PEDOT:PSS/absorber/C60/BCP/Ag | 242 |
| MA3Bi2I9 | 2.1 | 0.67 | 1.0 | 0.62 | 0.42 | ITO/TiO2/mp‐TiO2/absorber/Spiro‐OMeTAD/Ag | 240 |
| MA3Bi2I9 | 2.9 | 0.66 | 0.22 | 0.49 | 0.07 | ITO/PEDOT:PSS/absorber/PCBM/Ca/Al | 143 |
| MA3Bi2I9− xClx | 2.4 | 0.04 | 0.18 | 0.38 | 0.003 | FTO/TiO2/mp‐TiO2/absorber/Spiro‐OMeTAD/Au | 111 |
| (MA3Bi2I9)0.2(BIi3)0.8 | – | 0.57 | 0.27 | 0.50 | 0.08 | FTO/TiO2/mp‐TiO2/absorber/PTAA/PIDT‐DFBT/Ag | 128 |
| HDABiI5 | – | 0.40 | 0.12 | 0.43 | 0.027 | FTO/c‐TiO2/HDABiI5/mp‐TiO2/Spiro‐OMeTAD/Au | 253 |
| Cs3Bi2I9 | 2.1 | – | – | – | 8.0 | Glass/FTO/TiO2/Cs3Bi2I9/PTAA/Au | 141 |
| MA3Bi2I9+FPDI | 2.1 | 0.61 | 0.37 | 0.27 | 0.06 | ITO/MA3Bi2I9/Spiro‐OMeTAD/MoO3/Ag | 245 |
| AgBi2I7 | 1.87 | 0.56 | 3.30 | 0.67 | 1.22 | ITO/TiO2/mp‐TiO2/absorber/P3HT/Ag | 265 |
| Cs2AgBiBr6 | 2.21 | 0.98 | 3.93 | 0.63 | 2.43 | FTO/c‐TiO2/mp‐TiO2/absorber/Spiro‐OMeTAD/Au | 266 |
| CsBi3I6 | 1.77 | 0.31 | 3.4 | 0.38 | 0.40 | FTO/c‐TiO2/mp‐TiO2/absorber/P3HT/Ag | 143 |
| C6H5NBiI4 | 1.98 | 0.62 | 2.71 | 0.54 | 0.9 | FTO/c‐TiO2/mp‐TiO2/absorber/ZrO2/C | 242 |
| (H3NC6H12NH3)BiI5 | 2.1 | 0.40 | 0.12 | 0.43 | 0.03 | FTO/c‐TiO2/mp‐TiO2/absorber/Spiro‐OMeTAD/Au | 143 |
| Cs3Bi2I9 | 2.03 | 0.02 | 0.18 | 0.37 | 0.02 | FTO/TiO2/mp‐TiO2/absorber/P3HT/Ag | 143 |
| Cs3Bi2I9 | 2.2 | 0.85 | 2.15 | 0.6 | 1.09 | FTO/TiO2/mp‐TiO2/absorber/Spiro‐OMeTAD/Ag | 111 |
Figure 14.

SEM images of (MA)3Bi2I9 without and with different concentration of NMP additives. Reproduced with permission.244 Copyright 2017, Royal Society of Chemistry.
7.4. Antimony‐Based Perovskites
Antimony is another potential candidate for replacing lead in perovskite solar cells. Antimony has the ability to form +3 ions with valence electronic configuration similar to that of divalent Pb2+. The trivalent Sb3+ has a small ionic radius (76 pm) as compared to that of divalent metal cation Pb2+ (119 pm) and Sn2+ (110 pm), having comparable electronegativity of (2.05) in comparison to Pb (2.33) and Sn (1.96). Antimony‐based lead‐free perovskites form a 0D dimer or a 2D layered structure with the typical basic formula A3Sb2X9 where A is an organic or inorganic cation and X is a halogen.140 The choice of cationic or anionic species determines the structure and dimensions of antimony‐based perovskites used as light absorbers. In addition to it, the employed processing technique also effects the dimensions of the synthesized products. Cs3Sb2I9 has an inclination to a 0D dimer form if it is prepared by solution process whereas it prefers a 3D layered structure when prepared through a solid‐state or gas phase reaction. Saparov et al.140 carried out thin film preparation and characterization of Cs3Sb2I9 thin films as light absorber in perovskites solar cell. Cs3Sb2I9 film exists in two forms, viz., a 0D dimer form and a 2D layered form. The 0D dimer form of Cs3Sb2I9 is prepared through reactions of CsI and SbI3 in stoichiometric ratio of 3:2 in polar solvents. This film has an intense orange color and is stable under ambient air with an indirect bandgap of 2.06 eV whereas 2D layered films of Cs3Sb2I9 are obtained through a solid‐state or gas phase reactions, that is, by sequential deposition of CsI film through evaporation followed by annealing in SbI3 vapor. The layered films display red color with a direct bandgap of 2.05 eV, high absorption coefficient of 105 cm−1, and ionization energy of 5.6 eV with better stability in air. However, SPCE values of the perovskites solar device with layered forms of Cs3Sb2I9 as light absorber have minimal values of SPCE close to 1% with V OC of 0.30 V and a J SC below 0.1 mA cm−2 indicating a very low overall photovoltaic performance attributed to the presence of deep defects that promote nonradiative recombination. Boopathi et al.181 synthesized 0D dimer form of Cs3Sb2I9 as a light absorber and reported a SPCE of 0.84%, J SC of 2.91 mA cm−2, V OC of 0.60 V, and FF of 0.48 for Cs3Sb2I9 with addition of HI.181
A 2D layered perovskite was synthesized by using the mixture Cs+ and MA+ as the A‐site cation via solution process as opposite to reported by Saparov et al. where A‐site cation is substituted by a smaller cation Rb+, a 2D layered phase is achieved due to smaller radius of Rb+ (1.72 Å) as compared to that of Cs+ (1.88 Å) via solution processing through the reaction of RbI and SbI3. Using DFT calculations, the comparison of formation energies of 2D layered and 1D dimer forms of A3Sb2I9 (A‐Cs, Rb) reveals that the formation energy difference of 0.25 eV is higher for Rb‐based perovskites than that of cesium‐based counterparts having this difference equal to 0.1 eV thus clearly indicating the increased inclination of Rb3Sb2I9 for layered phase. The layered perovskites Rb3Sb2I9 achieved a SPCE of 0.66% with V OC of 0.55 V, J SC of 2.11 mA cm−2, and FF of 0.57.267 In addition, they show thermal stability up to 250 °C and no phase transition is reported in between −40 and 200 °C. The light absorption coefficient of Rb3Sb2I9 films is greater than 1 × 105 cm−1 with an indirect bandgap of 2.1 eV. A direct transition at 2.24 eV was calculated for Rb3Sb2I9 as compared to 2.05 eV for the bandgap of cesium. MA3Sb2I9 only forms a 0D dimer structure. The octahedral anionic metal halide [Sb2I9]3− surround the MA+ cations. Hebig et al. first prepared the flat and thin films of MA3Sb2I9 by spin‐coating process followed by toluene treatment. The obtained thin films show a peak absorption coefficient above 105 cm−1 and an optical bandgap of 2.14 eV. The fabricated planar perovskite cell achieved SPCE of 0.49%, V OC of 0.90 V, J SC of 1.0 mA cm−2, and FF of 0.55.147 Boopathi et al.181 synthesized 0D (MA)3Sb2I9 films for use as light absorbers in perovskite solar cells with HI as an additive. The addition of HI into the films resulted in an increase in light absorption in the visible wavelength regions about 400 nm. The XRD spectra studies revealed that the addition of HI leads to a better crystallinity, phase purity, and quality of the film. It reduces the bandgap thereby enhancing the light absorption toward higher wavelength regions. The achieved values of photovoltaic parameters with or without addition of HI are shown in Table 9. The nonsolvent treatment was investigated to enhance the surface morphology of Sn‐based dimer by using HI‐CB to enhance the heterogenous nucleation of Sb‐based perovskite used as light absorber.268 The interlayer of HI‐CB acted as a hydrophobic scaffold for the growth of (CH3NH3)3Sb2I9 crystals. The interlayer decreases the number of voids and enhances the quality of film. The fabricated films achieved a SPCE of 2.77%.268The DFT calculations have revealed that the most stable mixed metal organic–inorganic perovskite MA2SbI6 has a bandgap of 2.0 eV which is further confirmed by using XRD characterization of MA2SbI6 as a light absorber that has displayed an optical bandgap of 1.93 eV and good stability in air.269
A larger A‐site cation was used to synthesize high‐quality films of 2D layered phase in (CH3NH3)3Sb2ClxI9− x. The induction of methylammonium chloride into precursor solutions inhibits the formation of the undesirable 0D dimer phase leading to synthesis of high‐quality films of 2D layered phase that is favorable for application in lead‐free perovskite solar cells. These films achieved a SPCE of 2%.270 Similarly, Zuo and Ding synthesized a family of perovskite light absorbers (NH4)3Sb2IxBr9− x (0 ≤ x ≤ 9).116 These materials display good solubility in ethanol. The optical light absorption can be adjusted by adjusting the ratio of I and Br content. The absorption onset for films changes from 558 to 453 nm as x changes from 9 to 0. The single crystals of (NH4)3Sb2I9 showed a hole mobility of 12.3 cm2 V−1 s−1 and electron mobility of 12.3 cm2 V−1 s−1 achieving a V OC of 1.03 V and SPCE of 0.51% only.116 The use of methylammonium antimony sulfur diiodide (MASbSI2) as light absorber for lead‐free perovskite solar cells was first reported by Nie et al.271 The MASbSI2 is prepared through spin‐coating and thermal annealing of MAI solution on SbSI under mild temperature conditions. The fabricated MASbSI2 as light absorber achieved SPCE of 3.08% under the standard illumination condition of 100 mW cm−2. They achieved photovoltaic performance in MASbSI2 solar cells as of J SC (8.12 mA cm−2), V OC (0.65 V), FF (0.58), and SPCE of 3.08%. Unencapsulated cells stored in dark ambient conditions (humidity ≈60%, temperature 25 °C) retained 90% of their initial efficiency. The use of chalcogenide and halide mixed perovskite materials can be an effective strategy for fabrication of efficient, cheap, and stable solar cells. A mixed metal layered perovskite Cs4CuSb2Cl12 as a light absorber for perovskite solar cells has been reported.272 The layered perovskite Cs4CuSb2Cl12 is formed by incorporating Cu2+ and Sb2+ cations into layers that has a bandgap of 1 eV and conductivity is one order of magnitude greater than MAPbI3. Cs4CuSb2Cl12 has high photo and thermal stability and resistance to humidity. The achieved photovoltaic properties promise the excellent use of this material in optical light absorbing layer for perovskite solar cells.272
The normal (n‐i‐p) structured solar cells show better photovoltaic performance as compared to inverted structures. Baranwal et al. have proved it by making a comparison between the normal [n‐i‐p‐Tio2‐perovskite‐Spiro‐OMeTAD) and inverted [p‐i‐n‐NiO‐perovskite‐PCBM) structures.273 ABX6 compounds can form perovskite like 3D crystals frameworks like bromoantimonate (V) (N‐EtPY) (SbBr6) with short interhalide contacts.295 ASbBr2 is a black crystalline solid with an optical bandgap of 1.65 V that is much lower than that of conventional MAPbBr3 of 2.3 eV. The planar cells with standard architecture using P3HT as a HTM layer displayed better photovoltaic parameters as J SC (5.1 mA cm−2), V OC (1.285 V), FF (0.58), and SPCE of 3.8% whereas the inverted architecture using a double‐layer PDI as ETL films is fabricated by depositing first by spin‐coating from chlorobenzene solution followed by evaporation of additional layers of the material in vacuum and has shown J SC of 5.1 mA cm−2, V OC of 1.030 V, FF of 0.58, and SPCE of 3.1% only.274 The effect of substitution of antimony (Sb) with bismuth (Bi) in a 2D mixed layered perovskite (NH4)3(Sb1− xBix)2I9 as light absorber has been investigated extensively. The partial substitution of Sb with Bi did not change the structure of the crystal but enhanced the volume of the unit cell. The XRD patterns did not show any impurity phase with Bi addition but peaks shift toward lower angles as content of Bi increases showing an increase in unit cell size due to induction of bulkier bismuth cation. The films showed typical features of direct bandgaps due to strong absorption above 2.7 eV and indirect bandgaps because of absence of photoluminescence with long carrier lifetimes. The absorption coefficient increases due to increase in density of states in conduction band whereas bandgap reduces from 2.27 to 2.16 eV275 for 5% Bi film due to higher spin–orbit coupling .Bismuth pushes the conduction band downward as predicted by DFT calculations. It also shifts the valence band downward, thereby enhancing the ionization potential values from 5.78 to 5.9 eV for incorporation of 50% bismuth content. The urbach energies also showed a decrease with an increase in bismuth content. The carrier lifetimes do not follow a particular trend with increase in Bi incorporation in the perovskite film as 184 ± 8 ns (0% Bi), 94 ± 25 ns (20%Bi), 149 ± 12 ns (40% Bi), 91 ± 13 ns (50% Bi) as there is decrease in deep defects near the conduction band side due to addition of Bi but simultaneously there is an increase in defects near the valence band. The AC Hall measurements predicted the p‐type conduction band behavior for (NH4)3Sb2I9 with a carrier concentration of 3.95 × 1015 cm−3 and mobility of 0.5 ± 0.5 cm2 V−1 s−1. The carrier density is reduced by incorporating 10 and 20% of Bi owing to increase in mobility that got doubled to more than 1 cm2 V−1 s−1 thus the material undergoes a p‐to‐n transition for higher Bi contents (40%, 50%) that clearly indicates the changing nature of defects in the material. Therefore, the films show both p and n‐type regions. In order to increate p and n regions, electrical poling was used to adjust the load composition of the film by creating ionic drift. The unpoled (NH4)3Sb2I9 (p‐type) showed linear photocurrent voltage relationship. The device was negatively poled by applying a bias of −2 V µm−1 to electrode B under illumination by a blue LED (455 nm, power 1.4 mW mm−2) for 2 min.275 The V–I curves after negative poling indicates photovoltaic effect with V OC close to 200 mV which flipped to −0.2 V on ± poling.275 The material exhibited measurable photocurrent densities at short‐circuit conditions. The directions of dark and photocurrent densities were opposite resulting in a switch of current direction on illumination due to presence of opposite fields in the same compound. A negative voltage close to −0.6 V is required to achieve zero current condition in dark as opposed to +0.2 V required under illumination. Table 10 shows some photovoltaic parameters of antimony‐based lead‐free perovskites. The device configuration is shown in Figure 15 for the switchable photovoltaic device containing (NH4)3(Sb(1− x )Bix)2I9 perovskite material.275
Table 10.
