

Abstract
Objective
Studies have suggested a shared genetic aetiology between congenital long QT syndrome (LQTS) and diabetes, epilepsy and mental disorders. We investigated the prevalence of metabolic, neurological and psychiatric comorbidities in LQTS patients.
Methods
This retrospective cohort study was based on data from nationwide Danish registries, 2003–2017. LQTS patients were matched 1:5 with controls on sex and age.
Results
We matched 463 LQTS patients with 2315 controls from the background population. Mean age was 35.7 (SD 21.0) years, and 38% were males in both groups. LQTS patients had a higher prevalence of atrial fibrillation (6.5% vs 2.3%, p<0.001), diabetes (3.7% vs 1.8 %, p=0.011) and hearing loss (3.2% vs 1.7%, p=0.027). LQTS patients had a higher prevalence of psychiatric disorders overall (13.0% vs 9.1%, p=0.01) but the difference could not be attributed to a specific psychiatric disease subgroup. LQTS patients had a higher prevalence of neurological disorders (22.0% vs 13.2%, p<0.001), largely driven by epilepsy (6.7% vs 1.6%, p<0.001). In 20/27 (74%) of the LQTS patients, the epilepsy diagnosis did not reappear in the registries after the LQTS diagnosis was established.
Conclusions
In this nationwide cohort, patients with LQTS had a significantly increased burden of diabetes, neurological and psychiatric comorbidities, compared with the background population. The higher prevalence of neurological comorbidities was largely driven by epilepsy, despite a high rate of potentially misdiagnosed patients prior to LQTS diagnosis. Our data support that LQTS may be considered a multiorgan disease and suggest that patient management should be adjusted accordingly.
Keywords: long QT syndrome, comorbidity, diabetes, epilepsy, hearing loss
Key questions.
What is already known about this subject?
Small experimental and genetic studies have linked long QT syndrome (LQTS) with several extracardiac manifestations such as diabetes, epilepsy and psychiatric disorders, but this has not been tested in a real-life cohort.
What does this study add?
In our nationwide data, patients with LQTS had a significantly increased burden of diabetes, neurological and psychiatric comorbidities compared with an age and gender-matched background population.
How might this impact on clinical practice?
Our data support that LQTS may be considered a multiorgan disease and suggest that patient care should be adjusted accordingly.
Introduction
The congenital long QT syndrome (LQTS) is a rare inherited cardiac disease that may result in syncope and malignant arrhythmias. In approximately 75% of the LQTS patients, a disease-causing mutation can be identified in one of the three LQTS genes (KCNQ1, KCNH2 and SCN5A) coding for ion channels involved in the cardiac repolarisation.1
The initial LQTS cases described comprised cases of the Jervell-Lange-Nielsen syndrome, a rare KCNQ1 and KCNE1 associated LQTS subtype that causes congenital deafness, arrhythmia susceptibility, and sudden cardiac death.2 It has since become well known that mutations in the LQTS-associated genes can present with various cardiac phenotypes such as the Brugada syndrome, early onset of atrial fibrillation and cardiomyopathies.3 4 In addition, more extracardiac manifestations have become evident during recent years. Epilepsy and sudden unexpected death in epilepsy have in several cases been linked to KCNH2, and SCN5A mutations5–9 and an exploratory study found an increased epileptiform activity, primarily in LQTS patients with KCNH2 mutations.10 Mutations in the KCNH2 gene have also been associated with cerebral or mental disorders such as schizophrenia and impaired cognition.11 12 As a result, the concept of ‘cardiocerebral channelopathies’ has emerged.13
Similarly, studies have suggested a link between the LQTS causing genes and risks of metabolic disturbances. LQTS patients with KCNQ1 or KCNH2 mutations display an increased insulin release following glucose intake resulting in a clinically relevant increased risk of hypoglycaemia.14 15
Currently, it is undetermined whether such extracardiac manifestations are more frequently diagnosed in a real-world population of LQTS patients. This nationwide study investigated the burden of metabolic, psychiatric and neurological comorbidities in a Danish cohort of patients with LQTS, compared with the disease burden in the general population.
