

Abstract
Cryptosporidiosis is a human gastrointestinal disease caused by protozoans of the genus Cryptosporidium, which can be fatal in immunocompromised individuals. The essential enzyme, thymidylate synthase (TS), is responsible for de novo synthesis of deoxythymidine monophosphate. The TS active site is relatively conserved between Cryptosporidium and human enzymes. In previous work, we identified compound 1, (2-amino-4-oxo-4,7-dihydro-pyrrolo[2,3-d]pyrimidin-methyl-phenyl-L-glutamic acid), as a promising selective Cryptosporidium hominis TS (ChTS) inhibitor. In the present study, we explore the structure-activity relationship around 1 glutamate moiety by synthesizing and biochemically evaluating the inhibitory activity of analogues against ChTS and human TS (hTS). X-Ray crystal structures were obtained for compounds bound to both ChTS and hTS. We establish the importance of the 2-phenylacetic acid methylene linker in optimally positioning compounds 23, 24, and 25 within the active site. Moreover, through the comparison of structural data for 5, 14, 15, and 23 bound in both ChTS and hTS identified that active site rigidity is a driving force in determining inhibitor selectivity.
Keywords: SAR study, Antifolate, Cryptosporidium hominis, Thymidylate synthase, X-ray crystallography
Graphical Abstract

1. Introduction
Cryptosporidiosis is a human gastrointestinal illness caused by protozoans of the genus Cryptosporidium and is the second leading cause of diarrheal disease and deaths in children <2 years old across Africa and South Asia.[1–3] Additionally, outbreaks have occurred in industrialized countries, with major outbreaks occurring in Milwaukee and New York City.[4, 5] Among the several Cryptosporidium species that can cause human disease, Cryptosporidium hominis (C. hominis) and Cryptosporidium parvum (C. parvum) are responsible for 90 % of the human disease and share a high sequence identity (95–97%) at the genome level.[6–8] Cryptosporidium is capable of surviving without a host for many months at a wide range of temperatures.[9] Infection occurs when an individual ingest oocyst in contaminated food or water. Healthy individuals experience gastrointestinal distress that can last for two or more weeks. However, infection rates are higher and more severe in children, the elderly, and immunocompromised individuals, where it leads to severe and life-threatening wasting disease.[8–10] Currently, nitazoxanide is the only FDA-approved drug for the treatment of cryptosporidiosis.[10] However, the efficacy of nitazoxanide is variable in immunocompetent patients, limited in children, and ineffective in immunocompromised patients, indicating a pressing need for improved therapies.[1, 9, 10]
The essential enzymes, thymidylate synthase (TS) and dihydrofolate reductase (DHFR), have long been chemotherapeutic targets for cancer and infectious diseases.[11, 12] In humans and most organisms, these two enzymes exist as separate polypeptide chains, whereas in Cryptosporidium both activities are encoded into a single bifunctional enzyme TS-DHFR.[13, 14] TS-DHFR has been extensively characterized and shown to be a promising target for the development of inhibitors.[12, 15, 16] TS catalyzes the de novo synthesis of deoxythymidine monophosphate (dTMP) from deoxyuridine monophosphate (dUMP) and the cofactor 5,10-methylenetetrahydrofolate (CH2H4F).[11, 17] The dihydrofolate produced as the other product of the TS reaction is then reduced by DHFR utilizing the cofactor NADPH, to produce tetrahydrofolate and NADP+.[11, 18] The TS active site is highly conserved for both the C. hominis and the human enzymes, however there are two distinct, variant amino acids in the region of the binding pocket that interact with the glutamate moiety of folate.[19] In C. hominis TS (ChTS) these residues are A287 and S290, versus F80 and G83 in human TS (hTS), respectively.[11, 19] Our earlier kinetic studies with bifunctional ChTS-DHFR together with mutational and structural analyses identified this novel folate binding region containing the two variant residues, A287 and S290. These residues are critical to properly position the folate substrate for efficient interaction with the second substrate dUMP for methyl transfer during catalysis.[11, 20] In other species such as human, a bulky phenylalanine residue is located in this region. These studies suggested that this region might be exploited to build in increased inhibitor specificity and affinity for the ChTS-DHFR.
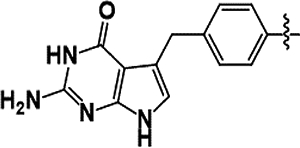
In previous work we identified compound 1 (2-amino-4-oxo-4,7-dihydro-pyrrolo[2,3-d]pyrimidin-methylphenyl-L-glutamic acid), originally designed as an anticancer agent, that proved to be a promising ChTS inhibitor (see Figure 1).[12, 16, 21] We reported the X-ray crystal structure of ChTS complexed with 1, in combination with inhibition activity of 1 demonstrating a 6-fold greater selectivity for the ChTS over the hTS enzyme.[12] This data suggested that these two variant active site residues might play an important role in determining selectivity. This concept was further supported by our recent work in which the X-ray crystal structure of 1 complexed with hTS was determined.[19] The variant ChTS residue S290 forms a hydrogen bond with the carboxylate of the glutamate moiety of 1 as well as hydrophobic interactions with A287, stabilizing this portion of compound 1 within the TS active site. In the case of hTS, a hydrogen bond with compound 1 glutamate moiety does not form, but rather hydrophobic interactions with F80 or interaction with solvent.
Figure 1.

Lead optimization guided by crystal structure of compound 1 bound to ChTS. A. Compound 1 bound in ChTS active site, hydrogen bond between γ-carboxylate with S290 denoted by black dotted line. B. Structure of compound 1 and scaffolds used for lead optimization.
