

Abstract
Retinal degeneration is characterized by the progressive destruction of retinal cells, causing the deterioration and eventual loss of vision. We explored whether the hormone prolactin provides trophic support to retinal cells, thus protecting the retina from degenerative pressure. Inducing hyperprolactinemia limited photoreceptor apoptosis, gliosis, and changes in neurotrophin expression, and it preserved the photoresponse in the phototoxicity model of retinal degeneration, in which continuous exposure of rats to bright light leads to retinal cell death and retinal dysfunction. In this model, the expression levels of prolactin receptors in the retina were upregulated. Moreover, retinas from prolactin receptor-deficient mice exhibited photoresponsive dysfunction and gliosis that correlated with decreased levels of retinal bFGF, GDNF, and BDNF. Collectively, these data unveiled prolactin as a retinal trophic factor that may regulate glial–neuronal cell interactions and is a potential therapeutic molecule against retinal degeneration.
Keywords: gliosis, prolactin, retinal degeneration, trophic factor
Introduction
Retinal degeneration results from the gradual destruction of retinal cells. Such deterioration occurs in aging retinas and accompanies a range of prominent human retinal disorders including age-related macular degeneration, glaucoma, and diabetic retinopathy (Zhang et al., 2012a). These disorders lead to some degree of visual disability, and their treatment remains an unmet need (Zhang et al., 2012a). Photoreceptors act as a fuse of sorts, since their irreversible loss has been identified as a cause of blindness in most degenerative retinopathies (Zack, 2000). Nevertheless, all retinal cells can contribute to disease activity as they eventually become dysfunctional or die.
Retinal homeostasis strongly relies on glial activity (Bringmann et al., 2006; Langmann, 2007). Glia help determine cell fate in the retina by contributing to the net secretion of proapoptotic and antiapoptotic signals, including growth factor and neurotrophins (Caffé et al., 1993; LaVail et al., 1998; Bringmann et al., 2006; Unterlauft et al., 2012). In addition to being neuroprotective, reactive glial cells can also damage neurons (Bringmann et al., 2006; Barres, 2008). Nevertheless, there is still no agreement on whether lessening gliosis, which is detected in every neurological disease and also in genetically determined and experimental models of inner and outer retinal degeneration (Bringmann et al., 2006; Grieshaber et al., 2012), is helpful or harmful (Barres, 2008). Though clear evidence emerged about a decade ago that the glia–neuron network is heavily involved in the survival of surrounding neurons in vivo (Bringmann et al., 2006; Barres, 2008), most aspects of its regulation remain to be elucidated. The identification of endogenous molecules able to modulate the function of this network will help promulgate retinal health. One such molecule may be prolactin, the hormone fundamental for lactation.
High levels of circulating prolactin in pregnant women are associated with increased electroretinogram (ERG) B-wave amplitude (De Luca Brunori et al., 1985). The primary role of prolactin in the retina, however, appears to be the regulation of its development, as prolactin gene deletion in zebrafish results in a thinner retinal inner nuclear layer (Huang et al., 2009). Also, prolactin promotes visual pigment synthesis in teleosts (Crim, 1975a,b; Allen and Cristy, 1978) and in amphibians (Crim, 1975b). In rodents and nonhuman primates, prolactin receptors are present in both outer and inner nuclear layers of the retina and also in the ganglion cell layer (Rivera et al., 2008). Pioneering studies in rats proposed that prolactin modulates photoreceptor susceptibility to light-induced degeneration (O'Steen and Kraeer, 1977). Additionally, antibodies or antisense oligonucleotides targeting prolactin provoke retinal neovascularization and vasodilation (Aranda et al., 2005). Together, these data identify various types of retinal cells as prolactin targets, but studies remain sparse.
Considering that prolactin has been recently reported to be neuroprotective in the hippocampus (Morales, 2011) and that it signals through pathways similar to those triggered by CNTF, which is one of the neurotrophic factors most studied for neuroprotection of the retina (Wen et al., 2012), we evaluated the hypothesis that prolactin mitigates retinal degeneration through trophic actions.
Materials and Methods
Ethics statement.
All experiments were approved by the Bioethics Committee of the Institute of Neurobiology from the National University of Mexico (clave NOM-062-ZOO-1999) in accordance with the rules and regulation of the Society for Neuroscience: Policies on the Use of Animals and Humans in Neuroscience Research. All efforts were made to minimize the number of animals used and their suffering.
Animals.
Male albino rats (Wistar, 250–300 g), and 28 male and 28 female albino mice (3 months old) were fed ad libitum and reared in normal cyclic light conditions (12 h light/dark cycle) with an ambient light level of ∼400 lux. Only rats were subjected to bright constant light exposure (BCL).
Rats.
Hyperprolactinemia was induced by implanting two anterior pituitary (AP) glands under the kidney capsule, as previously described (Adler, 1986), and sham rats were subjected to similar surgery without implantation. Rats were then transferred to cages that allow light to enter unimpeded. BCL was achieved using two cold white fluorescent light sources positioned 50 cm above the cages (40 W), at an intensity of ∼1200 lux at the cage floor. BCL exposure was maintained over a period of 48 h, after which time the animals were immediately returned to normal cyclic conditions. Ambient light-reared animals were used as negative controls. After a 24 h dark adaptation period, rats were evaluated by ERG or killed by CO2 inhalation and decapitation. Eyes were enucleated and processed for apoptosis or RNA extraction.
Mice.
Mice heterozygous for prolactin receptor (prlr+/−; Ormandy et al., 1997; 129Sv background) were crossbred, and prolactin receptor-null (prlr−/−) mice were selected by tail biopsy genotyping (Binart et al., 2000). Wild-type (7 females and 15 males), prlr+/− (10 females and 2 males), and prlr−/− (11 females and 11 males) mice were subjected to normal cyclic light. After a 24 h dark adaptation period, mice were evaluated by ERG or killed by CO2 inhalation and decapitation. Eyes were enucleated and processed for histology, apoptosis, immunostaining, or RNA extraction.
Serum prolactin.
Serum prolactin was measured in rats by the Nb2 cell bioassay, a standard procedure based on the proliferative response of the Nb2 lymphoma cells to prolactin as previously described (Tanaka et al., 1980).
ERG recordings.
Animals were anesthetized with 70% ketamine and 30% xylazine (1 μl/g body weight, intraperitoneally), and pupils were dilated with tropicamide-phenylephrin. ERG responses were recorded with a silver chloride ring electrode placed on the cornea. Two reference electrodes were positioned subcutaneously near the eye. The light stimulation included 0.7 ms flashes of increasing intensities (0.3, 0.6, 0.9, and 1.2 log cd.s/m2; PS33 Plus PhotoStimulator, GRASS Technologies). The bandpass was set at 10 Hz to 1 kHz (amplifier, A-M Systems). Sixteen responses were averaged, with flashes at 10 s intervals. Before ERG assessment under photopic conditions, animals were adapted to normal light (400 lux) for 20 min. The light stimulation under photopic conditions included 0.7 ms flashes of 1.2 log cd.s/m2 intensity. For quantitative analysis, the B-wave amplitude was measured between A- and B-wave peaks. For each animal, the ERG was recorded simultaneously in both eyes, and the recordings from the two eyes were averaged. Oscillatory potentials (OPs) were digitally isolated from the scotopic B-wave using a 100–500 Hz digital filter.
The amplitude of OPs was determined at maximal illumination intensity (1.2 log cd.s/m2), as the sum of the amplitudes of OP1–OP5 in rats and of OP1–OP4 in mice. Of note, separate analyses for male and female mice revealed no significant differences in ERG response (data not shown).
Apoptosis determination.
The apoptotic cell death detection ELISA (Roche Diagnostics) was used according to the manufacturer's instructions to quantify fragmented nucleosomal DNA associated with apoptotic cell death in harvested rat and mouse retinas.
Histology.
