

Abstract
Layer 1 neocortical GABAergic interneurons control the excitability of pyramidal neurons through cell-class-specific direct inhibitory and disynaptic disinhibitory circuitry. The engagement of layer 1 inhibitory circuits during behavior is powerfully controlled by the cholinergic neuromodulatory system. Here we report that acetylcholine (ACh) influences the excitability of layer 1 interneurons in a cell-class and activity-dependent manner. Whole-cell recordings from identified layer 1 interneurons of the rat somatosensory neocortex revealed that brief perisomatic application of ACh excited both neurogliaform cells (NGFCs) and classical-accommodating cells (c-ACs) at rest by the activation of nicotinic receptors. In contrast, under active, action potential firing states, ACh excited c-ACs, but inhibited NGFCs through muscarinic receptor-mediated, IP3 receptor-dependent elevations of intracellular calcium that gated surface-membrane calcium-activated potassium channels. These excitatory and inhibitory actions of ACh could be switched between by brief periods of NGFC action potential firing. Paired recordings demonstrated that cholinergic inhibition of NGFCs disinhibited the apical dendrites of layer 2/3 pyramidal neurons by silencing widespread, GABAB receptor-mediated, monosynaptic inhibition. Together, these data suggest that the cholinergic system modulates layer 1 inhibitory circuits in an activity-dependent manner to dynamically control dendritic synaptic inhibition of pyramidal neurons.
Keywords: axon, dendrite, ion-channel, neocortex, synaptic integration
Introduction
Acetylcholine (ACh) is a neuromodulator that influences many features of neocortical operation, including arousal state, attention, sensory processing, neuronal plasticity, and associative learning (Buzsaki et al., 1988; Metherate et al., 1992; Kilgard and Merzenich, 1998; Détári et al., 1999; Goard and Dan, 2009; Letzkus et al., 2011). Accordingly, the disruption of neocortical ACh signaling in human disease is associated with cognitive decline (Bartus et al., 1982; Coyle et al., 1983; Contestabile, 2011).
Cholinergic axons, which predominately originate from the basal forebrain in rodents, widely innervate all areas and layers of the neocortex (Mechawar et al., 2000; Arroyo et al., 2012; Kalmbach et al., 2012), with a particularly high density in neocortical layer 1 (Mechawar et al., 2000). Layer 1 is a unique neocortical layer that is devoid of excitatory neurons, but contains a rich plexus of axons, the apical dendrites of pyramidal neurons, and a relatively low density of GABAergic interneurons (Winer and Larue, 1989; Prieto et al., 1994). Layer 1 interneurons form cell-class specific circuits in the neocortex (Chu et al., 2003; Letzkus et al., 2011; Wozny and Williams, 2011; Arroyo et al., 2012; Jiang et al., 2013). One class of layer 1 interneurons, neurogliaform cells (NGFCs), provide a direct source of apical dendritic inhibition to layer 2/3 and layer 5 pyramidal neurons by the activation of GABAB receptors (Tamás et al., 2003; Oláh et al., 2009; Wozny and Williams, 2011; Palmer et al., 2012; Jiang et al., 2013), whereas other classes of layer 1 interneurons predominately disinhibit pyramidal neurons through disynaptic circuits (Christophe et al., 2002; Letzkus et al., 2011; Arroyo et al., 2012; Jiang et al., 2013). Activation of layer 1 interneurons by the basal forebrain cholinergic system has been found to be a key component of the neuronal circuitry underlying forms of associative learning (Letzkus et al., 2011). Consistent with this, synaptic ACh release or exogenous application excites layer 1 interneurons by the activation of nicotinic ACh receptors (nAChRs; Christophe et al., 2002; Letzkus et al., 2011; Arroyo et al., 2012). However, little attention has been paid to the cell-class specificity or activity dependence of the cholinergic control of layer 1 interneurons, despite their cell-type-specific network operations (Jiang et al., 2013). Notably, somatic aversive stimuli, which engage cholinergic mechanisms, differentially controls the excitability of layer 1 interneurons distributed across functionally unrelated neocortical areas, leading to either the transient enhancement or long-lasting inhibition of action potential output (Letzkus et al., 2011).
Here we investigate the cell-type-specific control of layer 1 interneurons and interneuronal circuits by ACh. We find that NGFCs, but not other classes of layer 1 interneurons, are transiently excited by ACh when quiescent, but are inhibited for long time periods when electrically active, revealing an activity-dependent signaling role of ACh, which controls the pattern of dendritic synaptic inhibition received by pyramidal neurons.
Materials and Methods
Brain-slice preparation.
Male Wistar rats (P24 to P31) were decapitated under deep isoflurane anesthesia in accordance with the guidelines of the Animal Ethics Committee of the University of Queensland. The brain was quickly removed from the skull and placed into ice-cold artificial CSF (aCSF) of the following composition (in mm): NaCl 125, NaHCO3 25, KCl 3, NaH2PO4 1.25, CaCl2 1, MgCl2 6, sodium pyruvate 3, and glucose 25, saturated with 95% O2 and 5% CO2. Coronal sections (300 μm) of the somatosensory cortex were made and brain-slices incubated at 34°C for 30 min and then stored at room temperature.
Whole-cell recordings.
Individual brain-slices were transferred to a recording chamber and perfused with a solution of the following composition (in mm): NaCl 125, NaHCO3 25, KCl 3, NaH2PO4 1.25, CaCl2 2, MgCl2 1, sodium pyruvate 3, and glucose 25, saturated with 95% O2 and 5% CO2 at 34–36°C. Single, double, and triple whole-cell recordings were made from the somata of identified somatosensory neocortical neurons imaged under video enhanced differential interference contrast microscopy. Pipettes were filled with a solution containing the following (in mm): potassium gluconate 135, NaCl 7, HEPES 10, Na2-ATP 2, Na-GTP 0.3, sodium phosphocreatine 10, MgCl2 2, and occasionally with AlexaFluor 594 0.01 (Invitrogen); pH was adjusted to 7.2–7.3 with KOH. All recordings were made with up to three identical current-clamp amplifiers (BVC 700A; Dagan) in “bridge” mode. When the calcium chelator 1,2-bis(2-aminophenoxy)-ethance-N,N,N′,N′-tetraacetic acid (BAPTA) was included in the intrapipette solution, the concentration of potassium gluconate was proportionally reduced. Pipettes had open-tip resistance of between 5 and 7 MΩ. Somatic whole-cell series resistance was between 15 and 30 MΩ. Voltage and current signals were low-pass filtered (DC to 10 kHz) and acquired at 30–50 kHz. Data were acquired and analyzed using Axograph software (AxographX).
Unitary inhibitory synaptic transmission was examined between up to three simultaneously recorded neurons. Monosynaptic connectivity was tested by the generation of one to three presynaptic action potentials, generated by the delivery of short positive current steps (50 ms, 0.4–1 nA; delivered at 0.25 Hz). If no connectivity was found, one of the recording electrodes was withdrawn and a new neuron recorded. To reduce ongoing excitatory synaptic transmission 6-cyano-7-nitroquinoxaline-2,3-dione (10 μm) was added to the bath solution.
Drug application.
Sharp micropipettes (∼100 MΩ) were filled with ACh (10–100 mm), connected to a Union-36 iontophoresis pump (Kation Scientific) and ACh applied (5–30 ms) close to the soma of recorded neurons (within 10–20 μm) using an ejection current (75–200 nA; retention current, 5–20 nA). In depletion-refilling experiments 1 m ACh was used, to ensure the adequate depletion of internal calcium stores. In some experiments ACh (100 μm, dissolved in aCSF) was applied close to the soma of recorded neurons by pressure application from pipettes (similar to those used for whole-cell recordings) connected to a Picospritzer 2 (Parker Instruments) pneumatic pump (1–2 bar for 20–30 ms). Antagonists and channel blockers were bath applied for 10–15 min before measurement of effects.
Calcium imaging.
Whole-cell somatic recordings were made from physiologically identified NGFCs with pipettes containing the calcium-sensitive dye Oregon Green BAPTA-6F (20–50 μm; Invitrogen) added to the electrode filling solution for 20 min before imaging with a Zeiss LSM 510 confocal microscope equipped with an Olympus 60× objective. Oregon Green BAPTA-6F was excited at 488 nm and the resulting fluorescence collected via a 510 nm emission filter. Line scans across the soma were acquired and analyzed offline with MATLAB (MathWorks). After background subtraction, ΔF/F was calculated.
Neuronal reconstruction.