In this present study, we describe a computational and structure-guided lead optimization strategy to explore the structure-activity-relationship (SAR) of compound 1 with the aim of identifying compounds which take advantage of the variant ChTS and hTS active site residues in an effort to increase potency and selectivity. A series of 26 compounds were designed and synthesized for this effort. The compounds were tested for inhibitory activity against both ChTS and hTS enzymes. A total of 12 X-ray crystal structures were determined for a number of these compounds bound in the ChTS active site to evaluate interactions that may be important for inhibitor potency. Additionally, we compare the X-ray crystal structures of four compounds bound in both ChTS and hTS active sites to understand inhibitor selectivity.
2. Results and Discussion
2.1. Lead optimization guided by computational and structural analyses
The three-dimensional structure of compound 1 bound the ChTS active site is shown in Figure 1(A) along with several scaffolds showing potential regions of the molecule to be targeted in the lead optimization efforts (B). Analysis of the crystal structure of compound 1 indicates that the accommodation of the pyrrolo[2,3-d]pyrimidine fragment of 1 in the ChTS active site is nearly ideal. The pyrrolo[2,3-d]pyrimidine π-stackes with the uracil ring of the FdUMP, with hydrogen bonds of normal lengths for all of the heteroatoms except N3 (its closest contact is 4.32 Å with the side-chain N of N319), and the central benzyl ring is well situated in a hydrophobic region and has an edge-to-face aryl-aryl interaction with F433. However, the amide linker moiety and its attachments are not making notable contacts. The α–carboxylate of the glutamate moiety has one oxygen 4.30 Å from the ammonium group of K284, while the γ–carboxylate forms a hydrogen bond with S290 (Figure 1A). Thus, our initial focus for analogues was on the eastern region of compound 1. Several possibilities were suggested by initial modeling based on the ChTS crystal structure with 1 (PDB: 40QE).[12] These include modifications which would retain the γ–carboxylate to preserve the hydrogen bond with S290 while substituting the α–carboxylic acid with an aryl ring to interact with hydrophobic pocket, changes to the amide linker, and additional functionalization of the aryl ring (Scaffold A, Fig. 1B). Modeling suggests that further elaboration of the aryl ring to substituted biphenyl compounds would also be viable (Scaffold B, Fig. 1B). Moreover, it has been shown that biphenyls are twisted 30° to provide some conformational variety.[22] This substitution pattern would feature a smaller group that projects towards K284, and a larger one for the opening between A287 and S290. The larger group could be used to control properties such as solubility as its terminus can project into the aqueous solvent. An example of a computed biphenyl structure is illustrated in Supplementary Figure S1.
2.2. Chemistry
A general outline of our synthetic strategy for the preparation of 2-amino-4-oxo-4,7-dihydro-pyrrolo[2,3-d]pyrimidin-methyl-phenyl-L-glutamic acid derivatives 2–26 is summarized in Schemes 1–3 and Experimental Section. A complete description of synthetic routes for the synthesis of the non-commercial amines is summarized in the Supplementary Information.
Scheme 1.

General scheme for the synthesis of compounds 2–9 and 11–25.
aReagents, conditions: (a) Et3N, DCM, r.t., o.n.; (b) MeI, NaH, DMF, r.t., o.n.; (c) Allyl alcohol, NaHCO3, Pd(OAc)2, Bu4NCl, 70 °C, o.n.; (d) 5,5-dibromo meldrum’s acid, HCl, THF, r.t., 18h; (e) 2,6-diaminopyrimidin-4-ol, NaOAc, MeOH/H2O, 45 °C, 2h; (f) 2N NaOH, MeOH/H2O, 60 °C, 1h; (g) NaN3, DMSO, 100 °C, o.n.
Scheme 3.

General scheme for the synthesis of compounds 26 and 27.
2.3. SAR Profiling using biochemical assays
A series of antifolate compounds were prepared to explore SAR based upon compound 1 and Scaffolds A and B (Figure 1B). The synthesized series of 26 antifolate compounds were biochemically evaluated against purified ChTS and hTS enzymes by monitoring the conversion of dUMP and CH2H4F to dTMP and H2F. Compound 1 was utilized as a positive antifolate control. TS inhibitory activity is summarized in (Tables 1 and 2 and Supplementary Table S1), and the SAR of these compounds with ChTS was investigated.
Table 1.
Compound 2–8 and 14–27 IC50 (μM) for ChTS
 |
|||||||
|---|---|---|---|---|---|---|---|
| Compounds | R1 | R2 | R3 | R4 | R5 | ChTS IC50 (μM) | ClogP |
| 2 | COO− | H | H | H | H | 20 ± 12 | 2.63 |
| 3 | H | COO− | H | H | H | 96 ± 10 | 2.47 |
| 4 | H | H | COO− | H | H | >100 | 2.49 |
| 5 | CH2COO− | H | H | H | H | 16 ± 1 | 1.96 |
| 6 | (CH2)2COO− | H | H | H | H | 114 ± 21 | 2.48 |
| 7 | Tetrazole | H | H | H | H | 159 ± 19 | 1.96 |
| 8 | CN | H | H | H | H | 123 ± 7 | 2.29 |
| 14 | COO− | H | H | Cl | H | 28 ± 6 | 3.28 |
| 15 | COO− | H | H | COO− | H | 18 ± 2 | 2.51 |
| 16 | COO− | H | H | OCH3 | H | 50 ± 8 | 2.66 |
| 17 | COO− | H | H | CN | H | 11 ± 1 | 2.36 |
| 18 | COO− | H | H | CH2OH | H | 181 ± 32 | 1.94 |
| 19 | COO− | H | H | CH2OCH3 | H | 68 ± 12 | 2.56 |
| 20 | COO− | H | H | CONH2 | H | 20 ± 4 | 1.42 |
| 21 | COO− | H | H | H | CH2OCH3 | >100 | 1.60 |
| 22 | COO− | H | OCH3 | H | H | 222 ± 77 | 2.66 |
| 23 | CH2COO− | H | H | OCH3 | H | 11 ± 1 | 1.99 |
| 24 | CH2COO− | H | H | CN | H | 9.1 ± 0.7 | 1.69 |
| 25 | CH2COO− | H | H | COO− | H | 10 ± 1 | 1.85 |
 |
|||||||
| 26 | O(CH2)2OCH3 | OCH3 | ~ ~ | ~ ~ | ~ ~ | 97 ± 44 | 3.42 |
| 27 | COO− | OCH3 | ~ ~ | ~ ~ | ~ ~ | >500 | 3.38 |
Table 2.
