

Abstract
Fragile X syndrome (FXS) is the most common inherited neurodevelopmental disorder with intellectual disability. Here, we show that the expression of tissue plasminogen activator (tPA) is increased in glial cells differentiated from neural progenitors of Fmr1 knock-out mice, a mouse model for FXS, and that tPA is involved in the altered migration and differentiation of these progenitors lacking FMR1 protein (FMRP). When tPA function is blocked with an antibody, enhanced migration of doublecortin-immunoreactive neurons in 1 d differentiated FMRP-deficient neurospheres is normalized. In time-lapse imaging, blocking the tPA function promotes early glial differentiation and reduces the velocity of nuclear movement of FMRP-deficient radial glia. In addition, we show that enhanced intracellular Ca2+ responses to depolarization with potassium are prevented by the treatment with the tPA-neutralizing antibody in FMRP-deficient cells during early neural progenitor differentiation. Alterations of the tPA expression in the embryonic, postnatal, and adult brain of Fmr1 knock-out mice suggest an important role for tPA in the abnormal neuronal differentiation and plasticity in FXS. Altogether, the results indicate that tPA may prove to be an interesting potential target for pharmacological intervention in FXS.
Keywords: depolarization, differentiation, FMRP, glia, neurons, stem cells
Introduction
Cellular models of monogenic disorders such as fragile X syndrome (FXS) have opened up novel possibilities to investigate mechanisms of brain development and pathophysiology underlying disorders of the CNS (Castrén, 2012). FXS is a common cause of inherited intellectual disability (Santoro et al., 2012). In addition to impairments of learning and memory, the FXS phenotype includes abnormalities of sensory processing, activity levels, communication, and social behaviors (Chonchaiya et al., 2009). A mutation in the Fragile X mental retardation 1 (FMR1) gene causes the loss of the functional FMR1 protein (FMRP) that leads to FXS (Verkerk et al., 1991). FMRP is an RNA-binding protein that interacts with a number of transcripts encoding presynaptic and postsynaptic proteins and regulates protein translation (Bagni and Greenough, 2005; Darnell et al., 2011). Maturation of glutamatergic signaling is affected in Fmr1 knock-out (KO) mice (Harlow et al., 2010). In particular, perturbations in signaling downstream of the metabotropic glutamate receptors (mGluRs) underlie circuit abnormalities in FXS (Huber et al., 2002; Dölen and Bear, 2008) and consequent alterations in the AMPA receptor function (Hu et al., 2008). Developmental changes in Fmr1 KO mouse brain are linked with disturbed neurogenesis and correlate with alterations of fate determination and proliferation capacity of neural progenitor cells (NPCs; Castrén et al., 2005; Bhattacharyya et al., 2008; Luo et al., 2010, Callan and Zarnescu, 2011; Guo et al., 2011). Alterations of brain-derived growth factor (BDNF)/TrkB signaling contribute to an aberrant phenotype of neurons differentiated from cortical FXS NPCs (Louhivuori et al., 2011; Castrén and Castrén, 2014). Reduced BDNF expression in double-transgenic FXS mouse with a deletion of one copy of the Bdnf gene normalizes the neural morphology and part of the behavioral phenotype (Uutela et al., 2012).
Tissue-type plasminogen activator (tPA) is a serine protease that is highly expressed by neuronal and glial cells in the CNS (Melchor and Strickland, 2005). tPA has been implicated in synaptic plasticity, learning, epilepsy, and excitotoxic cell death (Tsirka et al., 1995; Melchor and Strickland, 2005; Samson and Medcalf, 2006). Lack of tPA causes disturbances of neurite outgrowth and neuronal migration (Seeds et al., 1999). tPA cleaves the pro-enzyme plasminogen into active plasmin, but many of its functions may occur independently of plasminogen-dependent pathways (Melchor and Strickland, 2005; Samson and Medcalf, 2006). The plasminogenic activity of tPA is crucial for processing precursor form of BDNF (proBDNF) to active mature BDNF that is essential for neuronal maturation and plasticity (Pang et al., 2004). BDNF and glutamate stimulation induce tPA expression that is present in both neurons and glia (Hwang et al., 2011).
Here, we have explored the role of tPA in neurodevelopmental defects of the Fmr1 KO mouse. We found that tPA expression is altered in differentiating neuronal cells and developing and adult brain of Fmr1 KO mice. Activity-dependent processes are of major importance in neuronal maturation and plasticity and we show that tPA is involved in abnormal intracellular calcium responses to depolarization and neuronal migration during early neuronal differentiation of FMRP-deficient cells.
Materials and Methods
Mice.
Fmr1 KO mice (B6.129P2-Fmr1tm1/Cgr/J; Jackson Laboratory) were maintained on the C57BL/6JOlaHsd substrain. Male transgenic and wild-type littermates were used at embryonic and postnatal ages as indicated. Mice were housed in standard laboratory conditions in qualified animal facility in accordance with the National Institutes of Health guidelines. All animal experiments were performed according to the guidelines of the Guide for the Care and Use of Laboratory Animals and European Economic Community Council Directive. The studies were approved by the Experimental Animal Ethics Committee of the National Laboratory Animal Center, Finland.
Brain tissue.
Pregnant mice were killed by cervical dislocation and pups by decapitation under CO2 anesthesia. Cell cultures were prepared from brains of male pups at embryonic day 14 (E14). The postnatal day 7 (P7) and adult brains were collected freshly and fixed in 4% paraformaldehyde overnight at 4°C. For paraffin sectioning, fixed brains were washed twice with PBS, pH 7.4, and dehydrated through alcohol series: 70% ethanol for 2 h, 96% ethanol for 2 h, and 100% ethanol overnight at 4°C. Then brains were treated twice with xylene (Riedel-de Haën) for 2 h before immersion in paraffin overnight at 60°C. The next day, brains were molded into paraffin and cut into 20 μm coronal sections onto object glasses with a microtome (HM 355 MICROM; Thermo Scientific).
Cell cultures.