Mouse eyes were fixed in Davidson solution (40% formalin, 95% ethanol, glacial acetic acid, and distilled water), dehydrated, and embedded in paraffin for sectioning (8 μm thick) and H&E staining. To quantify the thickness of the retina, images were taken at equivalent retinal eccentricities from the optic nerve head. Layer thickness was measured in at least four areas from each retina in three animals per group.
RNA isolation and cDNA synthesis.
Retinas were dissected for mRNA analysis. All RNA was isolated using TRIzol (Life Technologies) according to the manufacturer's instructions. Total RNA was quantified. Contaminating genomic DNA was removed using 1 μl of RNase-free DNase I (Boehringer Mannheim)/10 μg of RNA at 37°C for 30 min. A 1 μg sample was reverse transcribed in a 20 μl reaction using the high-capacity cDNA reverse transcription kit (Applied Biosystems) in a Mastercycler (Eppendorf).
Quantitative PCR.
Primer sequences are documented in Table 1, and the conditions used for the PCRs were as follows: PCR products were detected and quantified with Máxima Green/ROX qPCR Master (Thermo Scientific) in a 10 μl final reaction volume containing template and 0.5 μm of each primer. Amplification, performed in a PCR CFX96 thermocycler (Bio-Rad), included a denaturation step of 10 min at 95°C, followed by 40 cycles of amplification (10 s at 95°C, 30 s at primer pair-specific annealing temperature, and 30 s at 72°C). The PCR data were analyzed by the 2-ΔΔCT method, and cycle thresholds normalized to the housekeeping gene hypoxanthine-guanine phosphoribosyltransferase were used to calculate the mRNA levels of interest.
Table 1.
Primers used for real-time RT-PCR
| mRNA | NCBI accession number | Direction | Sequence | Amplicon size (bp) | Primer efficiency (%) |
|---|---|---|---|---|---|
| m BNDF | EF125669.1 | F | ATTACCTTCCTGCATCTGTTG | 179 | 103.85 |
| R | TGTCCGTGGACGTTTACTTC | ||||
| r BDNF | NM_001270636.1 | F | TCCACCAGGTGAGAAGAGTGATG | 159 | 79.17 |
| R | TCACGCTCTCCAGAGTCCCATG | ||||
| m bFGF | NM_008006.2 | F | ACTTAGAAGCCAGCAGCCG | 154 | 99.84 |
| R | CCCGACGGCCGCGTGGAT | ||||
| r bFGF | NM_019305.2 | F | GTCTCCCGCACCCTATCC | 121 | 93.07 |
| R | ACAACGACCAGCCTTCCA | ||||
| m CNTF | NM_170786.2 | F | CTCTATCTGGCTAGCAAGGA | 146 | 72.81 |
| R | TCATCTCACTCCAGCGATCA | ||||
| r CNTF | NM_013166.1 | F | GGACCTCTGTAGCCGTTCTA | 161 | 101.35 |
| R | TCATCTCACTCCAACGATCA | ||||
| m GDNF | NM_010275.2 | F | AATGTCCAACTGGGGGTCTA | 180 | 133.16 |
| R | GCCGAGGGAGTGGTCTTC | ||||
| r GDNF | NM_019139.1 | F | AGAGGGAAAGGTCGCAGAG | 91 | 128.39 |
| R | AGCCCAAACCCAAGTCAGT | ||||
| m/r GFAP | NM_001131020.1/NM_017009.2 | F | AGGCAGAAGCTCCAAGATGA | 178/178 | 99.95 |
| R | GTTCTCGAACTTCCTCCTCA | ||||
| m HPRT | NM_013556.2 | F | GTAATGATCAGTCAACGGGGGAC | 177 | 98.39 |
| R | CCAGCAAGCTTGCAACCTTAACCA | ||||
| r HPRT | NM_012583.2 | F | GACCGGTTCTGTCATGTCG | 61 | 99.60 |
| R | ACCTGGTTCATCATCACTAATCAC | ||||
| m NGF | S62089.1 | F | GGACGCAGCTTTCTATACTG | 145 | 102.34 |
| R | TTCAGGGACAGAGTCTCCTT | ||||
| r NGF | NM_001277055.1 | F | TGCATAGCGTAATGTCCATGTTG | 149 | 159.97 |
| R | CTGTGTCAAGGGAATGCTGAA | ||||
| m PEDF | AF036164.1 | F | TCCAACTTCGGCTACGATCT | 143 | 103.8 |
| R | CGGTGAATGACAGACTCTGT | ||||
| r PEDF | NM_177927.2 | F | CCAACTCTTTGCAGGACATG | 78 | 97.67 |
| R | TCACAGGTTTGCCGGTAATC | ||||
| m rhodopsin | NM_145383.1 | F | ATTCACCACCACCCTCTACA | 155 | 94.80 |
| R | GCTTGCAGACCACCACGTA | ||||
| r prolactin R | NM_012630.2 | F | CCAGGAGAGTTCCGTTGAAA | 153 | 93.61 |
| R | GGTGGAAAGATGCAGGTCAT |
m, Mouse; r, rat; F, forward; R, reverse.
Statistical analysis.
All results were replicated in three or more independent experiments. Serum prolactin levels were presented as the mean ± SD, and all others as the mean ± SEM; all data showed normal distribution or equal variance according to D'Agostino–Pearson omnibus and Levene's tests, respectively. Statistical differences between two groups were determined by a two-tailed Student's t test, among three groups with one variable by one-way ANOVA followed by Bonferroni's post hoc comparison test, and among four groups with two variables by two-way ANOVA followed by Bonferroni's post hoc comparison test (SigmaStat version 7.0, SYSTAT). Differences in means with p < 0.05 were considered statistically significant.
Results
Hyperprolactinemia protects against retinal degeneration and photoresponsive dysfunction associated with light damage
Light damage has been used for >4 decades as an experimental model for human retinal degenerative diseases (Organisciak and Vaughan, 2010). We used a 48 h moderate BCL exposure in rats to evaluate the role of prolactin. Because systemic prolactin is taken up into the eye (O'Steen and Sundberg, 1982), we increased systemic levels of prolactin by placing AP implants under the kidney capsule of rats (Fig. 1A) and quantified apoptosis in BCL-exposed retinas by ELISA. BCL exposure induced a sixfold increase in retinal apoptosis (p < 0.0001; Fig. 1B). Hyperprolactinemia reduced by 25 ± 6% (p < 0.05) the levels of apoptosis in damaged retinas (Fig. 1B). Alone, hyperprolactinemia did not modify the basal levels of retinal apoptosis (Fig. 1B). Next, we addressed whether prolactin modifies the impact of light damage on retinal function. Figure 1C shows representative ERG recordings under scotopic conditions, corresponding to the massed light responses of rod and cone photoreceptors and of the ON bipolar cells. Quantitative analysis was performed at maximal illumination intensity (1.2 log cd.s/m2), given that no significant difference was observed at lower intensities (data not shown). Hyperprolactinemia did not prevent the reduction in amplitude of the A-wave registered in BCL-exposed retinas but did protect against the delay in A-wave implicit time caused by BCL (14 ± 3% of controls, p < 0.01; Fig. 1D). Also, hyperprolactinemia prevented the reduction in amplitude and the delay in implicit time of the B-wave registered in BCL-exposed retinas (23 ± 5% and 12 ± 3% of controls, respectively; p < 0.05; Fig. 1D). ERGs were then recorded under photopic conditions to test the responses of cones (Fig. 1E). BCL reduced and delayed the B-wave amplitude and implicit time by 48 ± 6% (of controls, p < 0.001) and 16 ± 5% (of controls, p < 0.01), respectively (Fig. 1F). Hyperprolactinemia did limit the reduction of B-wave amplitude and prevented the delay in B-wave implicit time induced by BCL (Fig. 1F). Alone, hyperprolactinemia did not modify any ERG parameters.
Figure 1.

