

Abstract
Cardiac fibrosis is a common feature in chronic hypertension patients with advanced heart failure, and endothelial-to-mesenchymal transition (EndMT) is known to promote Angiotensin II (Ang II)-mediated cardiac fibrosis. Previous studies have suggested a potential role for the transcription factor, ETS-1, in Ang II-mediated cardiac remodeling, however the mechanism are not well defined. In this study, we found that mice with endothelial Ets-1 deletion showed reduced cardiac fibrosis and hypertrophy following Ang II infusion. The reduced cardiac fibrosis was accompanied by decreased expression of fibrotic matrix genes, reduced EndMT with decreased Snail, Slug, Twist, and ZEB1 expression, as well as reduced cardiac hypertrophy and expression of hypertrophy-associated genes was observed. In vitro studies using cultured H5V cells further confirmed that ETS-1 knockdown inhibited TGF-β1-induced EndMT. This study revealed that deletion of endothelial Ets-1 attenuated Ang II-induced cardiac fibrosis via inhibition of EndMT, indicating an important ETS-1 function in mediating EndMT. Inhibition of ETS-1 could be a potential therapeutic strategy for treatment of heart failure secondary to chronic hypertension.
Keywords: Angiotensin II, Cardiac fibrosis, Endothelial-to-mesenchymal transition, ETS-1
INTRODUCTION
Cardiac fibrosis is a common occurrence in patients with chronic hypertension and advanced heart failure. Cardiac fibrosis is characterized by an increased number of fibroblasts in the myocardium and excessive deposition of the extracellular matrix (ECM) (1, 2). Myocardial fibrosis can lead to decreased ventricular compliance that contributes to diastolic and systolic dysfunction, ultimately leading to heart failure and possible premature death (1, 3).
The development of fibrosis in the heart was reported to follow a pathway similar to that of other organs, such as the liver, lung, kidney and solid tumors (4, 5). It is well known that myofibroblasts in fibrotic tissues are derived from the expansion and activation of resident tissue fibroblasts, tissue migration of bone marrow derived circulating fibrocytes, and the transition of epithelial cells into mesenchymal cells (4). Recently, endothelial to mesenchymal transition (EndMT) has emerged as a possible source of tissue myofibroblasts, and has been proven to contribute to cardiac fibrosis (6, 7). However, the molecular mechanism underlying the role of EndMT in cardiac fibrosis remain unclear.
The transcription factor ETS-1 (E26 avian leukemia oncogene 1, 5′ domain) is composed of 450 amino acids and contains the DNA-binding ETS domain which binds to the ETS-binding motif GGAA/T cis-acting element in target genes. It has been identified as a critical regulator of embryogenesis, angiogenesis and inflammation. Previous studies have demonstrated that ETS-1 is rapidly induced in endothelial cells and smooth muscle cells of mouse aorta that are subjected to chronic Ang II infusion, leading to vascular inflammation and remodeling (8). Furthermore, Ang II infusion results in an increase in heart size and ventricular wall thickness in mice, but these effects are significantly diminished in global Ets-1 knockout mice (8), suggesting that ETS-1 mediates Ang II-induced cardiac fibrotic effects (9). Our previous study has shown that ETS-1 is highly expressed in cardiac endothelial cells (10), but whether Ang II-mediated cardiac fibrosis is dependent on endothelial ETS-1 is unknown.
Previous studies have reported that ETS-1 participates in EMT in tumor initiation and metastasis, and renal fibrosis (11–13). In this study we hypothesize that ETS-1 regulates EndMT in the murine heart in response to chronic hypertension.
RESULTS
Endothelial-specific Ets-1 deletion mice exhibit normal cardiac structure and function
ETS-1 is expressed in endothelial cells, smooth muscle cells and fibroblasts in the heart. To assess the contribution of endothelial-specific ETS-1 in cardiac fibrosis, We crossed Tie2-Cre+ mice with Ets-1f/f mice to generate ETS-1 endothelial-specific knockout mice (Tie2-Cre+;Ets-1f/f). Western blot analysis confirmed that Ets-1 was specifically deleted in the endothelial cells of Tie2-Cre+;Ets-1f/f mice (Fig. 1A, B).
Fig. 1.