Compound 9–13 IC50 (μM) for ChTS
 |
|||
|---|---|---|---|
| Compound | ChTS IC50 (μM) | ClogP | |
| 9 | 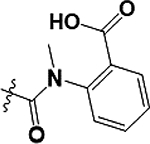 |
210 ± 81 | 2.87 |
| 10 | 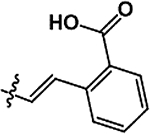 |
>100 | 3.60 |
| 11 | 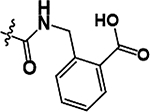 |
119 ± 13 | 1.81 |
| 12 | 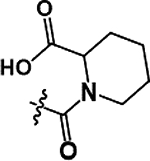 |
>500 | −0. 66 |
| 13 | 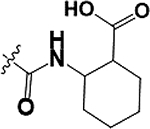 |
63 ± 8 | 1.90 |
As shown in Table 1 and Figure 2, the first compound synthesized, 2, involved replacement of the α-carboxylate of the glutamic acid moiety with an aryl ring containing a carboxyl acid at the ortho position to mimic the γ-carboxylate. Compound 2 had an IC50 = 20 μM. There was a ~ 5-fold loss of potency, when the carboxylate was in either the meta or para positions of the aryl ring, compounds 3 and 4, demonstrating a preference for the ortho position. The three-dimensional crystal structure for 2 in complex with the ChTS-DHFR protein was solved (see Supplementary Figure S2A). The structural analysis revealed that the ortho carboxylic acid was oriented toward S290, however the distance was too short to allow a hydrogen bond (see Figure 7, below). Accordingly, incorporation of a methylene linker to extend the ortho-carboxylate by one carbon unit from the aryl ring, in order to replicate the hydrogen bond observed in the crystal structure between 1 and S290, resulted in compound 5, which did result in a small increase in potency. The increased linker length allowed formation of the hydrogen bond with S290 as verified by the determination of the crystal structure of 5 bound to the ChTS active site (see Figure 7, below, and Supplementary Figure S2B). However, the addition of the longer ethylene linker resulted in a ~ 6-fold loss of potency for 6. Isosteric replacement of the carboxylate moiety with other functional groups that could form a hydrogen bond, such as a tetrazole or a cyano, also resulted in an ~ 8-fold and ~ 6-fold loss of potency, respectively, compound 7 and 8.
Figure 2.

Structure and ChTS IC50 values for compounds 2–8, modifications made to carboxylate (red). Coloration of compound 2 denotes modifications made during SAR, amide linker moiety (blue), aromaticity (pink), carboxylate (red), and substitutions to C-3, C-4, and C-5 positions (purple, green, and gold, respectively).
Figure 7.

Comparison of ChTS bound to 2 (light green/warm pink), ChTS bound to 5 (green/tan) and ChTS bound to 6 (dark green/cyan) with dUMP (yellow) (PDB: 6PF7, 6PF9, and 6PFB). A. Comparison of 2, 5, and 6 bound in ChTS active site, hydrogen bonds to 5 in black and to 6 in gray. B. Top view of benzoic acid, 2-phenylacetic acid, and 3-phenylpropanoic acid moiety for 2, 5, and 6, respectively.
As illustrated in Table 2 and Figure 3, in general modifications made to the amide linker moiety resulted in substantial decrease in potency. The addition of a methyl group to produce a tertiary amine resulted in an ~11-fold decrease in potency, compound 9. Removing the amide linker and replacing with an ethylene linker resulted in a greater than 5-fold decrease in potency, compound 10. Increasing the length of the linker region by inserting a methylene to elongate the linker region by one carbon unit resulted in a 6-fold loss in potency, compound 11. Compound 12 demonstrated a greater than 25-fold decrease in potency. Here the loss of aromaticity is combined with the creation of a piperidine ring, which shortens the amide linker moiety by insertion of a secondary amine into the ring structure. However, the loss of aromaticity alone resulted in a 3-fold loss of potency, compound 13.
Figure 3.

Structures and ChTS IC50 values for compounds 9–13, modifications made to the amide linker moiety (blue), and aromaticity (pink).
Based on the ortho substituted compound 2, a series of substituents were added to the C-4 position of the aryl ring to take advantage of the pocket surrounding K284 to improve potency as shown in Table 1 and Figure 4. Hydrophilic substituents in general were well tolerated, compounds 15, 17 and 20. While the addition of a carboxamide or carboxylate at C-4 maintained potency compared to 2, where the addition of a cyano at this position enhanced potency 2-fold in compound 17. This is confirmed by the crystal structure (Figure 9 and Supplementary Figure S3D), described below. However, the addition of a methoxy group or a hydroxymethyl group at the C-4 position resulted in a ~ 2.5-fold and ~ 9-fold decrease in potency, respectively, compound 16 and 18. The addition of a chloride substituent to the C-4 position resulted in a small loss of potency, compound 14. The addition of a methoxymethyl substituent to the C-4 position resulted in ~ 3.5-fold loss in potency for compound 19. Additionally, the addition of methoxymethyl or methoxy substituents to the aryl moiety at the C-3 and C-5 positions were not well tolerated, with a greater than 5-fold loss of potency, compound 21 and 22.