Methods
Study design and registries
This study was a retrospective, nationwide cohort study based on data from nationwide Danish registries, including the Danish National Prescription Registry, the National Population Register, and the Danish National Patient Register. By use of the unique social security number, given to all citizens on birth or immigration, we were able to link data from several registries at the individual level. Denmark has a freely available, tax-financed, public healthcare system for all citizens irrespective of income and social class. Only two per cent of healthcare activities are managed in private hospitals, and the demands for reporting to the central registries are the same as in the public hospital setting. Data on in-hospital diagnosis and procedures are consistently reported to the Danish National Patient Register on hospital discharge and following consultations in the outpatient clinic. This setup ensures a complete record of registered healthcare activities. Date of death is continuously reported to the National Population Register ensuring complete follow-up on vital status. All prescriptions dispensed from Danish pharmacies are collected in the Danish National Prescription Registry due to partial government reimbursement, which has been shown to accurately reflect patient’s history of filled prescriptions. The registries have been described in further detail elsewhere.16–18
Records were deidentified by central authorities prior to our analysis; names and addresses were removed, and social security numbers were encrypted. Efforts were made not to report data that may aid in the identification of individuals. Hence, Danish law prohibits reporting of low group numbers (n<3), which were replaced with ‘na’ throughout the paper or omitted entirely. The exact numbers are known to the investigators. Our study period went from 2003 until June 2017.
Identification of LQTS population and matched controls
We identified all patients diagnosed for the first time with congenital LQTS based on the WHO International Classification of Disease, 10th edition (ICD-10) DI472E code received in hospital or in an outpatient clinic. Patients were characterised and followed from the date of first diagnosis, henceforth referred to as index. If patients with congenital LQTS at index later received a diagnosis of acquired LQTS (DI472F), the patients were excluded from the analyses. Each LQTS patient was matched with five controls, forming the control group.
Validation of the LQTS diagnosis in Danish registries
To investigate the accuracy of the congenital LQTS ICD-10 code, we manually reviewed all charts of patients receiving a diagnosis of congenital LQTS (DI472E) at Rigshospitalet, Copenhagen, Denmark between November 2016 and March 2019. During this period, 123 patients with a DI472E diagnosis code were identified. After the manual review, we were able to verify the diagnosis in 120 out of 123 patients (positive predictive value=97.6%).
Comorbidities
Comorbidities were defined based on hospital discharge diagnoses, including both in-hospital admissions and hospital outpatient clinic visits, coded according to the ICD-10 at any time before and after index.
Cardiovascular comorbidities in focus were acute myocardial infarction (I20, I23-I25), venous thromboembolism (I26, I82, I80, excluding I820, I800 and I808), peripheral vascular disease (I70, I74), and atrial fibrillation (I48).
Metabolic comorbidities in focus were diabetes mellitus with complications (E10-E14, excluding E109, E119, E129, E139), hypoglycaemia (E100E, E110B, E159, E159, E161, E162) and hypothyroidism (E03). To strengthen the diabetes diagnosis, claimed prescription on antidiabetic drugs and insulin were analysed separately as a proxy for diabetes.
We defined a variable covering any neurological diseases (G00-G99) and further stratification based on major ICD-10 subgroups; inflammatory diseases of the central nervous system (G00-G09), systemic atrophies primarily affecting the central nervous system (G10-14), extrapyramidal and movement disorders (G20-26), other degenerative disease and demyelinating diseases (G30-G37), episodic and paroxysmal disorders (G40-47), nerve and plexus disorders (G50-59), polyneuropathies (G60-64), disease of myoneural junction and muscle (G70-73), cerebral palsy (G80-83), other disorders of the nervous system (G90-99). In addition, a variable covering hearing loss (H912, H918, H932, H933) was added and for practical reasons listed under neurological disorders.
The epilepsy diagnosis (G40-41) was singled out for further analysis. First, to address the possibility that epilepsy might be misdiagnosed as LQTS and vice versa due to similar symptomatic traits, we stratified all patients with an epilepsy diagnosis anytime, according to the index date. Four groups were defined including all patients receiving an epilepsy diagnosis prior to index (preindex), patients where the initial diagnosis did not reappear following index (dismissed), patients with a diagnosis following index without a diagnosis prior to index (acquired), and a summary group of patients with an epilepsy diagnosis anytime following index (postindex). It is noted that the groups are not mutually exclusive (see table 2 for a flow chart illustration). Second, we conducted separate analysis defining epilepsy based on antiepileptic drug use.
Table 2.