Neurons were filled with biocytin (0.5%, added to the pipette filling solution) and slices fixed overnight in 4% paraformaldehyde dissolved in 0.1 m sodium PBS, pH 7.4. Subsequently, slices were incubated for 6–12 h in PBS supplemented with 2% Triton X-100 and 0.2% streptavidin, AlexaFluor 488 conjugate (Invitrogen) at 4–6°C. After washing in PBS, slices were mounted on slides, covered with ProLong Gold (Invitrogen) and imaged with a Zeiss LSM 510 confocal microscope. Neuronal morphology was reconstructed from confocal image stacks using Neurolucida software (MBF Bioscience).
Results
Activity-dependent cholinergic control of layer 1 interneurons
Whole-cell current-clamp recording techniques were used to investigate the cholinergic control of layer 1 inhibitory interneuronal networks of the rat somatosensory cortex (Fig. 1). We focused our analysis on the two most numerous classes of layer 1 interneurons, NGFCs, and classical accommodating cells (c-ACs; Wozny and Williams, 2011), which could be readily identified morphologically and electrophysiologically (Table 1).
Figure 1.
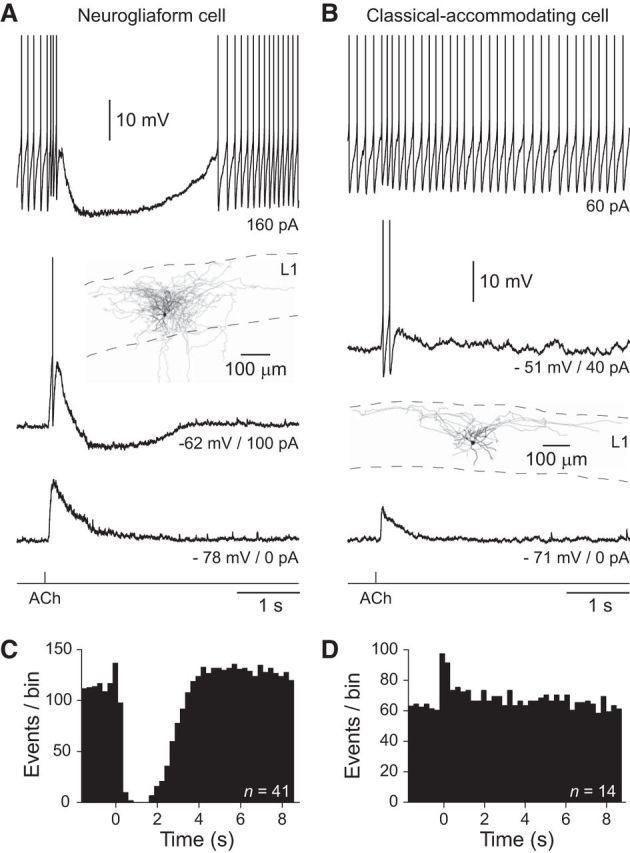
Cell-type-specific activity-dependent inhibition of layer 1 interneurons by Ach. A, The iontophoretic application of ACh (bottom trace, 100 mm, 20 ms) silences ongoing action potential firing in a NGFC. In contrast, ACh generates excitatory responses at the indicated, subthreshold holding potentials. The anatomical reconstruction of a NGFC is shown in the inset. The soma and dendrites are shown in black and the axon in gray. B, Excitation, but not inhibition of a c-AC by the iontophoretic application of ACh (bottom trace, 100 mm, 20 ms). Inset, The anatomical reconstruction of the c-AC. A, B, The action potential amplitude has been truncated for clarity. C, D, Summary of the control of ongoing action potential firing by ACh in NGFCs (C) and c-ACs (D). Data have been pooled from the indicated number of neurons to generate peristimulus time histograms of action potential firing (250 ms time bins). ACh was applied at time 0.
Table 1.
Electrophysiological and morphological properties of NGFCs and c-ACs
| NGFCs (n) | c-ACs (n) | |
|---|---|---|
| Resting membrane potential | −77.0 ± 0.5 mV (72) | −75.9 ± 0.73 mV (31) |
| Apparent input resistance | 109.3 ± 3.8 MΩ (72) | 123.2 ± 6.0 MΩ (31) |
| Action potential accommodation index | 0.42 ± 0.06 (72) | −0.42 ± 0.07 (34) |
| Action potential after-hyperpolarization | −24.9 ± 0.4 mV (68) | −15.2 ± 0.9 mV (28) |
| Dendritic field area | 0.013 ± 0.006 mm2 (5) | 0.041 ± 0.007 mm2 (7) |
| Axonal density | 0.191 ± 0.036 μm−1 (5) | 0.064 ± 0.014 μm−1 (7) |
Analysis revealed that interneuronal classes could be divided electrophysiologically by statistically significant differences in apparent input resistance (measured in response to a −20 pA current step; Students t test: T = 1.99, p = 0.0483), action potential accommodation index (calculated as the ratio of the first and last interspike interval, in response to positive current steps of magnitude set to the rheobase for the generation of repetitive action potential firing; Mann–Whitney test: U = 73.5, p < 0.0001), and the amplitude of the action potential after-hyperpolarization (measured from single action potential generated at rheobase; Students t test: T = 12.28, p < 0.0001). Morphological analysis revealed that the dendritic field area (Students t test: T = 2.76, p = 0.02) and axonal density (total axonal length/axonal field area; Students t test: T = 3.70, p = 0.0041) of interneuronal classes were significantly different. Values represent mean ± SEM.
To examine the cholinergic control of the excitability of layer 1 interneurons we focally applied ACh by iontophoresis (10–100 mm, 5–30 ms) to sites close to the soma (Fig. 1A,B). When recorded at resting membrane potential (RMP), the iontophoretic application of ACh transiently depolarized both NGFCs and c-ACs (Fig. 1A,B; NGFCs: amplitude = 7.4 ± 0.9 mV, half-width = 0.21 ± 0.04 s, n = 24, n = 24 slices, n = 16 animals; c-ACs: amplitude = 11.5 ± 1.5 mV, half-width = 0.18 ± 0.04 s, n = 13, n = 13 slices, n = 7 animals). ACh-evoked depolarizing responses were sufficient to transiently generate action potential firing in both NGFCs and c-ACs when the membrane was depolarized by the injection of tonic positive current through the recording electrode (Fig. 1A,B; NGFCs: −49.1 ± 2.2 mV, n = 15, n = 15 slices, n = 13 animals; c-ACs: −48.8 ± 2.2 mV, n = 12, n = 12 slices, n = 7 animals). Under conditions of repetitive action potential firing, however, ACh first transiently enhanced, and then silenced the neuronal output of NGFCs for long periods of time (Fig. 1A,C; 2.5 ± 0.18 s; n = 24, n = 24 slices, n = 16 animals), by the generation of large membrane hyperpolarization (−12.9 ± 0.8 mV). In contrast, ACh simply augmented the ongoing action potential firing rate, and did not lead to the inhibition of c-ACs (Fig. 1B,D). These cell-class and activity-dependent actions of ACh were reproduced when ACh was applied by pressure application to perisomatic sites (100 μm, 20–30 ms; NGFC excitation from resting membrane potential = 1.82 ± 1.24 mV; NGFC inhibition of repetitive action potential firing: 1.24 ± 0.31 s; n = 6, n = 6 slices, n = 5 animals; data not shown). Thus, under excited action potential firing states, ACh selectively and powerfully inhibits the neuronal output of NGFCs.
Pharmacology of cholinergic excitation and inhibition in NGFCs
To dissect the activity-dependent actions of ACh in NGFCs, we explored the receptor systems mediating excitatory and inhibitory responses (Fig. 2). The bath application of the broad-spectrum nAChR antagonist mecamylamine (10 μm) blocked cholinergic excitation of NGFCs, reducing the amplitude of ACh-evoked depolarizing potentials (Fig. 2A; amplitude control: 9.5 ± 2.2 mV; mecamylamine: 1.8 ± 0.7 mV; paired t test: T = 5.139; p = 0.0143, n = 4, n = 4 slices, n = 3 animals), but did not alter ACh-mediated inhibition of action potential firing (Fig. 2A,B). Consistent with previous findings (Arroyo et al., 2012; Bennett et al., 2012) we observed that ACh-evoked excitatory responses were attenuated by the non-α7 nAChR antagonist dihydro-β-erythroidine (DHβE, 500 nm; Fig. 2E,F; peak amplitude significantly different from control; paired t test: T = 10.72; p = 0.0001, n = 6, n = 6 slices, n = 5 animals). In the presence of DHβE, ACh evoked small amplitude depolarizing responses with fast kinetics that were sensitive to the broad-spectrum nAChR antagonist mecamylamine (10 μm; Fig. 2E,G; decay time constant significantly different from control; paired t test: T = 9.233; p = 0.0003, n = 6, n = 6 slices, n = 5 animals). The resistance of ACh-evoked inhibition to nAChR antagonists suggests that cholinergic inhibition of NGFCs may be mediated by the activation of muscarinic ACh receptors (mAChRs). To test this, we bath applied the specific M1 mAChR antagonist telenzepine (100 nm). Telenzepine blocked ACh-evoked inhibition of action potential firing in NGFCs, without altering cholinergic excitation (Fig. 2C,D; control: 3.46 ± 1.14 mV; telenzepine 3.36 ± 0.34 mV; paired t test: T = 0.09,997; p = 0.9267, n = 4, n = 4 slices, n = 4 animals). Consistent with previous work (Christophe et al., 2002; Letzkus et al., 2011; Arroyo et al., 2012), ACh-evoked excitation of c-ACs was mediated by the activation of nAChRs (amplitude control: 9.33 ± 1.68 mV; mecamylamine: 0.53 ± 0.21 mV; paired t test: T = 5.34; p = 0.013, n = 4, n = 4 slices, n = 2 animals; data not shown).