Antimony‐based lead‐free perovskites used as light absorber
| Light absorber | E g | J SC | V OC | FF | SPCE | Architecture | Ref. |
|---|---|---|---|---|---|---|---|
| Cs3Sb2I9 | 2.05 | <0.1 | 0.31 | – | <1.0 | FTO/C‐TiO2/absorber/PTAA/Au | 140 |
| Cs3Sb2I9 | 2.30 | 2.34 | 0.62 | 0.46 | 0.67 | ITO/PEDOT:PSS/absorber/PC61BM/C60/BCP/Al | 181 |
| Cs3Sb2I9+HI | 2.0 | 2.91 | 0.60 | 0.48 | 0.84 | ITO/PEDOT:PSS/absorber/PC61BM/C60/BCP/Al | 181 |
| Rb3Sb2I9 | 2.1 | 2.11 | 0.55 | 0.57 | 0.66 | FTO/C‐TiO2/mp‐TiO2/absorber/PolyTPD/Au | 267 |
| MA3Sb2I9 | 2.14 | 1.0 | 0.90 | 0.55 | 0.49 | ITO/PEDOT:PSS/absorber/PC61BM/nano‐ZnO/Al | 147 |
| MA3Sb2I9 | 2.20 | 3.81 | 0.64 | 0.45 | 1.11 | ITO/PEDOT:PSS/absorber/PC61BM/CO60/BCP/Al | 181 |
| MA3Sb2I9+HI | 1.95 | 5.41 | 0.62 | 0.60 | 2.04 | ITO/PEDOT:PSS/absorber/PC61BM/CO60/BCP/Al | 181 |
| (NH4)3Sb2IXBr9− x | 2.27 | 1.15 | 1.03 | 0.42 | 0.51 | ITO/PEDOT:PSS/absorber/PC61BM/Al | 116 |
| MASbSI2 | 2.03 | 8.12 | 0.65 | 0.58 | 3.08 | FTO/BL/mp‐TiO2/absorber/PCPD/TBT/PEDOT:PSS/Au | 271 |
| Cs4CuSb2Cl12 | 1.0 | – | – | – | 0.30 | – | 272 |
| (N‐EtPY)SbBr6 (standard) | 1.65 | 5.1 | 1.285 | 0.58 | 3.8 | ITO/C‐TiOX/absorber/P3HT/Au | 274 |
| (N‐EtPY)SbBr4 (inverted) | 1.65 | 5.1 | 1.030 | 0.58 | 3.1 | ITO/PEDOT:PSS/absorber/PD1/Ag | 274 |
| CH3Sc(NH2)2SbA3 | 2.41–3.34 | – | – | – | – | – | 275 |
Figure 15.
Schematic of the device configuration used for switchable photovoltaic study using (NH4)3(Sb(1− x )Bix)2I9 perovskite material. Reproduced with permission.275 Copyright 2018, Wiley‐VCH.
7.5. Copper‐Based Perovskites
The divalent Cu2+ cation is another suitable element for Pb2+ substitution as Cu2+ has nontoxic nature. Cu2+ has a small ionic radius (73 pm) as compared to Pb2+ (119 pm) and Sn2+ (110 pm). The divalent Cu2+ is more stable in air than Sn2+ and Ge2+.135, 136 Cu‐based perovskites usually form 2D layered perovskite structures owing to their smaller ionic radii with general formula (RNH3)2CuX4 where RNH3 + can be aliphatic or aromatic cation and X is a halogen. They can be easily prepared under suitable conditions by solution method. A 2D cupric perovskite solar cell [p‐F‐C6H5C2H4‐NH3]2CuBr4 and (CH3(CH2)3NH3)2‐CuBr4 with absorption range from 300 to 750 nm has been reported. The achieved SPCE values of the fabricated perovskite solar cell are 0.51 and 0.63%, respectively, with good air stability of less than 5% decrease of efficiencies after 1 d in air with humidity of 50% without encapsulation. The reported photovoltaic parameters of the fabricated device are shown in Table 10.135 The solar cells based on MA2CuClxBr4− x have been investigated in order to study the photovoltaic performance and stability of Cu‐based mixed halides. By tuning Cl/Br ratio, the optical absorption can be extended in the near‐infrared region. The small quantity of Cl− enhances the stability and crystallization of the perovskite material. Among all the investigated light absorbers, the highest SPCE of 0.17% is achieved using MA2CuCl2Br2 as light absorber. The minimal values of SPCE are attributed to reduction of Cu2+ and low absorption coefficient. The formation of Cu2+ ions was found to be responsible for the green photoluminescence of this material. (MA)2CuCl2Br2 and (MA)2CuCl0.5Br3.5 are found to be more stable under ambient conditions. The achieved values of photovoltaic parameters are shown in Table 10.136 It has been found that adding a small amount of CuBr2 into MAPbI3 remarkably enhances its morphology and efficiency but it is still under investigation whether Cu2+ can actually act as substitute for Pb2+.276
Li et al. investigated and characterized highly stable Cu‐based perovskite films C6H4NH2CuBr2I exhibiting extraordinary hydrophobic behavior with a contact angle of ≈90°. The XRD patterns of the perovskite films did not report any change even after 4 h of being immersed in water. The UV absorption of these films revealed their excellent absorption over the entire visible spectrum with low values of SPCE of ≈0.5% attributed to low absorption coefficient and heavy mass of holes.137 The other Cu‐based perovskite solar cells (CH3NH3)2CuCl4 and (CH3NH3)2CuCl2X2 [X = I, Br] were fabricated through grinding milling process by Elseman and team and on characterization by XRD reveals that (CH3NH3)2CuCl2 has monoclinic crystal structure and (CH3NH3)2CuCl2Br2 is crystallized with an orthorhombic structure. The tolerance factor and octahedral factor calculated for (CH3NH3)2CuCl4 were found to be 1.004 and 0.403, respectively, by assuming the ionic radius of methylammonium to be 18 pm. The calculated values are out of the optimum range of 0.8 < t < 0.9 and 0.42 < u < 0.895 for a stable 3D perovskite structure thus it crystallizes into 2D structures.277 It has been observed that the substitution of Cl− with I− or Br− has different effects on bond angles, unit cell dimensions, and ionic radius. The achieved photovoltaic parameters are depicted in Table 11 . The low SPCE values of (CH3NH3)2CuCl2Br2 are due to reduction of Cu2+ caused by the higher trap density. The chemical structures and the performance of Cu‐based perovskite solar cells (a) (CH3NH3)2CuCl4, (b) (CH3NH3)2CuCl2I2, and (c) (CH3NH3)2CuCl2Br2 powders, (d) current–voltage curve, and (e) EQE spectra of solar cells are shown in Figure 16 .277
Table 11.
Photovoltaic parameters of reported Cu‐based perovskites
| Light absorber | J SC [mA cm−2] | V OC | PCE | FF | E g | Architecture | Ref. |
|---|---|---|---|---|---|---|---|
| (CH3(CH2)3NH3)2CuBr4 | 1.78 | 0.88 | 0.63 | 0.40 | 1.76 | FTO/C‐TiO2/mp‐TiO2/absorber/Spiro‐OMeTAD/Ag | 135 |
| (p‐F‐C6H5C2H4‐NH3)2CuBr4 | 1.46 | 0.87 | 0.51 | 0.40 | 1.74 | FTO/TiO2/absorber/Spiro‐OMeTADLiTFSI/Ag | 135 |
| MA2CuCl2Br2 | 0.22 | 0.26 | 0.02 | 0.32 | 2.12 | FTO/C‐TiO2/mp‐TiO2/absorber/Spiro‐OMeTAD/Au | 136 |
| MA2CuCl0.5Br3.5 | 0.21 | 0.29 | 0.002 | 0.28 | 1.8 | FTO/C‐TiO2/mp‐TiO2/absorber/Spiro‐OMeTAD/Au | 136 |
| C6H4NH2CuBr2I | 6.20 | 0.20 | 0.46 | 0.46 | 1.64 | FTO/C‐TiO2/mp‐TiO2/absorber/ZrO2/C | 137 |
| (CH3NH3)2CuCl4 | 8.12 | 0.56 | 2.41 | 0.52 | 2.36 | Glass/FTO/TiO2/absorber/Spiro‐OMeTAD/Au | 277 |
| (CH3NH3)2CuCl2I2 | 6.78 | 0.54 | 1.75 | 0.47 | 1.90 | Glass/FTO/TiO2/absorber/Spiro‐OMeTAD/Au | 277 |
| (CH3NH3)2CuCl2Br2 | 3.35 | 0.58 | 0.99 | 0.50 | 1.04 | Glass/FTO/TiO2/absorber/Spiro‐OMeTAD/Au | 277 |
Figure 16.
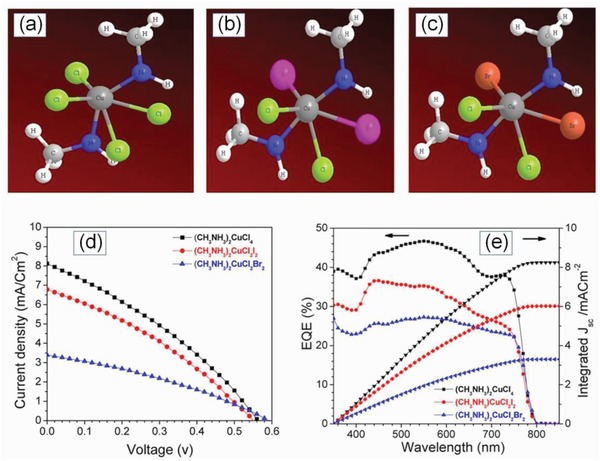
Chemical structures of perovskite solar cells using a) (CH3NH3)2CuCl4, b) (CH3NH3)2CuCl2I2, and c) (CH3NH3)2CuCl2Br2 powders. d) Current–voltage curve and e) EQE spectra of solar cells. Reproduced with permission.277 Copyright 2018, American Chemical Society.
7.6. Other Potential Candidates for Lead‐Free Perovskites
The alkaline earth metals Be2+, Mg2+, Ca2+, Sr2+, and Ba2+ have been investigated as an alternative to lead in lead‐free perovskite. However, the optical bandgap of Be2+ is too high to be used for PV applications. Mg2+ despite having a smaller ionic radius (72 pm) can replace Pb2+ (119 pm) to form a stable magnesium‐based perovskite.278, 279 The replacement of Pb2+ by Mg2+ results in a lead‐free magnesium‐based AMgX3 perovskite that exhibits low effective masses, direct optical absorption coefficients, and direct bandgap tunable within the visible region of electromagnetic spectrum depending upon the size of A‐site cation.278, 279 The bandgaps of magnesium‐based perovskite AMgI3 featured an increasing trend with A‐site cations such as FA+, MA+, and Cs+ having values 0.9, 1.5, and 1.7 eV, respectively.278 The study of photoluminescence properties of Eu2+‐doped CsMI3[M‐Mg, Ca, Sr] perovskite has revealed that the Eu2+‐doped CsMgI3 and CsSrI3 displayed a redshift emission with respect to Eu2+‐doped CsCaI3 perovskite. Eu2+‐doped CsMgI3 crystallizes in a distorted hexagonal CsNiCl3 structure whereas CsCaI3 crystallizes in an orthorhombic GdFeO3 structure and CsSrI3 crystallizes in a filled PuBr3 structure.280
The divalent Ca2+ has an ionic radius (100 pm) comparable to that of Pb2+ (119 pm)281, 282 whereas Sr2+ (118 pm) has a similar ionic radii to Pb2+ (119 pm).283 The divalent Ba2+ has an ionic radii of 135 pm larger than that of Pb2+ (119 pm).283 The DFT calculations have reported bandgaps of MACaI3, MASrI3, and MABaI3 to be 2.95, 3.6, and 3.3 eV, respectively.281, 283 However, MASrI3 and MABaI3 do have large bandgaps leading to light absorption in UV region.281, 283 The replacement of Pb2+ by Ca2+ and Sr2+ in MAPbI3 perovskite thin films has reported an increase in long carrier lifetime and fill factors of the devices reaching 0.85.284 Transition metals such as Ti, V, Mn, Ni, Pd, Fe, Cu, Zn, Cd, and Hg have been researched extensively for lead replacement in lead‐free perovskite. The crystals of CsNiX3 perovskite have been synthesized by hydrothermal method having BaNiO3 structure consisting of a face‐sharing NiX6 octahedral separated by CsX12 cuboctahedra.285 The 2D layered perovskite structure of bis(alkyl ammonium)metal(II) tetrahalide (CnH2 n −1NH3)2MX4 and (α,w) polymethylene diammonium metal (II) tetrahalide NH3(CH2)mNH3MX4 with M—Cd, Cu, Fe, Mn, or Pd and X—Br, Cl have been synthesized and a large single perovskite crystal has been obtained.286 The divalent Fe2+ has a smaller ionic radius (78 pm) as compared to Pb2+ (119 pm) that does not allow the formation of 3D perovskite structure. Iron‐based 2D layered perovskite (CH3NH3)2(FeCl4) exhibits a canted anti‐ferromagnetism in a magnetic field of strength greater than 2000 Oe and it also exhibits the phase transition from a high symmetry to a low symmetry.287 The magnetic susceptibility of (CH3NH3)2FeCl3Br perovskite depends upon the strength of applied magnetic field. Also, the size of halide anion has a direct effect on the canted spin.288 Just like Sn‐based perovskite, iron‐based perovskite is also unstable due to oxidation of Fe2+ to Fe3+.289 The divalent rare earth Eu2+‐based perovskite (C4H9NH3)2EuI4 has been synthesized through low‐temperature solid‐state reactions.290 The effect of doping of rare earth metal ions such as Eu2+, Tm2+, and Yb2+ in CsAX3(A‐Ca,Mg,Sr) perovskite has been investigated extensively.280, 291, 292 Another transition metal gold has been investigated for its potential in a perovskite framework. The optical properties of gold‐based 2D organic mixed AuI/AuIII layered perovskite have been reported with [AuI2]−/[AuI4]− layers supported by I3 − ions and appropriate organic dications.293 The [NH3(CH2)7NH3]2 [AuI I2] [AuIII I4](I3)2 and [NH3(CH2)8NH3]2 (AuI I2) (AuIII I4)(I3)2 perovskite exhibited a bandgap of 0.95 and 1.14 eV, respectively. The low bandgaps are attributed to the induced electronic interactions between [AuI I2]− and [AuIII I4]− units and I3 ions.293 Another transition metal tellurium‐based vacancy order perovskite Cs2TeI6 has been reported that consists of a face‐centered lattice of [TeI6]4− units with Cs2+ cations occupying the cuboctahedral position. This material do possess an indirect bandgap, electronic dispersion, and is intolerant to formation of defects that is not suitable for PV applications as per current research.210 Transition metal titanium‐based perovskite thin films Cs2TiBr6 have been prepared through low‐temperature‐based method having a bandgap of 1.8 eV that is comparable to eV of lead halide perovskite, balanced carrier diffusion lengths > 100 nm, and highly stable under environmental stresses. The fabricated device exhibited a SPCE of 3.3%. The incorporation of C60 interfacial layer between the Cs2TiBr6 light absorber thin films and TiO2 ETM resulted in a V OC of 1.02 V in a reverse scan direction and also enhanced other photovoltaic parameters. The thin films are highly stable under ambient conditions.294 By the application of split anion approach to MAPbI3, the replacement of Pb2+ with Bi3+ and I− with Se− or S− is done to maintain the charge neutrality thus resulting in lead‐free perovskite CH3NH3BiSeI2, and CH3NH3BeSI2 has been reported exhibiting a direct bandgap of 1.3–1.4 eV suitable for photovoltaic applications.295 Figure 17 shows the atomic structure and bandgap diagram of CH3NH3PbI3 and MABiSeI2 and a schematic illustrating the split‐anion approach to replace Pb in CH3NH3PbI3.295
Figure 17.
a,b) Atomic structures of CH3NH3PbI3 and CH3NH3BiSeI2 and a schematic illustrating the split‐anion approach to replacing Pb in CH3NH3PbI3. c,d) The calculated bandgaps of CH3NH3PbI3 and CH3NH3BiSeI2, respectively, using improved methods from PBE, HSE to HSE+SOC. The alignment of the band edge positions was obtained by assuming that the reference potentials from different methods are the same. Reproduced with permission.295 Copyright 2016, Royal Society of Chemistry.