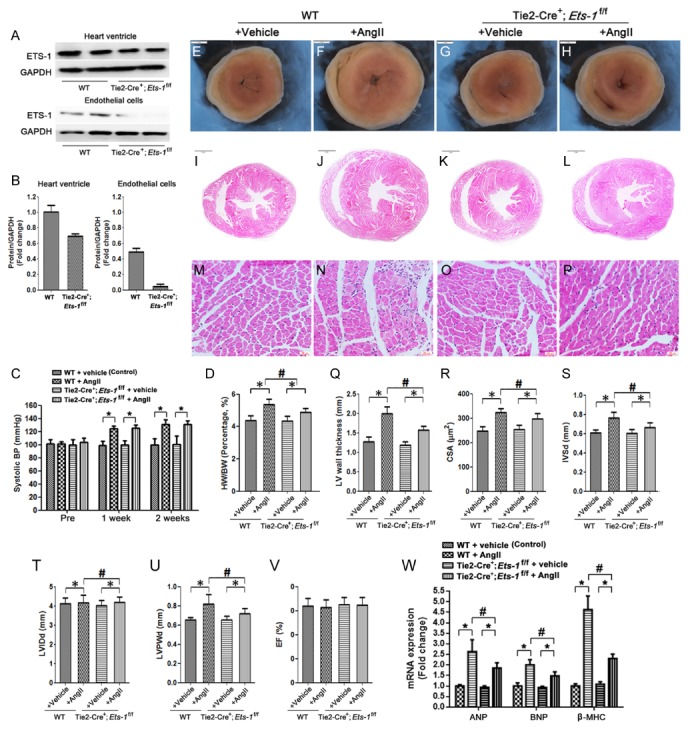
Endothelial deletion of Ets-1 attenuateds Ang II-induced cardiac hypertrophy. (A, B) Representative western blots and quantitative western blots analysis that showed ETS-1 expression in whole heart ventricle and isolated heart endothelial cells of Tie2-Cre+; Ets-1f/f mice compared with those of wild-type littermate control; (C) Systolic blood pressure measurement prior to and at 1 and 2 weeks after AngII infusion; (D) Statistical results of the heart weight/body weight (HW/BW) ratios (n = 6) at 2 weeks after AngII infusion; (E–H) H&E staining of the heart ventricular cross-sections (I–L); and cross sectional area (CSA) (M–P) at 2 weeks after AngII infusion. (Q) Statistical results of the left ventricular wall thickness at 2 weeks after AngII infusion and (R) cardiomyocyte CSA (n = 100 cells per section); (S–V) Echocardiography results at 2 weeks after AngII infusion (n = 6) (S: Interventricular septal thickness at diastole (IVSd), T: left ventricular internal diastolic diameter (LVIDd); U: left ventricular posterior wall thickness at diastole (LVPWd), V: Percent LV ejection fraction (EF)); (W) The mRNA expressions of ventricular ANP, BNP and β-MHC after 2 weeks of Ang II-infusion were detected by qRT-PCR (n = 6). Scale bar: (E–L) 1 mm; (M–P) 25 μm. *P < 0.05; #P < 0.05.
All Tie2-Cre+;Ets-1f/f mice survived to adulthood, with no deaths prior to the time that they were sacrificed. H&E staining revealed that Tie2-Cre+;Ets-1f/f mice had normal heart and aorta structure (data not shown). Echocardiography showed no significant difference in cardiac function between Tie2-Cre+; Ets-1f/f and littermate control mice (Fig. 1S–W).
Endothelial-specific Ets-1 deletion attenuateds cardiac hypertrophy induced by Ang II
Blood pressure of the mice was measured 3 days before and 7 and 14 days after Ang II infusion. No significant difference in systolic blood pressure was observed in Tie2-Cre+;Ets-1f/f mice compared with control mice (Fig. 1C), which was consistent with a previous study (8). This result indicated that endothelial deletion of Ets-1 did not influence blood pressure to affect cardiac remodeling.
To investigate the effect of endothelial deletion of Ets-1 on cardiac structure and function following Ang II infusion for 2 weeks, heart weight (HW) and body weight (BW) of mice were measured and echocardiography was performed. The results showed that the HW/BW ratio was significantly increased following Ang II infusion in wild-type control mice, but this increase was significantly attenuated in Tie2-Cre+;Ets-1f/f mice (Fig. 1D). H&E staining of cross-sections of the heart and cardiomyocyte cross-sectional areas (CSA) confirmed that endothelial deletion of Ets-1 significantly reduced the Ang II-induced increase in left ventricle wall thickness and size of cardiomyocytes (Fig. 1E–R).
Echocardiography analysis indicated that the left ventricular wall thickness in mice was remarkably increased, as shown by the increase in IVSd and LVPWd. Tie2-Cre+;Ets-1f/f mice exhibited a significantly reduced left ventricular wall thickness compared with the control mice. No significant change of LVEF was observed in all mice groups (Fig. 1S–V).
