

Abstract
We recently introduced RAP (reduced adjoining protonation) labelling as an easy to implement and cost-effective strategy to yield selectively methyl protonated protein samples. We show here that even though the amount of H2O employed in the bacterial growth medium is rather low, the intensities obtained in MAS solid-state NMR 1H,13C correlation spectra are comparable to spectra obtained for samples in which α-ketoisovalerate was employed as precursor. In addition to correlations for Leu and Val residues, RAP labelled samples yield also resonances for all methyl containing side chains. The labelling scheme has been employed to quantify order parameters, together with the respective asymmetry parameters. We obtain a very good correlation between the order parameters measured using a GlcRAP (glucose carbon source) and a α-ketoisovalerate labelled sample. The labelling scheme holds the potential to be very useful for the collection of long-range distance restraints among side chain atoms. Experiments are demonstrated using RAP and α-ketoisovalerate labelled samples of the α-spectrin SH3 domain, and are applied to fibrils formed from the Alzheimer’s disease Aβ1-40 peptide.
Subject terms: Biochemistry, Biophysics, Structural biology, Chemistry, Biophysical methods, NMR spectroscopy
Introduction
Methyl groups are valuable probes for structure and dynamics investigations of proteins and possess favorable relaxation properties, such as short T1 and long T2 times in deuterated microcrystalline protein samples. An efficient labelling scheme for selective Ileδ1 and Valγ1,2/Leuδ1,2 methyl labelling was introduced for solution-state1–5. In the solid-state, first pyruvate labelling was employed to yield high-resolution 1H,13C correlation spectra6. Pyruvate yields labelling of all methyl containing amino acids. However, as the precursor was commercially only available as 60% deuterated, methyl groups in the protein appeared as mixtures of CH3, CH2D, CHD2 and CD3 isotopomers, which compromised the overall sensitivity of the experiment. Later on, α-keto-acid and acetolactate7 labelling was employed to yield methyl relaxation rates8, side chain order parameters6 and methyl-methyl distance restraints9,10 in microcrystalline proteins in the solid-state.
We have subsequently observed that high resolution 1H,13C correlation spectra can be acquired in case the glucose employed in the bacterial growth medium is only 97% deuterated11. This triggered the development of RAP (reduced adjoining protonation) labelling schemes in which H2O was added to the deuterated minimal medium which contained 2H,13C glucose12. A similar approach (coined fractional deuteration and inverse fractional deuteration) was introduced later by others, in which the minimal medium consists of 1H,13C glucose and D2O, or 2H,13C glucose and H2O, respectively13,14. Alternatively, side chain protons can be introduced by addition of protonated 13C,15N labelled amino acids in the bacterial growth medium, which contains D2O and 2H,13C glucose otherwise. This approach was coined proton cloud labelling15. Finally, use of amino acid oxidases allows to generate keto-acids from amino acid mixtures. Transaminases in turn convert the keto-acids in specifically Cα protonated amino acids during bacterial growth16.
We compare here methyl order parameters, including dipolar coupling anisotropy values as well as asymmetry parameters, obtained from a selectively 13CHD2 Leu/Val methyl labelled α-spectrin SH3 sample and a randomly protonated SH3 sample using the RAP labelling scheme12,17, respectively. Surprisingly, we find that the intensities of even the 5% RAP labelled sample are rather high, and comparable to the intensities obtained for the α-keto-acid labelled sample. As expected, the order parameters for the two samples are rather similar. The RAP labelled sample, however, yields labelling of all methyl groups with similar enrichment of protons for all methyl bearing side chains. We believe that this labelling scheme will be employed widely in the future as it is easy to implement, allows to obtain assignments via HCCH type experiments18, as well as information on local dynamics and structure.
Results
1H,13C correlation spectroscopy
We recorded 1H-detected 2D 1H,13C HMQC spectra for the SH3 domain of α-spectrin comparing two different labelling schemes, namely 5% GlcRAP labelling17 (red) and selective Leu/Val 13CHD2 methyl labelling3 (blue, Fig. 1A). Both samples were crystallized in a 100% D2O buffer (details are given in the Methods part). We achieved high resolution for both samples spun at a MAS frequency of 50 kHz yielding 1H line widths on the order of ~22–25 Hz. Clearly, one major benefit of the 5% GlcRAP labelling is that all methyl groups (Ala, Ile, Leu, Met, Thr, Val) become observable at once with comparable resolution, while in the Leu/Val labelling only two out of six methyl-bearing amino acids can be detected (Fig. 1D).
Figure 1.

(A) 2D 1H,13C methyl correlation spectra of a 5% GlcRAP and a Leu/Val 13CHD2 sample of α-spectrin SH3 at an external magnetic field of 14.1 T (600 MHz), adjusting the MAS frequency to 50 kHz and the effective sample temperature to 20–25 °C. All samples were prepared in a 100% D2O buffer (details are given in the Methods part). The 1H, 13C HMQC spectrum of the 5% GlcRAP sample is shown (red) superimposed with the respective spectrum of the Leu/Val sample (blue). The 13C line width of the Leu/Val sample amounts to 25 Hz. The 1H line width for both samples is on the order of ~22–25 Hz. The methyl cross peaks in the 13C dimension of the 5% GlcRAP spectrum are split into doublets due to evolution of the 1JC,C scalar couplings (except for Met). (B) 1H,13C constant-time HSQC spectrum of the 5% GlcRAP sample using a constant-time delay . The 13C line width is ~25 Hz (or ~16 Hz if the indirect evolution period is doubled using mirror-image linear prediction). The apparent 13C methyl T2 time for both samples is roughly around ~30 ms. (C) 1H,13C HMQC spectrum of 5% GlcRAP labelled Aβ1-40 fibrils, recorded at an external magnetic field of 16.4 T (700 MHz) and a MAS frequency of 18 kHz. The 1H line width is on the order of ~130 Hz. (D) 1D projection of 2D 1H,13C HMQC spectra of all samples. All pulse sequences are given in Fig. S1. The figure was generated with Adobe Illustrator CS5 V.15.02.
Since the 5% GlcRAP sample was uniformly 13C labelled, evolution of 13C,13C scalar couplings affects the resolution in the methyl region, except for methionine methyl groups, which lack an adjacent carbon (Fig. 1A). In the Leu/Val sample only one carbon was isotopically enriched, which improved the methyl resolution in the indirect 13C dimension. However, appearance of splittings in the 13C dimension can be avoided by performing a constant-time HSQC experiment (Fig. S1B) at the expense of sensitivity. As shown in Fig. 1B, the application of a constant time sequence yields the same resolution for the 5% GlcRAP sample as for the Leu/Val sample. The 13C line widths are about 25 Hz for both samples. In a constant time experiment, magnetization does not decay as a function of the t1 evolution period, which is beneficial for mirror-image linear prediction19,20. Doubling the indirect t1 evolution period by linear prediction yielded a further improvement of the 13C resolution. The 13C line width thus amounts to ~16 Hz for the 5% GlcRAP sample. As shown previously21, 13C,13C splittings can be chemically removed using the [2]-GlyRAP or [1,3]-GlyRAP instead of the GlcRAP labelling scheme, respectively, since the former utilizes [2]- or [1,3]-13C glyercol instead of glucose as the carbon source during protein expression22. With this type of labelling, constant-time pulse sequence elements can be circumvented, which significantly enhances sensitivity as real time experiments can be used and at the same time yields high resolution comparable to Leu/Val labelling.