NPCs were propagated from the wall of lateral ventricles of wild-type and Fmr1 KO pups as described previously (Castrén et al., 2005). Cells were grown as free-floating aggregates referred to as neurospheres in Dulbecco's modified Eagle's medium F-12 nutrient mixture (DMEM/F-12) medium containing B27 supplement (both from Invitrogen Life Technologies), l-glutamine (2 mm), HEPES (15 mm), penicillin (100 U/ml), and streptomycin (100 U/ml) (all from Sigma-Aldrich) in the presence of basic fibroblastic growth factor (10 ng/ml) and epidermal growth factor (20 ng/ml) (both from PeproTech) in a 5% CO2-humidified incubator at +37°C. The culture medium was refreshed and growth factors were added three times per week. The cells were passaged by manual trituration at ∼2 week intervals. Neurospheres were differentiated in culture medium without mitogens and the neutralizing anti-murine tPA antibody (American Diagnostica) or normal IgG control antibody (Peprotech) was added to the cell culture medium (200 ng/ml) as indicated at the beginning of the differentiation.
Immunostaining.
Immunostaining of brain sections was performed as described previously (Tervonen et al., 2009). Briefly, the paraffin sections were sequentially deparaffinized 3 times in xylene for 10 min and thereafter rehydrated in 100% ethanol, 96% ethanol, 70% ethanol, and distilled water for 5 min each. For antigen retrieval, the sections were boiled in 10 mm citrate buffer, pH 6.0, for 15 min. Sections were permeabilized and nonspecific labeling was minimized by incubation with PBS containing 0.5% Triton X-100 and 20% normal goat serum (NGS) for 1 h at room temperature (RT). Thereafter, samples were incubated with the primary antibody overnight at +4°C. The primary antibodies were rabbit anti-PLAT (1:50; Proteintech), rabbit anti-PAI-1 (1:500; Abcam), chicken glial fibrillary acidic protein (GFAP, 1:1000; Abcam), and rabbit full-length-specific anti-TrkB (1:150; Santa Cruz Biotechnology). Secondary antibodies Alexa Fluor 488 anti-chicken IgG and Alexa Fluor 568 and 594 anti-rabbit IgG (1:500–1000 for brain sections and 1:500 for cells; Invitrogen) were applied for 1 h at RT. The cell nuclei were counterstained with 4′6-diaminodino-2-phenylindole (DAPI, 0.1 mg/ml, Boehringer Mannheim).
For immunocytochemistry of differentiated neurosphere cells, neurospheres were plated on cover glasses coated with 10 g/ml poly-d-lysine (Sigma-Aldrich) and 5 g/ml laminin (Invitrogen) in culture medium without mitogens and cells were differentiated for 24 h. Cells were fixed with 4% paraformaldehyde in PBS for 10 min at RT and permeabilized with 100% ice-cold methanol. Thereafter, nonspecific staining was blocked with PBS containing 20% NGS or 10% NGS and 0.2% Triton-X for 60 min at RT. Cells were then incubated with the following primary antibodies overnight at 4°C: anti-brain lipid binding protein (BLBP; 1:750; Millipore), anti-doublecortin (anti-DCX, 1:1000; Millipore), and anti-tPA (10 mg/ml; American Diagnostica). Secondary antibodies were applied for 45 min at RT. Secondary antibodies used were Alexa Fluor 488 (green) and Alexa Fluor 568 (red) antibodies (Invitrogen). The disappearance of all staining was seen in negative controls when primary antibodies were omitted. Samples were mounted using Vectashield mounting media containing DAPI for nuclear staining (blue; Vector Laboratories)
Image analysis.
The fluorescent stained brain sections and differentiated NPCs were viewed and images were obtained with the LSM 5 Pascal laser scanning confocal microscope (Zeiss) and analyzed with the image analysis program ImageJ. Image stack sizes were 1520 μm height × 1520 μm width × 2095 μm in z-dimension for the cortical parts of the brains, 595 μm width × 595 μm height × 1676 μm in the z-dimension for the hippocampus, and 632 μm width × 632 μm height × 134 μm in z-dimension for differentiating NPCs. The staining intensity in the hippocampal and cortical samples was measured as integrated density. The amount of tPA-positive NPCs were counted and compared with the total cell count (the amount of DAPI-positive cells) to get the relative amount of positive cells per sample.
Calcium imaging.
Calcium imaging was performed essentially as described previously (Kärkkäinen et al., 2009). For the experiments, 20–30 neurospheres were plated on poly-DL-ornithine-coated 25 mm cover glasses. After differentiation for 24 h in the presence or absence of tPA, antibody cells were incubated with 4 μm fura-2 acetoxymethylester (fura-2/AM, dissolved in 4 mm dimethyl sulfoxide; Sigma-Aldrich) at 37°C for 20 min in HEPES-buffered medium (HBM), pH 7.4, consisting of the following (in mm): 137 NaCl, 5 KCl, 0.44 KH2PO4, 4.2 NaHCO3, 2 CaCl2, 0.5 MgCl2, and 10 glucose. For high potassium chloride-induced depolarizations, NaCl was iso-osmotically replaced with 70 mm KCl. The cover glasses were attached to a perfusion chamber and perfused in HBM 2 ml/min at 37°C. The cells were excited by alternating wavelengths of 340 and 380 nm using narrow band excitation filters. Light emission was measured through a 430 nm dichroic mirror and a 510 nm barrier filter with an integrating charge-coupled device camera (Cohu). An image (ratio 340 nm/380 nm) was acquired each second and up to 100 cells in one neurosphere were recorded simultaneously.
Time-lapse imaging and cell tracking.
Cellular movement was monitored in a cell-culturing instrument combining phase-contrast microscopy, automation, and environmental control (Cell-IQ system; Chip-Man Technologies). The imaging system enables continuous monitoring of adherent cells in two plates in an integrated plate holder without the use of labels or dyes. Time-lapse image series of neurosphere cultures were captured after plating and removal of mitogens. Image analysis was performed using ImageJ freeware and motility was analyzed using MTJ track software.
Statistical analysis.
Data are expressed as the mean ± SEM and were analyzed for statistical significance using Student's t test or one-way ANOVA followed by the Bonferroni test as post hoc test. A p value < 0.05 was considered significant; Bonferroni correction p value < 0.05 false discovery rate.
Results
Increased tPA expression in NPCs derived from Fmr1 KO mice and its effects on the neuronal migration
We found that the number of tPA-immunoreactive cells was increased in the differentiated NPCs derived from the Fmr1 KO mice compared with wild-type controls after differentiation for 7 and 14 d (Fig. 1A). The tPA staining colocalized with the astrocytic marker GFAP after differentiation for 7 d (Fig. 1B). The tPA expression did not colocalize with the neuronal marker MAP2 (Fig. 1C). An increased tPA expression was also clearly seen in FMRP-deficient glial cells after differentiation for 3 d (Fig. 1D). The data suggested that tPA is involved in the pathological glial signaling that contributes to the abnormal neuronal maturation in FXS (Jacobs and Doering, 2010).