Analysis of epilepsy in relation to time of long QT syndrome (LQTS) diagnosis (index)
| LQTS | Controls | P value | Flow chart of LQTS patients with epilepsy |
|||
| N | % | N | % | |||
| Anytime;Epilepsy | 31 | 6.7 | 37 | 1.6 | <0.001 | 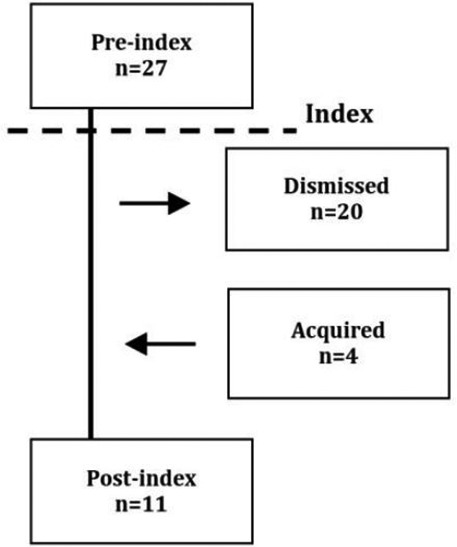 |
| Anytime;Antiepileptics | 20 | 4.3 | 23 | 1.0 | <0.001 | |
| Preindex; Epilepsy | 27 | 5.8 | 24 | 1.0 | <0.001 | |
| Preindex; Antiepileptics | 17 | 3.7 | 21 | 0.9 | <0.001 | |
| Dismissed†; Epilepsy | 20 | 4.3 | 23 | 1.0 | <0.001 | |
| Dismissed; Antiepileptics | 4 | 0.9 | 7 | 0.3 | 0.079 | |
| Acquired*; Epilepsy | 4 | 0.9 | 13 | 0.6 | 0.446 | |
| Acquired; Antiepileptics | 3 | 0.6 | 3 | 0.1 | 0.028 | |
| Postindex; Epilepsy | 11 | 2.4 | 14 | 0.6 | <0.001 | |
| Postindex; Antiepileptics | 16 | 3.5 | 17 | 0.7 | <0.001 | |
*Dismissed: only before index, not after.
†Acquired: not before index, only after.
We defined a variable covering any psychiatric disorder (F00-99) and further stratifications based on major ICD-10 subgroups that is, organic mental disorders (F00-09), mental disorders due to substance use (F10-19), schizophrenia (F20-29), affective disorders (F30-39), neurotic and somatoform disorders (F40-48), behavioural syndromes (F50-59), personality disorders (F60-69), mental retardation (F70-79), disorders of psychological development (F80-89) and behavioural disorders with onset in childhood (F90-98).
Statistics
LQTS patients were matched using a greedy match macro for SAS V.9.4, matching patients (1:5) on sex and birth year allowing for zero variation. Finally, we made sure that no controls died prior to the assigned index date of their respective LQTS case, to exclude any risk of immortality. Differences in patient characteristics were compared using Fishers exact test for continuous variables, and χ2 test for categorical variables. Follow-up time was compared as medians with IQR and a p value was estimated using the non-parametric Wilcoxon signed-rank test. ORs with 95% CIs were estimated using conditional logistic regression models.
Forest plot illustration was made using R V.3.4.1 (Single Candle). Data management and analyses were performed SAS V.9.4. Two-sided p values <0.05 were considered statistically significant.
Results
We identified 474 patients with a first time LQTS diagnosis. Subsequently, 11 patients were reclassified as acquired LQTS and excluded. The remaining 463 patients were matched with 2315 controls from the background population. Following successful matching, mean age ±SD was 35.7±21.0 years (LQTS; 35.6±20.9 years and controls; 35.8±21.0 years, p=0.84) and 38.0% were males in both groups (table 1). The follow-up time did not differ between groups (LQTS; median 4.4 years, IQR 2.2–7.8 years vs controls; 4.3 years, IQR 2.1–7.6 years, p=0.44). Also, the all-cause mortality was similar in the LQTS patients compared with controls during follow-up (4.2% vs 3.6%, respectively, p=0.49).
Table 1.