Figure 2.

Pharmacology of cholinergic excitation and inhibition of NGFCs. A, The nicotinic ACh receptor antagonist mecamylamine (10 μm, right) blocks subthreshold excitatory, but not suprathreshold inhibitory responses evoked by iontophoretic application of ACh (bottom trace, 100 mm, 20 ms). B, Summary of the control of action potential firing by ACh under control and in mecamylamine (10 μm). Peristimulus time histograms (250 ms time bins) of action potential firing from pooled NGFCs (n = 4 cells) under control conditions (top) and after the bath application of mecamylamine (bottom). ACh was applied at time 0. C, The muscarinic ACh receptor antagonist telenzepine (100 nm, right) blocks suprathreshold inhibitory, but not subthreshold excitatory responses evoked by iontophoretic application of ACh (bottom trace, 100 mm, 20 ms). A, C, The action potential amplitude has been truncated for clarity. D, Summary of the control of action potential firing by ACh under control and in telenzepine (n = 5). E, ACh-evoked excitatory responses in the presence of telenzepine (100 nm) are attenuated by DHβE (500 nm) and blocked by mecamylamine (10 μm). F, G, Summary of the actions of DHβE (500 nm) on the peak amplitude (F) and decay time constant (G) of ACh-evoked excitatory responses. Values represent mean ± SEM.
We next explored whether the cholinergic control of NGFCs was specific to layer 1 of the neocortex. NGFCs are distributed throughout the neocortex, but are predominately located in supragranular layers (Markram et al., 2004). The focal application of ACh to physiologically and morphologically identified NGFCs in layers 2 and 3 of the neocortex generated activity-dependent responses closely resembling those observed from layer 1 NGFCs (ACh depolarization from rest = 5.68 ± 1.49 mV; ACh inhibition of repetitive action potential firing: amplitude = −14.39 ± 1.11 mV, half-width = 1.58 ± 0.11 s; n = 16, n = 16 slices, n = 13 animals; data not shown), indicating that the activity-dependent cholinergic control of NGFCs is cell-class-specific, but not layer-specific.
Intracellular calcium signaling controls mAChR-mediated inhibition
M1 mAChRs are coupled with Gq pathways, phosphatidylinositol turnover, and the control of intracellular calcium in other neuronal systems (Caulfield and Birdsall, 1998). To test the involvement of these components in the cholinergic control of NGFCs we first investigated whether ACh-evoked inhibition was dependent on the level of free intracellular calcium. NGFCs were first recorded with pipettes filled with a standard intrapipette solution, and exhibited robust ACh-mediated silencing of NGFC neuronal output (Fig. 3A,B). Neurons were then repatched with pipettes filled with an intrapipette solution containing the fast calcium chelator BAPTA (Adler et al., 1991; 10 mm; Fig. 3A). The dialysis of NGFCs with BAPTA resulted in the blockade of the inhibitory actions of ACh, transforming the long ACh-evoked period of action potential silencing, to a weak enhancement of neuronal output (Fig. 3A,B; n = 8, n = 8 slices, n = 6 animals). In contrast, when NGFCs were repatched with pipettes containing normal intrapipette solution, ACh-evoked inhibition was not altered (Fig. 3C; n = 5, n = 5 slices, n = 2 animals).
Figure 3.
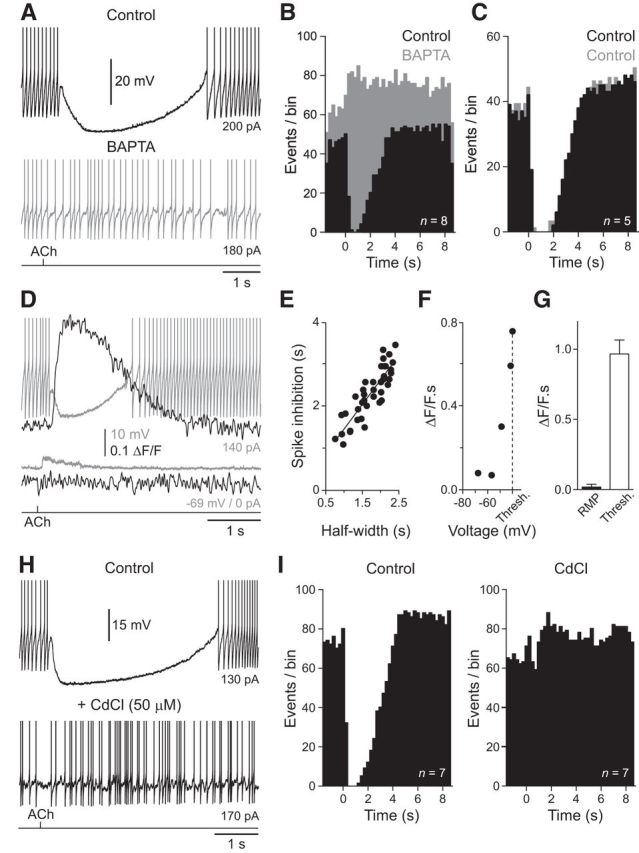
Intracellular calcium controls ACh-evoked inhibition of NGFCs. A, The iontophoretic application of ACh (bottom trace, 100 mm, 20 ms) silences ongoing action potential firing of a NGFC recorded with a pipette filled with normal internal solution. This inhibitory action was abolished when the NGFC was repatched with a pipette filled with an internal solution containing the calcium-chelator BAPTA (10 mm, gray trace). B, Summary of the abolition of the inhibitory actions of ACh by BAPTA (gray bars, n = 8 cells). Data have been pooled to generate peristimulus time histograms of action potential firing (250 ms time bins). ACh was applied at time 0. C, Repatching NGFCs with pipettes containing standard intrapipette solution does not disturb the inhibitory actions of ACh (gray bars, n = 5). D, Simultaneous voltage (gray traces) recording and Oregon green BAPTA-6F (ΔF/F black traces) imaging show that ACh-evoked inhibition of action potential firing is accompanied by a rise in intracellular calcium, whereas subthreshold excitatory responses are not. ACh was delivered by iontophoresis (bottom trace, 100 mm, 20 ms). E, Linear relationship between the time course of ACh-evoked inhibition of action potential firing and calcium signaling. Data are from multiple trials in 15 NGFCs evoked by ACh iontophoresis. The line represents a linear regression. F, Area of ACh-evoked Oregon Green BAPTA-6F signals (ΔF/F · s) as a function of holding voltage (mV) for the cell shown in D (Thresh. refers to action potential firing threshold).G, Summary of the area of ACh-evoked Oregon green BAPTA-6F signals (ΔF/F · s) at RMP and threshold (Thresh; n = 16 cells). Values represent mean ± SEM. H, The broad-spectrum calcium channel antagonist cadmium (50 μm) blocks the inhibitory actions of ACh. A, D, H, The action potential amplitude has been truncated for clarity. I, Summary of the abolition of the inhibitory actions of ACh by cadmium (right, n = 7 cells). Data have been pooled to generate peristimulus time histograms of action potential firing (250 ms time bins). ACh was applied at time 0.
To directly visualize the relationship between intracellular calcium and ACh-evoked inhibition of NGFC we used simultaneous electrophysiological and confocal imaging techniques. Calcium imaging revealed that the ACh-evoked inhibition of ongoing action potential firing was accompanied by a significant elevation of intracellular calcium, the time course of which was correlated with the period of action potential silencing (Fig. 3D,E; r2 = 0.7641; calcium-sensitive dye Oregon Green BAPTA-6F, 20–50 μm). In contrast, to the behavior observed during active states, the activation of AChRs from RMP evoked depolarizing potentials unaccompanied by measurable calcium signaling (Fig. 3D; RMP: 0.02 ± 0.02 ΔF/F · s; threshold: 0.97 ± 0.02 ΔF/F · s; paired t test: T = 9.513, p < 0.0001, n = 16, n = 16 slices, n = 10 animals). To explore the voltage dependency of this relationship we generated ACh responses across a wide voltage range, controlled by the tonic passage of current delivered through the recording electrode. Significant ACh-evoked elevation of intracellular calcium was only apparent at membrane potentials close to action potential initiation threshold (Fig. 3F,G; n = 16, n = 16 slices, n = 10 animals), suggesting that the time- and voltage-dependent recruitment of ion channels is necessary for the expression of ACh-evoked inhibition.