We then assessed whether endothelial deletion of Ets-1 affected the cardiac hypertrophy gene expression pattern following Ang II infusion for 2 weeks. Quantitative polymerase chain reaction (qPCR) results showed that mRNA levels of the cardiac hypertrophy markers ANP, BNP and β-MHC were up-regulated. Endothelial deletion of Ets-1 suppressed the Ang II-induced upregulation of ANP, BNP and β-MHC expression in the heart (Fig. 1W).
Endothelial-specific deletion of Ets-1 attenuates cardiac fibrosis induced by Ang II
Myocardial fibrosis is an early manifestation of hypertrophic cardiomyopathy (3). To investigate the effect of endothelial deletion of Ets-1 on cardiac fibrosis development following Ang II infusion for 2 weeks, both Masson’s Trichrome and Sirius Red stainings were performed. We found that Ang II infusion resulted in significant cardiac fibrosis in control mice, which was evident from the significant increase in perivascular and interstitial fibrosis, however, this effect was significantly attenuated in the Tie2-Cre+;Ets-1f/f mice compared with the control mice (Fig. 2A–J). Results from qPCR showed that the mRNA expression levels of ECM proteins, such as collagen I, collagen III, fibronectin and factors involved in ECM production, including TGF-β1, Connective Tissue Growth Factor (CTGF) in the heart were significantly reduced in Tie2-Cre+;Ets-1f/f mice (Fig. 2K). These data demonstrated that endothelial specific deletion of Ets-1 attenuated cardiac fibrosis induced by Ang II-infusion.
Fig. 2.
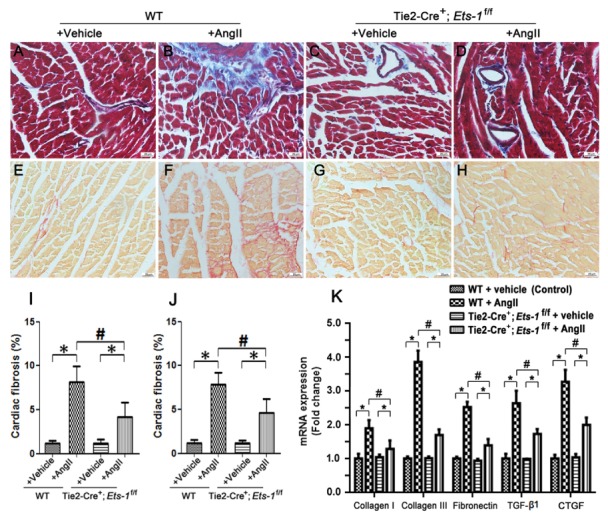
Endothelial deletion of Ets-1 attenuateds AngII-induced cardiac fibrosis. Left ventricular sections were stained by Masson’s Trichrome (A–D) and Sirius Red (E–H) in the indicated groups following AngII infusion for 2 weeks, with quantification of collagen assessed by Masson’s Trichrome Stain (I) and, Sirius Red (J); (K) Quantitative PCR analysis of mRNA levels of collagen I, collagen III, TGF-β1 and CTGF in the hearts of the indicated groups following AngII infusion for 2 weeks (n = 6). Scale bar: (A–H) 25 μm. *P < 0.05; #P < 0.05.
Endothelial deletion of Ets-1 inhibited the EndMT induced by Ang II
Because EndMT plays an important role in cardiac fibrosis (6), we investigated whether endothelial deletion of Ets-1 attenuated cardiac fibrosis via inhibition of EndMT. We performed ETS-1 and α-smooth muscle actin (α-SMA) co-immunofluorescence staining of heart sections and found increased ETS-1 expression in the heart endothelium and α-SMA+ cells in Ang II-infused control mice. however, this effect was alleviated in Tie2-Cre+;Ets-1f/f mice (Supplemental Fig. S1). Western blot analysis revealed significantly lower expression levels of α-SMA and fibroblast specific protein-1 (FSP-1) in the hearts of Tie2-Cre+;Ets-1f/f mice than those of the control mice, following Ang II infusion for 2 weeks (Fig. 3A, B). We also found, a concomitant increase in the expression level of CD31 that co-localized with α-SMA staining (Fig. 3C–N). These findings suggested that the loss of ETS-1 in the endothelium resulted in reduced transition of endothelial cells to activated fibroblasts.
Fig. 3.