Figure 4.

Structures and ChTS IC50 values for compounds 14–22, modifications made to the C-3 (purple), C-4 (green), and C-5 (gold) positions.
Figure 9.

Comparison of ChTS bound to 17 (green/navy) and ChTS bound to 24 (dark green/teal) with dUMP (yellow) (PDB: 6PFF, and 6PFH). A. Comparison of 17 and 24 bound in ChTS active site, hydrogen bonds to 17 in gray and to 24 in black. B. Zoom in view rotation of 24 2-phenylacetic acid moiety with respect to 17 benzoic acid moiety. C. Zoom in view of shift of pyrrolo[2,3-d]pyrimidine moiety of 17 and 24.
As shown in Table 1 and Figure 5, the combination of an ortho-phenylacetic acid moiety with a substituent at the C-4 position of the aryl ring resulted in ~ 2-fold increase in potency, compounds 23–25. These compounds were designed to form a hydrogen bond with S290 as well as interact with the pocket surrounding K284. These interactions are supported by the crystal structure (Figure 9 and Supplementary Figures S5 and S6), as described below.
Figure 5.

Structures and ChTS IC50 values for compounds 23–25, modifications made carboxylate (red), and C-4 (green) positions.
Finally, as illustrated in Table1 and Figure 6, elaboration of the aryl ring to a biphenyl, suggested by modeling to be favorable, resulted in a substantial drop in potency. The biphenyl moiety was poorly tolerated resulting in a loss of potency ~ 5-fold and greater than 25-fold for compounds 26 and 27 respectively.
Figure 6.

Structures and ChTS IC50 values for compounds 26–27.
2.4. X-ray crystal structures of antifolates complexed with ChTS
All 26 antifolate compounds were evaluated by biochemical assays to assess inhibition of ChTS catalytic activity. The compounds were then further evaluated with structural analyses. Co-crystal structures were obtained for 12 antifolate compounds: 2, 5, 6, 11, 14, 15, 16, 17, 20, 23, 24, and 25 with ChTS-DHFR. As further described in the Experimental section, structures of ChTS-DHFR complexed with antifolate compounds were obtained utilizing the same procedures. The resolution of the structures range from 2.5 to 3.3 Å and all twelve structures have been deposited in the Protein Data Bank. The co-crystal structures contained five protein molecules within the asymmetric subunit and crystallized as a C2 space group with a similar crystal lattice as reported for ChTS-DHFR complexed with 1.[12]
The antifolate compounds bind into the 5,10-methylenetetrahydrofolate (CH2H4F) pocket where the pyrrolo[2,3-d]pyrimidine and centralbenzyl moieties form similar interactions with ChTS active site as previously described for 1 (Supplementary Figures S2–4).[12] However, the pyrrolo[2,3-d]pyrimidine of compounds 2, 6, 15, 17, and 20 form fewer hydrogen bonding interactions with the ChTS active site (Supplementary Figure S2A, S2C, S3B, S3D, and S4A). Compounds 6, 15 and 17 do not form hydrogen bonding interactions with the backbone N of G430. Additionally, 15 and 17 do not form hydrogen bonding interactions with D426. All five compounds do not form a hydrogen bond with Y466.
The carboxylate group of the benzoic acid, 2-phenylacetic acid, and 3-phenylpropanoic acid moieties are directed towards S290. With an average distance of ~ 3.9 Å, the carboxylate group for compounds containing a benzoic acid moiety were not within hydrogen bonding distance to S290. The carboxylate group for compounds containing either 2-phenylacetic acid, or 3-phenylpropanoic acid moieties, with an average distance of ~3.3 Å, were within hydrogen bonding distance to S290. Compounds 2, 5, and 6, which contain a benzoic acid, 2-phenylacetic acid, and 3-phenylpropanoic acid moiety, respectively, illustrates the importance of linker length on inhibitor potency. Comparing the three compounds it is observed that the carboxylate of 2 is ~4.0 Å from S290, while the methylene linker of 5 extends the carboxylate within hydrogen bonding distance, ~ 3.3 Å, to S290 (Figure 7). The longer linker used in 6 enables the carboxylate to hydrogen bond with S290, however there is greater disorder due to the linker length resulting in the loss of two hydrogen bonds to the pyrrolo[2,3-d]pyrimidine moiety (Supplementary Figure S2C).
Compound 11 differs from 2 by the addition of a methylene to the amide linker moiety, which resulted in a 6-fold loss of potency. Comparing the two structures of 2 and 11, the carboxylate of both compounds is ~ 4.0 Å from S290 (Figure 8A). In Figure 8B, we observe the benzoic acid moiety shifts 1.4 Å into the pocket, while shifting the amide linker towards M519 by ~ 0.9 Å. The addition of the methylene to this region destabilizes the binding of 11 by increasing the compounds flexibility within the binding pocket.
Figure 8.

Comparison of ChTS bound to 2 (green/warm pink) and ChTS bound to 11 (dark green/lavender) with dUMP (yellow) (PDB: 6PF7, and 6PFA). A. Comparison of 2 and 11 bound in ChTS active site, carboxylate group for both compounds are similar distance from S290 B. Zoom in view of 11 benzoic acid moiety compared to 2.