Lead‐free perovskite with mixed chalcogen and halogen anion AB(Ch, X)3 where A: Ca or Ba, B: Sb or Bi, X: halogen, Ch: chalcogen has been investigated by using DFT calculations and solid‐state reactions that revealed their thermodynamically unstable nature, that is, their liability to decompose into ternary or binary secondary phases or form phases with nonperovskite structure. The synthesis of mixed perovskite with chalcogen and halogen anions has not been possible due to their unstable nature.296 The bandgaps of chalcogenide perovskite CaTiS3, BaZrS3, CaZrSe3, and CaHfSe3 with a distorted structure are within the suitable range for photovoltaic performance. Due to their suitable optical absorption properties, chalcogenide perovskite can be the potential candidates to combat the instability and toxicity issues.297 The synthesis of polycrystalline chalcogenide perovskite BaZrO3, CdZrO3, SrTiS3, SrZrS3, and BsZr(OxI3− x)3 has been reported by sulfonization of oxide perovskite by CS2. The BaZrS3 exhibited a distorted perovskite structure as evident from XRD pattern, with an optical absorption from UV to visible region. The perovskite material displayed photoluminescence in visible region and has an excellent stability in ambient air as compared to lead‐based halide perovskites.298 The quaternary halide double perovskite employing lanthanides (La3+, Ce3+, Pr3+, Nd3+, Sm3+, Eu3+, Gd3+, Dy3+, Er3+, Tm3+, Lu3+) and actinides (Pu3+, Am3+, Bk3+) has been investigated but no PV performance has been reported.299, 300 Table 12 summarizes the stability of lead‐free perovskites.
Table 12.
Stability of lead‐free perovskites
| Light absorber | SPCE | Stability | Ref. |
|---|---|---|---|
| MASnI3+SnF2 | 2.14% | 200 h under 1 sun degradation conditions (AM 1.5, 100 mW cm−2) | 194 |
| (FA)0.75(MA)0.25SnI3 | 8.12% | ≈80% of SPCE over a period of 400 h, stored in a glove box filled with nitrogen | 150 |
| MASnIBr1.8Cl0.2 | 3.1% | Average lifetime less than 100 ps | 191 |
| FASnI3+SnF2 pyrazine | 4.8% | Stable performances for over 100 d having 98% of its initial efficiency | 195 |
| FASnI3 | 6.23% | Stable efficiency of 6% for more than 100 s | 172 |
| en[FASnI3] | 7.14% | Unencapsulated device continues to have 96% of initial SPCE after 1000 h | 208 |
| (PEA)2(FA)8Sn9I28 | 5.94% | Unencapsulated devices display performance without significant decay in SPCE over 100 h | 156 |
| FASnI3+EDAI2 (1%) | 7.4% | Device stored in glove box displays maximum SPCE of 8.9% for over 1400 h with only slight reduction for storage beyond 2000 h | 203 |
| FASnI3 | 5.5% | Encapsulated devices exhibit stability over 1000 h under continuous 1 sun illumination encompassing UV region | 167 |
| CsSnIBr2+HPA | 3% | Exhibits stable efficiency for 77 d and power output within 9 h at high temperature up to 473 K | 193 |
| (PEA)2GeI4 | – | 2D structure is more stable than 3D MAGeI3 in air. | 234 |
| BA2MAn −1MnI3 n +1 | 1.94–2.53% | 2D structure is more stable as compared to 3D counterparts. | 235 |
| MA3Bi2I9 | 0.356% | Exhibits air and moisture stability for more than 60 d | 264 |
| MA3Bi2I9 + NMP | 0.31% | Unencapsulated device exhibits 88% of SPCE to relative 50–60% humidity for 30 d | 244 |
| MA3Bi2I9 | 0.26% | Exhibits stability for more than ten weeks under ambient conditions | 174 |
| Cs3Bi2I9 | 8% | Unencapsulated device displays initial SPCE for more than 500 h under 1 sun at 65 °C and relative humidity of 60–70% | 141 |
| MA3Bi2I9 (with FPDI ETM) | 0.06% | Exhibits limited degradation in SPCE after 17 d storage in ambient atmosphere | 245 |
| MA3Bi2I9 | 0.17% | Exhibits 56% of peak SPCE after 300 h exposure to air | 262 |
| Cs2AgBiBr6 | 2.43% | Unencapsulated device displays excellent stability to working conditions. | 266 |
| AgBi2I7 | 1.22% | Exhibits excellent stability for at least 10 d under ambient conditions | 265 |
| Cs2AgBiBr6 | – | Degrades after light exposure for two weeks | 300 |
| KBiI4 H2O | – | Exhibits considerable stability in air | 252 |
| Cs2BiAgBr6 | – | Exhibits stable SPCE in ambient conditions | 254 |
| Cs2AgBiBr6 | – | Exhibits degradation after a period of three weeks on exposure to ambient air and light | 256 |
| MA3Bi2I9 via (VASP) | 3.17% | Unencapsulated devices display stability for 60 d with 0.1%loss in SPCE | 247 |
| Cs3Sb2I9 | <1.00% | Increased stability under ambient air in comparison to MAPbI3 films stored in same condition | 140 |
| MA2AgSbI6 | – | Exhibits stability at room temperature in air with 20–60% humidity for 370 d | 269 |
| MA3Bi2I9 | 0.12% | Exhibits no degradation over a month in devices stored in dark in dry air with humidity less than 10% | 111 |
| Cs3Bi2I9 | 1.09% | ||
| AgBixI3 x +1 | 0.60% | Unencapsulated devices exhibited SPCE decreasing at a slow pace, 75% of efficiency even after weeks of storage in a N2 filled glove box under ambient light | 186 |
| 1 < x < 2.25 (Ag4Bi7I25) | |||
| MA3Bi2I9 | 0.19% | Displayed excellent stability for more than 400 d upon contact with 50% humidity and air at room temperature | 127 |
| MASbSI2 | 3.08% | Unencapsulated devices continues to have 90% of initial SPCE for a period of 15 d when stored in dark with 60% humidity at 25 °C | 271 |
| (p‐F‐C6H5C2H4‐NH3)2 CuBr4 (CH3(CH2)3NH3)2CuBr4 | 1.74% | Encapsulated device displays stability in air after 1 d in air with 50%of humidity | 135 |
| 1.76% | |||
| C6H4NH2CuBrI | 1.64% | Device exhibited hydrophobic behavior with a contact angle of 90° and unchanged XRD patterns after 4 h of water immersion | 137 |
| Cs2TiBr6 | 3.3% | Unencapsulated films displayed 94% of SPCE after 14 d at 70 °C, 30% RH and ambient light illumination retained 85% of efficiency | 294 |
| RbSn0.5Ge0.5I3 | – | The activation barrier for water penetration is 0.23 eV in a humid environment that is much higher than for MAPbI3 (0.09 eV). | 233 |
| CsGeI3 | 0.11% | Stable up to 350 °C | 144 |
| MAGeI3 | 0.20% | Stable up to 250 °C | |
| FAGeI3 | Stable up to 250 °C | ||
| MASnI3 | 5.8% | Continuous to have 80% of initial SPCE in first 12 h in a properly sealed nitrogen glove box | 106 |
| MA3Bi2I9 | 0.19% | Films display stability over 40 d on conditional exposure to 50% humidity level at room temperature | 127 |
| (NH4)3Sb2I9 | 0.51% | Films retained 80% of initial SPCE when stored in a glove box with O2 < 10 ppm and H2O < 0.1 ppm for 40 d but when in air with 50% humidity, the films lost their PV performance completely | 116 |
8. Recent Research on Lead‐Free Perovskites
In order to explore the potential material whose properties can be tailored to be used as a light absorber in a perovskite solar cell, a lot of research is being carried out at present. Research is going on to explore a perovskite material that is lead‐free, nontoxic, have low fabrication cost, easy fabrication technique, higher SPCE, and better stability in air, moisture, and heat. In an attempt to synthesize a low‐cost lead‐free perovskite solar cell, the CH3NH3SnBrxCl3− x crystals with a trigonal phase have been synthesized via aqueous solution based method by a reaction between HCl and H3PO2 without taking into account any protection against moisture. The synthesized crystals exhibit various low‐frequency vibrational modes of Sn—Cl and Sn—Br.261 Recent studies of lead‐free perovskite have shown that the hot antisolvent treatment of perovskite film increases its coverage and inhibits electrical shunting in photovoltaic device. Also, the average crystallite size increases due to annealing under a low partial pressure of dimethyl sulfoxide vapor. The topographical and electrical qualities of the perovskite film are enhanced facilitating the fabrication of tin‐based perovskite solar cell with a SPCE of over 7%.301 The effect of additives on the stability of lead‐free CsSnI3 perovskite films has been studied by using first principle based calculations. It has been reported that the additives effectively passivate the surface and enhance the stability of CsSnI3 films. The addition of SnBr2 as an additive in CsSnI3 films resulted in a SPCE of 4.3% with 100 h of stability.302
An additional additive formamidinium thiocyanate into quasi‐2D tin perovskite suppresses the oxidation of the material during film formation resulting in a highly crystalline structure with a coarser perovskite grain. The fabricated tin‐based perovskite solar cell reported a SPCE of 8.17% under a reverse scan and a steady‐state efficiency of 7.84%. The fabricated device retained 90% of its efficiency after 1000 h in a glove box filled with nitrogen.303 Another study on mixed tin‐germanium perovskite solar cell FA0.75MA0.25Sn1− xGexI3 has reported that most of the Ge atoms passivate the graded structure of tin perovskite. Upon doping with 5 wt% of Ge, the reported J SC (19.8 mA cm−2), FF (0.55), and SPCE (4.48%) have shown an increasing trend as compared to 0 wt% of Ge. On further increasing the doping of Ge, the photovoltaic parameters have shown a decreasing trend. The doping of Ge also enhances the stability in air as compared to the nondoped sample.304 A recent research on Mn and Ni‐doped CsGeI3 perovskite has revealed the effect of doping of Mn and Ni in CsGeI3 perovskite that has resulted in enhancement of optical absorption and photoconductivity in visible and UV light region. The optical absorption, dielectric constant, and photoconductivity of Mn‐doped CsGeI3 perovskite are larger than that of Ni‐doped counterpart. The Mn‐doped CsGe1− xMnxCl3 perovskite exhibited the potential properties that make it best among all the inorganic pure and metal‐doped CsGeI3 perovskite. Figure 18 shows the light absorption spectrum of pristine and metal‐doped (Ni, Mn) CsGeI3 perovskite as a function of: (a) photon‐energy‐dependent absorption coefficient, (b) wavelength‐dependent absorption coefficient, (c) reflectivity, (d) conductivity, (e) dielectric constant (real part), and (f) dielectric constant (imaginary part).305
Figure 18.
Light absorption spectrum of pristine and metal‐doped (Ni, Mn) CsGeI3 perovskite as a function of a) photon energy dependent absorption coefficient, b) wavelength‐dependent absorption coefficient, c) reflectivity, d) conductivity, e) dielectric constant (real part), and f) dielectric constant (imaginary part). Reproduced with permission.305 Copyright 2018, Royal Society of Chemistry.