Hyperprolactinemia prevents retinal cell apoptosis and photoresponsive dysfunction associated with light damage. A–F, Serum prolactin levels (A), retinal apoptosis (B), representative ERG responses under scotopic conditions (C), averaged amplitudes and implicit times of A- and B-waves under scotopic conditions (D), representative ERG responses under photopic conditions (E), and averaged amplitudes and implicit times of the photopic B-wave in sham and hyperprolactinemic (AP) rats exposed or not to BCL for 48 h (F). ERG analysis was performed on responses registered at maximal intensity stimulation (1.2 log cd.s/m2). Serum prolactin levels correspond to the mean ± SD of 4–14 animals per group. Other data correspond to the mean± SEM (n = 16–20 per group, three independent experiments; *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001, as indicated; two-way ANOVA with Bonferroni's post hoc test). n.s., Not significant; PRL, prolactin.
Next, we further assessed the neuroprotective role of prolactin in the retina. Because the A- and B-waves cannot be used to evaluate inner retinal function, we analyzed OPs, the high-frequency components of the ERG that are considered to partly reflect inner retinal activity and are known to be altered in retinal degenerative disorders (Wachtmeister, 1998; Walter et al., 1999; Rangaswamy et al., 2006). Representative traces of the five OP components are shown in Figure 2A. The reduction in the amplitudes of OPs caused by BCL (45 ± 8% of control, p < 0.01; Fig. 2B) was prevented in hyperprolactinemic animals. In addition, the prolonged latencies of OPs (15 ± 3% of control, p < 0.05; Fig. 2B) associated with BCL were eliminated by hyperprolactinemia. Alone, hyperprolactinemia did not modify OP amplitude or implicit time.
Figure 2.

Hyperprolactinemia protects against alterations of OPs caused by light damage. A, B, Representative OP recordings (A) and averaged OP amplitude and implicit time in response to the maximal light stimulus (1.2 log cd.s/m2) under scotopic conditions (B), in sham and hyperprolactinemic (AP) rats exposed or not exposed to BCL for 48 h. Data correspond to the mean ± SEM (n = 9–18 per group, three independent experiments; **p < 0.01 as indicated; two-way ANOVA with Bonferroni's post hoc test). OP1 to OP5 have been labeled as 1–5. n.s., Not significant.
High levels of circulating prolactin limit retinal gliosis and neurotrophin expression changes associated with light damage, which, in turn, upregulates the prolactin receptor in the retina
Because retinal neuron survival and function strongly depend on glial activity, we next evaluated whether prolactin modifies the gliosis that accompanies retinal injury. Gliosis, classically defined as the aggregation of glial cells at the sight of injury, is usually associated with increased synthesis of GFAP monomers that assemble into intermediate filaments in both astrocytes and Müller cells (Takamiya et al., 1988). Real-time RT-PCR analysis of the GFAP gene showed that hyperprolactinemia prevented the threefold increase in GFAP mRNA levels induced by BCL compared with levels in sham rats (p < 0.01; Fig. 3A). Alone, hyperprolactinemia drastically reduced (by 94 ± 0.1%; p < 0.01) the retinal content of GFAP transcript. Also, relative to unexposed animals, the BCL-induced increase in retinal GFAP mRNA levels is greater in hyperprolactinemic than in normoprolactinemic rats (20-fold vs 3-fold, respectively).
Figure 3.

High levels of circulating prolactin limit retinal gliosis and neurotrophin expression changes associated with light damage, which upregulates the prolactin receptor in the retina. A, Quantitative PCR (qPCR)-based quantification of GFAP mRNA levels in retinas from sham and hyperprolactinemic (AP) rats exposed or not exposed to BCL for 48 h. The data are presented as the mean ± SEM (n = 4–9 per group, three independent experiments; **p < 0.01 as indicated; two-way ANOVA with Bonferroni's post hoc test). B, qPCR-based quantification of bFGF, NGF, GDNF, PEDF, CNTF, and BDNF mRNA levels in retinas from sham and hyperprolactinemic (AP) rats exposed or not exposed to BCL for 48 h. The data are presented as the mean ± SEM (n = 4–9 per group, three independent experiments; *p < 0.05, **p < 0.01, and ***p < 0.001 as indicated; two-way ANOVA with Bonferroni's post hoc test). C, qPCR-based quantification of prolactin receptor (PRL-R) mRNA levels in retinas from rats exposed or not exposed to BCL for 48 h. The data are presented as the mean ± SEM (n = 4–9 per group, three independent experiments; ***p < 0.001 as indicated, Student's t test).
The response of glia to acute retinal damage is known to be mediated by several neurotrophic factors, including bFGF, GDNF, BDNF, CNTF, NGF, and pigment epithelium-derived factor (PEDF; Harada et al., 2000; Bringmann et al., 2006; Wen et al., 2012). Because prolactin promotes and reduces retinal cell survival and gliosis, respectively, we therefore studied whether prolactin upregulates the synthesis of retinal neuroprotective factors. Figure 3B shows that light damage decreased the bFGF mRNA levels by 74 ± 2% of controls (p < 0.01); increased the NGF, GDNF, PEDF, and CNTF mRNA levels by 180 ± 3% (p < 0.01), 240 ± 7% (p < 0.01), 320 ± 13% (p < 0.01), and 171 ± 8% (p < 0.05) of controls, respectively; but did not alter BDNF levels. Hyperprolactinemia fully blocked the BCL-mediated reduction and increase of bFGF and NGF retinal mRNA levels, respectively, observed in BCL-exposed retinas of normoprolactinemic animals. Additionally, the GDNF, PEDF, and BDNF mRNA levels in BCL-exposed retinas from hyperprolactinemic rats were upregulated by twofold (p < 0.05), fourfold (p < 0.01), and approximately threefold (p < 0.01), respectively, compared with values observed in the damaged retinas of normoprolactinemic animals. The CNTF mRNA levels in BCL-exposed retinas from hyperprolactinemic rats were not different from those in damaged retinas from normoprolactinemic animals. In the absence of BCL, hyperprolactinemia increased the bFGF, GDNF, PEDF, CNTF, and BDNF mRNA levels by approximately threefold (p < 0.05), approximately threefold (p < 0.05), approximately sevenfold (p < 0.001), threefold (p < 0.01), and approximately threefold (p < 0.05; relative to the control values), respectively, but did not alter NGF levels.
The long and short isoforms of the rat prolactin receptor differ in the length of their cytoplasmic tail. Both are determinant of the responsiveness of cells to prolactin and are present in the retina (Rivera et al., 2008). Moreover, the expression of these receptors is regulated by stress-related stimuli in certain regions of the CNS (Babenko et al., 2012; Benatti et al., 2012). Therefore, we next evaluated whether light damage regulates the expression of prolactin receptor in retinas using primers that recognize both isoforms. The prolactin receptor mRNA levels in retinas from rats exposed to BCL were fivefold greater than the levels in controls (p < 0.001; Fig. 3C).
prlr−/− mice show photoresponsive dysfunction
Next, we studied the retinas of mice with a genetic deletion of prlr (Ormandy et al., 1997). The histology of prlr−/− retinas is similar to that in wild-type mice (prlr+/+; Fig. 4A) and prlr+/− mice (data not shown). The retina is composed of cone and rod photoreceptor outer segments and of three cellular layers: the outer nuclear layer, comprising cone and rod somata; the inner nuclear layer, comprising horizontal, bipolar, and amacrine interneurons, and Müller glial cells; and the ganglion cell layer, containing retinal ganglion cells and displaced amacrine cells. These cell layers are separated from each other by synaptic layers: the outer plexiform layer comprises processes of and synapses among photoreceptors, horizontal cells, and bipolar cells; while the inner plexiform layer is constituted by processes of and synapses among bipolar, amacrine, and retinal ganglion cells (Wässle, 2004; Sanes and Zipursky, 2010). prlr−/− and prlr+/+ mice showed retinal layers of comparable width (Fig. 4B). Under scotopic conditions (Fig. 4C), the amplitudes of the A- and B-wave in prlr−/− retinas was reduced at all light intensities by an average of 37 ± 3% and 35 ± 4% (p < 0.05), respectively, compared with values in prlr+/+ mice (Fig. 4D). The B-wave in prlr+/− retinas was decreased (by 21 ± 4% of wild-type, respectively, p < 0.05) but only at the most intense illuminations. Under photopic conditions (Fig. 4E), prlr−/− retinas showed decreased B-wave amplitude (35 ± 7% lower than prlr+/+, p < 0.05) at maximal illumination intensity (1.2 log cd.s/m2; Fig. 4F). No difference in implicit times was found.