C-4 substituents to either benzoic acid or 2-phenylacetic acid moieties were directed into a small pocket adjacent to K284. Cyano, carboxylate, or carboxamide C-4 substituents, 15 and 25, 17 and 24, or 20, respectively, where within hydrogen bonding distance of the ammonium of K284, while the C-4 substituent of 20 was also within hydrogen bonding distance of the backbone carbonyl of I515 (Supplementary Figure S3B, S3D, S4A, S4C and S4D). Compounds 23, 24, and 25, which combine a C-4 substituent, in this case a methoxy, cyano or carboxylate, respectively, with the 2-phenylacetic acid moiety, demonstrate the greatest ChTS potency for this series of compounds. A structural comparison of compounds 23, 24, and 25 with the benzoic acid moiety counterparts 16, 17, and 15, respectively, reveals subtle differences in the way these compounds interact with the ChTS active site. First the carboxylate group of compounds 23, 24, and 25 is within hydrogen bonding distance of S290, while the carboxylate of the benzoic acid counterparts is an average ~ 3.8 Å away from this residue (Figure 9A and Supplementary Figure S5A and S6A). This hydrogen bond with S290 coupled with the C-4 substituents interactions within the pocket containing K284 stabilizes the 2-phenylacetic acid moiety within the ChTS active site. Secondly, the addition of the methylene linker in the 2-phenylacetic acid moiety of compounds 23, 24, and 25 produces ~ 22.0° clockwise rotation of this moiety with respect to their benzoic acid moiety counter parts (Figure 9B and Supplementary Figure S5B and S6B). This small rotation appears to transfer to the pyrrolo[2,3-d]pyrimidine moiety resulting in an average shift of ~ 0.3 Å into the pocket, stabilizing the hydrogen bonding interactions between the pyrrolo[2,3-d]pyrimidine and ChTS active site (Figure 9C and Supplementary Figure S5C and S6C).
2.5. X-ray crystal structures of antifolates complexed with hTS
All antifolate compounds where evaluated for structural analysis with hTS, however, co-crystal structures were obtained for antifolate compounds 5, 14, 15, and 23. A full description of the procedures used to obtain structures of hTS complexed with the antifolate compounds can be found in the Experimental section. The resolution of the structures range from 2.39 to 2.84 Å and all four structures have been deposited in the Protein Data Bank. The co-crystal structures contained four protein molecules within the asymmetric subunit, with 14 and 23, 5, and 15 crystalizing in a C2, P212121, and P41212 space groups, respectively. In all structures, density for the antifolate compounds was observed in all four chains of the asymmetric unit except for the structure for 15, where density was only observed in chains B and C.
The antifolate compounds bind into the CH2H4F pocket where the pyrrolo[2,3-d]pyrimidine and central benzyl moieties form similar interactions with the hTS active site as previously described for 1.[19] However, the pyrrolo[2,3-d]pyrimidine of compounds 5, 14, and 23 form fewer hydrogen bonding interactions with the hTS active site (Supplementary Figure S7A, S7B, and S7D). Compound 5 does not form hydrogen bonding interactions with the backbone carbonyl of A312, but this could be an artifact due to a lack of density to visualize the neighboring residue V313. Compound 14 and 23 do not form hydrogen bonding interactions with Q112. The carboxylate group of the benzoic acid, and 2-phenylacetic acid moieties are directed towards solvent, while C-4 substituents are directed towards the pocket containing residue K77. Additionally, the aryl ring of both the benzoic acid, and 2-phenylacetic acid moieties formed hydrophobic interactions with F80 (Supplementary Figure S7). Interestingly, unlike the positioning of the amide linker moiety for these compounds bound in ChTS, the amide linker moiety for all four compounds bound in the opposite orientation where the carbonyl points into the pocket near F225 (Supplementary Figure S7).
Comparing the structures of 5, 14, 15, and 23 in both the ChTS and hTS active sites provides a possible explanation for the observed trend in inhibitory selectivity the antifolate compounds display for both enzymes (Supplementary Table S1). As previously reported in our work with R-and S- enantiomers of 1, we observed that the ChTS active site is more rigid than the hTS active site.[19] Similarly here the hTS residue F225 rotates ~ 75° clockwise, as compared to its corresponding ChTS residue F433 for all four compound structures (Figure 10A and Supplementary Figure S8A, S9A, and S10A). The space afforded by the rotation of F225 allows the central benzyl moiety to shift back into the pocket towards F225 by ~ 0.8 Å, while providing space for the compound’s amide linker moiety carbonyl (Figure 10B and Supplementary Figure S8B, S9B, and S10B). The reverse orientation of the amide linker moiety appears to position the benzoic acid, and 2-phenylacetic acid moieties aryl rings towards F80, allowing for a more optimal position to hydrophobically interact with F80. The positioning of the compounds towards F80 provides space for the sidechain of L221 to rotate ~180° into the pocket as compared to its ChTS counterpart L429 (Figure 10C and Supplementary Figure S8C, S9C, and S10C). Taken together, hTS structural data and IC50 values indicate that with few exceptions the hTS active site is capable of accommodating most alterations in the compounds used in this SAR.
Figure 10.

Comparison of hTS bound to 14 (blue/dark red) and ChTS bound to 14 (green/purple) with dUMP (yellow) (PDB: 6PF8, and 6PF3). A. Comparison of 14 bound in hTS and ChTS active site. B. Zoom in view hTS residue F225 and ChTS residue F433 position upon binding 14. C. Zoom in view hTS residue L221 and ChTS residue L429 position upon binding 14.