In a recent study, lead‐free bismuth‐based perovskite CH3NH3BiX3 (X3‐I2Te, I2S, I2Se) as a light absorber has been investigated by using first principle calculations. The study has confirmed that CH3NH3BiX3 (X3‐I2Te, I2S, I2Se) perovskites are nontoxic in nature exhibiting a high optical absorption in the visible region. These properties pave the way for use of such bismuth‐based perovskite as a light absorber as an alternative to lead‐based CH3NH3PbI3 perovskite in photovoltaic applications.306 In another study, lead‐free mixed chalcogen halide perovskite material MABiI2S have been synthesized and characterized for its physical and optical properties that revealed a low bandgap of 1.52 eV suitable for optical absorption in the visible spectrum. The fabricated material exhibited an absorption up to over 1000 nm.307 The concentration of perovskite solution (0.15–0.30 m) has an effect on the crystallization in MA3Bi2I9 films. Also, the speed of rotation during spin‐coating process determines the layer coverage. The SPCE of the fabricated cells enhances from 0.004 to 0.17% after processing. The fabricated device has exhibited a V OC of 0.72 V after 48 h.308 Lead‐free inorganic AgBiI4 as a light absorber has been prepared by solution method of thin films. The AgBiI4 films have been fabricated by 0.6 m solution and annealed at 150 °C. The films exhibited a better morphology with a thermal stability and photostability than that of MAPbI3. The fabricated PSC exhibited 2.1% efficiency. The devices displayed long‐term stability and maintained 96% of initial SPCE even after 100 h at relative humidity of 26%.309 Lead‐free copper halide perovskite Cs3Cu2I5 have been reported with a 0D structure exhibiting a blue emission (≈445 nm) with a high quantum yield of 90 and 60% for single crystals and thin films. The 0D electronic nature of Cs3Cu2I5 is attributed to a large exciton binding energy of 49 meV and blue emission is demonstrated using solution method Cs3Cu2I5 thin films.310 Zinc‐based lead‐free CsZnCl2I perovskite 3D thin films have been reported that were deposited at 100 °C by aerosol‐assisted chemical vapor deposition method. The fabricated film displayed absorption peaks at 325 nm excitation covering the entire visible spectrum range.311 In another study, perovskite solar cells based on transition metal Ti, Ni, and Cd‐doped BiFeO3 as a light absorber with graphene electrode have been investigated. The V OC of pure BiFeO3, Ti, Ni, Cd‐doped BiFeO3 have been reported to be 0.49, 0.77, 0.56, and 0.49 V, respectively. A study of formation of thin films of pure and doped perovskites through three different processes—spinning, dipping, and spray process—has been carried out that revealed that Ti‐based BiFeO3 in spinning process have given the best results.312 Lead‐free Ti‐based perovskites have been investigated for their photovoltaic behavior.313 Transition metal palladium‐based lead‐free perovskite Cs2PdBr6 nanocrystals have been reported with an average particle diameter of 2.8 nm and a thickness of 1–2 units cells exhibiting a narrow bandgap of 1.69 eV and outstanding stability toward light humidity and heat. The fast anion exchange method has been employed to synthesize Cs2PdI6 nanocrystals.314
Lead‐free (1−x) (K0.44Na0.52Li0.04)(Nb0.91Ti0.05Sb0.04)O3‐xSmAlO3 [x = 0, 0.001, 0.004, 0.004, 0.008] ceramics have been synthesized by a solid‐state sintering method. The effect of doping of SmAlO3 on the phase structure and electrical properties of all the perovskite composition for reported values of x have been investigated thoroughly. From the study of XRD analysis, all the investigated composition reported a perovskite structure at the suitable sintering temperature. The enhanced electrical properties were obtained at the sintering temperature of 1180 °C.315 Lead‐free multiferroic (1−x) KNbO3‐(x)CoFe2O4 composites have been synthesized by employing solid‐state reaction method with x (0, 0.25, 0.5, 0.75, 1.0) mol. The careful study of XRD reveals that KNbO3 perovskite belong to an orthorhombic system, spinal CoFe2O4 belong to cubic system, and other compositions of x belong to mixed phase of KNbO3 and CoFe2O4. The high‐resolution SEM analysis has shown that the morphology of KNbO3 and CoFe2O4 was modified by CoFe2O4 content. The composite 0.5KNbO3 0.5 CoFe2O4 displayed a high value of coercivity and 0.5KNbO3 0.5CoFe2O4 and 0.75KNbO3 0.75 CoFe2O4 displayed an enhanced value of dielectric constant.316 At present, a lot of research is going on lead‐free double perovskite materials to explore their potential as a light absorber in perovskite solar device. Double perovskite A2B′B″X6[A‐Cs, MA, B′‐Bi, Sb, B″‐Cu, Ag, X‐Cl, Br, I] have been investigated for their structural, optical, and stability properties.317 The vapor‐assisted method has been employed to synthesize double perovskite Cs2AgBiB6 thin films with better morphology. The better quality of Cs2AgBiB6 films has a photoluminescence lifetime of 117 ns. The fabricated n‐i‐p perovskite solar cell has reported a SPCE of 1.37% with a better stability of 90% after 240 h of storage under ambient conditions.318 The diffusion of X halide anion in lead‐free double perovskite Cs2AgBiX6 [X‐Cl, Br], Cs2AgSbCl6, Cs2AgInCl6 has been investigated by using first principle calculations. The calculated values of formation energy of X‐site vacancies are related to electronic configuration of B‐site cations. The double perovskite Cs2AgInCl6 is having lowest vacancy formation energy due to unfilled s‐orbital of In3+. The hysteresis loss in Cs2AgBiBr6 solar cells is attributed to the lowest energy barrier for X‐site migration.319 Double perovskite lead‐free layered Cs4CuSb2Cl12 have been reported with a bandgap of 1 eV prepared by grinding of precursor salts at ambient conditions. A long range magnetic ordering is displayed by the synthesized perovskite at room temperature that plays a pivotal role in controlling the electronic properties of double perovskite Cs4CuSb2Cl16.320 By using first principle calculations, lead‐free double perovskite Cs2NaBX6[B‐Sb, Bi, X‐Cl, Br, I] have been synthesized and characterized for their electronic and optical properties. The simple solution method has been employed to prepare a layered MA3Bi2I9 perovskite and a composite layer of bismuth tri iodide (BiI3). By employing SEM and XRD techniques, the morphology of the active layer has been investigated that has a direct influence on performance of the perovskite device.321 The high‐temperature solid‐state reaction method has been employed to prepare polycrystalline material of double perovskite Dy2NiMnO6 with a monoclinic structure. The high‐temperature condition of the material is attributed to the presence of oxygen vacancies making it viable to use at different temperatures.322
9. Conclusion
The research in tin‐based perovskites MASnX3 has revealed a direct bandgap of 1.20–1.35 eV, electron mobility of 2320 cm2 V−1 s−1, hole carrier mobility of 322 cm2 V−1 s−1, and long charge carrier diffusion length of more than 500 nm. The alteration of Br−/I− ratio in MASnI3− x Brx has resulted in large value of V OC (0.88 V) in MASnBr3 and J SC (12.33 mA cm−2) in MASnIBr2. The absorption band can be tuned by altering the composition of halide anions in MASnX3 perovskites. However, Sn‐based perovskites suffer from degradation in air due to oxidation of Sn2+ into Sn4+. The incorporation of additives has resulted in reduced oxidation and better stability in air. The A‐site cation has a significant effect on photovoltaic performance. The use of diethylammonium (en) and FA+ at the A‐site of ASnX3 has resulted in wider bandgaps and improved stability. An efficiency of 8% has been achieved for (FA)0.75(MA)0.25SnI3 with a V OC (0.61 V) and bandgap (1.33 eV). Germanium‐based perovskites do have an optical bandgap of 1.63 eV, excellent hole and electron conducting behavior, and better stability in air. Using DFT calculations, it has been reported that with increase of size of halide anion, the bandgaps have decreasing values of 3.7, 2.81, and 1.61 eV, respectively. The replacement of the iodide content in AGeI3 by bromide results in enhanced photovoltaic performance and stability to a slight extent. Mixed Ge‐based perovskite RbSn0.5Ge0.5I3 exhibits a better optical absorption and effective masses for higher carrier mobility and good stability in water. By engineering the size of A‐site cation, its doping with another suitable cation and size of halide anion, it is possible to fabricate a Ge‐based perovskite as an efficient light absorber. Although, bismuth‐based perovskite (MA)3Bi2I9 has displayed low values of solar power conversion efficiencies up to 1.64 eV up to now, yet they have exhibited excellent stability in ambient air at room temperature and against exposure to humidity .The morphology of (MA)3Bi2I9 films can be enhanced by addition of various concentration of NMP into the precursor solution that not only controlled the rate of crystallization but also enhanced the efficiency and stability in a relative humidity of 50–60%. The concentration of perovskite solution and substrate temperature also impacts its efficiency and stability. The wide bandgaps of lead‐free perovskites can be engineered to a narrow bandgap by incorporating triiodide into P(4‐methyl piperidinium)3Bi2I9(MP‐Bi2I9) that exhibited a bandgap of 1.58 eV in comparison to 1.5 eV of MAPbI3. The various deposition methods have a direct influence on morphology of films. Cs3Bi2I9 has displayed an efficiency of 8% with a pure crystalline phase and stability. Bismuth‐based double perovskite like Cs2AgBiBr6 exhibited an indirect bandgap of 2.19 eV. The DFT calculations have further revealed that the family of 3D double perovskites have optical bandgap in the visible range and low carrier effective masses. Bismuth‐based perovskites can be thoroughly investigated for enhancement in their efficiency as these materials have excellent stability in ambient air and in relative humidity. In antimony‐based perovskites, the size of cationic or anionic species and the employed processing technique determine the structure. When A‐site cation Cs+ is replaced by a smaller cation Rb+, a 2D layered phase is achieved with a formation energy difference of 0.25 eV in comparison to Cs‐based counterparts. The addition of additive in 0D (MA)3Sb2I9 films has resulted in enhanced light absorption in the visible wavelength regions up to 400 nm. The use of chalcogenide and mixed perovskite materials can be an effective strategy for formation of efficient, cheap, and stable solar cells. Cs4CuSb2Cl12, besides having photo and thermal stability and resistance to humidity, have exhibited excellent photovoltaic properties. There has been a significant effect on photovoltaic parameters on substitution of Sb with Bi in 2D mixed layered perovskites (NH4)3(Sb1− xBix)2I9. By proper substitution of Bi into antimony‐based perovskites, it is possible to fabricate light harvesters with high efficiency and stability. Copper‐based perovskites usually form 2D layered structure owing to their smaller ionic radii. By proper tuning of Cl−/Br− ratio, the optical absorption of Cu‐based perovskites can be extended in the near‐infrared region. The (MA)2CuCl2Br2 and (MA)2CuCl0.5Br3.5 have reported better stability under ambient conditions. The divalent cations of alkaline earth metals like Mg2+, Ca2+, and Sr2+ can be effective replacement for lead. The perovskite solar cells based on transition metals Ti, Ni, and Cd‐doped BiFeO3 as a light absorber have displayed V OC values of 0.77, 0.56, and 0.49 V, respectively. By suitable selection of A and B‐site cations and halide anions, their alteration in composition and synthesis method, it is possible to fabricate lead‐free perovskites with maximum efficiency and stability without any toxic influence on environment.
Conflict of Interest
The authors declare no conflict of interest.
Kour R., Arya S., Verma S., Gupta J., Bandhoria P., Bharti V., Datt R., Gupta V., Potential Substitutes for Replacement of Lead in Perovskite Solar Cells: A Review. Global Challenges 2019, 3, 1900050 10.1002/gch2.201900050
References
- 1. Tanaka H., Misono M., Curr. Opin. Solid State Mater. Sci. 2001, 5, 381. [Google Scholar]
- 2. Yi Z., Ladi N. H., Shai X., Li H., Shen Y., Wang M., Nanoscale Adv. 2019, 1, 1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. He T., Huang Q., Ramirez A., Wang Y., Regan K., Rogado N., Hayward M. A., Haas M. K., Slusky J. S., Inumara K., Zandbergen H. W., Ong N. P., Cava R. J., Nature 2001, 411, 54. [DOI] [PubMed] [Google Scholar]
- 4. Goldschmidt V. M., Die Naturwiss. 1926, 14, 477. [Google Scholar]
- 5. Liu X., Hong R., Tian C., J. Mater. Sci.: Mater. Electron. 2009, 20, 323. [Google Scholar]
- 6. Kronmuller H., Parkin S., Handbook of Magnetism and Advanced Magnetic Materials, Wiley, Hoboken, NJ: 2007. [Google Scholar]
- 7. Buttner R. H., Maslen E. N., Acta Crystallogr., Sect. B: Struct. Sci. 1992, 48, 764. [Google Scholar]
- 8. Szuromi P., Grocholski B., Science 2017, 358, 732. [DOI] [PubMed] [Google Scholar]
- 9. Saparov B., Mitzi D. B., Chem. Rev. 2016, 116, 4558. [DOI] [PubMed] [Google Scholar]
- 10. Kieslich G., Sun S., Cheetham A. K., Chem. Sci. 2014, 5, 4712. [Google Scholar]
- 11. Kieslich G., Sun S., Cheetham A. K., Chem. Sci. 2015, 6, 3430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Shannon R. D., Acta Crystallogr., Sect. A: Found. Adv. 1976, 32, 751. [Google Scholar]
- 13. Chen Q., De Marco N., Wang Y., Song Y., Chen C. C., Zhao H., Hong Z., Zhou H., Yang Y., Nano Today 2015, 10, 355. [Google Scholar]
- 14. Li Z., Yang M., Park J. S., Wei S. H., Berry J. J., Zhu K., Chem. Mater. 2016, 28, 284. [Google Scholar]
- 15. Wang Q., Phung N., Girolamo D. D., Vivoc P., Abate A., Energy Environ. Sci. 2019, 12, 865. [Google Scholar]
- 16. Lee J. W., Kim D. H., Kim H. S., Seo S. W., Cho S. M., Park N. G., Adv. Energy Mater. 2015, 5, 1501310. [Google Scholar]
- 17. McMeekin D. P., Sadoughi G., Rehman W., Eperon G. E., Saliba M., Hörantner M. T., Haghighirad A., Sakai N., Korte L., Rech B., Johnston M. B., Herz L. M., Snaith H. J., Science 2016, 351, 151. [DOI] [PubMed] [Google Scholar]
- 18. Shah S., Thin Film Share to Decline to 7% in 2015—The Lowest so Far!, http://www.greenworldinvestor.com/2015/03/24/thin‐film‐share‐to‐declineto‐7‐in‐2015‐the‐lowest‐so‐far/ (accessed: June 2017).
- 19. Ruhle S., Sol. Energy 2016, 130, 139. [Google Scholar]
- 20. Bullis K., Record‐Breaking Solar Cell Points the Way to Cheaper Power, MIT Technology Review, 2014. [Google Scholar]
- 21. Dimroth F., Tibbits T. N. D., Niemeyer M., Predan F., Beutel P., Karcher C., Oliva E., Siefer G., Lackner D., Fuß‐Kailuweit P., Bett A. W., Krause R., Drazek C., Guiot E., Wasselin J., Tauzin A., Signamarcheix T., IEEE J. Photovoltaics 2016, 6, 343. [Google Scholar]
- 22. Zyg L., “Solar cell sets world record with a stabilized efficiency of 13.6%,” https://Phys.org (accessed: June 2015).
- 23. Essig S., Allebe C., Remo T., Geisz J. F., Steiner M. A., Horowitz K., Barraud L., Ward J. S., Schnabel M., Nat. Energy 2017, 2, 17144. [Google Scholar]
- 24. Green M. A., Emery K., Hishikawa Y., Warta W., Dunlop E. D., Prog. Photovoltaics 2013, 21, 1. [Google Scholar]
- 25. Parida B., Iniyan S., Goic R., Renewable Sustainable Energy Rev. 2011, 15, 1625. [Google Scholar]
- 26. Mathew S., Yella A., Gao P., Humphry‐Baker R., Curchod B. F. E., Ashari‐Astani N., Tavernelli I., Rothlisberger U., Nazeeruddin Md. K., Grätzel M., Nat. Chem. 2014, 6, 242. [DOI] [PubMed] [Google Scholar]
- 27. Perovskite Offers Shot at Cheaper Solar Energy , http://www.wsj.com/articles/perovskite‐offers‐shot‐at‐cheaper‐solar‐energy‐1411937799 (accessed: May 2015).