To investigate the ion channels involved in the inhibitory actions of ACh we generated ACh responses in the presence of the broad-spectrum calcium channel antagonist cadmium (50 μm). The bath application of cadmium abolished ACh-evoked silencing of action potential firing in NGFCs, and replaced long duration hyperpolarizing responses by the slow enhancement of neuronal output (Fig. 3H,I; n = 7, n = 7 slices, n = 3 animals). In contrast, the sodium channel blocker tetrodotoxin did not prevent mACHR-mediated inhibition of NGFCs (TTX, 1 μm; Fig. 4). Notably, we found that large amplitude hyperpolarizing responses and associated calcium signals could be readily evoked in the absence of action potential firing when NGFCs were held at depolarized membrane potentials (Fig. 4C; membrane potential in TTX = −34.7 ± 2.5 mV; hyperpolarization = −14.4 ± 3.58 mV; n = 4, n = 4 slices, n = 4 animals).
Figure 4.

Calcium release from internal stores controls cholinergic inhibition of NGFCs. A, Blockade of ACh-evoked silencing of action potential firing by thapsigargin (10 μm). Action potential amplitude has been truncated for clarity. B, Peristimulus time histogram of action potential firing under control and after bath application of thapsigargin (bottom; 250 time bins; n = 4 cells). ACh was applied at time 0. C, Average voltage (Vm) and Oregon Green BAPTA-6F signals (ΔF/F) evoked by the iontophoretic application of ACh in TTX (1 μm; top) are reduced by bath application of 2-APB (bottom). Single traces are shown in gray. D, E, Summary of the effects of 2-APB on ACh-evoked voltage responses (D) and Oregon green BAPTA-6F signals (E; n = 4). Values represent mean ± SEM.
We next examined whether calcium release from intracellular stores was also a necessary component of ACh-evoked inhibition of NGFCs. To test this we emptied intracellular calcium stores pharmacologically by applying the endoplasmic reticulum calcium ATPase thapsigargin (Treiman et al., 1998). Bath application of thapsigargin (10 μm) completely blocked ACh-evoked inhibition of action potential firing in NGFCs, and unmasked a pronounced excitatory response (Fig. 4A,B; n = 4, n = 4 animals). As calcium release from intracellular stores is controlled by Ryanodine-sensitive and IP3-sensitive receptors (IP3Rs; Berridge, 2002), and M1 mAChRs are linked to phosphatidylinositol turnover (Caulfield and Birdsall, 1998), we tested the role of IP3R activation in mAChR signaling in NGFCs. The bath application of the IP3R inhibitor 2-aminoethoxydiphenylborane (2-APB, 100 μm), in the presence of TTX (1 μm) decreased the voltage integral of ACh-evoked hyperpolarizing responses (control = −24.3 ± 5.6 mV · s; 2-APB = −0.6 ± 2 mV · s; paired t test: T = 3.575; p = 0.0374, n = 4, n = 4 animals) and associated calcium signals (Fig. 4C–E; control = 0.9 ± 0.2 ΔF/F · s; 2-APB = 0.2 ± 0.1 ΔF/F · s; ratio paired t test: T = 5.361; p = 0.0127, n = 4, n = 4 animals). Together these data suggest that the inhibitory actions of ACh are mediated by the time- and voltage-dependent refilling of intracellular stores by calcium entry during repetitive action potential firing.
To examine the impact of the physiological refilling of intracellular calcium stores we used simultaneous calcium imaging and electrophysiological recording to demonstrate that a period of repetitive action potential firing was required to refill intracellular calcium stores to allow the expression of both ACh-evoked inhibition and calcium signaling (Fig. 5A; n = 5, n = 5 slices, n = 4 animals). When refilling did not take place, by returning the membrane potential of NGFCs to RMP following a conditioning ACh inhibitory response (Fig. 5B, depleted), we observed that the inhibitory actions of ACh and ACh-evoked rises in intracellular calcium progressively increased during test responses, generated at variable times after the reintroduction of repetitive action potential firing (Fig. 5C,D). In contrast, when intracellular calcium stores were refilled by a period of action potential firing immediately following the conditioning ACh response (Fig. 5B, refilling), both the inhibitory actions of ACh and ACh-evoked calcium signaling were time-independent after the reintroduction of repetitive action potential firing (Fig. 5C,D). To directly demonstrate the physiological refilling of intracellular stores is responsible for the switching between net excitatory and inhibitory ACh responses, we iontophoretically applied ACh to NGFCs held at subthreshold membrane potentials resulting in the generation of depolarizing responses (Fig. 6A,B; voltage integral = 3.8 ± 0.65 mV · s; membrane potential = −56.5 ± 1.4 mV; n = 13, n = 13 slices, n = 9 animals). A train of 100 action potentials was then generated, and the ACh response re-examined, revealing the generation of a net inhibitory response (Fig. 6A,B; voltage integral = −8.9 ± 1.77 mV · s). Thus, the physiological filling of calcium stores is a critical determinant of the inhibitory actions of ACh in NGFCs.
Figure 5.
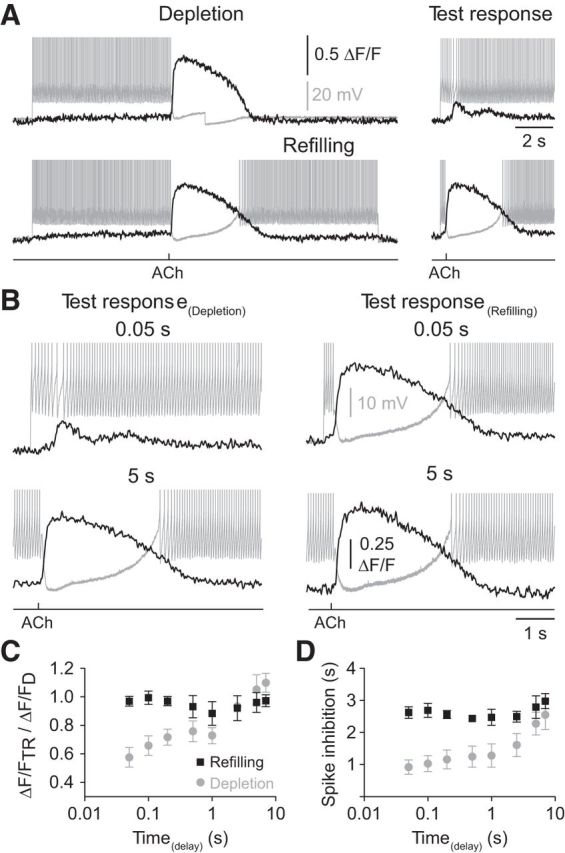
Depletion and refilling of internal calcium stores controls cholinergic inhibition of NGFCs. A, Illustration of the experimental paradigm. ACh was iontophoretically applied (1 m, 20 ms) after 7 s of repetitive action potential firing and evoked a pronounced hyperpolarization (gray trace) together with an Oregon green BAPTA-6F signal (black trace). To investigate the role of action potential-dependent calcium store refilling, the membrane voltage was either stepped back to resting membrane potential (depletion) or the NGFCs were allowed to fire action potentials for a further 7 s (refilling). In both cases, the inhibitory actions of ACh were re-examined at times following the reintroduction of repetitive action potential firing (test responses). B, In the depleted case (left), an ACh test response generated 50 ms after the reintroduction of repetitive action potential firing did not silence neuronal output (gray trace) and evoked a small amplitude Oregon green BAPTA-6F signal (black trace). In contrast, after 5 s of repetitive action potential firing the inhibitory actions of ACh were restored. In the refilling case (right), the inhibitory actions of ACh were time-independent. A, B, The action potential amplitude has been truncated for clarity. C, D, Time-dependent recovery of ACh-evoked Oregon green BAPTA-6F signals (C; gray symbols) and the time course of ACh-evoked silencing of action potential firing following depletion (D; gray symbol), but time-independent Oregon green BAPTA-6F signals (C; black symbols) and inhibition following refilling (D; black symbols; n = 5 NGFCs). Values represent mean ± SEM.
Figure 6.