3. Conclusions
We have designed, synthesized and evaluated a series of anti-cryptosporidium agents based on the chemical structure of lead compound 1, where the glutamate moiety is replaced by a benzoic acid. Structure-activity relationship analysis indicates (1) ChTS prefers an ortho-phenylacetic acid moiety over an ortho-benzoic acid or ortho-phenylpropanoic acid moieties (2) hydrophilic C-4 substituents on the 2-phenylacetic acid moieties increase potency (3) changes to the amide linker moiety are not tolerated. X-ray crystal structures of compounds 23, 24, and 25 bound to the ChTS active site reveal the key interactions that are necessary for compound potency, as well as the role that the 2-phenylacetic acid methylene linker plays in optimally positioning these compounds into the active site. Comparison of structural data for compounds 5, 14, 15, and 23 bound in both ChTS and hTS identified that active site structural rigidity is a driving factor in determining inhibitor selectivity. In summary, the results presented here provide a platform for further efforts to develop new anti-cryptosporidium agents.
4. Experimental section
4.1. General Information
NMR spectra were recorded on Agilent DD2 600 (600 MHz), DD2 500 (500 MHz) and DD2 400 (400 MHz) instruments. Column chromatography was carried out using CombiFlash over redisep column cartridges employing Merck silica gel (Kieselgel 60, 63–200 μm). Pre-coated silica gel plates F-254 were used for thinlayer analytical chromatography. Mass determinations were performed using electrospray ionization on a Waters Micromass ZQ (LC-MS) and on an Agilent Technologies 6890N (GC-MS). HRMS (ESI-TOF) analyses were performed on Waters Xevo QTOF equipped with Z-spray electrospray ionization source. HRMS values agree within ± 0.4 % of the theoretical values. The purity of all final synthesized compounds was determined by reverse phase HPLC, using Waters 2487 dual λ absorbance detector with a Waters 1525 binary pump and a Phenomenex Luna 5μ C18(2) 250 × 4.6 mm column. Samples were run at 1mL/min using gradient mixtures of 5–100% of water with 0.1% trifluoroacetic acid (TFA) (A) and 10:1 acetonitrile:water with 0.1% TFA (B) for 22 min followed by 3 min at 100% B. Additional details are provided in Supplementary Information.
4.2. General procedure for the synthesis of intermediates 29a-u. Method a.
Intermediates 29a-u have been obtained through general method a. To a solution of corresponding amine (28a-u) (1eq) in anhydrous dichloromethane (3.16 mL/eq) and dry triethylamine (1.10 eq), was added 4-iodobenzoylchloride (1.10 eq). The reaction was stirred overnight at room temperature. Solvent was evaporated under reduced pressure and the desired intermediates purified by flash column chromatography.[23]
4.2.1. methyl 1-(4-iodobenzoyl)piperidine-2-carboxylate (29a)
(hexanes to ethyl acetate). White powder, 0.54 g, 1.44 mmol, 37% yield. 1H NMR (400 MHz, CDCl3) δ 7.79 – 7.73 (m, 2H), 7.21 – 7.16 (m, 2H), 5.51 – 5.44 (m, 1H), 3.78 (s, 3H), 3.62 – 3.56 (m, 1H), 3.31 – 3.18 (m, 1H), 2.37 – 2.31 (m, 1H), 1.78 – 1.74 (m, 3H), 1.43 – 1.35 (m, 2H). LC-MS (ESI) m/z 374.1 [M+H]+.
4.2.2. methyl 2-(4-iodobenzamido)cyclohexane-1-carboxylate (29b)
(hexanes to hexanes/ dichloromethane 1:1). White powder, 0.54 g, 1.39 mmol, 32% yield. 1H NMR (400 MHz, CDCl3) δ 7.80 – 7.74 (m, 2H), 7.52 – 7.46 (m, 2H), 7.29 (s, 1H), 4.30 (qd, J = 9.0, 7.9, 4.4 Hz, 1H), 3.72 (s, 3H), 2.91 (q, J = 4.6 Hz, 1H), 2.24 – 2.13 (m, 1H), 1.83 – 1.51 (m, 5H), 1.51 – 1.40 (m, 1H), 1.33 – 1.20 (m, 1H). LC-MS (ESI) m/z 388.1 [M+H]+.
4.2.3. methyl 2-(4-iodobenzamido)benzoate (29c)
(hexanes/dichloromethane 8:2 to dichloromethane). White powder, 0.66 g, 1.73 mmol, 87% yield. 1H NMR (400 MHz, CDCl3) δ 12.07 (s, 1H), 8.90 (d, J = 8.5 Hz, 1H), 8.12 – 8.06 (m, 1H), 7.91 – 7.86 (m, 2H), 7.80 – 7.74 (m, 2H), 7.65 – 7.58 (m, 1H), 7.17 – 7.11 (m, 1H), 3.97 (s, 3H). LC-MS (ESI) m/z 382.0 [M+H]+.
4.2.4. methyl 3-(4-iodobenzamido)benzoate (29d)
(hexanes to ethyl acetate). White powder, 0.97 g, 2.55 mmol, 68% yield. 1H NMR (400 MHz, CDCl3) δ 8.16 – 8.09 (m, 1H), 8.05 – 8.01 (m, 1H), 7.92 (s, 1H), 7.88 – 7.80 (m, 3H), 7.63 – 7.58 (m, 2H), 7.46 (t, J = 7.9 Hz, 1H), 3.92 (s, 3H). LC-MS (ESI) m/z 382.1 [M+H]+.