We applied the 5% GlcRAP labelling scheme, furthermore, to amyloid fibrils, formed by the Alzheimer’s Aβ1-40 peptide23–25. We recorded 2D 1H,13C HMQC spectra for a 5% GlcRAP labelled Aβ1-40 fibril sample as shown in Fig. 1C. Even though the resolution in the 1H dimension is less compared to spectra of the microcrystalline SH3 domain, which is presumably due to the low spinning frequency of 18 kHz, fibril polymorphism and lower sample homogeneity, there is still a good spectral dispersion of the methyl groups. Solid-state NMR investigations of Aβ fibrils usually rely on 13C spectroscopy26,27. However, using the 13C chemical shift information of the methyl group does not allow to disperse the Ileδ1 signals, whereas the methyl groups appear resolved in the 1H dimension (Fig. 1C). We note, that Aβ1-40 contains only two isoleucine residues, while clearly three Ileδ1 resonances were detected, revealing the polymorphic nature of the fibril. In addition, the spectral sensitivity is significantly enhanced by 1H detection.
5% GlcRAP versus selective Leu/Val methyl labelling
In the following, we compared signal-to-noise ratios for the 5% GlcRAP and the Leu/Val labelled SH3 domain obtained in 2D 1H,13C HMQC experiments (Fig. 2A). Overall, the gain in sensitivity for Leu/Val labelling over 5% GlcRAP is up to a factor of ~3. We further compared the sensitivity to the proton concentration at a given methyl site for the respective labelling scheme. The average proton concentration at a methyl site in a Leu/Val 13CHD2, a 5% GlcRAP and an uniformly protonated sample is 16.7% (p = (1/3) × (1/2)), ~3.5% (averaged) and 100%, respectively. The proton incorporation rate for 5% GlcRAP has been experimentally estimated previously by solution-state NMR12. Due to the higher concentrations of protons in Leu/Val samples, we, thus, expect a signal-to-noise gain of a factor of ~5 over the 5% GlcRAP labelled sample. The lower than expected gain can be caused by several factors: (i) differences in the 1H dipolar network causing dipole-mediated line broadening, (ii) sample inhomogeneity, (iii) varying degrees of H2O impurities, (iv) different amounts of protein in the two rotors, (v) low incorporation of α-keto-acid precursors into the protein and (vi) pH differences.
Figure 2.

(A) Experimental signal-to-noise ratios extracted from 2D 1H,13C HMQC spectra of 5% GlcRAP (red) and selectively Leu/Val 13CHD2 labelled α-spectrin SH3 (blue). The experiments were run under comparable conditions. The experimental time to record a spectrum for each sample was ~40 min (details are given in the Methods part). (B) In silico calculated 1H,1H effective dipolar couplings for methyl groups based on the 1 μs MD relaxed crystal structure of the SH3 domain (note the logarithmic y-scale). Calculations were carried out for the proton spin diluted structures according to the 5% GlcRAP labelling scheme (red bars), the Leu/Val 13CHD2 labelling scheme (blue) and for the uniformly protonated structure (grey). Crosses depict the upper 2σ confidence interval. (C) The same as (B) incorporating all exchangeable protons. The figure was generated with Adobe Illustrator CS5 V.15.02.
The latter is expected to only have a small effect on the sensitivity of methyl groups as compared to the strongly pH-dependent exchangeable protons, such as amides. On the other hand, the sample homogeneity has a severe impact on sensitivity, however, we obtained similar apparent linewidths for both samples. Varying H2O-concentrations of the crystal solvent among both samples correlates to the protonation state at exchangeable sites and can, in principle, add to the 1H dipolar network. Both samples were lyophilized two times in 100% D2O, yet their H2O contents slightly varied due to different storage times. Based on 1D 1H spectra, we estimate the amide concentration to be around ~3% (~6%) for the Leu/Val (5% GlcRAP) sample at the time of measurement. A higher concentration of exchangeable protons can reduce the T1 relaxation time of amides by about ~10%28 and to a smaller extent also of methyl groups, which would increase the signal-to-noise ratio per unit time in favor of the 5% GlcRAP sample. We packed both rotors using the same ultracentrifuge device and, on the basis of previous experience29, we estimate the error in the amount of material in both samples to be on the order of 5%. In Fig. 2A, we assumed error bars of 10%. To assess the incorporation level of the α-keto-acid precursor molecule into the Leu/Val labelled SH3 protein, we compared the experimental to the theoretical mass (Fig. S2). Both masses agree well, which indicates a full incorporation of the precursor molecule.
We further investigated the 1H dipolar coupling network emerging from both labelling schemes. We estimated, therefore, the effective dipolar coupling based on the crystal structure of the SH3 domain for both schemes as well as for an uniformly protonated sample (details given in the Methods part). For an approximate validation of our models, we compared the theoretical mass calculated for the ensemble of the in silico structures to the experimentally measured protein mass and obtained a high correlation with a very small bias towards larger theoretical masses (Fig. S2). This indicates that in the measured sample the proton concentration was slightly higher than in the model due to experimental errors and unaccounted isotope impurities throughout the protein expression procedure, respectively. A deviation from the expected proton concentration within both samples will, of course, also contribute to some extent to the observed signal-to-noise differences (Fig. 2A).
In Fig. 2B the averaged effective 1H,1H dipolar coupling is shown, based on in silico structures. The mean value is larger for the Leu/Val sample compared to the 5% GlcRAP sample, respectively, and largest for the uniformly protonated sample as expected. We note, that the predominant protonated methyl isotopomer species is 13CHD2 for both the Leu/Val and 5% GlcRAP sample, respectively12. Other methyl isotopomers, which exhibit potentially large intra-methyl 1H,1H dipolar couplings, namely 13CH2D and 13CH3, can, in principle, be also populated in 5% GlcRAP samples, but are ~5 to ~20 fold less frequent than 13CHD2. Here, we included all possible isotopomers in the in silico models based on the experimentally derived statistics for proton incorporation12. Even though, the average effective dipolar coupling for the 5% GlcRAP sample is very small, we find large couplings within the 2σ (95.45% confidence) confidence limit. This is because infrequently there are singular structures, which contain higher protonated isotopomers, that add to the variance of the averaged effective dipolar coupling in 5% GlcRAP samples.
For calculation of the effective dipolar coupling we employed a very simple descriptor assuming static structures, which can, in principle, cause a systematic offset of the coupling value. Despite a potential offset, the dipolar couplings are approximately uniform for all methyl sites in the 5% GlcRAP sample due to the underlying random nature of the labelling scheme, whereas for the Leu/Val sample the coupling values strongly fluctuate. For the latter, the dipolar coupling even exceeds the upper confidence limit of the 5% GlcRAP sample for several residues (Fig. 2B, crosses), despite the absence of higher protonated isotopomers. This is mainly due to the spatial bundling of Leu and Val residues in the hydrophobic core region of the protein and a concomitant increase of the local proton density as Leu/Val methyl groups are overall the only proton-bearing residues in this labelling scheme. The situation is clearly worse for larger proteins with extensive, methyl-rich hydrophobic regions. It is worth noting, that probing all methyl groups (Ala, Ile, Leu, Met, Thr, Val) with one sample using selective methyl labelling30 would require the addition of further costly precursor molecules during protein expression. This would significantly add to the proton density as shown in Fig. S3, and, by far, exceed the favorable spectroscopic conditions found in 5% GlcRAP samples, which inherently contain all 1H-detectable methyl groups.
We further investigated the possibility to combine the 5% GlcRAP and the Leu/Val labelling scheme with full back-substitution of exchangeable protons (Fig. 2C). Evidently, due to the implicit low proton concentration in the 5% GlcRAP sample the effective 1H,1H dipolar couplings only mildly increase upon 100% proton back-substitution as opposed to the Leu/Val sample, making the former more suitable for proton detection of aliphatic and amide resonances using the same sample.
Structural restraints
In the following, we compared the 5% GlcRAP and Leu/Val labelling in terms of accessibility of long-range structural restraints, which are crucial for biomacromolecular structure determination. Using in silico models of the SH3 domain (vide supra), we calculated the frequency of encountering 1H,1H pairs within 5 Å for both labelling schemes as well for an uniformly protonated sample without and with full backsubstitution of exchangeable protons (Fig. 3A–E). We find, that the 5% GlcRAP labelled sample (Fig. 3A) shows essentially the same pattern of 1H,1H contacts as compared to the uniformly protonated sample (Fig. 3E) despite the considerably lower proton concentration, unlike the Leu/Val labelled sample, which only shows a very limited number of contacts (Fig. 3C), even though at a higher frequency compared to the 5% GlcRAP labelled sample. The 5% GlcRAP labelled sample additionally allows to sequentially connect residues as indicated by the contacts in parallel to the diagonal (Fig. 3A).