Figure 4.
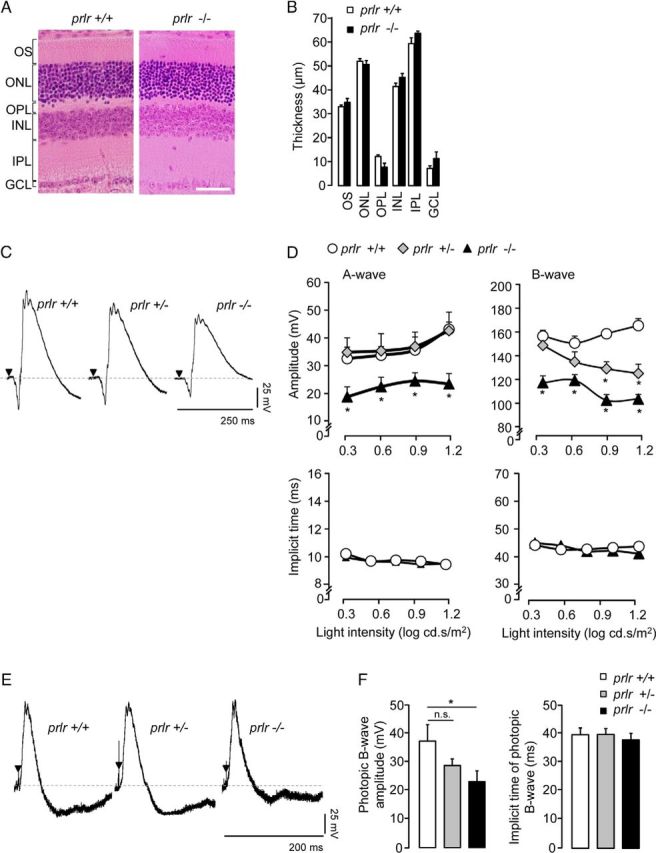
prlr−/− mice show photoresponsive dysfunction. A, B, Representative H&E-stained retinas (A) and averaged thickness of each layer of the retina (B) from prlr+/+ and prlr−/− mice. Four sections per retina from each of six animals per group were analyzed in three separate experiments. Scale bar, 50 μm. C, Representative scotopic ERG response in prlr+/+, prlr+/−, and prlr−/− mice at 1.2 log cd.s/m2. D, Averaged amplitudes and implicit times of the scotopic A- and B-waves in prlr+/+, prlr+/−, and prlr−/− mice at increasing stimulus intensities. Data are presented as the mean ± SEM (n = 3–13 per group, seven independent experiments; *p < 0.05 vs values in prlr+/+ mice; one-way ANOVA with Bonferroni's post hoc test). E, Representative photopic ERG response in prlr+/+, prlr+/−, and prlr−/− mice. F, Averaged amplitude and implicit time of the photopic B-wave in prlr+/+, prlr+/−, and prlr−/− mice at 1.2 log cd.s/m2. Data are presented as the mean ± SEM (n = 3 per group, three independent experiments; *p < 0.05 as indicated; one-way ANOVA with Bonferroni's post hoc test). n.s., Not significant; ONL, outer nuclear layer; INL, inner nuclear layer; IPL, inner plexiform layer; OPL, outer plexiform layer; OS, outer segment; GCL, ganglion cell layer.
We next analyzed OPs as sensitive indicators of inner retinal function (Fig. 5A). The amplitudes of OPs in prlr−/− and prlr+/− retinas were reduced by an average of 28 ± 4% and 28 ± 3%, respectively, compared with wild-type values (p < 0.05; Fig. 5B). The amplitudes of OPs did not differ between prlr+/− and prlr−/− retinas. The implicit times of OPs were not modified (data not shown).
Figure 5.

prlr−/− mice show altered OPs. A, Representative OPs in prlr+/+, prlr+/−, and prlr−/− mice in response to the maximal light stimulus (1.2 log cd.s/m2). OP1–OP4 have been labeled from 1 to 4. B, Averaged OP amplitude in prlr+/+, prlr+/−, and prlr−/− mice at 1.2 log cd.s/m2. Data correspond to the mean ± SEM (n = 3 per group, three independent experiments; *p < 0.05 vs values for prlr−/− mice; one-way ANOVA with Bonferroni's post hoc test).
Retinas from prlr−/− mice display increased GFAP levels and decreased expression of bFGF, GDNF, and BDNF
Alterations of ERG parameters can reflect retinal degeneration. Analysis of whole retinas showed that prlr deletion did not affect the levels of retinal apoptosis (Fig. 6A). In addition, we quantified the mRNA levels of rhodopsin, which are considered to correlate, though indirectly, with photoreceptor health. The rhodopsin mRNA levels in prlr−/− retinas were reduced by ∼2.5-fold (p < 0.05) compared with rhodopsin mRNA from prlr+/+ retinas (Fig. 6B).
Figure 6.

Retinas from prlr−/− mice show decreased expression levels of bFGF, GDNF, and BDNF, and a tendency to increased gliosis. A, Retinal apoptosis from prlr+/+ and prlr−/− mice. B, Quantitative PCR (qPCR)-based quantification of rhodopsin mRNA levels in retinas from prlr+/+, prlr+/−, and prlr−/− mice. Apoptosis data correspond to the mean ± SEM (n = 4 per group, two independent experiments. qPCR data correspond to the mean ± SEM (n = 4–9 per group; *p < 0.05 vs values for prlr+/+ mice; one-way ANOVA with Bonferroni's post hoc test). C, qPCR-based quantification of GFAP mRNA levels in retinas from prlr+/+, prlr+/−, and prlr−/− mice. The data are presented as the mean ± SEM (n = 4–9 per group, three independent experiments; *p < 0.05 vs values for prlr+/+ mice; one-way ANOVA with Bonferroni's post hoc test). D, qPCR-based quantification of bFGF, GDNF, BDNF, CNTF, PEDF, and NGF mRNA levels in retinas from prlr+/+, prlr+/−, and prlr−/− mice. The data are presented as the mean ± SEM (n = 4–9 per group, three independent experiments; **p < 0.01 and ***p < 0.001 vs values in prlr+/+ mice; one-way ANOVA with Bonferroni's post hoc test). n.s., Not significant.
Having demonstrated that increasing prolactinemia limits gliosis and neurotrophin expression changes upon retinal injury, we then investigated whether increased gliosis and reduced overall synthesis of neurotrophins correlate with ERG alterations in prlr−/− mice. The GFAP mRNA levels in prlr−/− retinas were approximately twofold (p < 0.05) greater than those in prlr+/+ and prlr+/− retinas (Fig. 6C). Moreover, the bFGF, GDNF, and BDNF mRNA levels were reduced in prlr−/− retinas by 67 ± 4%, 75 ± 1%, and 63 ± 5% (p < 0.01), respectively, compared with values from prlr+/+ retinas (Fig. 6D). The expression levels of CNTF and PEDF were the same in the retinas of prlr−/− and prlr+/+ mice. However, NGF mRNA levels in prlr−/− retinas were approximately eightfold greater (p < 0.001) than those in prlr+/+ mice.
Discussion
Retinal homeostasis strongly relies on glial activity, and Müller cells are a major source of neurotrophin release in response to neural death (Bringmann et al., 2006). Postmitotic photoreceptor cells cannot compensate for losses during aging and retinal degenerative disorders including age-related macular degeneration, retinitis pigmentosa, glaucoma, and diabetic retinopathy (Nag and Wadhwa, 2012). In these diseases, loss of neurotrophic support leads to neural death, and excessive light exposure can also enhance the progression and severity of neural damage (Cruickshanks et al., 1993). Therefore, natural promoters of the neurotrophic activity in the retina are potential therapeutic agents against degeneration. Here, we have identified prolactin signaling as necessary for proper photoresponse and as a molecular mechanism underlying neurotrophic support in the retina.