4.2.5. methyl 4-(4-iodobenzamido)benzoate (29e)
(hexanes to ethyl acetate). White powder, 0.29 g, 0.76 mmol, 20% yield. 1H NMR (400 MHz, CDCl3) δ 8.09 – 8.05 (m, 2H), 7.88 – 7.85 (m, 2H), 7.75 – 7.70 (m, 2H), 7.63 – 7.59 (m, 2H), 3.92 (s, 3H). LC-MS (ESI) m/z 382.1 [M+H]+.
4.2.6. methyl 2-(2-(4-iodobenzamido)phenyl)acetate (29f)
(hexanes/dichloromethane 8:2 to dichloromethane). White powder, 1.15 g, 2.91 mmol, 78% yield. 1H NMR (500 MHz, CDCl3) δ 9.78 (s, 1H), 8.02 (d, J = 8.1 Hz, 1H), 7.90 – 7.84 (m, 2H), 7.80 – 7.74 (m, 2H), 7.39 – 7.35 (m, 1H), 7.27 – 7.22 (m, 1H), 7.18 – 7.13 (m, 1H), 3.76 (s, 3H), 3.68 (s, 2H). LC-MS (ESI) m/z 396.0 [M+H]+.
4.2.7. methyl 3-(2-(4-iodobenzamido)phenyl)propanoate (29g)
(hexanes to ethyl acetate). White powder, 6.54 g, 15.99 mmol, 84% yield. 1H NMR (400 MHz, CDCl3) δ 9.75 (s, 1H), 7.93 – 7.80 (m, 5H), 7.31 – 7.24 (m, 1H), 7.23 – 7.13 (m, 2H), 3.67 (s, 3H), 2.91 (dd, J = 7.1, 4.7 Hz, 2H), 2.79 (dd, J = 7.1, 4.6 Hz, 2H) LC-MS (ESI) m/z 410.1 [M+H]+.
4.2.8. N-(2-cyanophenyl)-4-iodobenzamide (29h)
(hexanes to ethyl acetate). White powder, 5.82 g, 16.7 mmol, 84% yield. 1H NMR (400 MHz, CDCl3) δ 8.56 (d, J = 8.4 Hz, 1H), 8.35 (s, 1H), 7.93 – 7.87 (m, 2H), 7.70 – 7.61 (m, 4H), 7.26 – 7.20 (m, 1H). LC-MS (ESI) m/z 349.1 [M+H]+.
4.2.9. methyl 2-((4-iodobenzamido)methyl)benzoate (29i)
(hexanes to hexanes/ethyl acetate 6:4). White powder, 0.53 g, 1.35 mmol, 75% yield. 1H NMR (400 MHz, CDCl3) δ 7.99 (dd, J = 7.8, 1.4 Hz, 1H), 7.77 – 7.71 (m, 2H), 7.64 (dd, J = 7.7, 1.4 Hz, 1H), 7.60 (t, J = 6.8 Hz, 1H), 7.56 – 7.46 (m, 3H), 7.37 (td, J = 7.6, 1.4 Hz, 1H), 4.78 (d, J = 6.5 Hz, 2H), 3.95 (s, 3H). LC-MS (ESI) m/z 396.1 [M+H]+.
4.2.10. methyl 4-chloro-2-(4-iodobenzamido)benzoate (29j)
(hexanes/dichloromethane 8:2 to dichloromethane). White powder, 0.96 g, 2.31 mmol, 62% yield. 1H NMR (400 MHz, CDCl3) δ 12.10 (s, 1H), 9.01 (d, J = 2.0 Hz, 1H), 8.02 (d, J = 8.6 Hz, 1H), 7.93 – 7.84 (m, 2H), 7.80 – 7.71 (m, 2H), 7.11 (dd, J = 8.6, 2.1 Hz, 1H), 3.97 (s, 3H). LC-MS (ESI) m/z 416.1 [M+H]+.
4.2.11. dimethyl 2-(4-iodobenzamido)terephthalate (29k)
(hexanes to hexanes/ethyl acetate 8:2). White powder, 0.43 g, 0.98 mmol, 98% yield. 1H NMR (400 MHz, CDCl3) δ 12.03 (s, 1H), 9.52 (d, J = 1.6 Hz, 1H), 8.15 (d, J = 8.3 Hz, 1H), 7.92 – 7.87 (m, 2H), 7.81 – 7.75 (m, 3H), 4.00 (s, 3H), 3.96 (s, 3H). LC-MS (ESI) m/z 440.1 [M+H]+.
4.2.12. methyl 2-(4-iodobenzamido)-4-methoxybenzoate (29l)
(hexanes to hexanes/ethyl acetate 8:2). White powder, 0.59 g, 1.43 mmol, 95% yield. 1H NMR (400 MHz, DMSO-d6) δ 11.93 (s, 1H), 8.32 (s, 1H), 8.02 – 7.99 (m, 2H), 7.87 (d, J = 7.9 Hz, 1H), 7.76 – 7.68 (m, 2H), 6.82 (d, J = 7.9 Hz, 1H), 3.88 (s, 3H), 3.86 (s, 3H). LC-MS (ESI) m/z 412.1 [M+H]+.
4.2.13. methyl 4-cyano-2-(4-iodobenzamido)benzoate (29m)
(hexanes to hexanes/ethyl acetate 8:2). White powder, 0.56 g, 1.34 mmol, 92% yield. 1H NMR (400 MHz, DMSO-d6) δ 11.41 (s, 1H), 8.72 (d, J = 1.6 Hz, 1H), 8.10 (d, J = 8.1 Hz, 1H), 8.02 – 8.00 (m, 2H), 7 7.74 – 7.71 (m, 3H), 3.89 (s, 3H). LC-MS (ESI) m/z 407.1 [M+H]+.