Endothelial deletion of Ets-1 ameliorated AngII-induced EndMT. (A, B): Representative western blot and quantitative analysis that showed the mean CD31, α-SMA and FSP-1 levels in the cardiac homogenates from the Tie2-Cre;Ets-1f/f and WT control mice 2 weeks after AngII infusion (n = 6); (C–N): Myocardial sections were co-immunostained to detect CD31 (green)/α-SMA (red) and imaged via fluorescence microscopy. (O): qRT-PCR analysis of the mRNA expression levels of Snail, Slug, Twist2 and ZEB1 in the myocardium 2 weeks after AngII infusion (n = 6). Scale bar: (C–N) 25 μm. *P < 0.05; #P < 0.05.
To further elucidate the effect of endothelial deletion of Ets-1 on EndMT in the heart during Ang II infusion, we analyzed examined the transcription factors involved in EndMT in the heart including Snail (Snail1), Slug (Snail2), Twist1, and ZEB1. Expression levels of these factors were slightly decreased in Tie2-Cre+;Ets-1f/f mice compared with wild-type control mice following Ang II infusion for 2 weeks (Fig. 3O).
Endothelial knockdown of Ets-1 inhibited TGF-β1-induced EndMT of H5V cells
To further confirm the role of ETS-1 in EndMT, we knocked down Ets-1 in the H5V cells (cardiac endothelial cells) by using short hairpin RNA (shRNA) (Fig. 4A–C). We also treated H5V cells with 10 ng/ml of TGF-β1, an inducer of EndMT. After treatment with TGF-β1 for 48 hours, the cellular morphology changed from a cobblestone-like structure to a fusiform structure, with large junctions loosely connected to each other (Fig. 4D–K). Knockdown of Ets-1 significantly suppressed the TGF-β1-induced morphology changes (Fig. 4J, K), which was confirmed by the reversal of the TGF-β1-mediated decrease in CD31 expression and increase in the expression of the mesenchymal markers FSP-1 and α-SMA in H5V cells lacking ETS-1 (Fig. 4D–K).
Fig. 4.
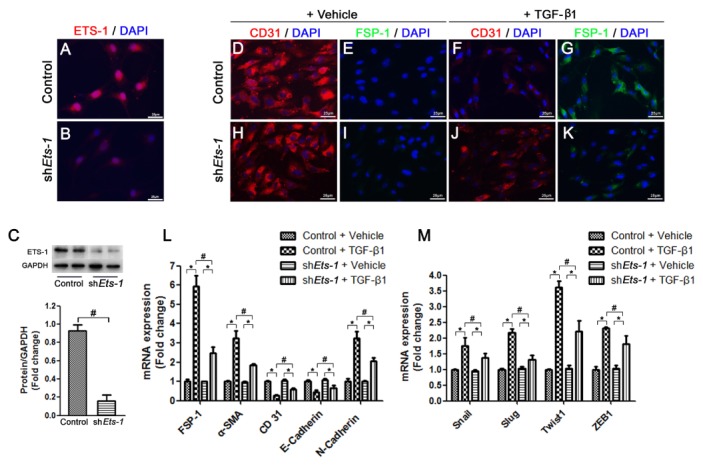
Knockdown of Ets-1 alleviated TGF-β1 induced EndMT in H5V cells. (A, B) shEts-1 H5V cells were immunostained to detect ETS-1 (Red) and imaged via fluorescence microscopy. (C) Representative western blots and quantitative analysis that showed ETS-1 protein levels in shEts-1 H5V cells; (D–K) H5V cells were co-immunostained to detect CD31 (Red) and FSP-1 (Green), and imaged via fluorescence microscopy; (L) qRT-PCR analysis of the mRNA expression levels of FSP-1, α-SMA, CD31, E-Cadherin and N-Cadherin in H5V cells at 2 days after TGF-β1 treatment (n = 6); (M) qRT-PCR analysis of the mRNA expression levels of Snail, Slug, Twist1 and ZEB1 in H5V cells at 2 days after TGF-β1 treatment (n = 6). Scale bar: (A, B), (D–K) 25 μm. *P < 0.01; #P < 0.01.
We further investigated the mRNA levels of endothelial and mesenchymal markers by using qPCR in TGF-β1-treated H5V cells. The expression of α-SMA, FSP-1 and N-cadherin were increased and expression levels of CD31 and E-cadherin were decreased in H5V cells after treatment with TGF-β1 for 2 days. Reduced mRNA expression of mesenchymal markers and an increase in the mRNA expression of endothelial markers showed that knockdown of Ets-1 can alleviate TGF-β1-induced EndMT in H5V cells (Fig. 4L).