This is the first report showing that the inactivation of prlr has a phenotype in adult retinal function. Mice lacking the prlr gene have an overall reduced ERG response, independent of the lighting conditions, implicating prolactin signaling as necessary for retinal function and consequently, visual perception. Targeted disruption of the prolactin gene in zebrafish did not compromise early eye formation, but the inner retinal neurons were insufficiently differentiated (Huang et al., 2009).
Consistent with previous studies (for review, see Organisciak and Vaughan, 2010), we show that BCL reduced the amplitudes and prolonged the latencies of ERG waves and OPs, in association with retinal cell apoptosis and glia activation. This BCL model recapitulates essential attributes of alterations observed in experimental retinal degeneration but also in retinal diseases (Jones et al., 2003; Marc et al., 2008; Bringmann et al., 2009). Most significantly, both BCL exposure and prlr deletion resulted in reduced ERG and OP magnitudes, effects that were mostly prevented by increasing systemic prolactin. Prolactin treatment also opposed the BCL-induced delays in ERG and OPs, and it reduced retinal cell apoptosis. Though prlr−/− retinas do not exhibit increased levels of apoptosis, previous findings showed that prolactin promotes neuronal cell survival (Morales, 2011) and decreases apoptosis in non-neuronal cells (Tessier et al., 2001; Bailey et al., 2004). In particular, prolactin upregulates the anti-apoptotic protein Bcl-2 (Leff et al., 1996), whose expression is reduced in retina and brain by stresses like exposure to damaging light (Grosche et al., 1995). Because Bcl-2 overexpression inhibits photoreceptor degeneration (Quiambao et al., 2001), increasing prolactinemia may limit retinal cell death by activating the Bcl-2 pathway. Further, our data may be explained by the incorporation of systemic prolactin into the retina (O'Steen and Sundberg, 1982), and it shows that prolactin limits the BCL proapoptotic effect at concentrations (0.06–0.16 μg/ml) close to those (0.2–0.3 μg/ml) circulating during pregnancy and lactation (Ben-Jonathan et al., 2008).
Abnormal ERG parameters provide information about the origin of retinal dysfunction. Photopic ERG reflects cone response while scotopic ERG includes both cone and rod responses if light intensity is sufficient. The 1.2 log cd.s/m2 stimulus evoked large ERG responses in the photopic recordings; therefore, it was intense enough to stimulate cones under scotopic conditions. In the prlr−/− eyes, cone function is compromised, because we found reduced scotopic A-wave amplitude at all illumination intensities in association with reduced photopic B-wave amplitude. Consistent with a defect in cone photoreceptor activity, the scotopic B-wave is reduced. Additionally, prlr−/− mice have decreased B-wave amplitudes under both scotopic and photopic conditions, which are known to involve Müller glia activity (Bringmann et al., 2006). We also found that the magnitude of summed OPs is reduced in the prlr−/− eyes. This indicates that the inner retinal activity is impaired, but there is no significant change in the number of retinal cells and processes since prlr−/− and prlr+/+ mice showed retinal layers of comparable width. Although the specific cellular origins of the OPs must still be established, the circuitry associated with the OPs in the mature retina includes “ON,” “OFF,” and feedback pathways (Dong et al., 2004), and many retinal cells influence OP characteristics (Wachtmeister, 1998). Collectively, these findings indicate that prolactin signaling helps maintain the activity of most retinal cells and implicate prolactin as a natural retinal trophic factor.
Photoreceptor degeneration alters the neuron–glial network to modify the production of neurotrophic factors required for an environment that minimizes damage (Harada et al., 2002). Our data support this notion by showing that BCL exposure increases the retinal expression of neuroprotective factors GDNF, PEDF, CNTF, and NGF, while decreasing that of bFGF. These neurotrophin changes help explain the presence of apoptosis in the BCL-exposed retinas. Neurotrophic rescue of photoreceptors is mediated by the action of neurotrophic factors like NGF, BDNF, CNTF, PEDF, and GDNF on Müller cells and microgliocytes that, in response, produce bFGF and GDNF, which act directly on the photoreceptor to promote survival (Fontaine et al., 1998; Wexler et al., 1998; Harada et al., 2000; Wahlin et al., 2000; Bringmann and Reichenbach, 2001). For example, blocking NGF signaling increases the levels of Müller cell-derived bFGF, thereby preventing light-induced photoreceptor death (Harada et al., 2000, 2002; Zack, 2000). We found that increasing prolactinemia prevented the BCL-induced increase and decrease of NGF and bFGF, respectively. Hyperprolactinemia also enhanced the BCL-induced expression of the prosurvival factors GDNF, PEDF, and BDNF, consistent with its anti-apoptotic effect. Moreover, the differences in growth factor mRNA production between prlr−/− and BCL-exposed animals may explain the lack of retinal cell apoptosis and layer disorganization in prlr−/− retinas. We assume that the reduced rhodopsin mRNA levels in the prlr−/− retinas do not reflect retinal cell degeneration, although this issue requires further studies. Despite decreases in bFGF, GDNF, and BDNF mRNA levels in prlr−/− retinas, CNTF and PEDF levels were maintained, and NGF level was increased compared with the wild-type, indicating that prlr−/− mice may have compensatory mechanisms responsible for retinal cell survival. The discrepancies between growth factor mRNA levels in hyperprolactinemic and prlr−/− animals may be attributed to the timing and duration of prolactin excess (the entire lifespan including early development in prlr−/− mice vs transient adult onset in AP-implanted rats), as previously reported (Ding et al., 2013). These data further indicate that in our two models, while the mRNA levels of certain growth factors (e.g., bFGF, GDNF, and BDNF) are primarily regulated by prolactin, others (NGF, PEDF, and CNTF) may be regulated by prolactin as well as other factors. The fact that bFGF, GDNF, BDNF, CNTF, PEDF, and NGF protect against retinal degeneration (Wahlin et al., 2000; Wen et al., 2012; Kolomeyer et al., 2013) supports the neuroprotective effects of prolactin. Previous investigations did not observe the protective effect of prolactin against photic damage (Olafson and O'Steen, 1976; O'Steen and Kraeer, 1977), but they did not assess retinal function.
Our working model proposes that prolactin contributes to retinal homeostasis and neuroprotection by strengthening the neurotrophic activity of retinal glial cells and limiting their gliosis. Indeed, GFAP mRNA levels are threefold lower in hyperprolactinemic animals than in normoprolactinemic animals exposed to BCL. Relative to unexposed animals, the BCL-induced increase in retinal GFAP mRNA levels is higher in hyperprolactinemic than in normoprolactinemic rats (20- vs 3-fold, respectively). However, this difference is due to the much lower GFAP expression found in nonexposed hyperprolactinemic rats. BCL exposure upregulates prolactin receptor in the retina. The signaling events that govern the retinal synthesis of prolactin receptor are unknown, although upregulation of the prolactin receptor may result directly from local photoreceptor death, as already proposed for the receptor of the neuroprotective leukemia inhibitory factor (Rattner and Nathans, 2005; Joly et al., 2008). Alternatively, or perhaps concurrently, prolactin receptor synthesis may be stimulated by the upregulation of TNF-α and IL-1β occurring in retinal glia under degenerative pressure (Busik et al., 2008; Zhang et al., 2012b). These proinflammatory cytokines increase prolactin receptor expression (Corbacho et al., 2003, 2004), and also prolactin expression in brain tissue (De Bellis et al., 2005) and damaged brain (Chioléro et al., 1988; DeVito et al., 1995; Agha et al., 2004; Möderscheim et al., 2007). Notably, prolactin promotes reactive gliosis (Möderscheim et al., 2007; Simard and Rivest, 2007; Azcoitia et al., 2010), stimulates astrocyte proliferation (DeVito et al., 1995), and may target microgliocytes (Möderscheim et al., 2007); prolactin induces inducible nitric oxide (NO) synthase expression and increases NO production in brain (Popeski et al., 1999; Raso et al., 1999; Dogusan et al., 2001; Vega et al., 2010), and NO produced at a lesion has been proposed as a stop signal for microglia to accumulate (Chen et al., 2000). While the role of increasing prolactinemia in retinal gliosis requires further research, our results show that hyperprolactinemia reduces GFAP expression per se and can limit the gliosis that accompanies retinal injury.