Characteristics and comorbidities for patients with long QT syndrome (LQTS) and their matched controls
| Demographics | LQTS (463) | Controls (2315) | P value | ||
| N | % | N | % | ||
| Age at diagnosis mean, (SD) | 35.6 | (±20.9) | 35.8 | (±21.0) | 0.836 |
| Sex, male | 176 | 38.0 | 880 | 38.0 | 1.000 |
| Cardiovascular comorbidities | |||||
| Atrial fibrillation | 30 | 6.5 | 53 | 2.3 | <0.001 |
| Venous thromboembolism | 10 | 2.2 | 41 | 1.8 | 0.569 |
| Peripheral vascular disease | 4 | 0.9 | 13 | 0.6 | 0.446 |
| Acute myocardial infarction | 14 | 3.0 | 34 | 1.5 | 0.019 |
| Metabolic comorbidities | |||||
| Diabetes mellitus | 17 | 3.7 | 42 | 1.8 | 0.011 |
| Hypothyroidism | 5 | 1.1 | 32 | 1.6 | 0.604 |
| Neurological comorbidities | |||||
| Any neurological disease | 102 | 22.0 | 285 | 12.3 | <0.001 |
| Episodic and paroxysmal disorders | 63 | 13.6 | 145 | 6.3 | <0.001 |
| Nerve and plexus disorders | 30 | 6.5 | 97 | 4.2 | 0.031 |
| Polyneuropathies | 7 | 1.5 | 26 | 1.1 | 0.481 |
| Myoneural junction | 3 | 0.6 | 5 | 0.2 | 0.113 |
| Cerebral palsy | 3 | 0.6 | 7 | 0.3 | 0.257 |
| Hearing loss | 15 | 3.2 | 39 | 1.7 | 0.027 |
| Psychiatric comorbidities | |||||
| Any psychiatric disorder | 60 | 13.0 | 210 | 9.1 | 0.010 |
| Organic mental disorders | 22 | 4.8 | 73 | 3.2 | 0.084 |
| Mental disorders due to substance use | 29 | 6.3 | 98 | 4.2 | 0.056 |
| Affective disorders | 9 | 1.9 | 40 | 1.7 | 0.747 |
| Neurotic and somatoform disorders | 14 | 3.0 | 50 | 2.2 | 0.258 |
Cardiovascular comorbidities
Compared with controls, LQTS patients had a significantly higher prevalence of atrial fibrillation (6.5% vs 2.3%, p<0.001) and acute myocardial infarction (3.0% vs 1.5%, p=0.019) (see figure 1). No difference was found comparing the prevalence of venous thromboembolism (2.2% vs 1.8%, p=0.57) and peripheral vascular disease (0.9% vs 0.6%, p=0.45).
Figure 1.

Comparison of comorbidities between long QT syndrome (LQTS) patients and their age and gender matched controls (1:5). Forrest plot depicts conditional logistic regression with ORs and 95% CIs. Background population is used as reference, and an OR exceeding 1 represents an increased risk of the investigated comorbidity for LQTS patients. In the subanalysis, epilepsy is compared in relation to time of LQTS diagnosis (index). *Composite of the ICD-10 codes H912, H918, H932 H933 and not included in the any neurological disorders category. **Epilepsy included in episodic disorders.
Neurological comorbidities
LQTS patients had an overall higher prevalence of neurological disorders (22.0% vs 12.3%, p<0.001), driven by an increased risk of episodic and paroxysmal disorders (13.6% vs 6.3%, p<0.001) and nerve and plexus disorders (6.5% vs 4.2%, p=0.031). No difference was observed for polyneuropathies (1.5% vs 1.1%, p=0.48), myoneural junction disorders (0.6% vs 0.2%, p=0.11) and cerebral palsy (0.6% vs 0.2%, p=0.26). LQTS patients had significantly more hearing loss (3.2% vs 1.7%, p=0.027).
The epilepsy diagnosis, a subset of the episodic and paroxysmal disorders diagnosis group, was more prevalent among LQTS patients anytime according to index (6.7% vs 1.6%, p<0.001) as well as preindex (5.8% vs 1.0%, p<0.001) and postindex (2.4% vs 0.6%, p<0.001) (table 2). LQTS patients more often received an epilepsy diagnosis that was dismissed after index (4.3% vs 1.0%, p<0.001), which was the case in 20 of 27 LQTS patients (74%) diagnosed with epilepsy prior to index of the initially diagnosed LQTS patients. None of the 20 patients with a dismissed epilepsy diagnosis filled a prescription on antiepileptic drugs postindex.
In a separate sensitivity analysis, defining epilepsy only based on antiepileptic drug use, we also found epilepsy to be more prevalent among LQTS patients overall (4.3% vs 1.0%, p<0.001), which was also found both preindex (3.7% vs 0.9%, p>0.001) and postindex (3.7% vs 0.9%, p>0.001).
Psychiatric comorbidities
LQTS patients had a higher prevalence of psychiatric diseases overall (13.0% vs 9.1%, p=0.010). We observed a trend towards higher prevalence for two of the psychiatric disease subgroups among LQTS patients compared with controls, although none of them reached statistical significance; organic mental disorders (4.8% vs 3.2%, p=0.084), mental disorders due to substance use (6.3% vs 4.2%, p=0.056), affective disorders (1.9% vs 1.7%, p=0.75) and anxiety (3.0% vs 2.2%, p=0.26).
Metabolic comorbidities
LQTS patients were more often diagnosed with diabetes mellitus (3.7% vs 1.8% p=0.011), with no difference for hypothyroidism (1.1% vs 1.4%, p=0.60) (table 1). Hypoglycaemia was present in less than three LQTS patients, and data were censored.