Figure 3.

(A–E) 1H,1H contact map for the 1 μs MD relaxed crystal structure of the SH3 domain of α-spectrin for an upper 1H,1H distance limit of 5 Å. The maps were calculated for the (A) 5% GlcRAP and (C) Leu/Val labelling scheme, respectively, as well as for an (E) uniformly protonated sample. Full proton back-substitution was also considered for the former in (B) and (D). (F) The probability P to detect at least one 1H,1H contact for N adjoining sites assuming a likelihood of p = 0.035 to find a proton at a given site. (G) Methyl region of a 1H,1H 2D projection extracted from a 1H-detected 3D H(H)CH RFDR spectrum (τmix = 8 ms) using the 5% GlcRAP labelled SH3 domain12. The spectrum was recorded at an external magnetic field of 14.1 T (600 MHz) and a MAS frequency of 20 kHz. The H2O content in the microcrystalline sample was < 10%. Clearly, a number of 1H,1H contacts were detected including Hmet-Hmet, Hmet-Hali, Hmet-solvent and Hmet-HN. The figure was generated with Adobe Illustrator CS5 V.15.02.
Including all exchangeable protons significantly elevates the frequency of 1H,1H contacts for the 5% GlcRAP sample (Fig. 3B), which drastically increases the probability to observe cross peaks that could serve as restraints to interconnect proximal residues. Similarly, for the Leu/Val sample the frequency of finding interconnecting 1H,1H contacts substantially increases (Fig. 3D) and resembles the 5% GlcRAP contact map. We note, however, that the Leu/Val sample with full proton backsubstitution shows considerably larger 1H,1H dipolar couplings compared to the 5% GlcRAP sample (Fig. 2C), indicating spectroscopic benefits for the latter.
The low proton concentration in the 5% GlcRAP sample affects the measurement of structural restraints that rely on the observation of 1H,1H contacts (Fig. 3A). To first order, with p being the likelihood of finding a proton at a given site yields a probability P = p2 to observe a cross peak for two structurally proximal protons. This probability P is vanishing if p is small as for methyl groups in 5% GlcRAP labelled samples (p ≈ 0.035). However, if we consider multiple proximal proton sites for a particular molecule within the ensemble, then the likelihood to find at least one cross peak among those sites increases beyond p2. For N proximal proton sites, there are cross peak combinations. The probability P to find a cross peak between one or more pairs is given by (Fig. 3F).
To experimentally validate the in silico results, we recorded a 1H-detected 3D H(H)CH RFDR spectrum using the 5% GlcRAP labelled SH3 domain12. The 1H,1H 2D projection is shown in Fig. 3G. Evidently, a large number of 1H,1H contacts is observable, including Hmet-Hmet, Hmet-Hali, Hmet-solvent and Hmet-HN, respectively. Furthermore, due to the low proton concentration per molecule, dipolar truncation is less pronounced in the 5% GlcRAP labelled sample, which allows the measurement of long 1H,1H distances, even up to 9 Å12.
Methyl dipole tensor
So far, we showed that the 5% GlcRAP labelling scheme produces samples, that yield sufficient signal-to-noise in 1H-detected solid-state NMR experiments and enables the detection of all methyl groups (Ala, Ile, Leu, Met, Thr, Val), unlike for the selectively methyl-labelled Leu/Val sample (only Leu, Val). Additionally, structural restraints can be obtained (vide supra). In the following, we determine dipolar order parameters and motional dependent tensor asymmetries employing REDOR experiments9 (Fig. S1C) for all methyl groups in α-spectrin SH3 using the 5% GlcRAP sample (Alaβ, Ileγ2,δ1, Metε, Thrγ2, Leuδ1,δ2, Valγ1,γ2), and for Leuδ1,δ2 and Valγ1,γ2 using the Leu/Val sample, respectively.
The REDOR experiment was shown to be a very powerful approach as it combines 1H-detection and accurate determination of the dipolar tensor with little sensitivity towards rf inhomogeneities and mismatches, offset effects and variations of the CSA properties of the involved nuclei31–34. Only the presence of remote protons can be an obstacle as REDOR is known to recouple the 1H,1H homonuclear dipolar coupling35,36. However, fast spinning frequencies ≥50 kHz renders the experiment essentially insensitive to remote protons, when applied to perdeuterated and amide back-exchanged proteins37. We note, that the 5% GlcRAP labelling scheme yields incorporation of a comparably small concentration of protons into the protein together with an approximately uniform distribution of 1H,1H effective dipolar couplings throughout the protein (Fig. 2B). This fact reduces systematic errors in quantitative experiments that are sensitive to remote protons, such as the REDOR experiment employed here. On the other hand, for Leu/Val samples the effective dipolar coupling varies significantly as a function of the methyl position. In extreme cases, different proton densities for each methyl group might have to be taken into account in the numerical simulations, which can complicate the data analysis and additionally requires a high-resolution structure of the system under study.
Experimental REDOR dephasing curves are shown in Fig. 4 for the Leu/Val and 5% GlcRAP sample, respectively. The dephasing curves for the common Leuδ1,δ2 and Valγ1,γ2 residues show very similar trends for both samples. However, the additional methyl order parameters for Ala, Ileγ2,δ1, Met and Thr, which became accessible using the 5% GlcRAP sample, allow to probe the amplitude of motion in nearly all protein regions. Ala11β displayed the largest squared order parameter (S2 = 0.80) among all methyls and reflects the dynamics of the protein backbone.
Figure 4.

1H-detected REDOR dephasing curves, using a selectively Leu/Val 13CHD2 methyl labelled and a 5% GlcRAP labelled sample of α-spectrin SH3. The external magnetic field was 14.1 T (600 MHz). The effective temperature of the sample was adjusted to 20–25 °C. The MAS frequency was set to 50 kHz. In the experiment, a ζ-delay of 4 µs was employed. The methyl order parameter S2 (S2axis in Eq. (4)) and the asymmetry η were determined as described in the Methods section. The values are given at the bottom of each plot. In the first two rows, the dephasing curves were fitted by simultaneously minimizing χ2 for both methyl sites (γ1/γ2 for Val and δ1/δ2 for Leu) to improve the fitting convergence. This yielded one order and asymmetry parameter for each Val and Leu residue. However, individual fits for γ1/γ2 (Val) and δ1/δ2 (Leu) were plotted in red and blue to indicate that proR and proS methyl groups show very similar dynamics. The figure was generated with Adobe Illustrator CS5 V.15.02.
The most significant asymmetric dynamics was detected for Val23, Ile30 and Val46, respectively. The dynamic property of Val23 with η = 0.60 ± 0.03 was also observed by 2H line shape analysis reporting the same asymmetry parameter (η = 0.59 ± 0.01)38,39. However, the methyl groups of Val46 could not be analyzed before by 2H experiments as they are too mobile to yield sufficient sensitivity during the 2H,13C cross polarization magnetization transfer. For Ile30δ1 a very low order parameter was obtained (S2 = 0.27), while the dipolar coupling asymmetry was very high (η = 0.60 ± 0.02). Interestingly, the dephasing curve could not be fitted reasonably well by simply assuming an asymmetric tensor, similar to results obtained for ubiquitin9. This hints to the presence of a more complex motion on a microsecond timescale. In principle, the dephasing curve could be fitted adequately by assuming contributions from multiple dipolar coupling tensors with different anisotropies and asymmetries.