Activity-dependent cholinergic inhibition of NGFCs is mediated by SK potassium channels. A, Action potential firing transforms ACh signaling. The top trace shows the transformation of ACh signaling from excitatory to inhibitory following a period of action potential (AP) firing (100 APs evoked at 17 Hz). ACh was applied iontophoretically (bottom trace, 100 mm, 20 ms). The middle voltage trace shows the sequential generation of excitatory responses by ACh in the absence of AP firing. B, Pooled data describing the voltage integral of ACh responses evoked before (open bar) and after (gray bar) AP firing. Values represent mean ± SEM. C, ACh-evoked silencing of AP firing is prevented by the blockade of SK potassium channels with apamin (100 nm). ACh delivered by iontophoresis (bottom trace, 100 mm, 20 ms). A, C, The action potential amplitude has been truncated for clarity. D, Summary of the control of ongoing AP firing under control and in the presence of apamin (right). Data have been pooled to generate peristimulus time histograms of AP firing (250 ms time bins). ACh was applied at time 0.
ACh-evoked inhibition is mediated by calcium-activated potassium channels
The intricate control of ACh-evoked calcium signaling in NGFCs suggests that the activity-dependent inhibitory actions of ACh are mediated by calcium-dependent effector mechanisms. We therefore explored whether calcium-activated potassium channels are involved in the inhibition of NGFCs. Previous evidence has indicted intracellular calcium activates SK potassium channels in central neurons (Adelman et al., 2012). To investigate the impact of SK channels on ACh-evoked inhibition in NGFCs, we applied the specific SK-channel inhibitor apamin (Kohler et al., 1996). Bath application of apamin (100 nm) completely and irreversibly blocked the inhibitory actions of ACh, converting the powerful and sustained silencing of action potential firing to an excitatory response (Fig. 6C,D; n = 6, n = 6 slices, n = 6 animals). Our results therefore indicate that the activity-dependent cholinergic inhibition of NGFCs is mediated by the M1 mAChR-dependent release of calcium from intracellular stores, which gates surface-membrane SK channels.
Activity-dependent modulation of synaptic inhibition
The class- and activity-dependent control of the excitability of layer 1 interneurons by ACh suggests that cholinergic modulation may influence the inhibitory synaptic outflow of layer 1. To examine this, we made paired recordings between identified classes of layer 1 interneurons and layer 2/3 pyramidal neurons. Single action potential firing of NGFCs inhibited layer 2/3 pyramidal neurons by the generation of slowly rising and decaying unitary IPSPs (uIPSPs) (Fig. 7A,B; uIPSP amplitude: −0.58 ± 0.1 mV, half-width 237 ± 21 ms; n = 19, n = 19 slices, n = 14 animals). uIPSPs were found to be mediated by the activation of GABAA and GABAB receptors, with pharmacological analysis showing that the GABAB receptor-dependent component underlay >65% of the area of uIPSPs (Fig. 7A,C). During repetitive action potential firing, slow NGFC-evoked uIPSPs summated to produce synaptic inhibition that silenced the current-evoked ongoing action potential firing of postsynaptic layer 2/3 pyramidal neurons (Fig. 7D,E). To investigate the impact of ACh on the synaptic output of layer 1 interneurons, we first verified that the activation of nAChRs could drive uIPSPs in postsynaptic layer 2/3 pyramidal neurons. The local perisomatic delivery of ACh to the soma of NGFCs held at subthreshold membrane potentials drove uIPSPs in pyramidal neurons with properties indistinguishable from those evoked by direct somatic current injection (Fig. 7F,G; current-evoked uIPSP amplitude: −0.99 ± 0.37 mV; ACh-evoked uIPSP amplitude: −0.98 ± 0.38 mV; paired t test: T = 0.064; p = 0.95, n = 5, n = 5 slices, n = 4 animals). Similarly, cholinergic- or current-evoked action potential firing of NGFCs directly inhibited c-ACs (uIPSP amplitude: −0.85 ± 0.12 mV; n = 5 connections, n = 5 slices, n = 5 animals, data not shown). In contrast, when nAChRs were blocked by bath application of mecamylamine (10 μm), ACh inhibited current-evoked action potential firing in NGFCs and consequently the postsynaptic inhibition of layer 2/3 pyramidal neurons (Fig. 7H,I). In c-ACs, held at subthreshold membrane potentials (−53.91 ± 1.71 mV), ACh consistently evoked action potential firing, which drove inhibition in postsynaptic targets (ACh-evoked uIPSP amplitude: −0.54 ± 0.10 mV; n = 8, n = 8 animals; data not shown). Notably, we found that c-ACs frequently inhibited NGFCs (uIPSP amplitude: −0.70 ± 0.12 mV; n = 5 connected pairs, n = 5 animals), but rarely layer 2/3 pyramidal neurons (n = 1 connection). A finding consistent with previous reports (Christophe et al., 2002; Wozny and Williams, 2011), and the confinement of the axonal arbor of c-ACs to layer 1, which together suggest that c-ACs may selectively innervate layer 1 interneurons.
Figure 7.
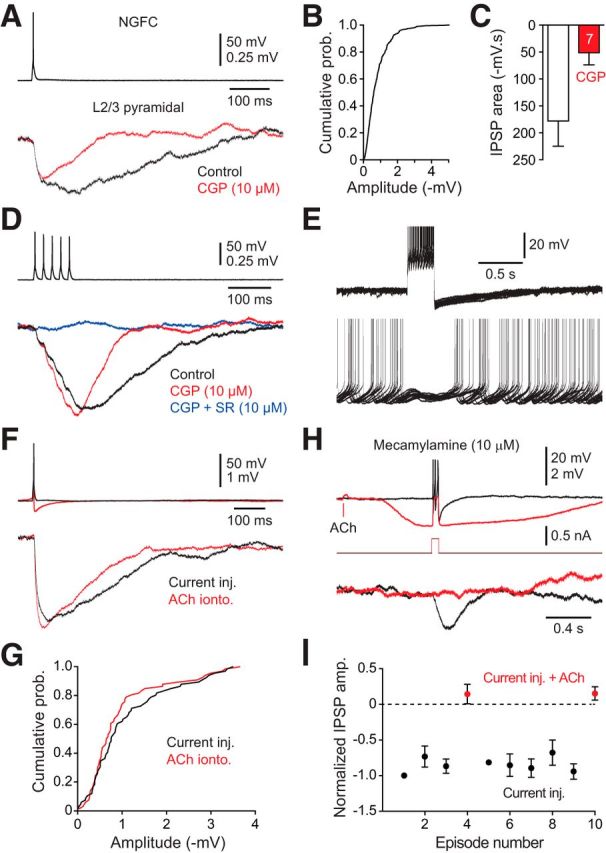
NGFCs drive powerful postsynaptic inhibition. A, Current-evoked single action potential firing of a NGFC evokes unitary IPSPs in a postsynaptic layer 2/3 pyramidal neuron that are substantially inhibited by a GABAB receptor antagonist (red trace; CGP 55845, 10 μm). B, Cumulative probability distribution of the peak amplitude of single action potential evoked unitary IPSPs in NGFC–layer 2/3 pyramidal neuron pairs (n = 437 IPSPs, n = 19 connections). C, Summary of the actions of CGP 55845 (10 μm) on the area of single action potential-evoked unitary IPSPs (n = 7 connections). D, Repetitive current-evoked NGFC action potential firing evokes summating IPSPs in a postsynaptic layer 2/3 pyramidal neuron that are sensitive to GABA receptor antagonist (CGP 55845, 10 μm; SR-95531, 10 μm). E, Current-evoked NGFC IPSPs silence action potential firing in a postsynaptic layer 2/3 pyramidal neuron (n = 14 overlain traces). Tonic action potential firing of the pyramidal neuron was evoked by the delivery of positive current (130 pA). F, ACh-evoked depolarization drives single action potential firing of NGFCs and postsynaptic IPSPs in layer 2/3 pyramidal neurons (red traces), with the same properties as current-evoked action potentials (black traces). ACh was delivered by iontophoresis (100 mm, 20 ms). G, Cumulative probability distribution of ACh- and current-evoked unitary IPSPs (n = 60 IPSPs, n = 5 connections). H, Muscarinic receptor-mediated ACh inhibition silences current-evoked action potential firing and the inhibitory synaptic output of NGFCs. Control responses show current-evoked NGFC action potential firing (top black trace) which drives postsynaptic inhibition (bottom black trace). The iontophoretic delivery of ACh (100 mm, 20 ms) in the presence of the nACHR antagonist mecamylamine (10 μm) leads to the generation of a hyperpolarization, which inhibits current-evoked action potential firing and postsynaptic inhibition (red traces). E, H, The action potential amplitude has been truncated for clarity. I, Pooled data describing the normalized peak amplitude of IPSPs evoked by bursts of action potentials in NGFC—layer 2/3 pyramidal neuron pairs (black symbols), which were silenced by the iontophoretic delivery of ACh (red symbols; 100 mm, 20 ms). Data have been pooled from n = 4 connections. C, I, Values represent mean ± SEM.