LQTS patients were also more often prescribed antidiabetic medication overall (5.2% vs 1.9%, p<0.001) and insulin (1.9% vs 0.6%, p=0.006) (table 3). Prior to index, the increased risk of diabetes (based on in-hospital diagnostic codes) was not statistically significantly higher in LQTS patients compared with controls (2.2% vs 1.2%, p=0.10), whereas LQTS patients more often filled prescription for antidiabetic medication (4.5% vs 1.7%, p<0.001) and insulin (1.7% vs 0.6%, p=0.008). In 21/24 (87.5%) of the LQTS patients, the antidiabetic treatment was prescribed prior to index (table 3).
Table 3.
Analysis of diabetes before and after long QT syndrome (LQTS) diagnosis
| LQTS | Controls | P value | |||
| N | % | N | % | ||
| Anytime;Diabetes | 17 | 3.7 | 42 | 1.8 | 0.011 |
| Anytime;Antidiabetic drugs | 24 | 5,2 | 45 | 1,9 | <0.001 |
| Anytime;Insulin | 9 | 1.9 | 15 | 0,6 | 0.006 |
| Preindex; Diabetes | 10 | 2.2 | 28 | 1.2 | 0.108 |
| Preindex;Antidiabetic drugs | 21 | 4,5 | 39 | 1,7 | <0.001 |
| Preindex;Insulin | 8 | 1,7 | 13 | 0,6 | 0.008 |
| Postindex; Diabetes | 12 | 2,6 | 26 | 1,1 | 0.013 |
| Postindex;Antidiabetic drugs | 22 | 4,8 | 43 | 1,9 | <0.001 |
| Postindex;Insulin | 8 | 1,7 | 14 | 0,6 | 0.013 |
Discussion
In this study, we included a nationwide cohort of patients with congenital LQTS, matched with relevant controls from the background population. Analysing the risks of extracardiac disorders, we found that LQTS patients had a significantly increased burden of diabetes, psychiatric and neurological comorbidities, in addition to the well-known risks of cardiac comorbidities. The risk of epilepsy seemed especially pronounced, even after taking into account the potentially misdiagnosed cases of epilepsy. Findings of this study support the multimorbid nature of LQTS and may be of importance for clinicians in order to manage both cardiac and extracardiac comorbidities from an early stage.
Cardiovascular comorbidities
Findings from this study confirms the well-known increased risk of atrial fibrillation in LQTS patients,4 but our data also show an increased risk of myocardial infarctions in the LQTS patients. An important caveat in interpreting these results is that LQTS patients will be offered regular visits at the cardiology department, introducing a potential bias towards higher cardiac disease prevalence among LQTS patients, when compared with the background population. Still, then one would also expect peripheral vascular disease and thromboembolic to be more prevalent, but they were not. The causal explanation for an increased risk of myocardial infarction remains uncertain but it may be hypothesised that the increased prevalence of diabetes among patients with LQTS may contribute to an increased risk of myocardial infarction. However, as the mean age of these patients is low and diabetes overall infrequent, the question remains open for further investigation.
Neurological comorbidities
Our finding that LQTS patients have an increased risk of neurological disorders, especially epilepsy, is important, as several research groups have previously indicated that epilepsy may share a molecular background with LQTS. A study by Auerbach et al demonstrated an increased risk of seizures in LQTS patients19 and Haugaa et al found that abnormal electroencephalograms were more common among LQTS patient when compared with their geno-negative relatives, though the abnormalities were not diagnostic for epilepsy.20 Also, a study found a higher prevalence of epilepsy among LQTS patients when compared with the lifetime prevalence in children.10 Our results support the idea that the relationship between the LQTS and epilepsy may extend beyond phenotypic mimicry.21 However, in 74% of our LQTS patients with an epilepsy diagnosis prior to the LQTS index, the epilepsy diagnosis did not reappear in the registries after diagnosing LQTS. This may suggest that in at least some cases, the epilepsy diagnosis have been revised following the LQTS diagnosis. Epileptiform activity has been observed in patients with verified LQTS, but it remains uncertain whether this is to be considered epilepsy, or whether it may be a secondary phenomenon due to transient cerebral hypoperfusion or head traumas caused by LQTS arrhythmias or vasovagal syncope, frequently observed in LQTS patients. Despite these uncertainties, which are not easily distinguished in registry data, we found that epilepsy was significantly more prevalent among LQTS patients.
Hearing loss, which was the extracardiac manifestation first characterised in LQTS, was also found to be more prevalent among our LQTS patients compared with the control population. Hearing loss was initially linked to the LQTS subtype Jervell-Lange-Nielsen syndrome, which may be the main driver for the findings of increased risks in this study. However, it may be worth considering, that studies have determined an incidence of 1.6–6 per million (with a Danish population of 5.7 million) and that only 27% of patients with putative KCNQ1 mutations suffers from overt hearing loss due to reduced penetrance.22 Consequently, we would expect 2–9 cases of Jervell-Lange-Nielsen syndrome with hearing loss in the Danish population, compared with the 15 LQTS patients in this study with a diagnosis of hearing loss. As these crude estimates remain indicative at best, and due to the lack of data on LQTS subtype and genotypes, as well as the unknown accuracy of the hearing loss diagnosis in our registries, further observational studies are warranted to address these associations in detail.