We also examined the changes in expression levels of transcription factors that play an important role in EndMT, such as Snail, Slug, Twist1 and ZEB1. Consistent with our in vivo findings, expression levels of all these transcription factors were decreased in the Ets-1 knockdown cells compared with scramble control shRNA cells after TGF-β1 treatment (Fig. 4M).
DISCUSSION
In this study, we found that deletion of Ets-1 in mouse endothelial cells had no basal effects but attenuated cardiac hypertrophy and fibrosis induced by chronic Ang II infusion. Our results are consistent with previous studies that showed that Ang II induced heart size and ventricular wall thickness, but these effects are significantly diminished in the global Ets-1 knockout mice (8). Our results provide further evidence for the hypothesis that expression of ETS-1 in endothelial cells plays a central role in the regulation of Ang II-induced cardiac hypertrophy and fibrosis.
Endothelial cells are critical for maintaining cardiovascular system function, and endothelial dysfunction result in cardiovascular disease. Organ fibrosis is characterized by EndMT, a process in which endothelial cells in small vessels undergo a transition to form fibroblasts that show active synthesis and secretion of numerous ECM factors (6). A recent study found that cardiac endothelial cells are converted to myofibroblasts via EndMT during cardiac fibrosis induced by pressure overload in mice, and cardiac function can be substantially improved by inhibiting the EndMT process. This indicates that EndMT plays an important role in cardiac fibrosis and in the development of heart failure (6). Thus, further exploration of EndMT and identification of the key factors that regulate EndMT may offer potential therapeutic targets for clinical treatment.
ETS-1 is a transcription factor that is critical for embryogenesis, angiogenesis and inflammation. Previous studies have reported that ETS-1 participates in the EMT process during tumor formation, and also regulates the EMT process of renal tubular epithelial cells under pathological conditions (11, 12). In this study, we found that EndMT was significantly decreased in Tie2-Cre+;Ets-1f/f hearts compared with the control mice following Ang II infusion for 2 weeks, which was characterized by the down-regulation of endothelial maker, CD31 and E-cadherin, as well as the acquisition of mesenchymal markers, including α-SMA, FSP-1 and N-cadherin in cardiac endothelial cells. Additionally, expression levels of the genes involved in EndMT promotion were decreased in the Ang II-infused Tie2-Cre+;Ets-1f/f mouse hearts. In vitro studies further revealed that Ets-1 knockdown in cardiac endothelial cells (H5V cells) suppressed EndMT induced by TGF-β1. Taken together, our results suggest a critical role for ETS-1 in regulating EndMT in the heart, specifically by promoting cardiac fibrosis in response to Ang II infusion.
The TGF-β1/Smads signaling pathways along with some downstream transcription factors, such as Snail, Slug, Twist and ZEB family of proteins, play an important regulatory role in cardiac EndMT (14). Previous studies have demonstrated that ETS-1 regulates TGF-β1/Smads pathways, is an agonist of the profibrotic effects of TGF-β1, and facilitates Smad2/3 binding to the promoters of genes involved in these pathways (15, 16). Additional studies have reported that ETS-1 transactivates Twist1 promoter activity in cells (17) and activates the ZEB1 promoter via interactions with putative ETS-1-binding sites (18, 19). In this study, our mouse model revealed that deletion of Ets-1 in endothelial cells amelioriated the Ang II-induced cardiac fibrosis response in vivo. The Ang II/TGF-β1-induced increase in the expression of Snail, Slug, Twist1 and ZEB1 were inhibited by the knockdown ETS-1 in endothelial cells, both in vivo and in vitro. All together, our results indicate that loss of ETS-1 can suppress EndMT of cardiac endothelial cells via its effect on TGF-β1 signaling pathways. Therefore, the ETS-1 signaling in the endothelium might be a potential therapeutic target for preventing cardiac fibrosis in conditions such as chronic systemic hypertension.
MATERIALS AND METHODS
Animals
We generated endothelial-specific Ets-1 knockout mice (Tie2-Cre+; Ets-1f/f) by crossing Ets-1flox/flox mice with Tie2-Cre+ mice that specifically expressed Cre in endothelial cells. The mice were bred into a C57/B6 genetic background. Male mice were used in all our experiments. Mice were housed in the Laboratory Animal Facility at Shanghai Jiao Tong University School of Medicine, China. All animal experiments were approved by the Animal Care Committee of Shanghai Jiao Tong University and in compliance with the animal management rules of the Chinese Ministry of Health and the National Institute of Health (NIH) guidelines.