Together with the presence of prolactin receptor in nonhuman primate retina (Rivera et al., 2008), these findings may suggest a role for prolactin in the human retina. High levels of circulating prolactin are associated with increased ERG B-wave amplitude in pregnant women (De Luca Brunori et al., 1985) and protect against diabetic retinopathy (Larinkari et al., 1982; Moloney and Drury, 1982; Bhatnagar et al., 2009). The present study provides a rationale to further investigate hormonal regulation of neuron–glia interactions under physiopathological conditions.
Footnotes
This study was supported by the Universidad Nacional Autónoma de México (UNAM) and the National Council of Science and Technology of Mexico (CONACYT) Grant 176393. E.A. was supported by CONACYT 99210 Fellowship as well as the UNAM posgrado program. We thank F. López-Barrera, G. Nava, D. Mondragón, A. Prado, M. García, and Elsa Nydia Hernández Ríos for their technical assistance; and D.D. Pless for critically editing this manuscript.
The authors declare no competing financial interests.
References
- Adler RA. The anterior pituitary-grafted rat: a valid model of chronic hyperprolactinemia. Endocr Rev. 1986;7:302–313. doi: 10.1210/edrv-7-3-302. [DOI] [PubMed] [Google Scholar]
- Agha A, Rogers B, Mylotte D, Taleb F, Tormey W, Phillips J, Thompson CJ. Neuroendocrine dysfunction in the acute phase of traumatic brain injury. Clin Endocrinol (Oxf) 2004;60:584–591. doi: 10.1111/j.1365-2265.2004.02023.x. [DOI] [PubMed] [Google Scholar]
- Allen DM, Cristy M. Thiourea does not block visual pigment responses to prolactin in trout. Vision Res. 1978;18:859–860. doi: 10.1016/0042-6989(78)90129-3. [DOI] [PubMed] [Google Scholar]
- Aranda J, Rivera JC, Jeziorski MC, Riesgo-Escovar J, Nava G, López-Barrera F, Quiróz-Mercado H, Berger P, Martínez de la Escalera G, Clapp C. Prolactins are natural inhibitors of angiogenesis in the retina. Invest Ophthalmol Vis Sci. 2005;46:2947–2953. doi: 10.1167/iovs.05-0173. [DOI] [PubMed] [Google Scholar]
- Azcoitia I, Santos-Galindo M, Arevalo MA, Garcia-Segura LM. Role of astroglia in the neuroplastic and neuroprotective actions of estradiol. Eur J Neurosci. 2010;32:1995–2002. doi: 10.1111/j.1460-9568.2010.07516.x. [DOI] [PubMed] [Google Scholar]
- Babenko O, Golubov A, Ilnytskyy Y, Kovalchuk I, Metz GA. Genomic and epigenomic responses to chronic stress involve miRNA-mediated programming. PLoS One. 2012;7:e29441. doi: 10.1371/journal.pone.0029441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey JP, Nieport KM, Herbst MP, Srivastava S, Serra RA, Horseman ND. Prolactin and transforming growth factor-beta signaling exert opposing effects on mammary gland morphogenesis, involution, and the Akt-forkhead pathway. Mol Endocrinol. 2004;18:1171–1184. doi: 10.1210/me.2003-0345. [DOI] [PubMed] [Google Scholar]
- Barres BA. The mystery and magic of glia: a perspective on their roles in health and disease. Neuron. 2008;60:430–440. doi: 10.1016/j.neuron.2008.10.013. [DOI] [PubMed] [Google Scholar]
- Benatti C, Valensisi C, Blom JM, Alboni S, Montanari C, Ferrari F, Tagliafico E, Mendlewicz J, Brunello N, Tascedda F. Transcriptional profiles underlying vulnerability and resilience in rats exposed to an acute unavoidable stress. J Neurosci Res. 2012;90:2103–2115. doi: 10.1002/jnr.23100. [DOI] [PubMed] [Google Scholar]
- Ben-Jonathan N, LaPensee CR, LaPensee EW. What can we learn from rodents about prolactin in humans? Endocr Rev. 2008;29:1–41. doi: 10.1210/er.2007-0017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhatnagar A, Ghauri AJ, Hope-Ross M, Lip PL. Diabetic retinopathy in pregnancy. Curr Diabetes Rev. 2009;5:151–156. doi: 10.2174/157339909788920929. [DOI] [PubMed] [Google Scholar]
- Binart N, Helloco C, Ormandy CJ, Barra J, Clément-Lacroix P, Baran N, Kelly PA. Rescue of preimplantatory egg development and embryo implantation in prolactin receptor-deficient mice after progesterone administration. Endocrinology. 2000;141:2691–2697. doi: 10.1210/endo.141.7.7568. [DOI] [PubMed] [Google Scholar]
- Bringmann A, Reichenbach A. Role of Muller cells in retinal degenerations. Front Biosci. 2001;6:E72–E92. doi: 10.2741/Bringman. [DOI] [PubMed] [Google Scholar]
- Bringmann A, Pannicke T, Grosche J, Francke M, Wiedemann P, Skatchkov SN, Osborne NN, Reichenbach A. Muller cells in the healthy and diseased retina. Prog Retin Eye Res. 2006;25:397–424. doi: 10.1016/j.preteyeres.2006.05.003. [DOI] [PubMed] [Google Scholar]
- Bringmann A, Iandiev I, Pannicke T, Wurm A, Hollborn M, Wiedemann P, Osborne NN, Reichenbach A. Cellular signaling and factors involved in Muller cell gliosis: neuroprotective and detrimental effects. Prog Retin Eye Res. 2009;28:423–451. doi: 10.1016/j.preteyeres.2009.07.001. [DOI] [PubMed] [Google Scholar]
- Busik JV, Mohr S, Grant MB. Hyperglycemia-induced reactive oxygen species toxicity to endothelial cells is dependent on paracrine mediators. Diabetes. 2008;57:1952–1965. doi: 10.2337/db07-1520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caffé AR, Söderpalm A, van Veen T. Photoreceptor-specific protein expression of mouse retina in organ culture and retardation of rd degeneration in vitro by a combination of basic fibroblast and nerve growth factors. Curr Eye Res. 1993;12:719–726. doi: 10.3109/02713689308995767. [DOI] [PubMed] [Google Scholar]
- Chen A, Kumar SM, Sahley CL, Muller KJ. Nitric oxide influences injury-induced microglial migration and accumulation in the leech CNS. J Neurosci. 2000;20:1036–1043. doi: 10.1523/JNEUROSCI.20-03-01036.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chioléro R, Lemarchand T, Schutz Y, de Tribolet N, Felber JP, Freeman J, Jéquier E. Plasma pituitary hormone levels in severe trauma with or without head injury. J Trauma. 1988;28:1368–1374. doi: 10.1097/00005373-198809000-00011. [DOI] [PubMed] [Google Scholar]
- Corbacho AM, Macotela Y, Nava G, Eiserich JP, Cross CE, Martínez de la Escalera G, Clapp C. Cytokine induction of prolactin receptors mediates prolactin inhibition of nitric oxide synthesis in pulmonary fibroblasts. FEBS Lett. 2003;544:171–175. doi: 10.1016/S0014-5793(03)00499-X. [DOI] [PubMed] [Google Scholar]
- Corbacho AM, Valacchi G, Kubala L, Olano-Martín E, Schock BC, Kenny TP, Cross CE. Tissue-specific gene expression of prolactin receptor in the acute-phase response induced by lipopolysaccharides. Am J Physiol Endocrinol Metab. 2004;287:E750–E757. doi: 10.1152/ajpendo.00522.2003. [DOI] [PubMed] [Google Scholar]