Furthermore, dipolar order parameters were obtained for the N-terminal residues Met1ε and Thr4γ2. These residues are not visible in the X-ray structures of α-spectrin SH3 due to dynamical disorder, which is also indicated by very low squared order parameters of 0.00–0.01 (Fig. 4). Residues with such low order parameters typically evade detection in cross-polarization experiments under MAS due to very low magnetization transfer efficiencies40–42. However, the significant downscaling of the dipolar coupling enhances the coherence lifetime and gives rise to cross-peaks using the INEPT magnetization scheme, which is based on scalar couplings. Similarly, the 1H,15N backbone dipolar tensors of the highly dynamic N-terminal residues Thr4 and Gly5 were detectable in the perdeuterated and proton back-exchanged SH3 domain using INEPT-based REDOR experiments37.
The fitted dipolar coupling anisotropies and asymmetries for Leu and Val methyl groups of both samples are summarized in Fig. 5. The linear correlation coefficients are 0.96 and 0.85 for the anisotropies and asymmetries, respectively. This reveals that both parameters are highly correlated if the two samples are compared, despite the different distribution of remote protons (Fig. 2B), which in turn indicates that the residual 1H,1H dipolar couplings sensed by the methyl protons in both samples sufficiently averages at a MAS frequency of 50 kHz. However, we note, that for the majority of methyl groups the dipolar coupling anisotropies found in the Leu/Val sample are systematically larger compared to the 5% GlcRAP sample (Fig. 5B). For the former, this indicates a stronger interference of extraneous protons with the REDOR dipolar coupling measurement, which is an effect previously described by others34. This is also in line with the in silico data (Fig. 2B), which shows larger 1H,1H dipolar couplings for the Leu/Val sample.
Figure 5.
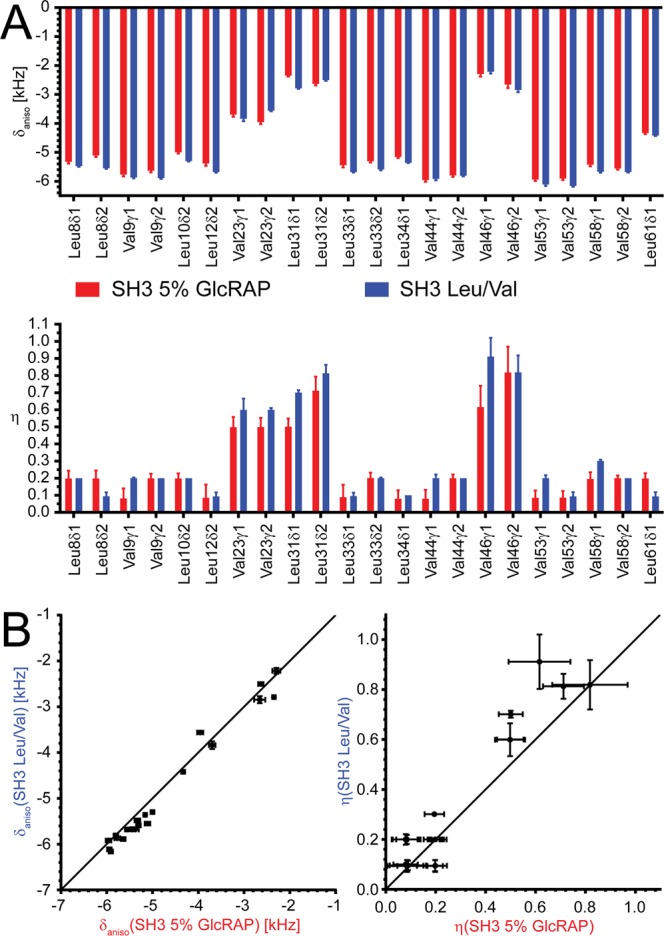
(A) Dipolar coupling anisotropies δaniso (top) and asymmetries η (bottom), using the 5% GlcRAP (red) and the selectively Leu/Val 13CHD2 methyl labelled sample (blue) of α-spectrin SH3, respectively. All values were determined by fitting REDOR dephasing curves represented in Fig. 4. (B) The anisotropies and asymmetries of both samples are linearly correlated, setting the slope to 1.0 and the y-intercept to 0.0. The linear correlation coefficient R2 is 0.96 and 0.85 for δaniso and η, respectively. The figure was generated with Adobe Illustrator CS5 V.15.02.
Discussion
In this work, we studied two different types of labelling schemes and samples to yield high resolution methyl 1H,13C correlation spectra in the solid-state. The experiments are applied to the microcrystalline SH3 domain of α-spectrin and disease-related amyloid fibrils of the Aβ1-40 peptide, respectively. We focus in particular on selective 13CHD2 methyl labelling of Leu and Val residues using α-keto-acid precursors and 5% GlcRAP labelling. In the first, either one of the two proR or proS methyl groups per residue per molecule is 13CHD2 labelled, while in the second, all methyl groups are stochastically protonated. The degree of protonation can be adjusted over the amount of H2O in the M9 minimal expression medium12. Here, we used an expression medium containing 5% H2O and 95% D2O, respectively, yielding an average methyl proton concentration of about ~3.5%. On the other hand, the proton concentration at either one of the methyl groups in the selectively Leu/Val 13CHD2 labelled sample is 16.7%.
Despite the low occurrence of methyl protons in the 5% GlcRAP sample, we were able to record high-sensitivity and high-resolution 2D 1H,13C methyl group correlations, when combining fast MAS frequencies and 1H-detection. This is particularly critical for the investigation of biological systems, for which the sample amount is generally limited, irrespective of the sample type, which can comprise of microcrystalls, aggregates or fibrils, such as the herein studied amyloid fibrils of the Aβ1-40 peptide. The recently reported structural models of Aβ determined by solid-state NMR spectroscopy relied on 13C detection, which suffers from low sensitivity43,44. Here, we show that 1H,13C correlation of Aβ1-40 fibrils can be measured with high sensitivity, even at very low MAS frequencies <20 kHz, owing to the largely deuterated background in the 5% GlcRAP sample and 1H detection. Moreover, the addition of the highly sensitive 1H chemical shift information allows to considerably enhance the resolution and determine structural restraints, despite the low proton concentration.
To first order, the proton concentration correlates with the sensitivity in the 1H-detected experiments. However, at the same time, sensitivity is reduced due to enhanced dipole-mediated line broadening29, which in turn requires much faster spinning frequencies at the magic angle45. It has been shown recently that MAS frequencies beyond 300 kHz are necessary to yield >80% of the theoretical achievable sensitivity in 13CH3 selectively methyl labelled protein samples. This is way beyond the current technical limit, which is around ~100 kHz. For the GlcRAP labelling, a drastic increase of the proton concentration generally results in significant population of highly protonated methyl isotopomers, namely 13CH2D and 13CH3, which not only add to line broadening, but also obscure the spectral resolution due to crowding29. Therefore, an optimal GlcRAP sample for detection of methyl groups maintaining sensitivity and resolution can only be achieved at rather low protonation levels. It was shown before, that even using ≥99% D2O in the expression medium (corresponding to a 0% GlcRAP sample) yielded detectable proton signals of methyl groups, originating from ≤1% proton impurities of the D2O and ≤3% residual protonation of the [2H,13C] glucose11. We find, however, that the optimal trade-off between sensitivity and resolution for methyl correlation spectra is found for ~5% GlcRAP labelling and MAS frequencies ≥50 kHz29. In these samples, all the remaining aliphatic sites, such as methine and methylene side chains, backbone and aromatic residues, become protonated and are as well detectable at high resolution using the same sample12,17.
One particular benefit of the 5% GlcRAP labelling scheme over the selective methyl labelling scheme is that at once all methyl groups (Alaβ, Ileγ2,δ1, Leuδ1,2, Metε, Thrγ2, Valγ1,2) become accessible in the first at low cost, while in the latter a multitude of precursor molecules has to be employed during expression to achieve full methyl labelling30, which, despite the high costs, introduces a significantly higher level of protonation into the sample. On the other hand, having randomly distributed protons in a deuterated matrix, such as in 5% GlcRAP samples, largely reduces the 1H,1H dipolar network and makes this type of samples particularly attractive for unbiased quantification of dynamics.