- 28. Kojima A., Teshima K., Shirai Y., Miyasaka T., J. Am. Chem. Soc. 2009, 131, 6050. [DOI] [PubMed] [Google Scholar]
- 29. Egger D. A., Edri E., Cahen D., Hodes G., J. Phys. Chem. Lett. 2015, 6, 279. [DOI] [PubMed] [Google Scholar]
- 30. Zhang M., Lyu M., Yu H., Yun J. H., Wang Q., Wang L., Chem. ‐ Eur. J. 2015, 21, 434. [DOI] [PubMed] [Google Scholar]
- 31. Shen H., Duong T., Wu Y., Peng J., Jacobs D., Wu N.,Weber K., White T., Catchpole K., Sci. Technol. Adv. Mater. 2018, 19, 53. [Google Scholar]
- 32. Liu M., Johnston M. B., Snaith H. J., Nature 2013, 501, 395. [DOI] [PubMed] [Google Scholar]
- 33. Zhou H., Chen Q., Li G., Luo S., Song T., Duan H. S., Hong Z., You J., Liu Y., Yang Y., Science 2014, 345, 542. [DOI] [PubMed] [Google Scholar]
- 34. Jeon N. J., Lee H. G., Kim Y. C., Seo J., Noh J. H., Lee J., Seok S., J. Am. Chem. Soc. 2014, 136, 7837. [DOI] [PubMed] [Google Scholar]
- 35. Heo J. H., Song D. H., Han H. J., Kim S. Y., Kim J. H., Kim D., Shin H. W., Ahn T. K., Wolf C., Lee T. W., Im S. H., Adv. Mater. 2015, 27, 3424. [DOI] [PubMed] [Google Scholar]
- 36. Green M. A., Bein T., Nat. Mater. 2015, 14, 559. [DOI] [PubMed] [Google Scholar]
- 37. Stranks S. D., Snaith H. J., Nat. Nanotechnol. 2015, 10, 391. [DOI] [PubMed] [Google Scholar]
- 38. Green M. A., Ho‐Baillie A. H., Snaith H. J., Nat. Photonics 2014, 8, 506. [Google Scholar]
- 39. Stranks S. D., Eperon G. E., Grancini G., Menelaou C., Marcelo, Alcocer J. P., Leijtens T., Herz L. M., Petrozza A., Snaith H. J., Science 2013, 342, 341. [DOI] [PubMed] [Google Scholar]
- 40. Song K., Wu Y., Chen X., He Y., Liu L., Chen G., Liu R., AIP Adv. 2018, 8, 035114. [Google Scholar]
- 41. Kim H. S., Lee C. R., Im J. H., Lee K. B., Moehl T., Marchioro A., Moon S. J., Baker R. H., Yum J. H., Moser J. E., Grätzel M., Park N. G., Sci. Rep. 2012, 2, 591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Noh J. H., Im S. H., Heo J. H., Mandal T. N., Seok S. I., Nano Lett. 2013, 13, 1764. [DOI] [PubMed] [Google Scholar]
- 43. Ball J. M., Lee M. M., Hey A., Snaith H. J., Energy Environ. Sci. 2013, 6, 1739. [Google Scholar]
- 44. Burschka J., Pellet N., Moon S.‐J., Humphry‐Baker R., Gao P., Nazeeruddin M. K., Grätzel M., Nature 2013, 499, 316. [DOI] [PubMed] [Google Scholar]
- 45. Abrusci A., Stranks S. D., Docampo P., Yip H.‐L.,Jen A. K.‐Y., Snaith H. J., Nano Lett. 2013, 13, 3124. [DOI] [PubMed] [Google Scholar]
- 46. Kojima A., Teshima K., Shirai Y., Miyasaka T., presented at 210th ECS Meeting Abstract, 2006. [Google Scholar]
- 47. Kojima A., Teshima K., Shirai Y., Miyasaka T., presented at 210th ECS Meeting Abstract, 2008. [Google Scholar]
- 48. Im J. H., Lee C. R., Lee J. W., Park S. W., Park N. G., Nanoscale 2011, 3, 4088. [DOI] [PubMed] [Google Scholar]
- 49. Bach U., Lupo D., Comte P., Moser J. E., Weissortel F., Salbeck J., Spreitzer H., Gratzel M., Nature 1998, 395, 583. [Google Scholar]
- 50. Burschka J., Dualeh A., Kessler F., Baranoff E., Cevey‐Ha N. L., Yi C. Y., Nazeeruddin M. K., Gratzel M., J. Am. Chem. Soc. 2011, 133, 18042. [DOI] [PubMed] [Google Scholar]
- 51. Lee M. M., Teuscher J., Miyasaka T., Murakami T. N., Snaith H. J., Science 2012, 338, 643. [DOI] [PubMed] [Google Scholar]
- 52. Burschka J., Pellet N., Moon S. J., Baker R. H., Gao P., Nazeeruddin M. K., Gratzel M., Nature 2013, 499, 316. [DOI] [PubMed] [Google Scholar]
- 53. Liu X., Chueh C. C., Zhu Z., Jo S. B., San Y., Jen A. K. Y., J. Mater. Chem. A 2016, 4, 15294. [Google Scholar]
- 54. You J., Meng L., Song T.‐B., Guo T.‐F., (Michael) Yang Y., Chang W.‐H., Hong Z., Chen H., Zhou H., Chen Q., Liu Y., De Marco N., Yang Y., Nat. Nanotechnol. 2016, 11, 75. [DOI] [PubMed] [Google Scholar]
- 55. Yang W. S., Park B. W., Jung E. H., Jeon N. J., Kim Y. C., Lee D. U., Shin S. S., Seo J., Kim E. K., Noh J. H., Seok S. I., Science 2017, 356, 1376. [DOI] [PubMed] [Google Scholar]
- 56. Dong X., Fang X., Lv M., Lin B., Zhang S., Ding J., Yuan N., J. Mater. Chem. A 2015, 3, 5360. [Google Scholar]
- 57. Han Y., Meyer S., Dkhissi Y., Weber K., Pringle J. M., Bach U., Spiccia L., Cheng Y. B., J. Mater. Chem. A 2015, 3, 8139. [Google Scholar]
- 58. Yang J., Siempelkamp B. D., Liu D., Kelly T. L., ACS Nano 2015, 9, 1955. [DOI] [PubMed] [Google Scholar]
- 59. Leijtens T., Eperon G. E., Pathak S., Abate A., Lee M. M., Snaith H. J., Nat. Commun. 2013, 4, 2885. [DOI] [PubMed] [Google Scholar]
- 60. Ono L. K., Raga S. R., Wang S., Kato Y., Qi Y., J. Mater. Chem. A 2015, 3, 9074. [Google Scholar]
- 61. Habisreutinger S. N., Leijtens T., Eperon G. E., Stranks S. D., Nicholas R. J., Snaith H. J., Nano Lett. 2014, 14, 5561. [DOI] [PubMed] [Google Scholar]
- 62. Snaith H. J., Abate A., Ball J. M., Eperon G. E., Leijtens T., Noel N. K., Stranks S. D., Wang J. T., Wojciechowski K., Zhang W. J., J. Phys. Chem. Lett. 2014, 5, 1511. [DOI] [PubMed] [Google Scholar]
- 63. Park N. G., Gratzel M., Miyasaka T., Zhu K., Emery K., Nat. Energy 2016, 1, 16152. [Google Scholar]
- 64. Yuan Y., Huang J., Acc. Chem. Res. 2016, 49, 286. [DOI] [PubMed] [Google Scholar]
- 65. Liu J., Li N., Jia J., Dong J., Qiu Z., Iqbal S., Cao B., Sol. Energy 2019, 181, 285. [Google Scholar]
- 66. Da P., Zheng G., Nano Res. 2017, 10, 1471. [Google Scholar]
- 67. Jeng J. Y., Chiang Y. F., Lee M. H., Peng S. R., Guo T. F., Chen P., Wen T. C., Adv. Mater. 2013, 25, 3727. [DOI] [PubMed] [Google Scholar]
- 68. Peng H., Sun W., Li Y., Ye S., Rau H., Yan W., Zhou H., Bian Z., Huang C., Nano Res. 2016, 9, 2960. [Google Scholar]
- 69. Bai Y., Meng S., Yang S., Adv. Energy Mater. 2018, 8, 1701883. [Google Scholar]
- 70. Liu T., Chen K., Hu Q., Zhu R., Gong Q., Adv. Energy Mater. 2016, 6, 1600457. [Google Scholar]
- 71. Atabaev T. S., Mater. Today: Proc. 2017, 4, 4919. [Google Scholar]
- 72. Liu Y., Ji S., Li S., He W., Wang K., Hu H., Ye C., J. Mater. Chem. A 2015, 3, 14902. [Google Scholar]
- 73. Jeon N. J., Noh J. H., Yang W. S., Kim Y. C., Ryu S., Seo J., Seok S. I., Nature 2015, 517, 476. [DOI] [PubMed] [Google Scholar]
- 74. Yang W. S., Noh J. H., Jeon N. J., Kim Y. C., Ryu S., Seo J., Seok S., Science 2015, 348, 1234. [DOI] [PubMed] [Google Scholar]
- 75. Bi D., Tress W., Dar M. I., Gao P., Luo J., Renevier C., Schenk K., Abate A., Giordano F., Baena J.‐P. C., Decoppet J.‐D., Zakeeruddin S. M., Nazeeruddin M. K., Grätzel M., Hagfeldt A., Sci. Adv. 2016, 2, e1501170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Snaith H. J., J. Phys. Chem. Lett. 2013, 4, 3623. [DOI] [PubMed] [Google Scholar]
- 77. Wehrenfennig C., Eperon G. E., Johnston M. B., Snaith H. J., Herz L. M., Adv. Mater. 2014, 26, 1584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Eperon G. E., Stranks S. D., Menelaou C., Johnston M. B., Herz L. M., Snaith H. J., Energy Environ. Sci. 2014, 7, 982. [Google Scholar]
- 79. Sadhanala A., Deschler F., Thomas T. H., Dutton S. E., Goedel K. C., Hanusch F. C., Lai M. L., Steiner U., Bein T., Docampo P., Cahen D., Friend R. H., J. Phys. Chem. Lett. 2014, 5, 2501. [DOI] [PubMed] [Google Scholar]
- 80. Sadhanala A., Kumar A., Pathak S., Rao A., Steiner U., Greenham N. C., Snaith H. J., Friend R. H., Adv. Electron. Mater. 2015, 1, 1500008. [Google Scholar]
- 81. De Wolf S., Holovsky J., Moon S. J., Loper P., Niesen B., Ledinsky M., Haug F. J., Yum J. H., Ballif C., J. Phys. Chem. Lett. 2014, 5, 1035. [DOI] [PubMed] [Google Scholar]
- 82. Giustino F., Snaith H. J., ACS Energy Lett. 2016, 1, 1233. [Google Scholar]
- 83. Xing G., Mathews N., Sun S., Lim S. S., Lam Y. M., Grätzel M., Mhaisalkar S., Sum T. C., Science 2013, 342, 344. [DOI] [PubMed] [Google Scholar]
- 84. Dong Q., Fang Y., Shao Y., Mulligan P., Qiu J., Cao L., Huang J., Science 2015, 347, 967. [DOI] [PubMed] [Google Scholar]
- 85. Miyata A., Mitioglu A., Plochocka P., Portugall O., Wang J. T. W., Stranks S. D., Snaith H. J., Nicholas R. J., Nat. Phys. 2015, 11, 582. [Google Scholar]
- 86. Stoumpos C. C., Malliakas C. D., Kanatzidis M. G., Inorg. Chem. 2013, 52, 9019. [DOI] [PubMed] [Google Scholar]
- 87. Kamat P. V., Bisquert J., Buriak J., ACS Energy Lett. 2017, 2, 904. [Google Scholar]
- 88. Wang A., Yan X., Zhang M., Sun S., Yang M., Shen W., Pan X., Wang P., Deng Z., Chem. Mater. 2016, 28, 8132. [Google Scholar]
- 89. de Quilettes D. W., Vorpahl S. M., Stranks S. D., Nagaoka H., Eperon G. E., Ziffer M. E., Snaith H. J., Ginger D. S., Science 2015, 348, 683. [DOI] [PubMed] [Google Scholar]
- 90. Filip M. R., Eperon G. E., Snaith H. J., Giustino F., Nat. Commun. 2014, 5, 5757. [DOI] [PubMed] [Google Scholar]
- 91. Pazos‐Outon L. M. P., Szumilo M., Lamboll R., Richter J. M., Crespo‐Quesada M., Abdi‐Jalebi M., Beeson H. J., Vrućinić M., Alsari M., Snaith H. J., Ehrler B., Friend R. H., Deschler F., Science 2016, 351, 1430. [DOI] [PubMed] [Google Scholar]
- 92. World Health Organisation , Exposure to Lead: A Major Public Health Concern, 2010, http://www.who.int/ipcs/features/lead.pdf (accessed: July 2017).
- 93. World Health Organisation , International Program on Chemical Safety: Ten Chemicals of Major Public Health Concern, 2010, http://www.who.int/ipcs/assessment/public_health/chemicals_phc/en/.