Figure 1.

Expression of tPA in cortical progenitors derived from Fmr1 KO mice. A, Number of tPA-immunoreactive cells was increased in differentiated neurospheres generated from Fmr1 KO mice compared with wild-type (WT) controls after differentiation for 7 and 14 d. Data are expressed as means ± SEM. *Significant difference from control (p < 0.001; n = 3–5/each group, Student's t test). B, tPA (green) immunoreactivity colocalized with GFAP (yellow) in 7 d differentiated neurospheres derived from wild-type and Fmr1 KO mouse brain. C, tPA immunoreactivity (red) was not seen in MAP2-positive neurons (green). DAPI staining show nuclei (blue). D, Glial tPA expression (red) in FMRP-deficient neurospheres after differentiation for 3 d. The arrow denotes radial glia; the arrowhead denotes colocalization of tPA with GFAP (green). Scale bars, 50 μm.
NPCs grown in neurosphere cultures mimic cellular processes during early brain development (Martino et al., 2009). In the neurosphere model, differentiating cells migrate out from the neurosphere cell cluster and show neural cell morphology and immunoreactivity to neuronal and radial glial (RG) markers. In time-lapse imaging using the Cell-IQ system, the migration of cells in differentiating neurospheres displays temporal profiles (Jansson et al., 2012). We compared the migration of wild-type and FMRP-deficient cells after plating and removal of mitogens in images captured over 24 h at 15 min intervals We found that neuron-like cells in FMRP-deficient neurospheres migrated longer radial distances from the edge of neurosphere cell cluster than neuron-like cells in wild-type neurospheres after differentiation for 24 h (Fig. 2A), although the variation of the phasic behavior of cellular movement was high. The migration of neuron-like FMRP-deficient cells did not differ from wild-type controls after treatment with a neutralizing tPA antibody (Xin et al., 2010) (Fig. 2A). In immunocytochemical analysis, we observed that neurons expressing DCX, a microtubule-associated protein found selectively in migrating newborn neurons (Gleeson et al., 1999), traveled significantly longer distances in FMRP-deficient neurospheres compared with wild-type controls (Fig. 2B,C). Blockage of tPA with the neutralizing antibody normalized the migration distance of DCX-immunopositive FMRP-deficient cells (Fig. 2C). A control IgG did not have any effects on the neuronal migration, confirming the specificity of the effects of the tPA antibody (data not shown).
Figure 2.

Effects of tPA on the migration of differentiating FMRP-deficient cells. A, In time-lapse imaging using the Cell-IQ system, the distance migrated by neurons from the edge of neurosphere cell cluster was significantly longer in neurospheres derived from Fmr1 KO mice than in wild-type (WT) controls after differentiation for 24 h (Student's t test, *p = 0.014) and the treatment with the neutralizing tPA antibody (KO + tPA ab) prevented the difference. The data are from 3–6 neurospheres in 3 experiments. B, Images of a representative neurosphere stained with DCX (red) and the radial glia marker BLBP (green) and the double staining (DCX + BLBP) used in the immunocytochemical analysis of neuronal migration in neurospheres differentiated for 24 h. The arrow denotes the outmost migrated DCX-immunopositive neuron and the dashed line the edge of the neurosphere. Scale bar, 50 μm. C, The average distance moved by the outmost migrated DCX-immunopositive neurons was significantly increased in FMRP-deficient neurospheres compared with wild-type controls and blockage of tPA with a neutralizing antibody normalized the migration defect (ANOVA, Bonferroni test, n = 15, *p = 0.013, **p = 0.008, ns = 0.955). D, The distance reached by radial glia in neurospheres derived from wild-type (WT) and Fmr1 KO mice did not differ during the differentiation for first 24 h. Treatment with the tPA antibody (KO + tPA ab) enhanced significantly the length of the processes after differentiation for 3, 6, 9, and 24 h compared with untreated controls in neurospheres derived from Fmr1 KO mice (Student's t test; *p < 0.05). Data are from wild-type neurospheres (n = 15) and neurospheres derived from Fmr1 KO mice (untreated n = 18 and treated with the tPA antibody n = 15) in three experiments. E, Interkinetic nuclear migration in radial glia was analyzed in time-lapse image sequences. F, Velocity of the interkinetic nuclear migration was reduced (Student's t test, *p = 0.041) in differentiated neurospheres derived from Fmr1 KO mice after treatment with the neutralizing tPA antibody (KO + tPA ab), but the velocity of the nuclear migration did not differ between nontreated wild-type (WT) neurospheres and neurospheres treated with the tPA antibody (WT + tPA ab). The data are from 3 experiments (n = 4–6 cells/experiment). Values are means ± SEM.
In the time-lapse imaging, thick processes of RG start to extend from the neurospheres during the first hours of differentiation (Jansson et al., 2012). The length of RG extensions did not differ between neurospheres derived from wild-type and FMRP-deficient mice during first 24 h of differentiation assessed in 3 h intervals (Fig. 2D). The treatment with the tPA antibody increased significantly the distance reached by FMRP-deficient RG compared with nontreated RG after differentiation for 24 h (Fig. 2D). Blockage of tPA enhanced the distance reached by FMRP-deficient RG already at the earlier time points of differentiation (after 3, 6, and 9 h), indicating that tPA-mediated mechanisms regulated the differentiation of neuronal cells from very early stages of differentiation. RG cells attached to the neurosphere cell cluster display cell cycle-dependent nuclear movement referred to as interkinetic nuclear migration (INM; for review, see Kawauchi et al., 2013). We observed that the velocity of the INM in differentiating RG was significantly reduced in FMRP-deficient neurospheres by treatment with the tPA antibody, suggesting that altered INM contributed to the restoration of the migration defect (Fig. 2E,F).