We reported previously, that RAP labelling allows to measure structural and dynamical parameters, such as 1H,1H distance restraints12 and aliphatic R1 relaxation rates21, respectively. Furthermore, others determined methyl order and asymmetry parameters for selectively Ileδ1/Leuδ1,δ2/Valγ1,γ2 13CHD2 methyl labelled microcrystals of ubiquitin9. Here, we report the measurement of methyl dipole tensors of the SH3 domain of α-spectrin by fitting REDOR dephasing curves. As reported recently34,37, the REDOR sequence is particularly sensitive to remote protons. Therefore, a 5% GlcRAP labelled sample at fast spinning is optimally suited for this type of NMR experiment. We were able to measure dipolar tensors for all methyl groups with high accuracy. When comparing the order parameters and asymmetries to the Leu/Val sample, we found comparable values within the experimental error, which indicates that at MAS frequencies ≥50 kHz remote protons have very little impact on the REDOR dephasing curve in both samples. However, dipolar coupling anisotropies measured for the 5% GlcRAP sample are slightly smaller compared to the Leu/Val sample, which indicates a weaker interference of remote protons during REDOR measurements for the former. This also reflected by the presented in silico data. Moreover, for low spinning rates the effect of remote protons becomes especially challenging for Leu/Val labelled samples as, on average, methyl protons in these samples experience larger 1H,1H effective dipolar couplings compared to the 5% GlcRAP sample. We further note, that there might be considerable clustering of methyl groups in hydrophobic core regions, depending on the biomolecular system, which can appreciably enhance the effective dipolar coupling in these molecular patches. This is not expected for the 5% GlcRAP labelling scheme due to its intrinsic random nature of protonation.
Using the 5% GlcRAP sample, we were able to determine methyl dipole tensors for all methyl sites, including for Ala, Ileγ2,δ1, Met, Thr. Alanine residues are particularly interesting in terms of dynamics, as the methyl group instantaneously relates to backbone motions, since it is directly connected to the Cα atom46. This way, the spectroscopic advantages of methyl groups, such as high sensitivity and resolution, can be exploited to probe the backbone dynamics with improved experimental accuracy and precision.
The dipolar asymmetry parameter contains information about the anisotropy of the side chain motion on the timescale of 1/δaniso47,48. Analytical equations to link the methyl order and asymmetry parameters to yield a motional model were given elsewhere9. We detected significant asymmetry for Val23 and Val46. The dynamic property of Val23 is also observed by 2H line shape analysis, as reported earlier38,39. Even though a slightly faster timescale can be probed by 2H, we obtained here the same values for the asymmetry parameters. However, the methyl groups of Val46 could not be analyzed by 2H experiments, as the methyl groups are too mobile to yield sufficient sensitivity during the 2H,13C cross polarization magnetization transfer. On the contrary, scalar based transfers were used here for the REDOR experiments, which particularly yield high sensitivity for mobile residues. Val46 is most probably undergoing slow dynamics on the µs time scale since it is not detectable by CP experiments.
In conclusion, the 5% GlcRAP labelling scheme enables 1H-detection of all methyl groups as shown for microcrystalline and fibrillar samples, and further allows the determination of structural restraints as well as methyl dipolar order parameters and asymmetries, which are crucial for understanding molecular dynamics. The labelling comes at relatively small cost compared to selectively methyl labelled samples and ensures high resolution and sensitivity, even at slow MAS frequencies <20 kHz. In addition, the 5% GlcRAP labelling scheme can be easily combined with backsubstitution of exchangeable protons with an only mild impact on the 1H,1H dipolar coupling network.
Methods
Sample preparation
The SH3 domain of chicken α-spectrin was produced, as described earlier49. Two different labelling schemes were employed in this study, (a) random protonation, following the RAP (reduced adjoining protonation) approach12,17, and (b) selective Leu/Val 13CHD2 methyl labelling3. For the RAP sample, hereinafter referred to as 5% GlcRAP, protein expression was carried out using 15NH4Cl, u-[2H, 13C] glucose as the sole nitrogen and carbon source, respectively. All buffers used for the M9 medium contained 5% H2O and 95% D2O.
For the selectively Leu/Val 13CHD2 methyl labelled sample - referred to as Leu/Val sample - protein expression was carried out using 15NH4Cl and u-[2H, 12C] glucose. We added 100 mg/mL of the precursor (two times in 100% D2O lyophilized 2-keto-3-methyl-13C,d2-3-d1-4-13C,d2-butyrate) one 1 hour prior to induction.
Both purified samples were lyophilized two times in 100% D2O at pH 3.5 prior to crystallization. Approximately ~3 mg microcrystals were packed into a 1.3 mm rotor by ultracentrifugation (~20 min, ~135.000 × g) employing an ultracentrifuge device50. The rotor caps were sealed by gluing, as described earlier29. Based on previous experience in packing 1.3 mm MAS rotors using the same setup and device29, we estimate the deviation in the amount of material in the rotor to be on the order of 5%. In the analysis presented above, we assumed an error of 10%.
5% GlcRAP labelled Aβ1-40 fibrils (with Met at position 0) were produced as described previously, using fibril seeding in a 100% D2O buffer51. For isotope labelling we followed the same protocol as for the SH3 domain. The final fibrils (~10 mg) were packed into a 3.2 mm rotor using a tabletop centrifuge.
MALDI-TOF
The labelling efficiency of α-spectrin SH3 samples (5% GlcRAP, Leu/Val and others) were monitored via MALDI-TOF and compared to the expected mass for the respective labelling scheme (vide infra). We considered impurities of the uniformly deuterated and 13C-enriched carbon source (3% residual protonation and 1% 12C). An excellent agreement between the experimental and simulated mass was achieved (Fig. S2).
NMR spectroscopy
NMR experiments with the SH3 domain of α-spectrin were carried out at 50 kHz MAS using a Bruker BioSpin Avance spectrometer operating at a 1H Larmor frequency of 600 MHz (14.09 T), equipped with a commercial 1.3 mm triple-resonance probe. The effective sample temperature was adjusted to ∼20–25 °C using the chemical shift difference between the solvent resonance and Leu8δ2. Experiments with the Aβ1-40 peptide were performed at a 1H Larmor frequency of 700 MHz (16.4 T) and a MAS frequency of 18 kHz, using a commercial 3.2 mm triple-resonance probe. The employed rf fields on the 1H and 13C channels for hard pulses were 119 kHz (109 kHz) and 100 kHz (62.5 kHz) at 600 MHz (700 MHz). Low-power 2H and 13C decoupling of 2.5 kHz was applied, using the WALTZ-16 decoupling scheme52.
High-resolution 1H,13C correlations were obtained for all samples via 2D HMQC spectroscopy and 2H decoupling during the 13C evolution period (Fig. S1A)12. No special care was taken for solvent suppression in these experiments. To spectroscopically eliminate splittings in the indirect 13C dimension due to the 13C,13C one-bond J-coupling in the 5% GlcRAP sample, we recorded 2D constant-time HSQC spectra (Fig. S1B)53. The spectral resolution in the indirect dimension was further enhanced by mirror-image linear prediction19.
1H,13C REDOR experiments were performed using the pulse sequence depicted in Fig. S1C. The rf fields for 1H and 13C π pulses were 125 and 100 kHz, respectively, during the REDOR recoupling element. The total REDOR dephasing period was a multiple of the rotor period, τr, and equal to with n ≥ 0. The REDOR shift ζ defines the timing of the 1H π pulse with respect to half of the rotor period, which was set here to 4.0 μs. Following the definition for ε described previously34 yielded a value of 0.4 setting ζ equal to 4.0 μs at 50 kHz sample spinning. A z-filter delay Δzf of 5 ms and a recycle delay of 2.1 s (3 s) was employed in experiments using the 5% GlcRAP (Leu/Val) labelled SH3 domain. Reference experiments to account for the signal decay during the recoupling period were scattered evenly (every 0.12 ms) over the total dephasing time of 1.92 ms by setting the rf field of the 1H π pulse to 0 Hz. The reference points in between were interpolated from a linear correlation. The bulk 1H T1 for both SH3 samples was ~1.4 s.