Psychiatric comorbidities
In our cohort, LQTS patients had an increased risk of psychiatric disorder. A trend was found toward LQTS may have more mental disorders due to substance use and as LQTS patients may live in fear of life-threatening events,23 it could be hypothesised that this represent a subclinical proxy for anxiety and depression. Nevertheless, this finding is important as especially cocaine may elicit the arrhythmic phenotype of LQTS and therefore should be avoided.24 25
Our results show no association between LQTS and schizophrenia nor mental retardation, despite that studies on the molecular level have demonstrated a possible association between isoforms of the hERG channel (especially the KCNH2-3.1 isoform) and the development of schizophrenia and impaired cognition. However, this isoform is primarily expressed in cerebral neurons and only sparse in cardiac cells.11 26
Metabolic comorbidities
In our study cohort, we observed an increased prevalence of diabetes, as well as higher prescription rates of antidiabetic medication, among LQTS patients compared with the background population, which has not previously been reported in a real-life clinical LQTS population. These analyses were decided on a priori, as the LQTS gene, KCNQ1, has previously been associated with type 2 diabetes in genome-wide association studies.27 28 These observations led to clinical studies demonstrating increased insulin release and temporary insulin resistance in LQTS patients with both KCNQ115 and KCNH2 mutations,14 which might provide a possible explanation for our findings. Still, LQTS patients will inevitably have more blood test done compared with the background population—leading to a higher probability of being diagnosed with diabetes. However, in our cohort, 88% of the patients started antidiabetic treatment before the patients were diagnosed with LQTS, suggesting a limited bias.
In our data, less than three LQTS patients had a hypoglycaemia diagnosis, but as hypoglycaemic symptoms may often be misinterpreted as dizziness or mood changes, and only rarely arrhythmias, they may be undiagnosed in some cases. Still, this potential extracardiac manifestation may be important to consider in future studies, as the hyperinsulinaemia studies14 15 raised concern about hypoglycaemia in LQTS patients. Considering additional interactions, a recent study by Poterucha et al suggested that beta-receptor blocking agents could cause hypoglycaemia in children with LQTS.29 However, the effect of beta-receptor blocking agents has earlier been tested in healthy adults, showing no hypoglycaemic effect, suggesting that the hypoglycaemia in LQTS patients is independent of beta-blocker therapy.30
Limitations
This study was based on data from the well-characterised nationwide registries. Further, we specifically validated the LQTS diagnosis for the purpose of this study and found a high positive predictive value of 98% for the LQTS diagnostic code. Also, to minimise the risk of bias due to inclusion of acquired LQTS in our full cohort, patients were systematically excluded if diagnosed with acquired LQTS after index.
Still, due to the study’s retrospective design, we were only able to determine associations and not causality. Based on the available data, the LQTS patients could not be subgrouped based on genotype, the severity of disease or length of QT-interval. Also, only diagnoses given in a hospital setting (in-hospital and outpatient clinics) were available for analysis. To strengthen specific diagnoses, epilepsy and diabetes, we also investigated use of antiepileptic drugs and antidiabetic drugs as a proxy for these comorbidities. However, minor comorbidities and complications managed in a primary care setting, such as mild depression and others, may have been missed.
Our findings could be biassed by the fact that LQTS over time may have more contacts with the healthcare system with a subsequent higher risk of additional diagnosis, compared with the background population. However, our subanalyses on diabetes showed that in most cases antidiabetic treatment was initiated before index and since only a few comorbidities were more frequent among LQTS patients, this does not seem to be a major issue. Nevertheless, the increased prevalence of comorbidities reflects the diagnostic odyssey many patients with LQTS go through before finally obtaining the LQTS diagnosis. Finally, as LQTS is a rare disease, our data are also limited by a low number of patients, and our results would preferably be tested in larger cohorts.
Conclusion
Patients with LQTS had a significantly increased burden of diabetes, and neurological and psychiatric comorbidities compared with an age and gender-matched background population. The increased burden of neurological diagnoses was largely driven by an increased risk of epilepsy. However, after patients had been diagnosed with LQTS, the epilepsy diagnosis did not reappear in 74% of the patients. This finding suggests that, at least in some cases, the epilepsy diagnosis may have been revised following the LQTS diagnosis. Although, we found that LQTS patients have a higher burden of psychiatric comorbidities, the difference could not be attributed to a specific subgroup of psychiatric diagnoses. Our data support that LQTS may be considered a multiorgan disease and suggest that patient care should be adjusted accordingly.