tPA modulates intracellular Ca2+ responses to depolarization in differentiating cells
Spontaneous activity regulates a variety of developmental processes (Moody and Bosma, 2005). Activity is of fundamental importance to maturation of ion channels and the transition between embryonic and mature signaling properties. We explored tPA-mediated effects on intracellular calcium ([Ca2+]i) responses to depolarization with high extracellular potassium ([K+]e). We performed Fura2-AM-based [Ca2+]i recordings in wild-type and FMRP-deficient cortical progenitor cultures derived from embryonic mouse brains. For analysis, the cell migration area starting from the edge of the neurosphere and reaching to the outermost migrated cells was divided into 3 zones (zones 1–3) and [Ca2+]i responses in individual cells migrated out from the neurosphere cell cluster were recorded (Fig. 3A). Depolarization with 75 mm [K+]e induced changes in [Ca2+]i in a large majority of cells after differentiation for 24 h (Fig. 3B). The responses to depolarization with 17 and 75 mm [K+]e were significantly augmented in zones 2 and 3 under basal conditions in cells derived from Fmr1 KO mice compared with wild-type controls (Fig. 3C,D). Repeated exposure to potassium caused an enhanced [Ca2+]i response in FMRP-deficient cells (data not shown). As shown in Figure 3E, after treatment with the tPA antibody, these enhanced [Ca2+]i responses to 75 mm [K+]e were reduced in FMRP-deficient cells to wild-type levels. Wild-type responses were not affected by the treatment with the tPA antibody. Respectively, the [Ca2+]i responses to 17 mm [K+]e were normalized after treatment with the tPA antibody in FMRP-deficient cells (data not shown). Treatment with the control IgG antibody did not have any effects on the enhanced [Ca2+]i responses to 17 or 75 mm [K+]e in FMRP-deficient cells. The results indicate that responses to depolarization are enhanced in FMRP-deficient neuronal cells during early differentiation and that the tPA-mediated mechanisms contribute to the changes in the activity-dependent processes.
Figure 3.

Effects of tPA on [Ca2+]i responses to depolarization with potassium activation in 1 d differentiated neurospheres derived from Fmr1 KO and wild-type mice. A, Fura-2 fluorescence image of the migration area that was categorized into 3 zones (zones 1–3). The cells in the migration area were divided to three groups based on their migration length, which correlated with the zones. B, Representative cell response after stimulation with [K+]e (75 mm) during the period indicated. C, D, Average calcium amplitude of the cells in each zone and its corresponding location in reference to the neurospheres derived from wild-type (WT) and Fmr1 KO male mice after stimulation with 17 mm (C) and with 75 mm [K+]e (D). WT(n) = 273 (n = 9) and KO(n) = 327 (n = 11) after exposure to 17 mm [K+]e; WT(n) = 332 (n = 9) and KO(n) = 349 (n = 11) after exposure to 75 mm [K+]e. E, Average calcium amplitude of [Ca2+]i) responses to 75 mm [K+]e in wild-type cells under basal condition (WT) and after treatment with the neutralizing tPA antibody (WT + tPA ab) compared with the amplitude of cells derived from Fmr1 KO mice under basal condition (KO) and after treatment with the neutralizing tPA antibody (KO + tPA ab). Data are from 4–5 neurospheres (n = 50–100 cells) in each group. Values are means ± SEM. *p < 0.05; **p < 0.01; ***p < 0.001.
Altered tPA expression in the developing brain of Fmr1 KO mice
In agreement with the altered tPA expression in NPCs derived from Fmr1 KO mouse embryos, we observed abnormal pattern of tPA expression in the embryonic brain of Fmr1 KO mice at E17 (Fig. 4A). The specificity of the tPA staining was confirmed by negative controls without primary antibody and, as shown in Figure 4B, the unique pattern of tPA expression clearly differed from that of tropomyosin-related kinase B (TrkB) receptors as a control staining. The expression pattern of tPA was significantly altered in brain of Fmr1 KO male mice compared with wild-type controls at P7, as shown in Figure 4C. The number of tPA-positive cells was increased (1.8-fold) in cortical layers I-III, but decreased (0.7-fold) in layers IV-V in the Fmr1-KO mouse brain (Fig. 4C,D). The number of cells with tPA expression was also increased (3.2-fold) in the dentate gyrus of the hippocampus of Fmr1 KO mouse, whereas the number of cells with PAI-1 expression was decreased (0.7-fold), indicating that the inhibitory mechanisms for tPA expression were suppressed (Fig. 4E,F).
Figure 4.

Altered tPA expression in the developing brain of Fmr1 KO mouse. A, Pattern of anti-tPA immunostaining of a sagittal brain section from the neocortex of Fmr1 KO mouse differed from that of wild-type (WT) mouse at E17. B, As a control of the specificity of the tPA staining, immunostaining with anti-TrkB receptors that shows clearly different expression pattern than that of tPA. C, Anti-tPA immunostaining at P7. An asterisk denotes the lateral ventricle. D, Bar graph showing that, at P7, the proportion of tPA-expressing cells is increased in layers I-III and decreased in layers IV-V of Fmr1 KO mouse brain compared with wild-type controls (WT). E, F, Number of tPA-immunopositive cells was increased (E) and the number of PAI-1-immunopositive cells was significantly decreased (F) in the dentate gyrus of Fmr1 KO mouse hippocampus compared with controls at P7. n(WT) = 3–4; n(Fmr1 KO) = 4–5. CP, cortical plate; IZ, intermediate zone; SVZ/VZ, subventricular/ventricular zone. Scale bars, 100 μm in A and 50 μm in B. Values are means ± SEM. *p < 0.05; **p < 0.01; ***p < 0.001.
Altered tPA expression in adult brain of Fmr1 KO mice
To determine whether alterations of tPA expression persist in the mature brain, we examined the tPA expression in adult brain of Fmr1 KO mice by immunohistochemistry. We found that the expression of tPA was increased in the somatosensory and visual cortex and in the dentate gyrus of the hippocampus in the FMRP-deficient transgenics compared with wild-type controls (Fig. 5A–D). The expression of tPA was increased, particularly in the cortical layer IV, the main input layer of the cortex, which might contribute to the plasticity changes in FXS. The expression of tPA colocalized with the expression of glial fibrillary acidic protein (GFAP) in the cortex (Fig. 5C) and in the hippocampus (Fig. 5D). In the hippocampus, however, tPA expression was not found in all GFAP-positive cells as in the cortex (Fig. 5D).
Figure 5.