Numerical simulations
All numerical simulations were performed with the software package SIMPSON54. For REDOR experiments (Fig. S1C), we simulated recoupling curves (S) assuming experimentally applied rf fields for 1H and 13C π pulses and a ζ delay of 4 μs. We employed a two spin system and generated a matrix of dipolar coupling anisotropies (100 Hz steps) and asymmetries (0.1 steps). The respective REDOR reference curves (S0) were simulated with zero rf field on the 1H channel. The typical REDOR dephasing curve () was finally determined by . Rf inhomogeneities were not taken into account.
Data analysis
The experimental REDOR spectra were processed in Topspin v3.2 and peak volumes were extracted by box integration, using in-house Python scripts. The experimental error was set to two times the standard deviation of the noise amplitude and uncertainties of the fitting parameters were estimated by 1000 Monte Carlo runs. In the reference experiment S0, the dipolar coupling was not reintroduced, unlike in the recoupling experiment S. The relaxation compensated REDOR dephasing curves were calculated according to similar to the simulations (vide supra). We performed a two parameter fit, considering the dipolar coupling anisotropy (δaniso) as well as the asymmetry (η). The experimental curves were fit against a grid, in which χ2 was minimized to find the best fit value for both parameters. In the case of Leu and Val residues, both methyl groups, δ1/δ2 and γ1/γ2, were fit simultaneously.
Determination of the methyl dipolar tensor and order parameter
The heteronuclear 1H,13C dipolar coupling is a powerful probe for fast dynamical processes on the sub-ms timescale. The dipolar coupling can be expressed in Cartesian coordinates in the principal axis frame by a traceless second rank tensor9
| 1 |
Here, is the asymmetry parameter, and is the dipolar coupling anisotropy, which is defined as (in units of [Hz])
| 2 |
where S is the order parameter, is the magnetic constant, is the reduced Planck constant, and are the gyromagnetic rations of nuclei i and j (here 1H and 13C), respectively, and the internuclear distance9,54–56. Both the asymmetry parameter and the order parameter can vary between zero and one. In the absence of motion, S is equal to one and equal to zero. We employed a rigid-limit 1H,13C bond length for methyl groups of 1.115 Å yielding an anisotropy of −21794 Hz57,58. We calculated the experimentally derived order parameter by dividing the determined dipolar coupling anisotropy by the rigid-limit value.
At room temperature, methyl groups steadily undergo fast rotations around their local threefold axis with correlation times on the picosecond timescale. The amplitude of this fast, axially symmetric rotation is characterized by an order parameter 58–61,
| 3 |
yielding for an ideal tetrahedral geometry (). Ideal tetrahedral geometry was assumed for all methyl-bearing amino acids, noting that methyl groups are usually slightly distorted from ideal geometry61,62. Due to the fast timescale of the methyl rotation, which is much shorter than 1/δaniso, the dipolar coupling anisotropy is averaged by this rotational motion. Further averaging is induced by motions of the symmetry axis of the methyl group, which are related to the axis order parameter . Thus, a generalized order parameter for the threefold methyl axis can be defined as
| 4 |
In silico calculation of 1H,1H dipolar couplings
We used a structural ensemble of 32 monomers of the SH3 domain as a template, which was relaxed by MD simulation for 1 μs21, and determined the averaged effective 1H,1H dipolar coupling experienced by the methyl proton at site j in monomer l over all proximal protons k according to the simple descriptor , with N as the number of considered monomers throughout the calculation37,63. was set to zero if no proton was found at the specific methyl site. We employed a distance cut-off of 10 Å. Intra-methyl 1H,1H couplings were scaled by with yielding a scaling factor of 0.564.
In the calculations, the labelling scheme was taken into consideration. The population of stochastically incorporated protons in the 5% GlcRAP labelling scheme was based on the experimentally determined proton concentrations12, that were scaled to match the experimental mass derived by MALDI (vide supra). For the Leu/Val 13CHD2 labelled sample, we assumed an equal distribution of pro-R (50%) and pro-S (50%) 13CHD2 methyl groups, while either one of the two methyl groups at the same residue was considered to be labelled, as constrained by the utilized precursor molecule. Since both samples were re-buffered in 100% D2O, all exchangeable protons have been considered as 2H. For sufficient statistics, we generated 32000 in silico structures and calculated the effective dipolar coupling for each methyl group averaged over all structures. We used the corrected standard deviation of the mean with 1/(N-1).
Supplementary information
Acknowledgements
We acknowledge P. Schanda (IBS Grenoble) for helpful discussions. We are grateful to Bruker BioSpin for providing measurement time, especially to S. Wegner and G. Althoff for technical support, as well as to K. Szekely (ETH Zürich) for help with 1.3 mm rotor packing. We dedicate this manuscript to the late Dr. Frank Eisenmenger (FMP Berlin).
Author contributions
S.A. performed experiments and analyzed data; S.A., B.R. performed data interpretation, designed research and experimental plans; both authors contributed in writing the manuscript.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
is available for this paper at 10.1038/s41598-019-52383-3.
References
- 1.Gardner KH, Kay LE. Production and incorporation of N-15, C-13, H-2 (H-1-delta 1 methyl) isoleucine into proteins for multidimensional NMR studies. J. Am. Chem. Soc. 1997;119:7599–7600. doi: 10.1021/ja9706514. [DOI] [Google Scholar]
- 2.Goto NK, Gardner KH, Mueller GA, Willis RC, Kay LE. A robust and cost-effective method for the production of Val, Leu, Ile (delta 1) methyl-protonated N-15-, C-13-, H-2-labeled proteins. J. Biomol. NMR. 1999;13:369–374. doi: 10.1023/A:1008393201236. [DOI] [PubMed] [Google Scholar]
- 3.Tugarinov V, Kanelis V, Kay LE. Isotope Labeling Strategies for the Study of High-molecular-weight Proteins by Solution NMR Spectroscopy. Nat Protoc. 2006;1:749–754. doi: 10.1038/nprot.2006.101. [DOI] [PubMed] [Google Scholar]
- 4.Tugarinov V, Kay LE. Ile, Leu, and Val methyl assignments of the 723-residue malate synthase G using a new labeling strategy and novel NMR methods. J. Am. Chem. Soc. 2003;125:13868–13878. doi: 10.1021/ja030345s. [DOI] [PubMed] [Google Scholar]