Acknowledgments
This research was supported by the Danish Heart Foundation (Grant 17-R115-A7532-22065), fondsbørsvekselerer Henry Hansen og hustrus legat, the FUKAP foundation, A.P. Møller og Hustru Chastine Mc-Kinney Møllers Fond til Almene Formaal and Købmand Sven Hansen og Hustru Ina Hansens Fond.
Footnotes
Contributors: PM, MEJ, CA and JT contributed to the conception or design of the work and final approval of the version to be published. PM, MEJ, CJ contributed to the data collection. PM, MEJ, CA, JT, JKK, HB, CT-P and GG contributed to the data analysis and interpretation. PM and MEJ drafted the article. JT, CA, HB, PW, JT-H, CJ, GG, CT-P and JKK contributed to the critical revision of the article.
Funding: This study was funded by he FUKAP foundation, A.P. Møller og Hustru Chastine Mc-Kinney Møllers Fond til almene Formaal, Købmand Sven Hansen og Hustru Ina Hansens Fond, Fondsbørsvekselerer Henry Hansen og hustrus legat, The Danish Heart Foundation (Grant number:17-R115-A7532-22065).
Competing interests: None declared.
Patient consent for publication: Not required.
Provenance and peer review: Not commissioned; internally peer reviewed.
Data availability statement: All data relevant to the study are included in the article or uploaded as supplementary information.
References
- 1.Ackerman MJ, Priori SG, Willems S, et al. . HRS/EHRA expert consensus statement on the state of genetic testing for the channelopathies and cardiomyopathies. Europace 2011. [DOI] [PubMed] [Google Scholar]
- 2.Schwartz PJ, Spazzolini C, Crotti L, et al. . The Jervell and Lange-Nielsen syndrome: natural history, molecular basis, and clinical outcome. Circulation 2006. [DOI] [PubMed] [Google Scholar]
- 3.Wilde AAM, Brugada R. Phenotypical manifestations of mutations in the genes encoding subunits of the cardiac sodium channel. Circ. Res 2011. [DOI] [PubMed] [Google Scholar]
- 4.Johnson JN, Tester DJ, Perry J, et al. . Prevalence of early-onset atrial fibrillation in congenital long QT syndrome. Heart Rhythm 2008;5:704–9. 10.1016/j.hrthm.2008.02.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Johnson JN, Hofman N, Haglund CM, et al. . Identification of a possible pathogenic link between congenital long QT syndrome and epilepsy. Neurology 2009;72:224–31. 10.1212/01.wnl.0000335760.02995.ca [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tiron C, de Llano CT, Campuzano O, et al. . Further evidence of the association between LQT syndrome and epilepsy in a family with KCNQ1 pathogenic variant. Seizure 2015;25:65–7. 10.1016/j.seizure.2015.01.003 [DOI] [PubMed] [Google Scholar]
- 7.Partemi S, Cestèle S, Pezzella M, et al. . Loss-of-function KCNH2 mutation in a family with long QT syndrome, epilepsy, and sudden death. Epilepsia 2013;54:e112–6. 10.1111/epi.12259 [DOI] [PubMed] [Google Scholar]
- 8.Terra VC, Cysneiros R, Cavalheiro EA, et al. . Sudden unexpected death in epilepsy: from the lab to the clinic setting. Epilepsy Behav 2013. [DOI] [PubMed] [Google Scholar]
- 9.Tu E, Bagnall RD, Duflou J, et al. . Post-Mortem review and genetic analysis of sudden unexpected death in epilepsy (SUDEP) cases. Brain Pathol 2011;21:201–8. 10.1111/j.1750-3639.2010.00438.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Anderson JH, Bos JM, Cascino GD, et al. . Prevalence and spectrum of electroencephalogram-identified epileptiform activity among patients with long QT syndrome. Heart Rhythm 2014;11:53–7. 10.1016/j.hrthm.2013.10.010 [DOI] [PubMed] [Google Scholar]
- 11.Huffaker SJ, Chen J, Nicodemus KK, et al. . A primate-specific, brain isoform of kcnh2 affects cortical physiology, cognition, neuronal repolarization and risk of schizophrenia. Nat Med 2009;15:509–18. 10.1038/nm.1962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hashimoto R, Ohi K, Yasuda Y, et al. . The KCNH2 gene is associated with Neurocognition and the risk of schizophrenia. World J Biol Psychiatry 2013. [DOI] [PubMed] [Google Scholar]