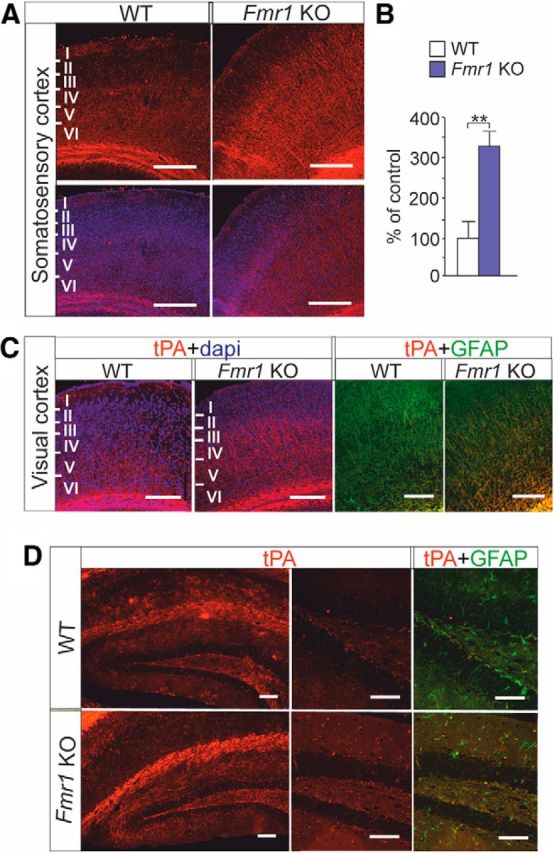
tPA expression in adult brain of Fmr1 KO mouse. A, Top, tPA immunoreactivity in red. Bottom, Merged images of tPA immunoreactivity in red and DAPI labeled nuclei in blue in the somatosensory cortex of adult wild-type (WT) and Fmr1 KO mouse. Scale bars, 200 μm. B, tPA immunoreactivity is significantly increased in the somatosensory cortex of Fmr1 KO mouse compared with that of WT controls. C, Merged images of anti-tPA immunostaining (red) combined with DAPI-labeled nuclei (blue) and anti-tPA immunostaining (red) combined with anti-GFAP (green) in sagittal brain sections from visual cortex of WT and Fmr1 KO mouse. D, tPA immunoreactivity (red) in the hippocampus. tPA expression (red) and its colocalization with GFAP (green) is shown in the dentate gyrus in a higher magnification. Scale bars, 200 μm. Data are expressed as means ± SEM. **p < 0.01, Student's t test.
Discussion
Multiple functions are attributed to tPA in the CNS, where the tPA expression is found in vascular endothelial cells and in both neurons and glia (Kalderon et al., 1990; Ware et al., 1995). Mesenchymal stem cells can induce astrocytes to secrete tPA (Xin et al., 2010) and tPA is expressed in activated glial cells in regions of inflammatory damage and gliosis (Teesalu et al., 2002). The function of tPA in synaptic plasticity is associated with the tPA-driven conversion of plasminogen to plasmin, which activates proBDNF to its mature form (Pang et al., 2004). Plasmin-independent entzymatic activity of tPA potentiates glutamatergic signaling (Nicole et al., 2001; Fernández-Monreal et al., 2004). Furthermore, tPA has been shown to display antiapoptotic activity and protection against ischemic damage or zinc toxicity independently of its proteolytic activity (Kim et al., 1999; Yi et al., 2004). Here, we have explored the role of tPA in developmental abnormalities found in the brain of Fmr1 KO mice. We observed that the expression of tPA is increased in glial cells differentiated from neural progenitors derived from Fmr1 KO mice compared with wild-type controls. In agreement with the upregulated tPA expression in the immature cultured cells, tPA expression was also increased in the supragranular layers of postnatal brain lacking FMRP. The [Ca2+]i responses to depolarization were enhanced in FMRP-deficient progenitors and treatment with an antibody against tPA normalized the augmentation of the [Ca2+]i responses. The increased immunoreactivity of tPA in the cortex and hippocampus of adult brain of Fmr1 KO mice extends the role of tPA to perturbations in mature brain. Together, our results demonstrate that tPA-mediated processes are involved in the aberrances of neuronal maturation and plasticity in FXS.
Activity-dependent Ca2+ signaling promotes neurogenesis and changes in free [Ca2+]i modulates the speed and direction of cell migration, cell proliferation, and programmed cell death (D'Ascenzo et al., 2006; Brustein et al., 2013). We found that activity-dependent [Ca2+]i responses were significantly enhanced in FMRP-lacking neural progenitors differentiated for 24 h. Blocking the tPA function with an antibody did not significantly affect the responses in wild-type cells, but reduced the responses to depolarization in FMRP-deficient cells to the wild-type levels. Ca2+ influx through functional voltage activated channels in neural progenitors has been shown to correlate with the neural progenitor differentiation toward the neuronal lineages and migration of neurons (Brustein et al., 2013). Enhanced migration of DCX-positive neurons during early differentiation of FMRP-deficient neurospheres and an increased production of neuroblasts in the expense of glia from FMRP-deficient embryonic cortical NPCs (Castrén et al., 2005) suggest alterations in the maturation and expression of ion channels in the absence of FMRP. The present results show that the treatment with the neutralizing tPA antibody normalizes the migration defect and [Ca2+]i responses to depolarization in FMRP-deficient neurospheres under our culturing conditions, indicating that tPA-mediated mechanisms play a role in aberrances observed in FMRP-deficient NPCs.
Neuronal migration is a fundamental mechanism for the generation of brain circuitries and wiring of the brain. Integrated radial and tangential migration position cells of different origin into specific coordinates during development of cerebral cortex. FMRP has been implicated in the regulation of neuronal migration (Moro et al., 2006; Tervonen et al., 2009), but the role of FMRP in the control of the migration of different neuronal subtypes and maintaining the homeostasis during brain development is not well understood. Nuclei of neural progenitors show salutatory movement that is associated with Ca2+ transients and correlate with the speed of cell migration (Pearson et al., 2005). There is evidence that INM is involved in cell fate determination of neural progenitors. Perturbation of INM by downregulation of centrosome- and microtubule-associated proteins has been shown to disrupt maintenance of the neural progenitor pool and enhance neurogenesis at the expense of neural progenitor cells (Xie et al., 2007; Del Bene et al., 2008). Mutations in LIS1 and DCX genes disturb the regulation of nucleokinesis during neuronal migration and result in neurological syndromes with migration defects of cortical neurons. Treatment with the tPA antibody retarded the velocity of INM in FMRP-deficient progenitors and normalized the radial movement of DCX-expressing cells in differentiated FMRP-deficient neurospheres, indicating that the increased glial tPA expression was associated with alterations in the migration of FMRP-deficient neurons.