- Crim JW. Prolactin-induced modification on visual pigments in the eastern red-spotted newt, Notophthalmus viridescens. Gen Comp Endocrinol. 1975a;26:233–242. doi: 10.1016/0016-6480(75)90140-9. [DOI] [PubMed] [Google Scholar]
- Crim JW. Prolactin-thyroxine antagonism and the metamorphosis of visual pigments in Rana catesbeiana tadpoles. J Exp Zool. 1975b;192:355–362. doi: 10.1002/jez.1401920309. [DOI] [PubMed] [Google Scholar]
- Cruickshanks KJ, Klein R, Klein BE. Sunlight and age-related macular degeneration. The Beaver Dam Eye Study. Arch Ophthalmol. 1993;111:514–518. doi: 10.1001/archopht.1993.01090040106042. [DOI] [PubMed] [Google Scholar]
- De Luca Brunori I, Moggi G, Fornaro P, Teti G, Murru S, Gadducci A, Castrogiovanni P, Perossini M. Evaluation of the central dopaminergic activity in gestational hyperprolactinaemia by means of the electroretinographic technique. Clin Exp Obstet Gynecol. 1985;12:13–15. [PubMed] [Google Scholar]
- De Bellis A, Bizzarro A, Pivonello R, Lombardi G, Bellastella A. Prolactin and autoimmunity. Pituitary. 2005;8:25–30. doi: 10.1007/s11102-005-5082-5. [DOI] [PubMed] [Google Scholar]
- DeVito WJ, Stone S, Shamgochian M. Prolactin induced expression of glial fibrillary acidic protein and tumor necrosis factor-alpha at a wound site in the rat brain. Mol Cell Endocrinol. 1995;108:125–130. doi: 10.1016/0303-7207(94)03465-6. [DOI] [PubMed] [Google Scholar]
- Ding J, Sackmann-Sala L, Kopchick JJ. Mouse models of growth hormone action and aging: a proteomic perspective. Proteomics. 2013;13:674–685. doi: 10.1002/pmic.201200271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dogusan Z, Hooghe R, Verdood P, Hooghe-Peters EL. Cytokine-like effects of prolactin in human mononuclear and polymorphonuclear leukocytes. J Neuroimmunol. 2001;120:58–66. doi: 10.1016/S0165-5728(01)00420-9. [DOI] [PubMed] [Google Scholar]
- Dong CJ, Agey P, Hare WA. Origins of the electroretinogram oscillatory potentials in the rabbit retina. Vis Neurosci. 2004;21:533–543. doi: 10.1017/S0952523804214043. [DOI] [PubMed] [Google Scholar]
- Fontaine V, Kinkl N, Sahel J, Dreyfus H, Hicks D. Survival of purified rat photoreceptors in vitro is stimulated directly by fibroblast growth factor-2. J Neurosci. 1998;18:9662–9672. doi: 10.1523/JNEUROSCI.18-23-09662.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grieshaber MC, Moramarco F, Schoetzau A, Flammer J, Orguel S. Detection of retinal glial cell activation in glaucoma by time domain optical coherence tomography. Klin Monbl Augenheilkd. 2012;229:314–318. doi: 10.1055/s-0031-1281857. [DOI] [PubMed] [Google Scholar]
- Grosche J, Härtig W, Reichenbach A. Expression of glial fibrillary acidic protein (GFAP), glutamine synthetase (GS), and Bcl-2 protooncogene protein by Muller (glial) cells in retinal light damage of rats. Neurosci Lett. 1995;185:119–122. doi: 10.1016/0304-3940(94)11239-F. [DOI] [PubMed] [Google Scholar]
- Harada T, Harada C, Nakayama N, Okuyama S, Yoshida K, Kohsaka S, Matsuda H, Wada K. Modification of glial-neuronal cell interactions prevents photoreceptor apoptosis during light-induced retinal degeneration. Neuron. 2000;26:533–541. doi: 10.1016/S0896-6273(00)81185-X. [DOI] [PubMed] [Google Scholar]
- Harada T, Harada C, Kohsaka S, Wada E, Yoshida K, Ohno S, Mamada H, Tanaka K, Parada LF, Wada K. Microglia–Müller glia cell interactions control neurotrophic factor production during light-induced retinal degeneration. J Neurosci. 2002;22:9228–9236. doi: 10.1523/JNEUROSCI.22-21-09228.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang X, Hui MN, Liu Y, Yuen DS, Zhang Y, Chan WY, Lin HR, Cheng SH, Cheng CH. Discovery of a novel prolactin in non-mammalian vertebrates: evolutionary perspectives and its involvement in teleost retina development. PLoS One. 2009;4:e6163. doi: 10.1371/journal.pone.0006163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joly S, Lange C, Thiersch M, Samardzija M, Grimm C. Leukemia inhibitory factor extends the lifespan of injured photoreceptors in vivo. J Neurosci. 2008;28:13765–13774. doi: 10.1523/JNEUROSCI.5114-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones BW, Watt CB, Frederick JM, Baehr W, Chen CK, Levine EM, Milam AH, Lavail MM, Marc RE. Retinal remodeling triggered by photoreceptor degenerations. J Comp Neurol. 2003;464:1–16. doi: 10.1002/cne.10703. [DOI] [PubMed] [Google Scholar]
- Kolomeyer AM, Sugino IK, Zarbin MA. Characterization of the effects of retinal pigment epithelium-conditioned media on porcine and aged human retina. Graefes Arch Clin Exp Ophthalmol. 2013;251:1515–1528. doi: 10.1007/s00417-013-2326-3. [DOI] [PubMed] [Google Scholar]
- Langmann T. Microglia activation in retinal degeneration. J Leukoc Biol. 2007;81:1345–1351. doi: 10.1189/jlb.0207114. [DOI] [PubMed] [Google Scholar]
- Larinkari J, Laatikainen L, Ranta T, Mörönen P, Pesonen K, Laatikainen T. Metabolic control and serum hormone levels in relation to retinopathy in diabetic pregnancy. Diabetologia. 1982;22:327–332. doi: 10.1007/BF00253576. [DOI] [PubMed] [Google Scholar]
- LaVail MM, Yasumura D, Matthes MT, Lau-Villacorta C, Unoki K, Sung CH, Steinberg RH. Protection of mouse photoreceptors by survival factors in retinal degenerations. Invest Ophthalmol Vis Sci. 1998;39:592–602. [PubMed] [Google Scholar]
- Leff MA, Buckley DJ, Krumenacker JS, Reed JC, Miyashita T, Buckley AR. Rapid modulation of the apoptosis regulatory genes, bcl-2 and bax by prolactin in rat Nb2 lymphoma cells. Endocrinology. 1996;137:5456–5462. doi: 10.1210/endo.137.12.8940371. [DOI] [PubMed] [Google Scholar]
- Marc RE, Jones BW, Watt CB, Vazquez-Chona F, Vaughan DK, Organisciak DT. Extreme retinal remodeling triggered by light damage: implications for age related macular degeneration. Mol Vis. 2008;14:782–806. [PMC free article] [PubMed] [Google Scholar]
- Möderscheim TA, Gorba T, Pathipati P, Kokay IC, Grattan DR, Williams CE, Scheepens A. Prolactin is involved in glial responses following a focal injury to the juvenile rat brain. Neuroscience. 2007;145:963–973. doi: 10.1016/j.neuroscience.2006.12.053. [DOI] [PubMed] [Google Scholar]
- Moloney JB, Drury MI. The effect of pregnancy on the natural course of diabetic retinopathy. Am J Ophthalmol. 1982;93:745–756. doi: 10.1016/0002-9394(82)90471-8. [DOI] [PubMed] [Google Scholar]
- Morales T. Recent findings on neuroprotection against excitotoxicity in the hippocampus of female rats. J Neuroendocrinol. 2011;23:994–1001. doi: 10.1111/j.1365-2826.2011.02141.x. [DOI] [PubMed] [Google Scholar]
- Nag TC, Wadhwa S. Ultrastructure of the human retina in aging and various pathological states. Micron. 2012;43:759–781. doi: 10.1016/j.micron.2012.01.011. [DOI] [PubMed] [Google Scholar]