Three-month-old male Tie2-Cre+;Ets-1f/f mice and the littermate control mice (Tie2-Cre−;Ets-1f/f) were subjected to chronic cardiac fibrosis generated by Ang II (A9525, Sigma, USA) infusion using an osmotic minipump (model 2002, Alzet, USA). Mice were randomly assigned into the following four groups (n = 6 per group): group 1, wild-type (WT) mice infused with vehicle (saline); group 2, Tie2-Cre+;Ets-1f/f mice treated with vehicle; group 3, WT mice treated with Ang II (1.4 mg/kg per day) for 2 weeks; and group 4, Tie2-Cre+;Ets-1f/f mice treated with Ang II (1.4 mg/kg per day) for 2 weeks.
Isolated cardiac endothelium cells
Heart ventricle tissue was minced into 1 mm3 pieces, and then digested using 2 mg/ml collagenase II (LS004174, Worthington, Bio Corp, USA). Tissue debris was removed by centrifugation to produce the single cell suspension. Single cells were magnetically labeled with anti-CD31 super paramagnetic polystyrene beads (11155D, Thermo Fisher Scientific Life Sciences, USA) at 4°C for 20 min and separated using a magnet (12303D, Thermo Fisher Scientific Life Sciences). CD31+ cells bound to the beads were washed out, collected and used for western blotting analysis.
Echorcardiography
Echocardiography was performed using a Vevo 2100 instrument (Fuji Film Visual Sonics) equipped with an MS-400 imaging transducer (18–38 MHz) as previously described (20). Mice were anaesthetized using 1–2% isoflurane that was administered via inhalation. Interventricular septal thickness at diastole (IVSd), left ventricular internal diastolic diameter (LVIDd); left ventricular posterior wall thickness at diastole (LVPWd) were measured. Percent left ventricular ejection fraction (EF) was calculated using M-mode measurements.
Blood pressure measurements
Systolic blood pressure was measured via a tail-cuff method using a non-invasive blood pressure instrument (BP-98A, Softron, China) at 3 days before and at 7 and 14 days after Ang II infusion as previously described (21).
Histology, Masson’s Trichrome stain and Sirius Red stain analysis
Mice were anesthetized by isoflurane inhalation and were sacrificed by CO2 asphyxiation. The hearts were dissected and fixed in 4% paraformaldehyde overnight at 4°C, and then embedded in paraffin. Longitudinal sections or transverse sections were cut at 10 μm thickness from paraffin blocks and stained with hematoxylin and eosin. Masson’s Trichrome stain and Sirius Red stain were performed as described previously (22).
Immunofluorescence stain
Immunofluorescence staining was performed on the cryosections as described previously (10). Briefly, tissues were fixed in 4% paraformaldehyde overnight at 4°C and then cryo-embedded in optimum cutting temperature compound (OCT) (Tissue Tek, USA). Transverse heart sections were cut at 10 μm thickness, and the cryosections were incubated with primary antibodies overnight at 4°C. After washing with 0.5% TritonX-100 in phosphate buffer saline, sections were incubated with secondary antibodies for 2 hours at room temperature. The following antibodies were used: CD31 (SC-1506, Santa Cruz Biotechnology, USA), α-SMA (ab5694, Abcam, USA), FSP-1 (ab27957, Abcam) and ETS-1 (SC-350; Santa Cruz Biotechnology).
Western blot analysis
Western blot analysis was performed as previously described (22). The blots were incubated overnight with the primary antibodies against ETS-1 (SC-350; Santa Cruz Biotechnology), CD31 (SC-1506, Santa Cruz Biotechnology), α-SMA (ab5694, Abcam), and FSP-1 (ab27957, Abcam) at 4°C overnight. Then, blots were washed and incubated with secondary antibodies.
Culture of H5V cells and TGF-β1 treatment
The mouse heart endothelium cells (H5V) were a gift from Dr. Xin Ma (23). The cells were cultured in 90% Dulbecco’s modified Eagle’s medium (DMEM) and 10% fetal bovine serum as described previously (23).
For TGF-β1 treatment, H5V cells were starved in a serum-free culture medium for 48 hours, and then administrated TGF-β1 (10 ng/ml) (14–8342, eBioscience, USA) for two consecutive days.