Dysregulated protein synthesis related to group 1 mGluR activation has been shown to underlie aberrant neuronal plasticity in FXS (Huber et al., 2002; Bear et al., 2004). Glutamate stimulation increases tPA expression and release from neurons via mGluR-dependent mechanisms (Shin et al., 2004). Astrocytes constitutively endocytose extracellular tPA that is released from astrocytes in a regulated manner (Cassé et al., 2012) and this astrocytic uptake/recycling of tPA buffers extracellular tPA concentration and reduces NMDA receptor-mediated neurotoxicity. Extracellular glutamate promotes accumulation of tPA in astrocytes and increased tPA expression in FMRP-deficient glial cells is likely linked with alterations found in the maturation of glutamatergic neurons and glutamate signaling in FXS (Mataga et al., 2002; Harlow et al., 2010). FMRP is expressed in cells of glial origin during development and glial defects have been shown to contribute to the aberrances of the neuronal differentiation and migration in FXS (Castrén et al., 2005; Pacey and Doering, 2007). How the absence of FMRP affects glial function and the heterogenous population of astrocytes is not known. The abnormal morphology and reduced survival of FXS neurons can be partially restored when neurons are cocultured with wild-type astrocytes, in agreement with glial defects in the absence of FMRP (Jacobs and Doering, 2010).
Alterations of the tPA expression in differentiating FMRP-deficient progenitors are consistent with abnormalities observed in the tPA expression in the developing brain of Fmr1 KO mice. Substantial changes take place in the laminar distribution of neurons during brain development and alterations of the relative proportion of neurons in infragranular and supragranular layers may lead to disturbances in the ranking of brain regions to hierarchical levels during the developmental remodeling of cortical connectivity (Wen et al., 2013). The supragranular layers mediate the ability of cortical neurons to adjust their synaptic efficiency in response to changes in sensory input (Huang et al., 1998). The number of the neuronal population is maintained unchanged in the superficial layers of Fmr1 mouse cortex compared with wild-type controls (Castrén et al., 2005; Tervonen et al., 2009; Saffary and Zhigang, 2011) and the increased tPA expression in the superficial layers could contribute to the FXS phenotype by enhancing the inhibitory effects of tPA on the glutamate-evoked apoptosis. The increased tPA expression in adult cortex of Fmr1 mouse indicates a role for tPA in the neuronal plasticity also in the mature brain. Spatiotemporal changes of the tPA function both in wild-type and FXS brain, however, remain to be studied. The proteolytic effects of tPA on the maturation of pro-BDNF link changes in tPA expression with alterations of BDNF/TrkB signaling in NPCs derived from Fmr1 KO mice. Further studies also elucidating the role of other substrates of tPA and both proteolytic and nonproteolytic functions of tPA in NPCs will establish whether tPA may prove to be an interesting potential target for pharmacological intervention in FXS.
Footnotes
This work was supported by the Academy of Finland, the Arvo and Lea Ylppö Foundation, and the Finnish Brain Research Foundation. We thank Outi Nikkilä at the Neuroscience Center of the University of Helsinki for help in mouse genotyping.
The authors declare no competing financial interests.
References
- Bagni C, Greenough WT. From mRNP trafficking to spine dysmorphogenesis: the roots of fragile X syndrome. Nat Rev Neurosci. 2005;6:376–387. doi: 10.1038/nrn1667. [DOI] [PubMed] [Google Scholar]
- Bear MF, Huber KM, Warren ST. The mGluR theory of fragile X mental retardation. Trends Neurosci. 2004;27:370–377. doi: 10.1016/j.tins.2004.04.009. [DOI] [PubMed] [Google Scholar]
- Bhattacharyya A, McMillan E, Wallace K, Tubon TC, Jr, Capowski EE, Svendsen CN. Normal neurogenesis but abnormal gene expression in human fragile X cortical progenitor cells. Stem Cells Dev. 2008;17:107–117. doi: 10.1089/scd.2007.0073. [DOI] [PubMed] [Google Scholar]
- Brustein E, Côté S, Ghislain J, Drapeau P. Spontaneous glycine-induced calcium transients in spinal cord progenitors promote neurogenesis. Dev Neurobiol. 2013;73:168–175. doi: 10.1002/dneu.22050. [DOI] [PubMed] [Google Scholar]
- Callan MA, Zarnescu DC. Heads-up: New roles for the fragile X mental retardation protein in neural stem and progenitor cells. Genesis. 2011;49:424–440. doi: 10.1002/dvg.20745. [DOI] [PubMed] [Google Scholar]
- Cassé F, Bardou I, Danglot L, Briens A, Montagne A, Parcq J, Alahari A, Galli T, Vivien D, Docagne F. Glutamate controls tPA recycling by astrocytes, which in turn influences glutamatergic signals. J Neurosci. 2012;32:5186–5199. doi: 10.1523/JNEUROSCI.5296-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castrén M. Neural stem cells. In: Denham R, editor. Results and problems in cell differentiation. Berlin Heidelberg: Springer; 2012. pp. 33–40. [DOI] [PubMed] [Google Scholar]
- Castrén ML, Castrén E. BDNF in fragile X syndrome. Neuropharmacology. 2014;76:729–736. doi: 10.1016/j.neuropharm.2013.05.018. [DOI] [PubMed] [Google Scholar]
- Castrén M, Tervonen T, Kärkkäinen V, Heinonen S, Castrén E, Larsson K, Bakker CE, Oostra BA, Åkerman KEO. Altered neuronal differentiation of neural stem cells in fragile X syndrome. Proc Natl Acad Sci U S A. 2005;102:17408–17449. doi: 10.1073/pnas.0508995102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chonchaiya W, Schneider A, Hagerman RJ. Fragile X: A family of disorders. Adv Pediatr. 2009;56:165–186. doi: 10.1016/j.yapd.2009.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darnell JC, Van Driesche SJ, Zhang C, Hung KY, Mele A, Fraser CE, Stone EF, Chen C, Fak JJ, Chi SW, Licatalosi DD, Richter JD, Darnell RB. FMRP stalls ribosomal translocation on mRNAs linked to synaptic function and autism. Cell. 2011;146:247–261. doi: 10.1016/j.cell.2011.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Ascenzo M, Piacentini R, Casalbore P, Budoni M, Pallini R, Azzena GB, Grassi C. Role of L-type Ca2+ channels in neural stem/progenitor cell differentiation. Eur J Neurosci. 2006;23:935–944. doi: 10.1111/j.1460-9568.2006.04628.x. [DOI] [PubMed] [Google Scholar]