Finally, we tested whether the cholinergic modulation of the inhibitory synaptic output of NGFCs could be controlled in an activity-dependent manner. To do this we first applied ACh to the soma of quiescent NGFCs, leading to the generation of subthreshold excitatory responses, which did not inhibit direct current-evoked action potential firing (Fig. 8A–D; test ACh response; n = 7 connections, n = 7 animals). Action potential firing was then evoked by the repeated delivery of positive current steps, which reliably drove postsynaptic inhibition of layer 2/3 pyramidal neurons, indicating that NGFC to layer 2/3 inhibitory synapses can sustain transmission during repetitive activation (Fig. 8A–D). Following a period of action potential firing, the application of ACh inhibited NGFCs, suppressing current-evoked action potential firing and synaptic release (Fig. 8A–D; conditioned ACh response). Thus, the activity-dependent cholinergic modulation of NGFCs dramatically impacts the inhibitory synaptic control of the apical dendrites of layer 2/3 pyramidal neurons.
Figure 8.
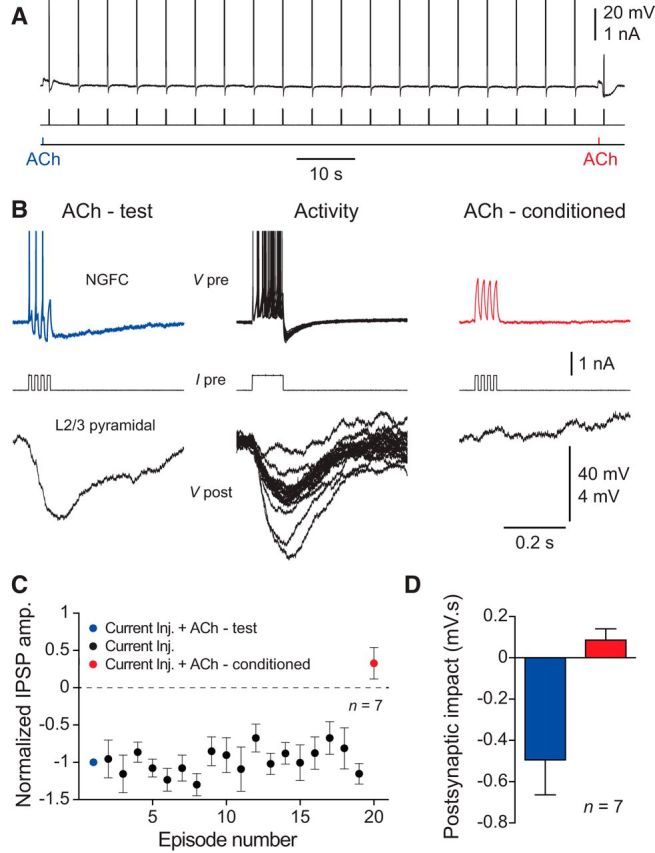
Activity-dependent silencing of the synaptic output of NGFCs by ACh. A, Continuous voltage record (top trace) showing the repeated generation of current-evoked (middle trace) action potential firing of a NGFC. At the beginning of the sequence ACh-evoked a small excitatory response (ACh delivered by iontophoresis, 100 mm, 20 ms, bottom trace, blue label, test response). In contrast, after the generation of 18 bursts of action potential firing, ACh evoked a hyperpolarization, which silenced current-evoked action potential firing (red label, test response). B, High gain representation of the voltage record shown in A, showing NGFC voltage responses (top traces), the pattern of driving current (middle traces), and the inhibitory synaptic output recorded from a layer 2/3 pyramidal neuron (bottom traces). C, Summary data showing the activity-dependent silencing of the synaptic output of NGFCs by ACh. IPSP amplitude has been normalized to the first response. Data have been pooled from n = 7 connections. D, Pooled data describing the inhibition of the inhibitory synaptic output of NGFCs by ACh. Postsynaptic impact refers to the area of the postsynaptic response measured from layer 2/3 pyramidal neurons over similar time periods for test (blue) and conditioned ACh responses (red). Data pooled from n = 7 connections. C, D, Values represent mean ± SEM.
Discussion
Previous work has shown that ACh excites layer 1 GABAergic interneurons through the activation of nAChRs (Christophe et al., 2002; Letzkus et al., 2011; Arroyo et al., 2012; Bennett et al., 2012). Our results demonstrate that the actions of ACh in this neocortical layer are more complex. We confirm previous observations that layer 1 interneurons are excited at resting membrane potential predominately by the activation of non-α7 nAChRs, suggesting that the local perisomatic iontophoretic application of ACh replicates the synaptic release of ACh (Arroyo et al., 2012; Bennett et al., 2012). However, as layer 1 interneurons fire trains of action potentials in vivo (Letzkus et al., 2011; Palmer et al., 2012; Jiang et al., 2013), we investigated the actions of ACh during the generation of action potential firing in vitro. Under these conditions, cholinergic signaling in layer 1 of the neocortex was interneuron class-dependent. ACh transiently augmented the action potential firing of c-ACs, but inhibited the firing of NGFCs for long time periods. This interneuron class-dependent pattern of cholinergic signaling is compatible with findings made from unidentified layer 1 interneurons in vivo (Letzkus et al., 2011); where aversive somatic stimulation, which engages cholinergic mechanisms, has been found to either transiently augment or inhibit for long time periods the ongoing action potential firing rate of extracellularly recorded layer 1 interneurons.
Mechanism of cholinergic inhibition of NGFCs
The inhibitory actions of ACh in NGFCs were mediated by the activation of M1 mAChRs, which triggered an IP3 receptor-dependent release of calcium from intracellular stores. The correlation between the time course of directly imaged M1 mAChR-evoked calcium release and the inhibition of action potential firing, together with the blockade of cholinergic inhibition by the antagonism of voltage-gated calcium channels, and the pharmacological and functional depletion of intracellular calcium stores, suggests that store calcium filling and release plays an essential role in the direct cholinergic inhibition of NGFCs. Our data indicates that M1 mAChR-triggered calcium release from intracellular stores, which may be augmented by the synergistic interaction between IP3 receptor activation and calcium entry (Stutzmann et al., 2003), forms a calcium signal which activates SK calcium-activated potassium channels.
SK calcium-activated potassium channels are exclusively gated by intracellular calcium (Adelman et al., 2012). Previous observations in hippocampal CA1 pyramidal neurons have, however, indicated that the activation of mAChRs leads to a reduction of SK potassium channel activity, through the second messenger tuning of the calcium sensitivity of these channels (Buchanan et al., 2010; Giessel and Sabatini, 2010). In contrast, the transient activation of mAChRs in neocortical pyramidal neurons and midbrain dopaminergic neurons has been found to gate the activation of SK channels through the elevation of intracellular calcium by its release from intracellular stores (Fiorillo and Williams, 2000; Gulledge and Stuart, 2005; Gulledge et al., 2009). These contrasting actions of ACh on SK channel activation may reflect the spatiotemporal profile of mAChR signaling. Consistent with this, the inhibitory actions of ACh in midbrain dopaminergic neurons are predominately expressed following the phasic activation of mAChRs (Fiorillo and Williams, 2000). Our results indicate that the phasic activation of AChRs in NGFCs powerfully activates SK calcium-activated potassium channels to produce inhibition, which dominates over nAChR-mediated excitation, to silence action potential firing.
Potential functional consequences
Neurogliaform cells are the source of GABAB receptor-mediated inhibition in the neocortex, controlling large assemblies of postsynaptic neurons by the release of GABA as a volume transmitter (Tamás et al., 2003; Oláh et al., 2009). Layer 1 NGFCs are therefore strategically placed in the neocortical microcircuit to influence apical dendritic integration in assemblies of layer 2/3 and layer 5 pyramidal neurons, because of the intersection of the axonal nest of NGFCs with the apical dendrites of pyramidal neurons. Direct electrical recording and imaging approaches have shown that active apical dendritic integration powerfully influences the action potential output of neocortical pyramidal neurons (Kim and Connors, 1993; Larkum et al., 1999; Williams and Stuart, 2002; Larkum et al., 2007; Harnett et al., 2013). Apical dendritic targeting interneurons have therefore been suggested to influentially control pyramidal neuron output, particular during correlative integrative operations (Larkum et al., 1999; Murayama et al., 2009; Jiang et al., 2013) that underlie the processing of top-down signals (Larkum, 2013) in the neocortex, conveyed by corticocortical connections that heavily innervate layer 1 (Cauller et al., 1998; Mao et al., 2011; Larkum, 2013). Consistent with a role of NGFCs in the control of top-down signaling, the electrical stimulation of layer 1, and the activation of corticocortical and nonspecific thalamocortical pathways drives NGFCs (Pérez-Garci et al., 2006; Cruikshank et al., 2012; Palmer et al., 2012; Lee et al., 2013). This provides a substrate for the powerful and long-lasting GABAB receptor-mediated inhibition of apical dendritic integration and calcium electrogenesis in pyramidal neurons (Pérez-Garci et al., 2006, 2013; Palmer et al., 2012).