Ets-1 shRNA plasmid (Ets-1 shRNA Sequence: CCGGCCT TGCAGACAGACTACTTCTCAAGAGAAAGTAGTCTGTCTGC AAGGTTTTTTG) and the lentiviral packaging vectors pCMV.DR8 and pMD2.G (Plasmid #12259, Addgene) were transfected in human embryonic kidney (HEK) 293T cells (ATCC, CRL-1573). The viral supernatants were harvested on day 2 and 3 after transfection, filtered with 0.4-μm filters and applied to H5V cells in the presence of 8 μg/ml polybrene (Santa Cruz Biotechnology) for 6–8 hours. The efficiency of infection was determined by western bloting (24).
Quantitative reverse-transcription PCR (qRT-PCR) analysis of mRNA expression
Total RNA was extracted from mouse left ventricle or cultured H5V cells using an RNeasy kit (9747, Takararr, Japan) according to the manufacturer’s recommendations. 500 μg of RNA was retro-transcribed to cDNA using cDNA reverse transcription kit (037A, Takararr). qRT-PCR was performed using in a QuantStudio DS Real-time fluorescence quantitative PCR System with SYBR Green Master Mix (420A, Takararr) and gene-specific primers. Primer sequences are listed in Supplemental Table S1. Standard and melting curves were measured for every plate and for every gene to ensure efficiency and specificity of the reaction. Ct value of glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was used as internal control and the 2–ΔΔCT method was used to analyze the relative expression levels of various genes.
Statistical analysis
Graphpad prism 6.01 was used for statistical analysis. All data were presented as mean ± standard deviation (SD). Shapiro-Wilk normality test was first applied for checking normality distribution of a variable. Student’s t-test was used for two-group comparisons. Multiple comparisons were tested using one-way ANOVA followed by the post hoc Dunnett’s test (Levene’s tests for equal variance). Differences were considered as statistically significant at P < 0.05.
Supplementary Information
ACKNOWLEDGEMENTS
This work was supported by the Natural Science Foundation of Shanghai (17ZR1423800) and the Science and Technology Commission of Shanghai Municipality (18140903402).
Footnotes
CONFLICTS OF INTEREST
The authors have no conflicting interests.
REFERENCES
- 1.Gourdie RG, Dimmeler S, Kohl P. Novel therapeutic strategies targeting fibroblasts and fibrosis in heart disease. Nat Rev Drug Discov. 2016;15:620–638. doi: 10.1038/nrd.2016.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Furtado MB, Nim HT, Boyd SE, Rosenthal NA. View from the heart: cardiac fibroblasts in development, scarring and regeneration. Development. 2016;143:387–397. doi: 10.1242/dev.120576. [DOI] [PubMed] [Google Scholar]
- 3.Ho CY, Lopez B, Coelho-Filho OR, et al. Myocardial fibrosis as an early manifestation of hypertrophic cardiomyopathy. N Engl J Med. 2010;363:552–563. doi: 10.1056/NEJMoa1002659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Piera-Velazquez S, Li Z, Jimenez SA. Role of endothelial-mesenchymal transition (EndoMT) in the pathogenesis of fibrotic disorders. Am J Pathol. 2011;179:1074–1080. doi: 10.1016/j.ajpath.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zeisberg EM, Potenta SE, Sugimoto H, Zeisberg M, Kalluri R. Fibroblasts in kidney fibrosis emerge via endothelial-to-mesenchymal transition. J Am Soc Nephrol. 2008;19:2282–2287. doi: 10.1681/ASN.2008050513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zeisberg EM, Tarnavski O, Zeisberg M, et al. Endothelial-to-mesenchymal transition contributes to cardiac fibrosis. Nat Med. 2007;13:952–961. doi: 10.1038/nm1613. [DOI] [PubMed] [Google Scholar]
- 7.Li Y, Lui KO, Zhou B. Reassessing endothelial-to-mesenchymal transition in cardiovascular diseases. Nat Rev Cardiol. 2018;15:445–456. doi: 10.1038/s41569-018-0023-y. [DOI] [PubMed] [Google Scholar]
- 8.Zhan Y, Brown C, Maynard E, et al. Ets-1 is a critical regulator of Ang II-mediated vascular inflammation and remodeling. J Clin Invest. 2005;115:2508–2516. doi: 10.1172/JCI24403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hao G, Han Z, Meng Z, et al. Ets-1 upregulation mediates angiotensin II-related cardiac fibrosis. Int J Clin Exp Pathol. 2015;8:10216–10227. [PMC free article] [PubMed] [Google Scholar]