- Del Bene F, Wehman AM, Link BA, Baier H. Regulation of neurogenesis by interkinetic nuclear migration through an apical-basal notch gradient. Cell. 2008;134:1055–1065. doi: 10.1016/j.cell.2008.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dölen G, Bear MF. Role for metabotropic glutamate receptor 5 (mGluR5) in the pathogenesis of fragile X syndrome. J Physiol. 2008;586:1503–1508. doi: 10.1113/jphysiol.2008.150722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernández-Monreal M, López-Atalaya JP, Benchenane K, Cacquevel M, Dulin F, Le Caer JP, Rossier J, Jarrige AC, Mackenzie ET, Colloc'h N, Ali C, Vivien D. Arginine 260 of the amino-terminal domain of NR1 subunit is critical for tissue-type plasminogen activator-mediated enhancement of N-methyl-D-aspartate receptor signaling. J Biol Chem. 2004;279:850–856. doi: 10.1074/jbc.M407069200. [DOI] [PubMed] [Google Scholar]
- Gleeson JG, Lin PT, Flanagan LA, Walsh CA. Doublecortin is a microtubule-associated protein and is expressed widely by migrating neurons. Neuron. 1999;23:257–271. doi: 10.1016/S0896-6273(00)80778-3. [DOI] [PubMed] [Google Scholar]
- Guo W, Allan AM, Zong R, Zhang L, Johnson EB, Schaller EG, Murthy AC, Goggin SL, Eisch AJ, Oostra BA, Nelson DL, Jin P, Zhao X. Ablation of Fmrp in adult neural stem cells disrupts hippocampus-dependent learning. Nat Med. 2011;17:559–565. doi: 10.1038/nm.2336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harlow EG, Till SM, Russell TA, Wijetunge LS, Kind P, Contractor A. Critical period plasticity is disrupted in the barrel cortex of FMR1 knockout mice. Neuron. 2010;65:385–398. doi: 10.1016/j.neuron.2010.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu H, Qin Y, Bochorishvili G, Zhu Y, van Aelst L, Zhu JJ. Ras signaling mechanisms underlying impaired GluR1-dependent plasticity associated with fragile X syndrome. J Neurosci. 2008;28:7847–7862. doi: 10.1523/JNEUROSCI.1496-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W, Armstrong-James M, Rema V, Diamond ME, Ebner FF. Contribution of supragranular layers to sensory processing and plasticity in adult rat barrel cortex. J Neurophysiol. 1998;80:3261–3271. doi: 10.1152/jn.1998.80.6.3261. [DOI] [PubMed] [Google Scholar]
- Huber KM, Gallagher SM, Warren ST, Bear MF. Altered synaptic plasticity in a mouse model of Fragile X mental retardation. Proc Natl Acad Sci U S A. 2002;99:7746–7750. doi: 10.1073/pnas.122205699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang IY, Sun ES, An JH, Im H, Lee SH, Lee JY, Han PL, Koh JY, Kim YH. Zinc-triggered induction of tissue plasminogen activator by brain-derived neurotrophic factor and metalloproteinases. J Neurochem. 2011;118:855–863. doi: 10.1111/j.1471-4159.2011.07322.x. [DOI] [PubMed] [Google Scholar]
- Jacobs S, Doering LC. Astrocytes prevent abnormal neuronal development in the fragile X mouse. J Neurosci. 2010;30:4508–4514. doi: 10.1523/JNEUROSCI.5027-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansson LC, Louhivuori L, Wigren HK, Nordström T, Louhivuori V, Castrén ML, Åkerman KE. BDNF increases the motility of a particular NMDA/GABA-responsive subset of neural progenitor cells. Neuroscience. 2012;224:223–234. doi: 10.1016/j.neuroscience.2012.08.038. [DOI] [PubMed] [Google Scholar]
- Kalderon N, Ahonen K, Fedoroff S. Developmental transition in plasticity properties of differentiating astrocytes: age-related biochemical profile of plasminogen activators in astroglial cultures. Glia. 1990;3:413–426. doi: 10.1002/glia.440030513. [DOI] [PubMed] [Google Scholar]
- Kärkkäinen V, Castrén ML, Louhivuori V, Akerman KE. Neurotransmitter responsiveness during early maturation of neural progenitor cells. Differentiation. 2009;77:188–198. doi: 10.1016/j.diff.2008.10.005. [DOI] [PubMed] [Google Scholar]
- Kawauchi T, Shikanai M, Kosodo Y. Extra-cell cycle regulatory functions of cyclin-dependent kinases (CDK) and CDK inhibitor proteins contribute to brain development and neurological disorders. Genes Cells. 2013;18:176–194. doi: 10.1111/gtc.12029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YH, Park JH, Hong SH, Koh JY. Nonproteolytic neuroprotection by human recombinant tissue plasminogen activator. Science. 1999;284:647–650. doi: 10.1126/science.284.5414.647. [DOI] [PubMed] [Google Scholar]
- Louhivuori V, Vicario A, Uutela M, Rantamäki T, Louhivuori LM, Castrén E, Tongiorgi E, Akerman KE, Castrén ML. BDNF and TrkB in neuronal differentiation of Fmr1-knockout mouse. Neurobiol Dis. 2011;41:469–480. doi: 10.1016/j.nbd.2010.10.018. [DOI] [PubMed] [Google Scholar]
- Luo Y, Shan G, Guo W, Smrt RD, Johnson EB, Li X, Pfeiffer RL, Szulwach KE, Duan R, Barkho BZ, Li W, Liu C, Jin P, Zhao X. Fragile x mental retardation protein regulates proliferation and differentiation of adult neural stem/progenitor cells. PLoS Genet. 2010;6:e1000898. doi: 10.1371/journal.pgen.1000898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martino S, di Girolamo I, Cavazzin C, Tiribuzi R, Galli R, Rivaroli A, Valsecchi M, Sandhoff K, Sonnino S, Vescovi A, Gritti A, Orlacchio A. Neural precursor cell cultures from GM2 gangliosidosis animal models recapitulate the biochemical and molecular hallmarks of the brain pathology. J Neurochem. 2009;109:135–147. doi: 10.1111/j.1471-4159.2009.05919.x. [DOI] [PubMed] [Google Scholar]