In awake and behaving animals, however, imaging techniques have revealed the generation of widespread calcium electrogenesis in the apical dendritic arbor of pyramidal neurons (Murayama and Larkum, 2009; Xu et al., 2012), suggesting that mechanisms exist to control the impact of NGFC-mediated GABAB receptor-dependent inhibition under physiological conditions. One possibility is that the coordinated activity of interneuronal circuits controls the excitability of NGFCs. Consistent with this we find that layer 1 c-ACs directly inhibit NGFCs, supporting previous findings that cholinergic excitation of interneuron targeting layer 1 interneurons drives disinhibitory circuits (Christophe et al., 2002; Letzkus et al., 2011; Arroyo et al., 2012).
We suggest that direct cholinergic inhibition may contribute to the inhibitory control of NGFCs. Notably, ACh levels in the neocortex are dependent on arousal state, and increase during attentive behavior (Giovannini et al., 2001), providing an attention-related signal for the cholinergic inhibition of NGFCs, and so the disinhibition of the apical dendrites of pyramidal neurons. The activity-dependent nature of the cholinergic modulation of NGFCs indicates that such disinhibition will only occur when NGFCs are engaged by network operations. Our results therefore suggest that the cholinergic inhibition of NGFCs, and so the silencing of direct feedforward monosynaptic GABAB receptor-mediated inhibition of the apical dendritic arbor of pyramidal neurons, will operate in parallel with the cholinergic excitation of other layer 1 inhibitory circuits that disinhibit proximal dendritic sites of pyramidal neurons through disynaptic pathways (Christophe et al., 2002; Letzkus et al., 2011; Arroyo et al., 2012). Further work will be required to investigate the concerted role that the cholinergic system plays in the modulation of these, and other interneuronal circuits (Lee et al., 2013; Pi et al., 2013), in particular those that control Somatostatin-positive cells, which provide feedback GABAA receptor-mediated apical dendritic inhibition of pyramidal neurons (Silberberg and Markram, 2007; Murayama et al., 2009). In summary, our results contribute to the description of the cholinergic modulation of interneuronal circuits (McCormick and Prince, 1986; Kawaguchi, 1997; Xiang et al., 1998; Porter et al., 1999), which in concert with the direct modulation of pyramidal neurons (Gulledge and Stuart, 2005; Gulledge et al., 2009) dynamical influence information processing in the neocortical network.
Footnotes
This work was supported by the Australian Research Council (FT100100502 and DP130101630) and the National Health and Medical Research Council (APP1004575). We thank J. Hales for help with morphological reconstructions.
The authors declare no competing financial interests.
References
- Adelman JP, Maylie J, Sah P. Small-conductance Ca2+-activated K+ channels: form and function. Annu Rev Physiol. 2012;74:245–269. doi: 10.1146/annurev-physiol-020911-153336. [DOI] [PubMed] [Google Scholar]
- Adler EM, Augustine GJ, Duffy SN, Charlton MP. Alien intracellular calcium chelators attenuate neurotransmitter release at the squid giant synapse. J Neurosci. 1991;11:1496–1507. doi: 10.1523/JNEUROSCI.11-06-01496.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arroyo S, Bennett C, Aziz D, Brown SP, Hestrin S. Prolonged disynaptic inhibition in the cortex mediated by slow, non-α7 nicotinic excitation of a specific subset of cortical interneurons. J Neurosci. 2012;32:3859–3864. doi: 10.1523/JNEUROSCI.0115-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartus RT, Dean RL, 3rd, Beer B, Lippa AS. The cholinergic hypothesis of geriatric memory dysfunction. Science. 1982;217:408–414. doi: 10.1126/science.7046051. [DOI] [PubMed] [Google Scholar]
- Bennett C, Arroyo S, Berns D, Hestrin S. Mechanisms generating dual-component nicotinic EPSCs in cortical interneurons. J Neurosci. 2012;32:17287–17296. doi: 10.1523/JNEUROSCI.3565-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berridge MJ. The endoplasmic reticulum: a multifunctional signaling organelle. Cell Calcium. 2002;32:235–249. doi: 10.1016/S0143416002001823. [DOI] [PubMed] [Google Scholar]
- Buchanan KA, Petrovic MM, Chamberlain SE, Marrion NV, Mellor JR. Facilitation of long-term potentiation by muscarinic M(1) receptors is mediated by inhibition of SK channels. Neuron. 2010;68:948–963. doi: 10.1016/j.neuron.2010.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buzsaki G, Bickford RG, Ponomareff G, Thal LJ, Mandel R, Gage FH. Nucleus basalis and thalamic control of neocortical activity in the freely moving rat. J Neurosci. 1988;8:4007–4026. doi: 10.1523/JNEUROSCI.08-11-04007.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caulfield MP, Birdsall NJ. International union of pharmacology: XVII. Classification of muscarinic acetylcholine receptors. Pharmacol Rev. 1998;50:279–290. [PubMed] [Google Scholar]
- Cauller LJ, Clancy B, Connors BW. Backward cortical projections to primary somatosensory cortex in rats extend long horizontal axons in layer I. J Comp Neurol. 1998;390:297–310. doi: 10.1002/(SICI)1096-9861(19980112)390:2<297::AID-CNE11>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- Christophe E, Roebuck A, Staiger JF, Lavery DJ, Charpak S, Audinat E. Two types of nicotinic receptors mediate an excitation of neocortical layer I interneurons. J Neurophysiol. 2002;88:1318–1327. doi: 10.1152/jn.2002.88.3.1318. [DOI] [PubMed] [Google Scholar]
- Chu Z, Galarreta M, Hestrin S. Synaptic interactions of late-spiking neocortical neurons in layer 1. J Neurosci. 2003;23:96–102. doi: 10.1523/JNEUROSCI.23-01-00096.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Contestabile A. The history of the cholinergic hypothesis. Behav Brain Res. 2011;221:334–340. doi: 10.1016/j.bbr.2009.12.044. [DOI] [PubMed] [Google Scholar]
- Coyle JT, Price DL, DeLong MR. Alzheimer's disease: a disorder of cortical cholinergic innervation. Science. 1983;219:1184–1190. doi: 10.1126/science.6338589. [DOI] [PubMed] [Google Scholar]
- Cruikshank SJ, Ahmed OJ, Stevens TR, Patrick SL, Gonzalez AN, Elmaleh M, Connors BW. Thalamic control of layer 1 circuits in prefrontal cortex. J Neurosci. 2012;32:17813–17823. doi: 10.1523/JNEUROSCI.3231-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Détári L, Rasmusson DD, Semba K. The role of basal forebrain neurons in tonic and phasic activation of the cerebral cortex. Prog Neurobiol. 1999;58:249–277. doi: 10.1016/S0301-0082(98)00084-7. [DOI] [PubMed] [Google Scholar]
- Fiorillo CD, Williams JT. Cholinergic inhibition of ventral midbrain dopamine neurons. J Neurosci. 2000;20:7855–7860. doi: 10.1523/JNEUROSCI.20-20-07855.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giessel AJ, Sabatini BL. M1 muscarinic receptors boost synaptic potentials and calcium influx in dendritic spines by inhibiting postsynaptic SK channels. Neuron. 2010;68:936–947. doi: 10.1016/j.neuron.2010.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giovannini MG, Rakovska A, Benton RS, Pazzagli M, Bianchi L, Pepeu G. Effects of novelty and habituation on acetylcholine, GABA, and glutamate release from the frontal cortex and hippocampus of freely moving rats. Neuroscience. 2001;106:43–53. doi: 10.1016/S0306-4522(01)00266-4. [DOI] [PubMed] [Google Scholar]