- 5.Tugarinov V, Kay LE. An isotope labeling strategy for methyl TROSY spectroscopy. J. Biomol. NMR. 2004;28:165–172. doi: 10.1023/B:JNMR.0000013824.93994.1f. [DOI] [PubMed] [Google Scholar]
- 6.Agarwal V, Diehl A, Skrynnikov N, Reif B. High resolution H-1 detected H-1,C-13 correlation spectra in MAS solid-state NMR using deuterated proteins with selective H-1,H-2 isotopic labeling of methyl groups. J. Am. Chem. Soc. 2006;128:12620–12621. doi: 10.1021/ja064379m. [DOI] [PubMed] [Google Scholar]
- 7.Gans P, et al. Stereospecific isotopic labeling of methyl groups for NMR spectroscopic studies of high‐molecular‐weight proteins. Angewandte Chemie International Edition. 2010;49:1958–1962. doi: 10.1002/anie.200905660. [DOI] [PubMed] [Google Scholar]
- 8.Agarwal V, Xue Y, Reif B, Skrynnikov NR. Protein Side-Chain Dynamics As Observed by Solution- and Solid-State NMR Spectroscopy: A Similarity Revealed. J. Am. Chem. Soc. 2008;130:16611–16621. doi: 10.1021/ja804275p. [DOI] [PubMed] [Google Scholar]
- 9.Schanda P, Huber M, Boisbouvier J, Meier BH, Ernst M. Solid-State NMR Measurements of Asymmetric Dipolar Couplings Provide Insight into Protein Side-Chain Motion. Angew. Chem., Int. Ed. 2011;50:11005–11009. doi: 10.1002/anie.201103944. [DOI] [PubMed] [Google Scholar]
- 10.Huber M, et al. A Proton-Detected 4D Solid-State NMR Experiment for Protein Structure Determination. ChemPhysChem. 2011;12:915–918. doi: 10.1002/cphc.201100062. [DOI] [PubMed] [Google Scholar]
- 11.Agarwal V, Reif B. Residual methyl protonation in perdeuterated proteins for multi-dimensional correlation experiments in MAS solid-state NMR spectroscopy. J. Magn. Reson. 2008;194:16–24. doi: 10.1016/j.jmr.2008.05.021. [DOI] [PubMed] [Google Scholar]
- 12.Asami S, Schmieder P, Reif B. High Resolution 1H-Detected Solid-State NMR Spectroscopy of Protein Aliphatic Resonances: Access to Tertiary Structure Information. J. Am. Chem. Soc. 2010;132:15133–15135. doi: 10.1021/ja106170h. [DOI] [PubMed] [Google Scholar]
- 13.Mance D, et al. An Efficient Labelling Approach to Harness Backbone and Side‐Chain Protons in 1H‐Detected Solid‐State NMR Spectroscopy. Angewandte Chemie International Edition. 2015;54:15799–15803. doi: 10.1002/anie.201509170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Medeiros‐Silva J, et al. 1H‐Detected Solid‐State NMR Studies of Water‐Inaccessible Proteins In Vitro and In Situ. Angewandte Chemie International Edition. 2016;55:13606–13610. doi: 10.1002/anie.201606594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sinnige T, Daniëls M, Baldus M, Weingarth M. Proton clouds to measure long-range contacts between nonexchangeable side chain protons in solid-state NMR. J. Am. Chem. Soc. 2014;136:4452–4455. doi: 10.1021/ja412870m. [DOI] [PubMed] [Google Scholar]
- 16.Movellan KT, et al. Alpha protons as NMR probes in deuterated proteins. J. Biomol. NMR. 2019;73:81–91. doi: 10.1007/s10858-019-00230-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Asami S, Reif B. Proton-Detected Solid-State NMR Spectroscopy at Aliphatic Sites: Application to Crystalline Systems. Acc Chem Res. 2013;46:2089–2097. doi: 10.1021/ar400063y. [DOI] [PubMed] [Google Scholar]
- 18.Asami S, Reif B. Assignment Strategies for Aliphatic Protons in the Solid-State in Randomly Protonated Proteins. J. Biomol. NMR. 2012;52:31–39. doi: 10.1007/s10858-011-9591-4. [DOI] [PubMed] [Google Scholar]
- 19.Zhu G, Bax A. Improved Linear Prediction for Truncated Signals of Known Phase. J. Magn. Reson. 1990;90:405–410. [Google Scholar]
- 20.Vandeven FJM, Philippens MEP. Optimization of Constant-Time Evolution in Multidimensional Nmr Experiments. J. Magn. Reson. 1992;97:637–644. [Google Scholar]
- 21.Asami S, Porter JR, Lange OF, Reif B. Access to Calpha Backbone Dynamics of Biological Solids by 13C T1 Relaxation and Molecular Dynamics Simulation. J. Am. Chem. Soc. 2015;137:1094–1100. doi: 10.1021/ja509367q. [DOI] [PubMed] [Google Scholar]
- 22.LeMaster DM, Kushlan DM. Dynamical mapping of E-coli thioredoxin via C-13 NMR relaxation analysis. J. Am. Chem. Soc. 1996;118:9255–9264. doi: 10.1021/ja960877r. [DOI] [Google Scholar]
- 23.Sipe JD. Amyloidosis. Annu. Rev. Biochem. 1992;61:947–975. doi: 10.1146/annurev.bi.61.070192.004503. [DOI] [PubMed] [Google Scholar]
- 24.Sacchettini JC, Kelly JW. Therapeutic strategies for human amyloid diseases. Nat. Rev. Drug Discov. 2002;1:267–275. doi: 10.1038/nrd769. [DOI] [PubMed] [Google Scholar]
- 25.Tycko R. Molecular structure of amyloid fibrils: insights from solid-state NMR. Q. Rev. Biophys. 2006;39:1–55. doi: 10.1017/S0033583506004173. [DOI] [PubMed] [Google Scholar]
- 26.Petkova AT, et al. Self-propagating, molecular-level polymorphism in Alzheimer’s beta-amyloid fibrils. Science. 2005;307:262–265. doi: 10.1126/science.1105850. [DOI] [PubMed] [Google Scholar]
- 27.Petkova AT, Yau WM, Tycko R. Experimental Constraints on Quaternary Structure in Alzheimer’s Beta-Amyloid Fibrils. Biochemistry. 2006;45:498–512. doi: 10.1021/bi051952q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Akbey U, et al. Optimum levels of exchangeable protons in perdeuterated proteins for proton detection in MAS solid-state NMR spectroscopy. J. Biomol. NMR. 2010;46:67–73. doi: 10.1007/s10858-009-9369-0. [DOI] [PubMed] [Google Scholar]
- 29.Asami S, Szekely K, Schanda P, Meier BH, Reif B. Optimal Degree of Protonation for (1)H Detection of Aliphatic Sites in Randomly Deuterated Proteins as a Function of the MAS Frequency. J. Biomol. NMR. 2012;54:155–168. doi: 10.1007/s10858-012-9659-9. [DOI] [PubMed] [Google Scholar]
- 30.Kerfah R, Plevin MJ, Sounier R, Gans P, Boisbouvier J. Methyl-specific isotopic labeling: a molecular tool box for solution NMR studies of large proteins. Curr Opin Struct Biol. 2015;32:113–122. doi: 10.1016/j.sbi.2015.03.009. [DOI] [PubMed] [Google Scholar]
- 31.Gullion T, Schaefer J. Rotational-Echo Double-Resonance Nmr. J. Magn. Reson. 1989;81:196–200. doi: 10.1016/j.jmr.2011.09.003. [DOI] [PubMed] [Google Scholar]
- 32.Gullion T, Schaefer J. Detection of Weak Heteronuclear Dipolar Coupling by Rotational-Echo Double-Resonance Nuclear Magnetic Resonance. Adv. Magn. Opt. Reson. 1989;13:57–83. doi: 10.1016/B978-0-12-025513-9.50009-4. [DOI] [Google Scholar]
- 33.Jain MG, et al. Sine-Squared Shifted Pulses for Recoupling Interactions in Solid-State NMR. J. Chem. Phys. 2017;146:244201. doi: 10.1063/1.4986791. [DOI] [PubMed] [Google Scholar]
- 34.Schanda P, Meier BH, Ernst M. Accurate Measurement of One-Bond H-X Heteronuclear Dipolar Couplings in MAS Solid-State NMR. J. Magn. Reson. 2011;210:246–259. doi: 10.1016/j.jmr.2011.03.015. [DOI] [PubMed] [Google Scholar]