- Mataga N, Nagai N, Hensch TK. Permissive proteolytic activity for visual cortical plasticity. Proc Natl Acad Sci U S A. 2002;99:7717–7721. doi: 10.1073/pnas.102088899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melchor JP, Strickland S. Tissue plasminogen activator in central nervous system physiology and pathology. Thromb Haemost. 2005;93:655–660. doi: 10.1160/TH04-12-0838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moody WJ, Bosma MM. Ion channel development, spontaneous activity, and activity-dependent development in nerve and muscle cells. Physiol Rev. 2005;85:883–941. doi: 10.1152/physrev.00017.2004. [DOI] [PubMed] [Google Scholar]
- Moro F, Pisano T, Bernardina BD, Polli R, Murgia A, Zoccante L, Darra F, Battaglia A, Pramparo T, Zuffardi O, Guerrini R. Periventricular heterotopia in fragile X syndrome. Neurology. 2006;67:713–715. doi: 10.1212/01.wnl.0000230223.51595.99. [DOI] [PubMed] [Google Scholar]
- Nicole O, Ali C, Docagne F, Plawinski L, MacKenzie ET, Vivien D, Buisson A. Neuroprotection mediated by glial cell line-derived neurotrophic factor: involvement of a reduction of NMDA-induced calcium influx by the mitogen-activated protein kinase pathway. J Neurosci. 2001;21:3024–3033. doi: 10.1523/JNEUROSCI.21-09-03024.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacey LK, Doering LC. Developmental expression of FMRP in the astrocyte lineage: implications for fragile X syndrome. Glia. 2007;55:1601–1609. doi: 10.1002/glia.20573. [DOI] [PubMed] [Google Scholar]
- Pang PT, Teng HK, Zaitsev E, Woo NT, Sakata K, Zhen S, Teng KK, Yung WH, Hempstead BL, Lu B. Cleavage of proBDNF by tPA/plasmin is essential for long-term hippocampal plasticity. Science. 2004;306:487–491. doi: 10.1126/science.1100135. [DOI] [PubMed] [Google Scholar]
- Pearson R, Lüneborg NL, Becker DL, Mobbs P. Gap junctions modulate interkinetic nuclear movement in retinal progenitor cells. J Neurosci. 2005;25:10803–10814. doi: 10.1523/JNEUROSCI.2312-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saffary R, Xie Z. FMRP Regulates the transition from radial glial cells to intermediate progenitor cells during neocortical development. J Neurosci. 2011;31:1427–1439. doi: 10.1523/JNEUROSCI.4854-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samson AL, Medcalf RL. A tissue-type plasminogen activator: A multifaceted modulator of neurotransmission and synaptic plasticity. Neuron. 2006;50:673–678. doi: 10.1016/j.neuron.2006.04.013. [DOI] [PubMed] [Google Scholar]
- Santoro MR, Bray SM, Warren ST. Molecular mechanisms of fragile X syndrome: a twenty-year perspective. Annu Rev Pathol. 2012;7:219–245. doi: 10.1146/annurev-pathol-011811-132457. [DOI] [PubMed] [Google Scholar]
- Seeds NW, Basham ME, Haffke SP. Neuronal migration is retarded in mice lacking the tissue plasminogen activator gene. Proc Natl Acad Sci U S A. 1999;96:14118–14123. doi: 10.1073/pnas.96.24.14118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin C, Kundel M, Wells DG. Rapid, activity-induced increase in tissue plasminogen activator is mediated by metabotropic glutamate receptor-dependent mRNA translation. J Neurosci. 2004;24:9425–9433. doi: 10.1523/JNEUROSCI.2457-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teesalu T, Kulla A, Asser T, Koskiniemi M, Vaheri A. Tissue plasminogen activator as a key effector in neurobiology and neuropathology. Biochem Soc Trans. 2002;30:183–189. doi: 10.1042/. [DOI] [PubMed] [Google Scholar]
- Tervonen TA, Louhivuori V, Sun X, Hokkanen ME, Kratochwil CF, Zebryk P, Castrén E, Castrén ML. Aberrant differentiation of glutamatergic cells in neocortex of mouse model for fragile X syndrome. Neurobiol Dis. 2009;33:250–259. doi: 10.1016/j.nbd.2008.10.010. [DOI] [PubMed] [Google Scholar]
- Tsirka SE, Gualandris A, Amaral DG, Strickland S. Excitotoxin-induced neuronal degeneration and seizure are mediated by tissue plasminogen activator. Nature. 1995;377:340–344. doi: 10.1038/377340a0. [DOI] [PubMed] [Google Scholar]
- Uutela M, Lindholm J, Louhivuori V, Wei H, Louhivuori LM, Pertovaara A, Akerman K, Castrén E, Castrén ML. Reduction of BDNF expression in Fmr1 knockout mice worsens cognitive deficits but improves hyperactivity and sensorimotor deficits. Genes Brain Behav. 2012;11:513–523. doi: 10.1111/j.1601-183X.2012.00784.x. [DOI] [PubMed] [Google Scholar]
- Verkerk AJ, Pieretti M, Sutcliffe JS, Fu YH, Kuhl DP, Pizzuti A, Reiner O, Richards S, Victoria MF, Zhang FP. Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell. 1991;65:905–914. doi: 10.1016/0092-8674(91)90397-H. [DOI] [PubMed] [Google Scholar]
- Ware JH, Dibenedetto AJ, Pittman RN. Localization of tissue plasminogen activator mRNA in adult rat brain. Brain Res Bull. 1995;37:275–281. doi: 10.1016/0361-9230(95)00008-3. [DOI] [PubMed] [Google Scholar]
- Wen JA, DeBlois MC, Barth AL. Initiation, labile, and stabilization phases of experience-dependent plasticity at neocortical synapses. J Neurosci. 2013;33:8483–8493. doi: 10.1523/JNEUROSCI.3575-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Z, Moy LY, Sanada K, Zhou Y, Buchman JJ, Tsai LH. Cep120 and TACCs control interkinetic nuclear migration and the neural progenitor pool. Neuron. 2007;56:79–93. doi: 10.1016/j.neuron.2007.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xin H, Li Y, Shen LH, Liu X, Wang X, Zhang J, Pourabdollah-Nejad DS, Zhang C, Zhang L, Jiang H, Zhang ZG, Chopp M. Increasing tPA activity in astrocytes induced by multipotent mesenchymal stromal cells facilitate neurite outgrowth after stroke in the mouse. PLoS One. 2010;5:e9027. doi: 10.1371/journal.pone.0009027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi JS, Kim YH, Koh JY. Infarct reduction in rats following intraventricular administration of either tissue plasminogen activator (tPA) or its non-protease mutant S478A-tPA. Exp Neurol. 2004;189:354–360. doi: 10.1016/j.expneurol.2004.05.032. [DOI] [PubMed] [Google Scholar]