- 10.Ye M, Coldren C, Liang X, et al. Deletion of ETS-1, a gene in the Jacobsen syndrome critical region, causes ventricular septal defects and abnormal ventricular morphology in mice. Hum Mol Genet. 2010;19:648–656. doi: 10.1093/hmg/ddp532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li C, Wang Z, Chen Y, et al. Transcriptional silencing of ETS-1 abrogates epithelial-mesenchymal transition resulting in reduced motility of pancreatic cancer cells. Oncol Rep. 2015;33:559–565. doi: 10.3892/or.2014.3613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shirakihara T, Saitoh M, Miyazono K. Differential regulation of epithelial and mesenchymal markers by deltaEF1 proteins in epithelial mesenchymal transition induced by TGF-beta. Mol Biol Cell. 2007;18:3533–3544. doi: 10.1091/mbc.e07-03-0249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Okano K, Hibi A, Miyaoka T, et al. Inhibitory effects of the transcription factor Ets-1 on the expression of type I collagen in TGF-beta1-stimulated renal epithelial cells. Mol Cell Biochem. 2012;369:247–254. doi: 10.1007/s11010-012-1388-6. [DOI] [PubMed] [Google Scholar]
- 14.Wei WY, Zhang N, Li LL, et al. Pioglitazone alleviates cardiac fibrosis and inhibits endothelial to mesenchymal transition induced by pressure overload. Cell Physiol Biochem. 2018;45:26–36. doi: 10.1159/000486220. [DOI] [PubMed] [Google Scholar]
- 15.Koinuma D, Tsutsumi S, Kamimura N, et al. Chromatin immunoprecipitation on microarray analysis of Smad2/3 binding sites reveals roles of ETS1 and TFAP2A in transforming growth factor beta signaling. Mol Cell Biol. 2009;29:172–186. doi: 10.1128/MCB.01038-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Czuwara-Ladykowska J, Sementchenko VI, Watson DK, Trojanowska M. Ets1 is an effector of the transforming growth factor beta (TGF-beta ) signaling pathway and an antagonist of the profibrotic effects of TGF-beta. J Biol Chem. 2002;277:20399–20408. doi: 10.1074/jbc.M200206200. [DOI] [PubMed] [Google Scholar]
- 17.Millien G, Cao Y, O’Hara CJ, et al. ETS1 regulates Twist1 transcription in a Kras (G12D)/Lkb1(−/−) metastatic lung tumor model of non-small cell lung cancer. Clin Exp Metastasis. 2018;35:149–165. doi: 10.1007/s10585-018-9912-z. [DOI] [PubMed] [Google Scholar]
- 18.Sinh ND, Endo K, Miyazawa K, Saitoh M. Ets1 and ESE1 reciprocally regulate expression of ZEB1/ZEB2, dependent on ERK1/2 activity, in breast cancer cells. Cancer Sci. 2017;108:952–960. doi: 10.1111/cas.13214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dave N, Guaita-Esteruelas S, Gutarra S, et al. Functional cooperation between Snail1 and twist in the regulation of ZEB1 expression during epithelial to mesenchymal transition. J Biol Chem. 2011;286:12024–12032. doi: 10.1074/jbc.M110.168625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ye M, Parente F, Li X, Perryman MB, et al. Gene-targeted deletion of OPCML and Neurotrimin in mice does not yield congenital heart defects. Am J Med Genet A. 2014;164A:966–974. doi: 10.1002/ajmg.a.36441. [DOI] [PubMed] [Google Scholar]
- 21.Sheng LJ, Ruan CC, Ma Y, et al. Beta3 adrenergic receptor is involved in vascular injury in deoxycorticosterone acetate-salt hypertensive mice. FEBS Lett. 2016;590:769–778. doi: 10.1002/1873-3468.12107. [DOI] [PubMed] [Google Scholar]
- 22.Lin JR, Zheng YJ, Zhang ZB, et al. Suppression of endothelial-to-mesenchymal transition by SIRT (Sirtuin) 3 alleviated the development of hypertensive renal injury. Hypertension. 2018;72:350–360. doi: 10.1161/HYPERTENSIONAHA.118.10482. [DOI] [PubMed] [Google Scholar]
- 23.He D, Jin J, Zheng Y, Bruce IC, Tam S, Ma X. Anti-angiogenesis effect of trichosanthin and the underlying mechanism. Biochem Biophys Res Commun. 2013;430:735–740. doi: 10.1016/j.bbrc.2012.11.080. [DOI] [PubMed] [Google Scholar]
- 24.Plotnik JP, Budka JA, Ferris MW, Hollenhorst PC. ETS1 is a genome-wide effector of RAS/ERK signaling in epithelial cells. Nucleic Acids Res. 2014;42:11928–11940. doi: 10.1093/nar/gku929. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.