- Olafson RP, O'Steen WK. Hormonal influences on photoreceptor damage: the pituitary gland and ovaries. Invest Ophthalmol. 1976;15:869–872. [PubMed] [Google Scholar]
- Organisciak DT, Vaughan DK. Retinal light damage: mechanisms and protection. Prog Retin Eye Res. 2010;29:113–134. doi: 10.1016/j.preteyeres.2009.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ormandy CJ, Camus A, Barra J, Damotte D, Lucas B, Buteau H, Edery M, Brousse N, Babinet C, Binart N, Kelly PA. Null mutation of the prolactin receptor gene produces multiple reproductive defects in the mouse. Genes Dev. 1997;11:167–178. doi: 10.1101/gad.11.2.167. [DOI] [PubMed] [Google Scholar]
- O'Steen WK, Kraeer SL. Effects of hypophysectomy, pituitary gland homogenates and transplants, and prolactin on photoreceptor destruction. Invest Ophthalmol Vis Sci. 1977;16:940–946. [PubMed] [Google Scholar]
- O'Steen WK, Sundberg DK. Patterns of radioactivity in the eyes of rats after injection of iodinated prolactin. Ophthalmic Res. 1982;14:54–62. doi: 10.1159/000265174. [DOI] [PubMed] [Google Scholar]
- Popeski N, Amir S, Woodside B. Changes in NADPH-d staining in the paraventricular and supraoptic nuclei during pregnancy and lactation in rats: role of ovarian steroids and oxytocin. J Neuroendocrinol. 1999;11:53–61. doi: 10.1046/j.1365-2826.1999.00291.x. [DOI] [PubMed] [Google Scholar]
- Quiambao AB, Tan E, Chang S, Komori N, Naash MI, Peachey NS, Matsumoto H, Ucker DS, Al-Ubaidi MR. Transgenic Bcl-2 expressed in photoreceptor cells confers both death-sparing and death-inducing effects. Exp Eye Res. 2001;73:711–721. doi: 10.1006/exer.2001.1083. [DOI] [PubMed] [Google Scholar]
- Rangaswamy NV, Zhou W, Harwerth RS, Frishman LJ. Effect of experimental glaucoma in primates on oscillatory potentials of the slow-sequence mfERG. Invest Ophthalmol Vis Sci. 2006;47:753–767. doi: 10.1167/iovs.05-1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raso GM, Meli R, Gualillo O, Pacilio M, Di Carlo R. Prolactin induction of nitric oxide synthase in rat C6 glioma cells. J Neurochem. 1999;73:2272–2277. doi: 10.1046/j.1471-4159.1999.0732272.x. [DOI] [PubMed] [Google Scholar]
- Rattner A, Nathans J. The genomic response to retinal disease and injury: evidence for endothelin signaling from photoreceptors to glia. J Neurosci. 2005;25:4540–4549. doi: 10.1523/JNEUROSCI.0492-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivera JC, Aranda J, Riesgo J, Nava G, Thebault S, López-Barrera F, Ramírez M, Martínez de la Escalera G, Clapp C. Expression and cellular localization of prolactin and the prolactin receptor in mammalian retina. Exp Eye Res. 2008;86:314–321. doi: 10.1016/j.exer.2007.11.003. [DOI] [PubMed] [Google Scholar]
- Sanes JR, Zipursky SL. Design principles of insect and vertebrate visual systems. Neuron. 2010;66:15–36. doi: 10.1016/j.neuron.2010.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simard AR, Rivest S. Neuroprotective effects of resident microglia following acute brain injury. J Comp Neurol. 2007;504:716–729. doi: 10.1002/cne.21469. [DOI] [PubMed] [Google Scholar]
- Takamiya Y, Kohsaka S, Toya S, Otani M, Tsukada Y. Immunohistochemical studies on the proliferation of reactive astrocytes and the expression of cytoskeletal proteins following brain injury in rats. Brain Res. 1988;466:201–210. doi: 10.1016/0165-3806(88)90045-4. [DOI] [PubMed] [Google Scholar]
- Tanaka T, Shiu RP, Gout PW, Beer CT, Noble RL, Friesen HG. A new sensitive and specific bioassay for lactogenic hormones: measurement of prolactin and growth hormone in human serum. J Clin Endocrinol Metab. 1980;51:1058–1063. doi: 10.1210/jcem-51-5-1058. [DOI] [PubMed] [Google Scholar]
- Tessier C, Prigent-Tessier A, Ferguson-Gottschall S, Gu Y, Gibori G. PRL antiapoptotic effect in the rat decidua involves the PI3K/protein kinase B-mediated inhibition of caspase-3 activity. Endocrinology. 2001;142:4086–4094. doi: 10.1210/endo.142.9.8381. [DOI] [PubMed] [Google Scholar]
- Unterlauft JD, Eichler W, Kuhne K, Yang XM, Yafai Y, Wiedemann P, Reichenbach A, Claudepierre T. Pigment epithelium-derived factor released by Muller glial cells exerts neuroprotective effects on retinal ganglion cells. Neurochem Res. 2012;37:1524–1533. doi: 10.1007/s11064-012-0747-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vega C, Moreno-Carranza B, Zamorano M, Quintanar-Stéphano A, Méndez I, Thebault S, Martínez de la Escalera G, Clapp C. Prolactin promotes oxytocin and vasopressin release by activating neuronal nitric oxide synthase in the supraoptic and paraventricular nuclei. Am J Physiol Regul Integr Comp Physiol. 2010;299:R1701–R1708. doi: 10.1152/ajpregu.00575.2010. [DOI] [PubMed] [Google Scholar]
- Wachtmeister L. Oscillatory potentials in the retina: what do they reveal. Prog Retin Eye Res. 1998;17:485–521. doi: 10.1016/S1350-9462(98)00006-8. [DOI] [PubMed] [Google Scholar]
- Wahlin KJ, Campochiaro PA, Zack DJ, Adler R. Neurotrophic factors cause activation of intracellular signaling pathways in Muller cells and other cells of the inner retina, but not photoreceptors. Invest Ophthalmol Vis Sci. 2000;41:927–936. [PubMed] [Google Scholar]
- Walter P, Widder RA, Lüke C, Königsfeld P, Brunner R. Electrophysiological abnormalities in age-related macular degeneration. Graefes Arch Clin Exp Ophthalmol. 1999;237:962–968. doi: 10.1007/s004170050331. [DOI] [PubMed] [Google Scholar]
- Wässle H. Parallel processing in the mammalian retina. Nat Rev Neurosci. 2004;5:747–757. doi: 10.1038/nrn1497. [DOI] [PubMed] [Google Scholar]
- Wen R, Tao W, Li Y, Sieving PA. CNTF and retina. Prog Retin Eye Res. 2012;31:136–151. doi: 10.1016/j.preteyeres.2011.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wexler EM, Berkovich O, Nawy S. Role of the low-affinity NGF receptor (p75) in survival of retinal bipolar cells. Vis Neurosci. 1998;15:211–218. doi: 10.1017/S095252389815201X. [DOI] [PubMed] [Google Scholar]
- Zack DJ. Neurotrophic rescue of photoreceptors: are Muller cells the mediators of survival? Neuron. 2000;26:285–286. doi: 10.1016/S0896-6273(00)81160-5. [DOI] [PubMed] [Google Scholar]
- Zhang K, Zhang L, Weinreb RN. Ophthalmic drug discovery: novel targets and mechanisms for retinal diseases and glaucoma. Nat Rev Drug Discov. 2012a;11:541–559. doi: 10.1038/nrd3745. [DOI] [PubMed] [Google Scholar]
- Zhang M, Xu G, Liu W, Ni Y, Zhou W. Role of fractalkine/CX3CR1 interaction in light-induced photoreceptor degeneration through regulating retinal microglial activation and migration. PLoS One. 2012b;7:e35446. doi: 10.1371/journal.pone.0035446. [DOI] [PMC free article] [PubMed] [Google Scholar]