- 94. Needleman H., Annu. Rev. Med. 2004, 55, 209. [DOI] [PubMed] [Google Scholar]
- 95. Babayigit A., Ethirajan A., Muller M., Conings B., Nat. Mater. 2016, 15, 247. [DOI] [PubMed] [Google Scholar]
- 96. Nordberg G. F., Fowler B. A., Nordberg M., Handbook on the Toxicology of Metals, Elsevier, Amsterdam: 2015. [Google Scholar]
- 97. Patrick L., Med. Rev. 2006, 11, 2. [PubMed] [Google Scholar]
- 98. Berhe T. A., Su W. N., Chen C. H., Pan C. J., Cheng J. H., Chen H. M., Tsai M. C., Chen L. Y., Dubale A. A., Hwang B. J., Energy Environ. Sci. 2016, 9, 323. [Google Scholar]
- 99. Leijtens T., Eperon G. E., Noel N. K., Habisreutinger S. N., Petrozza A., Snaith H. J., Adv. Energy Mater. 2015, 5, 1500963. [Google Scholar]
- 100. Kosasih F. U., Ducati C., Nano Energy 2018, 47, 243. [Google Scholar]
- 101. Hailegnaw B., Kirmayer S., Edri E., Hodes G., Cahen D., J. Phys. Chem. Lett. 2015, 6, 1543. [DOI] [PubMed] [Google Scholar]
- 102. Gupta S., Bendikov T., Hodes G., Cahen D., ACS Energy Lett. 2016, 1, 1028. [Google Scholar]
- 103. Sha W. E., Ren X., Chen L., Choy W. C., Appl. Phys. Lett. 2015, 106, 221104. [Google Scholar]
- 104. Bisquert J., J. Phys. Chem. Lett. 2013, 4, 2597. [Google Scholar]
- 105. Tsarev S., Boldyreva A. G., Luchkin S. Yu., Elshobaki M., Afanasov M. I., Stevenson K. J., Troshinab P. A., J. Mater. Chem. A 2018, 6, 21389. [Google Scholar]
- 106. Hao F., Stoumpos C. C., Cao D. H., Chang R. P. H., Kanatzidis M. G., Nat. Photonics 2014, 8, 489. [Google Scholar]
- 107. Takahashi Y., Hasegawa H., Takahashi Y., Inabe T., J. Solid State Chem. 2013, 205, 39. [Google Scholar]
- 108. Herz L. M., ACS Energy Lett. 2017, 2, 1539. [Google Scholar]
- 109. Koh T. M., Krishnamoorthy T., Yantara N., Shi C., Leong W. L., Boix P. P., Grimsdale A. C., Mhaisalkar S. G., Mathews N., J. Mater. Chem. A 2015, 3, 14996. [Google Scholar]
- 110. Shum K., Chen Z., Qureshi J., Yu C., Wang J. J., Pfenninger W., Vockic N., Midgley J., Kenney J. T., Appl. Phys. Lett. 2010, 96, 221903. [Google Scholar]
- 111. Park B. W., Phitippe B., Zhang S., Rensmo H., Boschiloo G., Johansson E. M. J., Adv. Mater. 2015, 27, 6806. [DOI] [PubMed] [Google Scholar]
- 112. Ghosh B., Wu B., Mulmudi H. K., Guet C., Weber K., Sum T. C., Mhaisalkar S., Mathews N., ACS Appl. Mater. Interfaces 2018, 10, 35000. [DOI] [PubMed] [Google Scholar]
- 113. Zhao Y. Q., Wang X., Liu B., Yu Z.‐L., He P.‐B., Wan Q., Cai M. Q., Yu H. L., Org. Electron. 2018, 53, 50. [Google Scholar]
- 114. Saidaminov M. I., Abdelhady A. L., Murali B., Alarousu E., Burlakov V. M., Peng W., Dursun I., Wang L., He Y., Maculan G., Goriely A., Wu T., Mohammed O. F., Bakr O. M., Nat. Commun. 2015, 6, 7586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Xiao Z., Zhou Y., Hosono H., Kamiya T., Phys. Chem. Chem. Phys. 2015, 17, 18900. [DOI] [PubMed] [Google Scholar]
- 116. Zuo C., Ding L., Angew. Chem. 2017, 129, 6628. [Google Scholar]
- 117. Ma L., Hao F., Stoumpos C. C., Phelan B. T., Wasielewski M. R., Kanatzidis M. G., J. Am. Chem. Soc. 2016, 138, 14750. [DOI] [PubMed] [Google Scholar]
- 118. Wu B., Zhou Y., Xing G., Xu Q., Garces H. F., Solanki A., Goh T. W., Padture N. P., Sum T. C., Adv. Funct. Mater. 2017, 27, 1604818. [Google Scholar]
- 119. Chen Z., Yu C., Shum K., Wang J. J., Pfenninger W., Vockic N., Midgley J., Kenney J. T., J. Lumin. 2012, 132, 345. [Google Scholar]
- 120. Noel N. K., Stranks S. D., Abate A., Wehrenfennig C., Guarnera S., Haghighirad A.‐A., Sadhanala A., Eperon G. E., Pathak S. K., Johnston M. B., Petrozza A., Herza L. M., Snaith H. J., Energy Environ. Sci. 2014, 7, 3061. [Google Scholar]
- 121. Mitzi D. B., Feild C. A., Schlesinger Z., Laibowitz R. B., J. Solid State Chem. 1995, 114, 159. [Google Scholar]
- 122. Milot R. L., Eperon G. E., Green T., Snaith H. J., Johnston M. B., Herz L. M., J. Phys. Chem. Lett. 2016, 7, 4178. [DOI] [PubMed] [Google Scholar]
- 123. Chung I., Song J. H., Im J., Androulakis J., Malliakas C. D., Li H., Freeman A. J., Kenney J. T., Kanatzidis M. G., J. Am. Chem. Soc. 2012, 134, 8579. [DOI] [PubMed] [Google Scholar]
- 124. Kagan C. R., Mitzi D. B., Dimitrakopoulos C. D., Science 1999, 286, 945. [DOI] [PubMed] [Google Scholar]
- 125. Matsushima T., Hwang S., Sandanayaka A. S. D., Qin C., Terakawa S., Fujihara T., Yahiro M., Adachi C., Adv. Mater. 2016, 28, 10275. [DOI] [PubMed] [Google Scholar]
- 126. Bhatia1 A., Hautier G., Nilgiansku T., Miglio A., Rignanese G. M., Gonze X., Suntivich J., Chem. Mater. 2016, 28, 30. [Google Scholar]
- 127. Lyu M., Yun J.‐H., Cai M., Jiao Y., Bernhardt P. V., Zhang M., Wang Q., Du A., Wang H., Liu G., Wang L., Nano Res. 2016, 9, 692. [Google Scholar]
- 128. Abulikemu M., Chikh S. O., Miao X., Alarousu E., Murali B., Ndjawa G. O. N., Barbé J., El Labban A., Amassian A., Gobbo S. D., J. Mater. Chem. A 2016, 4, 12504. [Google Scholar]
- 129. Funabiki F., Toda Y., Hosono H., J. Phys. Chem. C 2018, 122, 10749. [Google Scholar]
- 130. Sanchez R. S., Mas‐Marza E., Sol. Energy Mater. Sol. Cells 2016, 158, 189. [Google Scholar]
- 131. Chen M., Ju M.‐G., Garces H. F., Carl A. D., Ono L. K., Hawash Z., Zhang Y., Shen T., Qi Y., Grimm R. L., Pacifici D., Zeng X. C., Zhou Y., Padture N. P., Nat. Commun. 2019, 10, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Abate A., Hollman D. J., Teuscher J., Pathak S., Avolio R., Errico G. D., Vitiello G., Fantacci S., Snaith H. J., J. Am. Chem. Soc. 2013, 135, 13538. [DOI] [PubMed] [Google Scholar]
- 133. Zhao X., Wang M., Mater. Today Energy 2018, 7, 208. [Google Scholar]
- 134. Cappel U. B., Daeneke T., Bach U., Nano Lett. 2012, 12, 4925. [DOI] [PubMed] [Google Scholar]
- 135. Cui X. P., Jiang K. J., Huang J. H., Zhang Q. Q., Su M. J., Yang L. M., Song Y. L., Zhou X. Q., Synth. Met. 2015, 209, 247. [Google Scholar]
- 136. Cortecchia D., Dewi H. A., Yin J., Bruno A., Chen S., Baikie T., Boix P. P., Gratzel M., Mhaisalkar S., Soci C., Mathews N., Inorg. Chem. 2016, 55, 1044. [DOI] [PubMed] [Google Scholar]
- 137. Li X., Zhong X., Hu Y., Li B., Sheng Y., Zhang Y., Weng C., Feng M., Han H., Wang J., J. Phys. Chem. Lett. 2017, 8, 1804. [DOI] [PubMed] [Google Scholar]
- 138. Zhao D., Sexton M., Park H.‐Y., Baure G., Nino J. C., So F., Adv. Energy Mater. 2015, 5, 1401855. [Google Scholar]
- 139. Ryu S., Noh J. H., Jeon N. J., Kim Y. C., Yang W. S., Seo J., Seok S. I., Energy Environ. Sci. 2014, 7, 2614. [Google Scholar]
- 140. Saparov B., Hong F., Sun J. P., Duan H. S., Meng W., Cameron S., Hill I. G., Yan Y., Mitzi D. B., Chem. Mater. 2015, 27, 5622. [Google Scholar]
- 141. Heo J. H., Lee M. H., Song D. H., Song C. E., Lee J. J., Hong K. H., Im S. H., Nanosci. Nanotechnol. Lett. 2018, 10, 591. [Google Scholar]
- 142. Ke W., Stoumpos C. C., Spanopoulos I., Mao L., Chen M., Wasielewski M. R., Kanatzidis M. G., J. Am. Chem. Soc. 2017, 139, 14800. [DOI] [PubMed] [Google Scholar]
- 143. Oz S., Hebig J.‐C., Jung E., Singh T., Lepcha A., Olthof S., Flohre J., Gao Y., German R., van Loosdrecht P. H. M., Meerholz K., Kirchartz T., Mathur S., Sol. Energy Mater. Sol. Cells 2016, 158, 195. [Google Scholar]
- 144. Krishnamoorthy T., Ding H., Yan C., Leong W. L., Baikie T., Zhang Z., Sherburne M., Li S., Asta M., Mathews N., Mhaisalkar S. G., J. Mater. Chem. A 2015, 3, 23829. [Google Scholar]
- 145. Shao S., Liu J., Portale G., Fang H. H., Blake G. R., Brink G. H., Koster L. A., Loi M. A., Adv. Energy Mater. 2018, 8, 1702019. [Google Scholar]
- 146. Liu X., Wang Y., Xie F., Yang X., Han L., ACS Energy Lett. 2018, 3, 1116. [Google Scholar]
- 147. Hebig J. C., Kühn I., Flohre J., Kirchartz T., ACS Energy Lett. 2016, 1, 309. [Google Scholar]
- 148. Wong K. W., Ip H. L. Y, Luo Y., Wong K. Y., Lau W. M., Low K. H., Chow H. F., Gao Z. Q., Yeung W. L., Chang C. C., Appl. Phys. Lett. 2002, 80, 2788. [Google Scholar]
- 149. Huang D., Goh T., Kong J., Zheng Y., Zhao S., Xu Z., Taylor A. D., Nanoscale 2017, 9, 4236. [DOI] [PubMed] [Google Scholar]
- 150. Zhao Z., Gu F., Li Y., Sun W., Ye S., Rao H., Liu Z., Bian Z., Huang C., Adv. Sci. 2017, 4, 1700204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Yu Y., Zhao D., Grice C. R., Meng W., Wang C., Liao W., Cimaroli A. J., Zhang H., Zhu K., Yan Y., RSC Adv. 2016, 6, 90248. [Google Scholar]
- 152. Liang P. W., Liao C. Y., Chueh C. C., Zuo F., Williams S. T., Xin X. K., Lin J., Jen A. K. Y., Adv. Mater. 2014, 26, 3748. [DOI] [PubMed] [Google Scholar]
- 153. Sun S., Salim T., Mathews N., Duchamp M., Boothroyd C., Xing G., Sum T. C., Lam Y. M., Energy Environ. Sci. 2014, 7, 399. [Google Scholar]
- 154. Xiao Z., Bi C., Shao Y., Dong Q., Wang Q., Yuan Y., Wang C., Gao Y., Huang J., Energy Environ. Sci. 2014, 7, 2619. [Google Scholar]
- 155. Seo J., Park S., Kim Y. C., Jeon N. J., Noh J. H., Yoon S. C., Seok S. I., Energy Environ. Sci. 2014, 7, 2642. [Google Scholar]
- 156. Liao Y., Liu H., Zhou W., Yang D., Shang Y., Shi Z., Li B., Jiang X., Zhang L., Quan L. N., Quintero‐Bermudez R., Sutherland B. R., Mi Q., Sargent E. H., Ning Z., J. Am. Chem. Soc. 2017, 139, 6693. [DOI] [PubMed] [Google Scholar]
- 157. Wang N., Zhou Y., Ju M. G., Garces H. F., Ding T., Pang S., Zeng X. C., Padture N. P., Sun X. W., Adv. Energy Mater. 2016, 6, 1601130. [Google Scholar]
- 158. Christians J. A., Fung R. C. M., Kamat P. V., J. Am. Chem. Soc. 2014, 136, 758. [DOI] [PubMed] [Google Scholar]
- 159. Marshall K. P., Walton R. I., Hatton R. A., J. Mater. Chem. A 2015, 3, 11631. [Google Scholar]
- 160. Minemoto T., Murata M., Curr. Appl. Phys. 2014, 14, 1428. [Google Scholar]
- 161. Zhou D., Zhou T., Tian Y., Zhu X., Tu Y., J. Nanomater. 2018. [Google Scholar]
- 162. Hao F., Stoumpos C. C., Guo P., Zhou N., Marks T. J., Chang R. P. H., Kanatzidis M. G., J. Am. Chem. Soc. 2015, 137, 11445. [DOI] [PubMed] [Google Scholar]
- 163. Marshall K. P., Walker M., Walton R. I., Hatton R. A., Nat. Energy 2016, 1, 16178. [Google Scholar]
- 164. Giacomo F. D., Fakharuddin A., Jose R., Brown T. M., Energy Environ. Sci. 2016, 9, 3007. [Google Scholar]
- 165. Chen Q., Zhou H., Hong Z., Luo S., Duan H.‐S., Wang H.‐H., Liu Y., Li G., Yang Y., J. Am. Chem. Soc. 2014, 136, 622. [DOI] [PubMed] [Google Scholar]