- 35.Goetz JM, Schaefer J. REDOR Dephasing by Multiple Spins in the Presence of Molecular Motion. J. Magn. Reson. 1997;127:147–154. doi: 10.1006/jmre.1997.1198. [DOI] [PubMed] [Google Scholar]
- 36.Chan JCC, Eckert H. Dipolar Coupling Information in Multispin Systems: Application of a Compensated REDOR NMR Approach to Inorganic Phosphates. J. Magn. Reson. 2000;147:170–178. doi: 10.1006/jmre.2000.2191. [DOI] [PubMed] [Google Scholar]
- 37.Asami S, Reif B. Comparative Study of REDOR and CPPI Derived Order Parameters by (1)H-Detected MAS NMR and MD Simulations. J. Phys. Chem. B. 2017;121:8719–8730. doi: 10.1021/acs.jpcb.7b06812. [DOI] [PubMed] [Google Scholar]
- 38.Hologne M, Chevelkov V, Reif B. Deuterated peptides and proteins in MAS solid-state NMR. Prog. Nucl. Magn. Reson. Spectrosc. 2006;48:211–232. doi: 10.1016/j.pnmrs.2006.05.004. [DOI] [Google Scholar]
- 39.Hologne M, Faelber K, Diehl A, Reif B. Characterization of dynamics of perdeuterated proteins by MAS solid-state NMR. J. Am. Chem. Soc. 2005;127:11208–11209. doi: 10.1021/ja051830l. [DOI] [PubMed] [Google Scholar]
- 40.Nowacka A, Bongartz NA, Ollila OH, Nylander T, Topgaard D. Signal Intensities in (1)H-(1)(3)C CP and INEPT MAS NMR of Liquid Crystals. J. Magn. Reson. 2013;230:165–175. doi: 10.1016/j.jmr.2013.02.016. [DOI] [PubMed] [Google Scholar]
- 41.Sackewitz M, et al. Structural and Dynamical Characterization of Fibrils from a Disease-Associated Alanine Expansion Domain using Proteolysis and Solid-State NMR Spectroscopy. J. Am. Chem. Soc. 2008;130:7172–7173. doi: 10.1021/ja800120s. [DOI] [PubMed] [Google Scholar]
- 42.Helmus JJ, Surewicz K, Surewicz WK, Jaroniec CP. Conformational Flexibility of Y145Stop Human Prion Protein Amyloid Fibrils Probed by Solid-State Nuclear Magnetic Resonance Spectroscopy. J. Am. Chem. Soc. 2010;132:2393–2403. doi: 10.1021/ja909827v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Colvin MT, et al. Atomic Resolution Structure of Monomorphic Abeta42 Amyloid Fibrils. J. Am. Chem. Soc. 2016;138:9663–9674. doi: 10.1021/jacs.6b05129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wälti MA, et al. Atomic-Resolution Structure of a Disease-Relevant Abeta(1-42) Amyloid Fibril. Proc. Natl. Acad. Sci. USA. 2016;113:E4976–4984. doi: 10.1073/pnas.1600749113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Xue K, et al. Magic-Angle Spinning Frequencies beyond 300 kHz Are Necessary To Yield Maximum Sensitivity in Selectively Methyl Protonated Protein Samples in Solid-State NMR. J. Phys. Chem. C. 2018;122:16437–16442. doi: 10.1021/acs.jpcc.8b05600. [DOI] [Google Scholar]
- 46.Henry GD, Weiner JH, Sykes BD. Backbone Dynamics of a Model Membrane-Protein - C-13 Nmr-Spectroscopy of Alanine Methyl-Groups in Detergent-Solubilized M13 Coat Protein. Biochemistry. 1986;25:590–598. doi: 10.1021/bi00351a012. [DOI] [PubMed] [Google Scholar]
- 47.Tritt-Goc J. Proton dipolar coupling tensors in barium nitroprusside trihydrate. J. Phys. Chem. Solids. 1995;56:935–942. doi: 10.1016/0022-3697(95)00011-9. [DOI] [Google Scholar]
- 48.Tritt-Goc J, Pi≈õlewski N, Haeberlen U. NMR chemical shift and asymmetric dipolar tensors of water protons in sodium nitroprusside (SNP) Chem. Phys. 1986;102:133–140. doi: 10.1016/0301-0104(86)85124-2. [DOI] [Google Scholar]
- 49.Chevelkov V, Rehbein K, Diehl A, Reif B. Ultrahigh Resolution in Proton Solid-State NMR Spectroscopy at High Levels of Deuteration. Angew. Chem., Int. Ed. 2006;45:3878–3881. doi: 10.1002/anie.200600328. [DOI] [PubMed] [Google Scholar]
- 50.Bockmann A, et al. Characterization of different water pools in solid-state NMR protein samples. J. Biomol. NMR. 2009;45:319–327. doi: 10.1007/s10858-009-9374-3. [DOI] [PubMed] [Google Scholar]
- 51.Dasari M, et al. Bacterial Inclusion Bodies of Alzheimer’s Disease beta-Amyloid Peptides Can Be Employed To Study Native-Like Aggregation Intermediate States. Chembiochem. 2011;12:407–423. doi: 10.1002/cbic.201000602. [DOI] [PubMed] [Google Scholar]
- 52.Shaka AJ, Keeler J, Frenkiel T, Freeman R. An Improved Sequence for Broad-Band Decoupling - Waltz-16. J. Magn. Reson. 1983;52:335–338. [Google Scholar]
- 53.Vuister GW, Bax A. Resolution Enhancement and Spectral Editing of Uniformly C-13-Enriched Proteins by Homonuclear Broad-Band C-13 Decoupling. J. Magn. Reson. 1992;98:428–435. [Google Scholar]
- 54.Bak M, Rasmussen JT, Nielsen NC. SIMPSON: A General Simulation Program for Solid-State NMR Spectroscopy. J. Magn. Reson. 2000;147:296–330. doi: 10.1006/jmre.2000.2179. [DOI] [PubMed] [Google Scholar]
- 55.Haeberlen, U. High resolution NMR in solids: selective averaging (Acad. Press, 1976).
- 56.Levitt, M. H. Spin dynamics: basics of nuclear magnetic resonance (Wiley, 2015).
- 57.Henry ER, Szabo A. Influence of Vibrational Motion on Solid-State Line-Shapes and Nmr Relaxation. J. Chem. Phys. 1985;82:4753–4761. doi: 10.1063/1.448692. [DOI] [Google Scholar]
- 58.Ishima R, Petkova AP, Louis JM, Torchia DA. Comparison of methyl rotation axis order parameters derived from model-free analyses of H-2 and C-13 longitudinal and transverse relaxation rates measured in the same protein sample. J. Am. Chem. Soc. 2001;123:6164–6171. doi: 10.1021/ja0104711. [DOI] [PubMed] [Google Scholar]
- 59.Woessner DE. Spin Relaxation Processes in a 2-Proton System Undergoing Anisotropic Reorientation. J. Chem. Phys. 1962;36:1-&. doi: 10.1063/1.1732274. [DOI] [Google Scholar]
- 60.Lipari G, Szabo A. Model-Free Approach to the Interpretation of Nuclear Magnetic-Resonance Relaxation in Macromolecules .2. Analysis of Experimental Results. J. Am. Chem. Soc. 1982;104:4559–4570. doi: 10.1021/ja00381a010. [DOI] [Google Scholar]
- 61.Nicholson LK, et al. Dynamics of Methyl-Groups in Proteins as Studied by Proton-Detected C-13 Nmr-Spectroscopy - Application to the Leucine Residues of Staphylococcal Nuclease. Biochemistry. 1992;31:5253–5263. doi: 10.1021/bi00138a003. [DOI] [PubMed] [Google Scholar]
- 62.Koetzle TF, Golic L, Lehmann MS, Verbist JJ, Hamilton WC. Precision neutron diffraction structure determination of protein and nucleic acid components. XV. Crystal and molecular structure of the amino acid L‐valine hydrochloride. J. Chem. Phys. 1974;60:4690–4696. doi: 10.1063/1.1680969. [DOI] [Google Scholar]
- 63.Zorin VE, Brown SP, Hodgkinson P. Origins of Linewidth in H1 Magic-Angle Spinning NMR. J. Chem. Phys. 2006;125:144508. doi: 10.1063/1.2357602. [DOI] [PubMed] [Google Scholar]
- 64.Kaikkonen A, Otting G. Residual dipolar (1)H-(1)H couplings of methyl groups in weakly aligned proteins. J. Am. Chem. Soc. 2001;123:1770–1771. doi: 10.1021/ja003674i. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.