- 13.Heron SE, Hernandez M, Edwards C, et al. . Neonatal seizures and long QT syndrome: a cardiocerebral channelopathy? Epilepsia 2010;51:293–6. 10.1111/j.1528-1167.2009.02317.x [DOI] [PubMed] [Google Scholar]
- 14.Hyltén-Cavallius L, Iepsen EW, Wewer Albrechtsen NJ, et al. . Patients With Long-QT Syndrome Caused by Impaired hERG-Encoded Kv11.1 Potassium Channel Have Exaggerated Endocrine Pancreatic and Incretin Function Associated With Reactive Hypoglycemia. Circulation 2017;135:1705–19. 10.1161/CIRCULATIONAHA.116.024279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Torekov SS, Iepsen E, Christiansen M, et al. . Kcnq1 long QT syndrome patients have hyperinsulinemia and symptomatic hypoglycemia. Diabetes 2014;63:1315–25. 10.2337/db13-1454 [DOI] [PubMed] [Google Scholar]
- 16.Sandegaard JL, Schmidt SAJ, Sørensen HT, et al. . The Danish national patient registry: a review of content, data quality, and research potential. Clin Epidemiol 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Thygesen SK, Christiansen CF, Christensen S, et al. . The predictive value of ICD-10 diagnostic coding used to assess Charlson comorbidity index conditions in the population-based Danish national Registry of patients. BMC Med Res Methodol 2011;11 10.1186/1471-2288-11-83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wallach Kildemoes H, Toft Sørensen H, Hallas J. The Danish national prescription registry. Scand J Public Health 2011. [DOI] [PubMed] [Google Scholar]
- 19.Auerbach DS, McNitt S, Gross RA, et al. . Genetic biomarkers for the risk of seizures in long QT syndrome. Neurology 2016;87:1660–8. 10.1212/WNL.0000000000003056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Haugaa KH, Vestervik TT, Andersson S, et al. . Abnormal electroencephalograms in patients with long QT syndrome. Heart Rhythm 2013;10:1877–83. 10.1016/j.hrthm.2013.09.070 [DOI] [PubMed] [Google Scholar]
- 21.Medford BA, Bos JM, Ackerman MJ. Epilepsy misdiagnosed as long QT syndrome: it can go both ways. Congenit Heart Dis 2014;9:E135–9. 10.1111/chd.12104 [DOI] [PubMed] [Google Scholar]
- 22.Giudicessi JR, Ackerman MJ. Prevalence and potential genetic determinants of sensorineural deafness in KCNQ1 homozygosity and compound heterozygosity. Circ Cardiovasc Genet 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Michael Giuffre R, Gupta S, Crawford SG, et al. . Fears and anxiety in children with long-QT syndrome compared to children with asthma. J Natl Med Assoc 2016. [DOI] [PubMed] [Google Scholar]
- 24.Singh N, Singh HK, Singh PP, et al. . Cocaine-Induced torsades de pointes in idiopathic long Q-T syndrome. Am J Ther 2001;8:299–302. 10.1097/00045391-200107000-00012 [DOI] [PubMed] [Google Scholar]
- 25.Fazio G, Vernuccio F, Grutta G, et al. . Drugs to be avoided in patients with long QT syndrome: focus on the anaesthesiological management. World J Cardiol 2013;5:87 10.4330/wjc.v5.i4.87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Imbrici P, Camerino DC, Tricarico D. Major channels involved in neuropsychiatric disorders and therapeutic perspectives. Front Genet 2013;4:76 10.3389/fgene.2013.00076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yasuda K, Miyake K, Horikawa Y, et al. . Variants in KCNQ1 are associated with susceptibility to type 2 diabetes mellitus. Nat Genet 2008;40:1092–7. 10.1038/ng.207 [DOI] [PubMed] [Google Scholar]
- 28.Unoki H, Takahashi A, Kawaguchi T, et al. . Snps in KCNQ1 are associated with susceptibility to type 2 diabetes in East Asian and European populations. Nat Genet 2008;40:1098–102. 10.1038/ng.208 [DOI] [PubMed] [Google Scholar]
- 29.Poterucha JT, Bos JM, Cannon BC, et al. . Frequency and severity of hypoglycemia in children with beta-blocker–treated long QT syndrome. Heart Rhythm 2015;12:1815–9. 10.1016/j.hrthm.2015.04.034 [DOI] [PubMed] [Google Scholar]
- 30.Toft-Nielsen M, Hvidberg A, Hilsted J, et al. . No effect of beta-adrenergic blockade on hypoglycaemic effect of glucagon-like peptide-1 (GLP-1) in normal subjects. Diabet Med 1996;13:544–8. [DOI] [PubMed] [Google Scholar]