- Goard M, Dan Y. Basal forebrain activation enhances cortical coding of natural scenes. Nat Neurosci. 2009;12:1444–1449. doi: 10.1038/nn.2402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gulledge AT, Stuart GJ. Cholinergic inhibition of neocortical pyramidal neurons. J Neurosci. 2005;25:10308–10320. doi: 10.1523/JNEUROSCI.2697-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gulledge AT, Bucci DJ, Zhang SS, Matsui M, Yeh HH. M1 receptors mediate cholinergic modulation of excitability in neocortical pyramidal neurons. J Neurosci. 2009;29:9888–9902. doi: 10.1523/JNEUROSCI.1366-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harnett MT, Xu NL, Magee JC, Williams SR. Potassium channels control the interaction between active dendritic integration compartments in layer 5 cortical pyramidal neurons. Neuron. 2013;79:516–529. doi: 10.1016/j.neuron.2013.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang X, Wang G, Lee AJ, Stornetta RL, Zhu JJ. The organization of two new cortical interneuronal circuits. Nat Neurosci. 2013;16:210–218. doi: 10.1038/nn.3305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalmbach A, Hedrick T, Waters J. Selective optogenetic stimulation of cholinergic axons in neocortex. J Neurophysiol. 2012;107:2008–2019. doi: 10.1152/jn.00870.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawaguchi Y. Selective cholinergic modulation of cortical GABAergic cell subtypes. J Neurophysiol. 1997;78:1743–1747. doi: 10.1152/jn.1997.78.3.1743. [DOI] [PubMed] [Google Scholar]
- Kilgard MP, Merzenich MM. Cortical map reorganization enabled by nucleus basalis activity. Science. 1998;279:1714–1718. doi: 10.1126/science.279.5357.1714. [DOI] [PubMed] [Google Scholar]
- Kim HG, Connors BW. Apical dendrites of the neocortex: correlation between sodium- and calcium-dependent spiking and pyramidal cell morphology. J Neurosci. 1993;13:5301–5311. doi: 10.1523/JNEUROSCI.13-12-05301.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Köhler M, Hirschberg B, Bond CT, Kinzie JM, Marrion NV, Maylie J, Adelman JP. Small-conductance, calcium-activated potassium channels from mammalian brain. Science. 1996;273:1709–1714. doi: 10.1126/science.273.5282.1709. [DOI] [PubMed] [Google Scholar]
- Larkum M. A cellular mechanism for cortical associations: an organizing principle for the cerebral cortex. Trends Neurosci. 2013;36:141–151. doi: 10.1016/j.tins.2012.11.006. [DOI] [PubMed] [Google Scholar]
- Larkum ME, Zhu JJ, Sakmann B. A new cellular mechanism for coupling inputs arriving at different cortical layers. Nature. 1999;398:338–341. doi: 10.1038/18686. [DOI] [PubMed] [Google Scholar]
- Larkum ME, Waters J, Sakmann B, Helmchen F. Dendritic spikes in apical dendrites of neocortical layer 2/3 pyramidal neurons. J Neurosci. 2007;27:8999–9008. doi: 10.1523/JNEUROSCI.1717-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Kruglikov I, Huang ZJ, Fishell G, Rudy B. A disinhibitory circuit mediates motor integration in the somatosensory cortex. Nat Neurosci. 2013;16:1662–1670. doi: 10.1038/nn.3544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letzkus JJ, Wolff SB, Meyer EM, Tovote P, Courtin J, Herry C, Lüthi A. A disinhibitory microcircuit for associative fear learning in the auditory cortex. Nature. 2011;480:331–335. doi: 10.1038/nature10674. [DOI] [PubMed] [Google Scholar]
- Mao T, Kusefoglu D, Hooks BM, Huber D, Petreanu L, Svoboda K. Long-range neuronal circuits underlying the interaction between sensory and motor cortex. Neuron. 2011;72:111–123. doi: 10.1016/j.neuron.2011.07.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markram H, Toledo-Rodriguez M, Wang Y, Gupta A, Silberberg G, Wu C. Interneurons of the neocortical inhibitory system. Nat Rev Neurosci. 2004;5:793–807. doi: 10.1038/nrn1519. [DOI] [PubMed] [Google Scholar]
- McCormick DA, Prince DA. Mechanisms of action of acetylcholine in the guinea-pig cerebral cortex in vitro. J Physiol. 1986;375:169–194. doi: 10.1113/jphysiol.1986.sp016112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mechawar N, Cozzari C, Descarries L. Cholinergic innervation in adult rat cerebral cortex: a quantitative immunocytochemical description. J Comp Neurol. 2000;428:305–318. doi: 10.1002/1096-9861(20001211)428:2<305::AID-CNE9>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- Metherate R, Cox CL, Ashe JH. Cellular bases of neocortical activation: modulation of neural oscillations by the nucleus basalis and endogenous acetylcholine. J Neurosci. 1992;12:4701–4711. doi: 10.1523/JNEUROSCI.12-12-04701.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murayama M, Larkum ME. Enhanced dendritic activity in awake rats. Proc Natl Acad Sci U S A. 2009;106:20482–20486. doi: 10.1073/pnas.0910379106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murayama M, Pérez-Garci E, Nevian T, Bock T, Senn W, Larkum ME. Dendritic encoding of sensory stimuli controlled by deep cortical interneurons. Nature. 2009;457:1137–1141. doi: 10.1038/nature07663. [DOI] [PubMed] [Google Scholar]
- Oláh S, Füle M, Komlósi G, Varga C, Báldi R, Barzó P, Tamás G. Regulation of cortical microcircuits by unitary GABA-mediated volume transmission. Nature. 2009;461:1278–1281. doi: 10.1038/nature08503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer LM, Schulz JM, Murphy SC, Ledergerber D, Murayama M, Larkum ME. The cellular basis of GABA(B)-mediated interhemispheric inhibition. Science. 2012;335:989–993. doi: 10.1126/science.1217276. [DOI] [PubMed] [Google Scholar]
- Pérez-Garci E, Gassmann M, Bettler B, Larkum ME. The GABAB1b isoform mediates long-lasting inhibition of dendritic Ca2+ spikes in layer 5 somatosensory pyramidal neurons. Neuron. 2006;50:603–616. doi: 10.1016/j.neuron.2006.04.019. [DOI] [PubMed] [Google Scholar]
- Pérez-Garci E, Larkum ME, Nevian T. Inhibition of dendritic Ca2+ spikes by GABAB receptors in cortical pyramidal neurons is mediated by a direct Gi/o-β-subunit interaction with Cav1 channels. J Physiol. 2013;591:1599–1612. doi: 10.1113/jphysiol.2012.245464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pi HJ, Hangya B, Kvitsiani D, Sanders JI, Huang ZJ, Kepecs A. Cortical interneurons that specialize in disinhibitory control. Nature. 2013;503:521–524. doi: 10.1038/nature12676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter JT, Cauli B, Tsuzuki K, Lambolez B, Rossier J, Audinat E. Selective excitation of subtypes of neocortical interneurons by nicotinic receptors. J Neurosci. 1999;19:5228–5235. doi: 10.1523/JNEUROSCI.19-13-05228.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prieto JJ, Peterson BA, Winer JA. Morphology and spatial distribution of GABAergic neurons in cat primary auditory cortex (AI) J Comp Neurol. 1994;344:349–382. doi: 10.1002/cne.903440304. [DOI] [PubMed] [Google Scholar]
- Silberberg G, Markram H. Disynaptic inhibition between neocortical pyramidal cells mediated by Martinotti cells. Neuron. 2007;53:735–746. doi: 10.1016/j.neuron.2007.02.012. [DOI] [PubMed] [Google Scholar]
- Stutzmann GE, LaFerla FM, Parker I. Ca2+ signaling in mouse cortical neurons studied by two-photon imaging and photoreleased inositol triphosphate. J Neurosci. 2003;23:758–765. doi: 10.1523/JNEUROSCI.23-03-00758.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamás G, Lorincz A, Simon A, Szabadics J. Identified sources and targets of slow inhibition in the neocortex. Science. 2003;299:1902–1905. doi: 10.1126/science.1082053. [DOI] [PubMed] [Google Scholar]
- Treiman M, Caspersen C, Christensen SB. A tool coming of age: thapsigargin as an inhibitor of sarco-endoplasmic reticulum Ca(2+)-ATPases. Trends Pharmacol Sci. 1998;19:131–135. doi: 10.1016/S0165-6147(98)01184-5. [DOI] [PubMed] [Google Scholar]
- Williams SR, Stuart GJ. Dependence of EPSP efficacy on synapse location in neocortical pyramidal neurons. Science. 2002;295:1907–1910. doi: 10.1126/science.1067903. [DOI] [PubMed] [Google Scholar]
- Winer JA, Larue DT. Populations of GABAergic neurons and axons in layer I of rat auditory cortex. Neuroscience. 1989;33:499–515. doi: 10.1016/0306-4522(89)90402-8. [DOI] [PubMed] [Google Scholar]
- Wozny C, Williams SR. Specificity of synaptic connectivity between layer 1 inhibitory interneurons and layer 2/3 pyramidal neurons in the rat neocortex. Cereb Cortex. 2011;21:1818–1826. doi: 10.1093/cercor/bhq257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang Z, Huguenard JR, Prince DA. Cholinergic switching within neocortical inhibitory networks. Science. 1998;281:985–988. doi: 10.1126/science.281.5379.985. [DOI] [PubMed] [Google Scholar]
- Xu NL, Harnett MT, Williams SR, Huber D, O'Connor DH, Svoboda K, Magee JC. Nonlinear dendritic integration of sensory and motor pathways produces an object localization signal. Nature. 2012;492:247–251. doi: 10.1038/nature11601. [DOI] [PubMed] [Google Scholar]