- 166. Yokoyama T., Cao D. H., Stoumpos C. C., Song T. B., Sato Y., Aramaki S., Kanatzidis M. G., J. Phys. Chem. Lett. 2016, 7, 776. [DOI] [PubMed] [Google Scholar]
- 167. Lee S. J., Shin S. S., Im J., Ahn T. K., Noh J. H., Jeon N. J., Seok S., Seo J., ACS Energy Lett. 2018, 3, 46. [Google Scholar]
- 168. Ito S., Tanaka S., Manabe K., Nishino H., J. Phys. Chem. C 2014, 118, 16995. [Google Scholar]
- 169. Kim H. S., Jang I. H., Ahn N., Choi M., Guerrero A., Bisquert J., Park N. G., J. Phys. Chem. Lett. 2015, 6, 4633. [DOI] [PubMed] [Google Scholar]
- 170. Zhu P., Chen C., Gu S., Lin R., Zhu J., Sol. RRL 2018, 2, 1700224. [Google Scholar]
- 171. Zhang M., Lyu M., Yun J. H., Noori M., Zhou X., Cooling N. A., Wang Q., Yu H., Dastoor P. C., Wang L., Nano Res. 2016, 9, 1570. [Google Scholar]
- 172. Liao W., Zhao D., Yu Y., Grice C. R., Wang C., Cimaroli A. J., Schulz P., Meng W., Zhu K., Xiong R. G., Yan Y., Adv. Mater. 2016, 28, 9333. [DOI] [PubMed] [Google Scholar]
- 173. Lee J. W., Lee T. Y., Yoo P. J., Gratzel M., Mhaisalkar S., Park N. G., J. Mater. Chem. A 2014, 2, 9251. [Google Scholar]
- 174. Singh T., Kulkarni A., Ikegami M., Miyasaka T., ACS Appl. Mater. Interfaces 2016, 8, 14542. [DOI] [PubMed] [Google Scholar]
- 175. Wojciechowski K., Stranks S. D., Abate A., Sadoughi G., Sadhanala A., Kopidakis N., Rumbles G., Li C. Z., Friend R. H., Jen A. K. Y., Snaith H. J., ACS Nano 2014, 8, 12701. [DOI] [PubMed] [Google Scholar]
- 176. Grancini G., Santosh Kumar R. S., Abrusci A., Yip H. L., Li C. Z., Jen A. K. Y., Adv. Funct. Mater. 2012, 22, 2160. [Google Scholar]
- 177. Cui J., Li P., Chen Z., Cao K., Li D., Han J., Shen Y., Peng M., Fu Y. Q., Wang M., Appl. Phys. Lett. 2016, 109, 171103. [Google Scholar]
- 178. Huang J., Zhang X., Zheng D., Yan K., Li C. Z., Yu J., Sol. RRL 2017, 1, 1600008. [Google Scholar]
- 179. Pelarda M. V., Hames B. C., Benito I. G., Almora O., Ontoria A. M., Sanchez R. S., Garcia Belmonte G., Martín N., Sero I. M., J. Phys. Chem. Lett. 2016, 7, 4622. [DOI] [PubMed] [Google Scholar]
- 180. Abrusci A., Stranks S. D., Docampo P., Yip H. L., Jen A. K. Y., Snaith H. J., Nano Lett. 2013, 13, 3124. [DOI] [PubMed] [Google Scholar]
- 181. Boopathi K. M., Karuppuswamy P., Singh A., Hanmandlu C., Lin L., Abbas S. A., Chang C. C., Wang P. C., Li G., Chu C. W., J. Mater. Chem. A 2017, 5, 20843. [Google Scholar]
- 182. Dong J., Zhao Y., Shi J., Wei H., Xiao J., Xu X., Luo J., Xu J., Li D., Luo Y., Meng Q., Chem. Commun. 2014, 50, 13381. [DOI] [PubMed] [Google Scholar]
- 183. Son D. Y., Im J. H., Kim H. S., Park N. G., J. Phys. Chem. C 2014, 118, 16567. [Google Scholar]
- 184. Xu Y., Liu T., Li Z., Feng B., Li S., Duan J., Ye C., Zhang J., Wang H., Appl. Surf. Sci. 2016, 388, 89. [Google Scholar]
- 185. Anwar F., Mahbub R., Satter S. S., Ullah S. M., Int. J. Photoenergy 2017. [Google Scholar]
- 186. Shao Z., Mercier T. L., Madic M. B., Pauporte T., Mater. Des. 2018, 141, 81. [Google Scholar]
- 187. Huang L. Y., Lambrecht W. R. L., Phys. Rev. B 2013, 88, 165203. [Google Scholar]
- 188. Dang Y., Zhou Y., Liu X., Ju D., Xia S., Xia H., Tao X., Angew. Chem., Int. Ed. 2016, 55, 3447. [DOI] [PubMed] [Google Scholar]
- 189. Mandadapu U., Vedanayakam S. V., Thyagarajan K., Reddy M. R., Jagdeeshbabu B., Int. J. Renewable Energy Res. 2017, 7, 1603. [Google Scholar]
- 190. Jung M. C., Raga S. R., Qi Y., RSC Adv. 2016, 6, 2819. [Google Scholar]
- 191. Tsai C. M., Mohanta N., Wang C. Y., Lin Y. P., Wang C. L., Diau E. W. G., Angew. Chem., Int. Ed. 2017, 56, 13819. [DOI] [PubMed] [Google Scholar]
- 192. Hoshi H., Shigeeda N., Dai T., Mater. Lett. 2016, 183, 391. [Google Scholar]
- 193. Li W., Li J., Li J., Fan J., Mai Y., Wang L., J. Mater. Chem. A 2016, 4, 17104. [Google Scholar]
- 194. Fujihara T., Terakawa S., Matsushima T., Qin C., Yahiro M., Adachi C., J. Mater. Chem. C 2017, 5, 1121. [Google Scholar]
- 195. Lee S. J., Shin S. S., Kim Y. C., Kim D., Ahn T. K., Noh J. H., Seo J., Seok S. I., J. Am. Chem. Soc. 2016, 138, 3974. [DOI] [PubMed] [Google Scholar]
- 196. Hao F., Stoumpos C. C., Chang R. P. H., Kanatzidis M. G., J. Am. Chem. Soc. 2014, 136, 8094. [DOI] [PubMed] [Google Scholar]
- 197. Yokoyama T., Cao D. H., Stoumpos C. C., Bin Song T. B., Sato Y., Aramaki S., Kanatzidis M. G., J. Phys. Chem. Lett. 2016, 7, 776. [DOI] [PubMed] [Google Scholar]
- 198. Liu X., Yang Z., Chueh C., Rajagopal A., Williams S. T., Sun Y., Jen A. K. Y., J. Mater. Chem. A 2016, 4, 17939. [Google Scholar]
- 199. Song T. B., Yokoyama T., Stoumpos C. C., Logsdon J., Cao D. H., Wasielewski M. R., Aramaki S., Kanatzidis M. G., J. Am. Chem. Soc. 2017, 139, 836. [DOI] [PubMed] [Google Scholar]
- 200. Wang F., Ma J., Xie F., Li L., Chen J., Fan J., Zhao N., Adv. Funct. Mater. 2016, 26, 3417. [Google Scholar]
- 201. Ferrara C., Patrini M., Pisanu A., Quadrelli P., Milanese C., Tealdi C., Malavasi L., J. Mater. Chem. A 2017, 5, 9391. [Google Scholar]
- 202. Ran C., Xi J., Gao W., Yuan F., Lei T., Jiao B., Hou X., Wu Z., ACS Energy Lett. 2018, 3, 713. [Google Scholar]
- 203. Jokar E., Chien C. H., Fathi A., Rameez M., Chang Y. H., Diau E. W. G., Energy Environ. Sci. 2018, 11, 2353. [Google Scholar]
- 204. Chen K., Wu P., Yang W., Su R., Luo D., Yang X., Tu Y., Zhu R., Gong Q., Nano Energy 2018, 49, 411. [Google Scholar]
- 205. Tsai C. M., Lin Y. P., Pola M. K., Narra S., Jokar E., Yang Y. W., Diau E. W. G., ACS Energy Lett. 2018, 3, 2077. [Google Scholar]
- 206. Ke W., Stoumpos C. C., Spanopoulos I., Chen M., Wasielewski M. R., Kanatzidis M. G., ACS Energy Lett. 2018, 3, 1470. [Google Scholar]
- 207. Ke W., Stoumpos C. C., Logsdon J. L., Wasielewski M. R., Yan Y., Fang G., Kanatzidis M. G., J. Am. Chem. Soc. 2016, 138, 14998. [DOI] [PubMed] [Google Scholar]
- 208. Ke W., Stoumpos C. C., Zhu M., Mao L., Spanopoulos I., Liu J., Kontsevoi O. Y., Chen M., Sarma D., Zhang Y., Wasielewski M. R., Kanatzidis M. G., Sci. Adv. 2017, 3, e1701293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209. Cao D. H., Stoumpos C. C., Yokoyama T., Logsdon J. L., Song T.‐B., Farha O. K., Wasielewski M. R., Hupp J. T., Kanatzidis M. G., ACS Energy Lett. 2017, 2, 982. [Google Scholar]
- 210. Maughan A. E., Ganose A. M., Bordelon M. M., Miller E. M., Scanlon D. O., Neilson J. R., J. Am. Chem. Soc. 2016, 138, 8453. [DOI] [PubMed] [Google Scholar]
- 211. Konstantakoua M., Stergiopoulos T., J. Mater. Chem. A 2017, 5, 11518. [Google Scholar]
- 212. Chen Z., Wang J. J., Ren Y., Yu C., Shum K., Appl. Phys. Lett. 2012, 101, 093901. [Google Scholar]
- 213. Song T. B., Yokoyama T., Aramaki S., Kanatzidis M. G., ACS Energy Lett. 2017, 2, 897. [Google Scholar]
- 214. Jiang J., Onwudinanti C. K., Hatton R. A., Bobbert P. A., Tao S., J. Phys. Chem. C 2018, 122, 17660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215. Kumar M. H., Dharani S., Leong W. L., Boix P. P., Prabhakar R. R., Baikie T., Shi C., Ding H., Ramesh R., Asta M., Graetzel M., Mhaisalkar S. G., Mathews N., Adv. Mater. 2014, 26, 7122. [DOI] [PubMed] [Google Scholar]
- 216. Qiu X., Cao B., Yuan S., Chen X., Qiu Z., Jiang Y., Ye Q., Wang H., Zeng H., Liu J., Kanatzidis M. G., Sol. Energy Mater. Sol. Cells 2017, 159, 227. [Google Scholar]
- 217. Sabba D., Mulmudi H. K., Prabhakar R. R., Krishnamoorthy T., Baikie T., Boix P. P., Mhaisalkar S., Mathews N., J. Phys. Chem. C 2015, 119, 1763. [Google Scholar]
- 218. Parrott E. S., Milot R. L., Stergiopoulos T., Snaith H. J., Johnston M. B., Herz L. M., J. Phys. Chem. Lett. 2016, 7, 1321. [DOI] [PubMed] [Google Scholar]
- 219. Song T. B., Yokoyama T., Logsdon J., Wasielewski M. R., Aramaki S., Kanatzidis M. G., ACS Appl. Energy Mater. 2018, 1, 4221. [Google Scholar]
- 220. Moghe D., Wang L., Traverse C. J., Redoute A., Sponseller M., Brown P. R., Bulovic V., Lunt R. R., Nano Energy 2016, 28, 469. [Google Scholar]
- 221. Maughan A. E., Kurzman J. A., Neilson J. R., Inorg. Chem. 2015, 54, 370. [DOI] [PubMed] [Google Scholar]
- 222. Kaltzoglou A., Antoniadou M., Perganti D., Siranidi E., Raptis V., Trohidou K., Psycharis V., Kontos A. G., Falaras P., Electrochim. Acta 2015, 184, 466. [Google Scholar]
- 223. Qiu X., Jiang Y., Zhang H., Qiu Z., Yuan S., Wang P., Cao B., Phys. Status Solidi RRL 2016, 10, 587. [Google Scholar]
- 224. Lee B., Stoumpos C. C., Zhou N., Hao F., Malliakas C., Yeh C. Y., Marks T. J., Kanatzidis M. G., Chang R. P. H., J. Am. Chem. Soc. 2014, 136, 15379. [DOI] [PubMed] [Google Scholar]
- 225. Saparov B., Sun J. P., Meng W., Xiao Z., Duan H. S., Gunawan O., Shin D., Hill I. G., Yan Y., Mitzi D. B., Chem. Mater. 2016, 28, 2315. [Google Scholar]
- 226. Lee B., Krenselewski A., Il Baik S. I., Seidman D. N., Chang R. P. H., Sustainable Energy Fuels 2017, 1, 710. [Google Scholar]
- 227. Kretsinger R. H., Uversky V. N., Permyakov E. A., Encyclopedia of Metalloproteins, Springer, New York: 2013. [Google Scholar]
- 228. Lu X., Zhao Z., Li K., Han Z., Wei S., Guo C., Zhou S., Wu Z., Guo W., Wu C. L., RSC Adv. 2016, 6, 86976. [Google Scholar]
- 229. Stoumpos C. C., Frazer L., Clark D. J., Kim Y. S., Rhim S. H., Freeman A. J., Ketterson J. B., Jang J. I., Kanatzidis M. G., J. Am. Chem. Soc. 2015, 137, 6804. [DOI] [PubMed] [Google Scholar]
- 230. Sun P. P., Li Q. S., Yang L. N., Li Z. S., Nanoscale 2016, 8, 1503. [DOI] [PubMed] [Google Scholar]
- 231. Tang L. C., Chang Y. C., Huang J. Y., Lee M. H., Chang C. S., Jpn. J. Appl. Phys. 2009, 48, 112402. [Google Scholar]
- 232. Kopacic I., Friesenbichler B., Hoefler S. F., Kunert B., Plank H., Rath T., Trimmel G., ACS Appl. Energy Mater. 2018, 1, 343. [Google Scholar]
- 233. Ju M. J., Dai J., Ma L., Zeng X. C., J. Am. Chem. Soc. 2017, 139, 8038. [DOI] [PubMed] [Google Scholar]
- 234. Cheng P., Wu T., Zhang J., Li Y., Liu J., Jiang L., Mao X., Lu R., Deng W., Han K., J. Phys. Chem. Lett. 2017, 8, 4402. [DOI] [PubMed] [Google Scholar]
- 235. Liang M., Ju M. G., Dai J., Zeng X. C., Nanoscale 2018, 10, 11314. [DOI] [PubMed] [Google Scholar]
- 236. Huang C. Y., Yan X. C., Cui G., Liu Z., Pang S., Xu H., CN Patent 103943368, 2014.
- 237. Hoefler S. F., Trimmel G., Rath T., Monatsh. Chem. 2017, 148, 795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238. Lee L. C., Huq T. N., Driscoll J. L. M., APL Mater. 2018, 6, 084502. [Google Scholar]
- 239. Lehner A. J., Fabini D H., Evans H. A., Hebert C. A., Smock S. R., Hu J., Wang H., Zwanziger J. W., Chabiny M. L., Seshadri R., Chem. Mater. 2015, 27, 7137. [Google Scholar]
- 240. Zhang X., Wu G., Gu Z., Guo B., Liu W., Yang S., Ye T., Chen C., Tu W., Chen H., Nano Res. 2016, 9, 2921. [Google Scholar]
- 241. Ran C., Wu Z., Xi J., Yuan F., Dong H., Lei T., He X., Hou X., J. Phys. Chem. Lett. 2017, 8, 394. [DOI] [PubMed] [Google Scholar]
- 242. Okano T., Suzuki Y., Mater. Lett. 2017, 191, 77. [Google Scholar]
- 243. Wenderott J. K., Raghav A., Shtein M., Green P. F., Satapathi S., Langmuir 2018, 34, 7647. [DOI] [PubMed] [Google Scholar]
- 244. Kulkarni A., Singh T., Ikegami M., Miyasaka T., RSC Adv. 2017, 7, 9456. [Google Scholar]
- 245. Huang J., Gu Z., Zhang X., Wu G., Chen H., J. Alloys Compd. 2018, 767, 870. [Google Scholar]
- 246. Sanders S., Stummler D., Pfeiffer P., Ackermann N., MRS Adv. 2018, 3, 3085. [Google Scholar]
- 247. Jain S. M., Phuyal D., Davies M. L., Philippe B., Castro C. D., Qiu Z., Kim J., Watson T., Tsoi W. C., Karis O., Rensmo H., Boschloo G., Edvinsson T., Durrani R., Nano Energy 2018, 49, 614. [Google Scholar]
- 248. Zhang W., Liu X., Li L., Sun Z., Han S., Wu Z., Luo J., Chem. Mater. 2018, 30, 4081. [Google Scholar]
- 249. Shin S. S., Baena J. P. C., Kurchin R. C., Polizzotti A., Yoo J. J., Wieghold S., Bawendi M. G., Chem. Mater. 2018, 30, 336. [Google Scholar]
- 250. Li H., Wu C., Yan Y., Chi B., Pu J., Li J., Priya S., ChemSusChem 2017, 10, 3994. [DOI] [PubMed] [Google Scholar]
- 251. Zhang Z., Li X., Xia X., Wang Z., Huang Z., Lei B., Gao Y., J. Phys. Chem. Lett. 2017, 8, 4300. [DOI] [PubMed] [Google Scholar]
- 252. Yelovik N. A., Mironov A. V., Bykov M. A., Kuznetsov A. N., Grigorieva A. V., Wei Z., Dikarev E. V., Shevelkov A. V., Inorg. Chem. 2016, 55, 4132. [DOI] [PubMed] [Google Scholar]
- 253. Filip M. R., Hillman S., Haghighirad A. A., Snaith H. J., Giustino F., J. Phys. Chem. Lett. 2016, 7, 2579. [DOI] [PubMed] [Google Scholar]
- 254. Fabian D. M, Ardo S., J. Mater. Chem. A 2016, 4, 6837. [Google Scholar]
- 255. Sun S., Tominaka S., Lee J. H., Xie F., Bristowe P. D., Cheetham A. K., APL Mater. 2016, 4, 031101. [Google Scholar]
- 256. McClure E. T., Ball M. R., Windl W., Woodward P. M., Chem. Mater. 2016, 28, 1348. [Google Scholar]
- 257. Slavney A. H., Hu T., Lindenberg A. M., Karunadasa H. I., J. Am. Chem. Soc. 2016, 138, 2138. [DOI] [PubMed] [Google Scholar]
- 258. Wei F., Deng Z., Sun S., Xie F., Kieslich G., Evans D. M., Carpenter M. A., Bristowe P. D., Cheetham A. K., Mater. Horiz. 2016, 3, 328. [Google Scholar]
- 259. Giorgi G., Yamashita K., Chem. Lett. 2015, 44, 826. [Google Scholar]
- 260. Miller N. C., Bernechea M., APL Mater. 2018, 6, 084503. [Google Scholar]
- 261. Volonakis G., Filip M. R., Haghighirad A. A., Sakai N., Wenger B., Snaith H. J., Giustino F., J. Phys. Chem. Lett. 2016, 7, 1254. [DOI] [PubMed] [Google Scholar]
- 262. Zhang L., Liu C., Wang L., Liu C., Wang K., Zou B., Angew. Chem., Int. Ed. 2018, 57, 11213. [DOI] [PubMed] [Google Scholar]
- 263. Khazaee M., Sardashti K., Sun J.‐P., Zhou H., Clegg C., Hill I. G., Jones J. L., Lupascu D. C., Mitzi D. B., Chem. Mater. 2018, 30, 3538. [Google Scholar]
- 264. Mali S. S., Kim H., Kim D., Hong C. K., ChemistrySelect 2017, 2, 1578. [Google Scholar]
- 265. Kim Y., Yang Z., Jain A., Voznyy O., Kim G. H., Liu M., Quan L. N., de Arquer F. P. G., Comin R., Fan J. Z., Sargent E. H., Angew. Chem., Int. Ed. 2016, 55, 958666. [DOI] [PubMed] [Google Scholar]
- 266. Greul E., Petrus M. L., Binek A., Docampo P., Bein T., J. Mater. Chem. A 2017, 5, 19972. [Google Scholar]
- 267. Harikesh P. C., Mulmudi H. K., Ghosh B., Goh T. W., Teng Y. T., Thirumal K., Lockrey M., Weber K., Koh T. M., Li S., Mhaisalkar S., Mathews N., Chem. Mater. 2016, 28, 7496. [Google Scholar]
- 268. Karoppuswamy P., Boopathy K. M., Mohaptra A., Chen H., Wong K. T., Wang P. C., Chu C., Nano Energy 2018, 45, 330. [Google Scholar]
- 269. Li Y. J, Wu T., Sun L., Yang R. X., Jiang L., Cheng P. F., Hao Q. Q., Wang T. J., Luc R. F., Deng W. Q., RSC Adv. 2017, 7, 35175. [Google Scholar]
- 270. Jiang F., Yang D., Jiang Y., Liu T., Zhao X., Ming Y., Luo B., Qin F., Fan J., Han H., Zhang L., Zhou Y., J. Am. Chem. Soc. 2018, 140, 1019. [DOI] [PubMed] [Google Scholar]
- 271. Nie R., Mehta A., Park B. W., Kwon H., Im J., J. Am. Chem. Soc. 2018, 140, 872. [DOI] [PubMed] [Google Scholar]
- 272. Vargas B., Ramos E., Gutierrez E. P., Alonso J. C., Ibarra D. S., J. Am. Chem. Soc. 2017, 139, 9116. [DOI] [PubMed] [Google Scholar]
- 273. Baranwal A. K., Masutani H., Sugita H., Kanda H., Kanaya S., Shibayama N., Sanehira Y., Ikegami M., Numata Y., Yamada K., Miyasaka T., Umeyama T., Imahori H., Ito S., Nano Convergence 2017, 4, 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274. Adonin S. A., Frolova L. A., Sokolov M. N., Shilov G. B., Korchagin D. V., Fedin V. P., Aldoshion S. M., Stevenson K. J., Troshin P. A., Adv. Energy Mater. 2018, 8, 1701140. [Google Scholar]
- 275. Harikesh P. C., Wu B., Ghosh B., John R. A., Lie S., Thirumal K., Wong L. H., Sum T. C., Mhaisalkar S., Mathews N., Adv. Mater. 2018, 30, 1802080. [DOI] [PubMed] [Google Scholar]
- 276. Jahandar M., Heo J. H., Song C. E., Kong K. J., Shin W. S., Lee J. C., Im S. H., Moon S. J., Nano Energy 2016, 27, 330. [Google Scholar]
- 277. Elseman A. M., Rashid M. M., Sajid S., Hassan Ali M. A., Elseman A. M., Shalan A. E., Sajid S., Rashad M. M., Hassan A. M., Li M., ACS Appl. Mater. Interfaces 2018, 10, 11699. [DOI] [PubMed] [Google Scholar]
- 278. Filip M. R., Giustino F., J. Phys. Chem. C 2016, 120, 166. [Google Scholar]
- 279. Choudhary K., Identification of Potential Replacement Materials for Lead in CH3NH3PbI3 using First Principle Calculations, 2015, eprint: arXiv:1505.01238. [Google Scholar]
- 280. Suta M., Wickleder C., J. Mater. Chem. C 2015, 3, 5233. [Google Scholar]
- 281. Uribe J. I., Ramirez D., Guillen J. M. O., Osorio J., Jaramillo F., J. Phys. Chem. C 2016, 120, 16393. [Google Scholar]
- 282. Pazoki M., Jacobsson T. J., Hagfeldt A., Boschloo G., Edvinsson T., Phys. Rev. B 2016, 93, 144105. [Google Scholar]
- 283. Jacobsson T. J., Pazoki M., Hagfeldt A., Edvinsson T., J. Phys. Chem. C 2015, 119, 25673. [Google Scholar]
- 284. Rey D. P. D., Forgacs D., Hutter E. M., Savenije T. J., Nordlund D., Schulz P., Berry J. J., Sessolo M., Bolink H. J., Adv. Mater. 2016, 28, 9839. [DOI] [PubMed] [Google Scholar]
- 285. Raw A. D., Ibers J. A., J. Solid State Chem. 2012, 192, 34. [Google Scholar]
- 286. Arend H., Huber W., J. Cryst. Growth 1978, 43, 213. [Google Scholar]
- 287. Han J., Nishihara S., Inoue K., Kurmoo M., Inorg. Chem. 2014, 53, 2068. [DOI] [PubMed] [Google Scholar]
- 288. Semary M. A., Mostafa M. F., Ahmed M. A., Solid State Commun. 1978, 25, 443. [Google Scholar]
- 289. Boix P. P., Agarwala S., Koh T. M., Mathews N., Mhaisalkar S. G., J. Phys. Chem. Lett. 2015, 6, 898. [DOI] [PubMed] [Google Scholar]
- 290. Mitzi D. B., Liang K., Chem. Mater. 1997, 9, 2990. [Google Scholar]
- 291. Grimm J., Suyver J. F., Beurer E., Carver G., Gudel H. U., J. Phys. Chem. B 2006, 110, 2093. [DOI] [PubMed] [Google Scholar]
- 292. Suta M., Urland W., Daul C., Wickleder C. P., Phys. Chem. Chem. Phys. 2016, 18, 13196. [DOI] [PubMed] [Google Scholar]
- 293. Castro L. M. C., Guloy A. M., Angew. Chem., Int. Ed. 2003, 42, 2771. [DOI] [PubMed] [Google Scholar]
- 294. Chen M., Ju M. G., Carl A. J. D., Zong Y., Grimm R. L., Gu J., Zeng X. C., Zhou Y., Padture N. P., Joule 2018, 2, 1. [Google Scholar]
- 295. Sun Y. Y., Shi J., Lian J., Gao W., Agiorgousis M. L., Zhang P., Zhang S., Nanoscale 2016, 8, 6284. [DOI] [PubMed] [Google Scholar]
- 296. Hong F., Saparov B., Meng W., Xiao Z., Mitzi D. B., Yan Y., J. Phys. Chem. C 2016, 120, 6435. [Google Scholar]
- 297. Sun Y. Y., Agiorgousis M. L., Zhang P., Zhang S., Nano Lett. 2015, 15, 581. [DOI] [PubMed] [Google Scholar]
- 298. Perera S., Hui H., Zhao C., Xue H., Sun F., Deng C., Gross N., Milleville C., Xu X., Watson D. F., Weinstein B., Sun Y.‐Y., Zhang S., Zeng H., Nano Energy 2016, 22, 129. [Google Scholar]
- 299. Morss L. R., Siegal M., Stenger L., Edelstein N., Inorg. Chem. 1970, 9, 1771. [Google Scholar]
- 300. Tran T. N., An N. M., Nguyen K. D., Nguyen T. D., Truong T. T., J. Sci.: Adv. Mater. Devices 2018, 3, 471. [Google Scholar]
- 301. Liu J., Ozaki M., Yakumaru S., Handa T., Nishikubo R., Kanemitsu Y., Saeki A., Murata Y., Murdey R., Wakamiya A., Angew. Chem., Int. Ed. 2018, 57, 13221. [DOI] [PubMed] [Google Scholar]
- 302. Heo J. H., Kim J., Kim H., Moon S. H., Im S. H., Hong K. H., J. Phys. Chem. Lett. 2018, 9, 6024. [DOI] [PubMed] [Google Scholar]
- 303. Kim H., Lee Y. H., Yoo J. H., Park T., Oh J. H., J. Mater. Chem. A 2018, 6, 18173. [Google Scholar]
- 304. Hayase S., Ito N., Kamarudin M. A. K., Shen Q., Ogomi Y., likubo S., Yoshino K., Minemoto T., Toyoda T., Proc. SPIE 2018, 107371F. [Google Scholar]
- 305. Rahamam M. Z., Hossain A. K. M. A., RSC Adv. 2018, 8, 33010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306. Benabdallah I., Boujnah M., Kenz A. E., Benyoussef A., Abatal M., Bassam A., J. Alloys Compd. 2019, 773, 796. [Google Scholar]
- 307. Zhang C., Teo S., Gao L., Kamata Y., MA T., Proc. SPIE 2018, 107240A. [Google Scholar]
- 308. Sanders S., Stummler D., Pfeiffer P., Ackermann N., Schimkat F., Simkus G., Heuken M., Baumann P. K., Vescan A., Kalisch H., Phys. Status Solidi A 2018, 215, 1800409. [Google Scholar]
- 309. Lu C., Zhang J., Sun H., Hou D., Gan X., Shang M. H., Li Y., Hu Z., Zhu Y., Han L., ACS Appl. Energy Mater. 2018, 1, 4485. [Google Scholar]
- 310. Jun T., Sim K., Iimura S., Sasase M., Kamioka H., Kim J., Hosono H., Adv. Mater. 2018, 30, 1804547. [DOI] [PubMed] [Google Scholar]
- 311. Aamir M., Khan M. D., Sher M., Revaprasadu N., Malik M. A., Akhter J., New J. Chem. 2018, 42, 17181. [Google Scholar]
- 312. Kumari K., Chakrabarti T., Jana A., Bhattachartjee D., Gupta B., Sarkar S. K., Opt. Mater. 2018, 84, 681. [Google Scholar]
- 313. Padture N., Proc. SPIE 2018, 107370R. [Google Scholar]
- 314. Zhou L., Liao J. F., Huang Z. G., Wang X. D., Xu Y. F., Chen H. Y., Kuang D. B., Su C. Y., ACS Energy Lett. 2018, 3, 2613. [Google Scholar]
- 315. Zhao Y., Wei D., Du J., Xu Z., J. Electroceram. 2019, 42, 74. [Google Scholar]
- 316. Raja S., Vadivel M., Babu R. R., Kumar S., Ramamurti K., Solid State Sci. 2018, 85, 60. [Google Scholar]
- 317. Meyer E., Mutukwa D., Zingwe N., Taziwa R., Metals 2018, 8, 667. [Google Scholar]
- 318. Wang M., Zeng P., Bai S., Gu J., Li F., Yang Z., Liu M., Sol. RRL 2018, 2, 1800217. [Google Scholar]
- 319. Lan C., Zhao S., Luo J., Fan P., Phys. Chem. Chem. Phys. 2018, 20, 24339. [DOI] [PubMed] [Google Scholar]
- 320. Singhal N., Chakraborty R., Ghosh P., Nag A., Chem. ‐ Asian J. 2018, 13, 2085. [DOI] [PubMed] [Google Scholar]
- 321. Zhang Z., Gao L., Ma T., Proc. SPIE 2018, 107370W. [Google Scholar]
- 322. Das R., Choudhary R. N. P., J. Mater. Sci.: Mater. Electron. 2018, 29, 19099. [Google Scholar]