4.2.14. methyl 4-(hydroxymethyl)-2-(4-iodobenzamido)benzoate (29n)
(hexanes to hexanes/ethyl acetate 8:2). White powder, 0.95 g, 2.31 mmol, 45% yield. 1H NMR (400 MHz, CDCl3) δ 12.11 (s, 1H), 9.03 (d, J = 1.7 Hz, 1H), 8.10 (d, J = 8.2 Hz, 1H), 7.91 – 7.86 (m, 2H), 7.78 – 7.74 (m, 2H), 7.18 (dd, J = 8.2, 1.7 Hz, 1H), 5.41 (s, 2H), 3.97 (s, 3H). LC-MS (ESI) m/z 412.1 [M+H]+.
4.2.15. methyl 2-(4-iodobenzamido)-4-(methoxymethyl)benzoate (29o)
(hexanes to hexanes/ethyl acetate 8:2). White powder, 0.26 g, 0.62 mmol, 61% yield. 1H NMR (400 MHz, DMSO-d6) δ 11.60 (s, 1H), 8.52 (d, J = 1.6 Hz, 1H), 8.02 – 7.98 (m, 3H), 7.75 – 7.71 (m, 2H), 7.19 (dd, J = 8.1, 1.6 Hz, 1H), 4.51 (s, 2H), 3.89 (s, 3H), 3.35 (s, 3H). LC-MS (ESI) m/z 426.1 [M+H]+.
4.2.16. methyl 4-carbamoyl-2-(4-iodobenzamido)benzoate (29p)
(hexanes to dichloromethane). White powder, 0.25 g, 0.58 mmol, 13% yield. 1H NMR (400 MHz, DMSO-d6) δ 11.39 (bs, 1H), 8.80 (d, J = 1.7 Hz, 1H), 8.16 (bs, 1H), 8.05 – 7.98 (m, 3H), 7.77 – 7.72 (m, 2H), 7.69 (dd, J = 8.2, 1.7 Hz, 1H), 7.60 (bs, 1H), 3.88 (s, 3H). LC-MS (ESI) m/z 425.1 [M+H]+.
4.2.17. methyl 2-((4-iodophenyl)amino)-3-(methoxymethyl)benzoate (29q)
(hexanes to hexanes/ethyl acetate 1:1). White powder, 0.48 g, 1.18 mmol, 62% yield. 1H NMR (400 MHz, DMSO-d6) δ 11.39 (bs, 1H), 8.80 (d, J = 1.7 Hz, 1H), 8.16 (bs, 1H), 8.05 – 7.98 (m, 3H), 7.77 – 7.72 (m, 2H), 7.69 (dd, J = 8.2, 1.7 Hz, 1H), 7.60 (bs, 1H), 3.88 (s, 3H). LC-MS (ESI) m/z 425.1 [M+H]+.
4.2.18. methyl 2-(4-iodobenzamido)-5-methoxybenzoate (29r)
(hexanes to ethyl acetate). White powder, 1.17 g, 2.85 mmol, 86% yield. 1H NMR (400 MHz, CDCl3) δ 11.80 (s, 1H), 8.82 (d, J = 9.3 Hz, 1H), 7.88 – 7.84 (m, 2H), 7.77 – 7.72 (m, 2H), 7.57 (d, J = 3.1 Hz, 1H), 7.18 (dd, J = 9.3, 3.1 Hz, 1H), 3.97 (s, 3H), 3.84 (s, 3H). LC-MS (ESI) m/z 412.1 [M+H]+.
4.2.19. methyl 2-(2-(4-iodobenzamido)-4-methoxyphenyl)acetate (29s)
(hexanes to hexanes/ethyl acetate 8:2). White powder, 0.26 g, 0.6 mmol, 30% yield. 1H NMR (400 MHz, CDCl3) δ 9.83 (s, 1H), 7.91 – 7.84 (m, 2H), 7.80 – 7.73 (m, 2H), 7.71 (d, J = 2.6 Hz, 1H), 7.12 (d, J = 8.5 Hz, 1H), 6.70 (dd, J = 8.5, 2.7 Hz, 1H), 3.83 (s, 3H), 3.75 (s, 3H), 3.61 (s, 2H). LC-MS (ESI) m/z 426.1 [M+H]+.
4.2.20. methyl 2-(4-cyano-2-(4-iodobenzamido)phenyl)acetate (29t)
(hexanes to ethyl acetate). White powder, 2.38 g, 5.66 mmol, 82% yield. 1H NMR (400 MHz, CDCl3) δ 9.96 (s, 1H), 8.45 (d, J = 1.2 Hz, 1H), 7.90 – 7.87 (m, 2H), 7.77 – 7.74 (m, 2H), 7.42 (dd, J = 7.9, 1.5 Hz, 1H), 7.35 (d, J = 7.9 Hz, 1H), 3.79 (s, 3H), 3.74 (s, 2H). LC-MS (ESI) m/z 421.1 [M+H]+.
4.2.21. methyl 3-(4-iodobenzamido)-4-(2-methoxy-2-oxoethyl)benzoate (29u)
(hexanes to ethyl acetate). White powder, 2.38 g, 5.66 mmol, 82% yield. 1H NMR (500 MHz, DMSO-d6) δ 1H NMR (500 MHz, DMSO-d6) δ 10.17 (s, 1H), 7.99 (d, J = 1.9 Hz, 1H), 7.96 – 7.92 (m, 2H), 7.82 (dd, J = 8.0, 1.8 Hz, 1H), 7.73 – 7.69 (m, 2H), 7.50 (d, J = 8.0 Hz, 1H), 3.86 (s, 3H), 3.85 (s, 2H), 3.51 (s, 3H). LC-MS (ESI) m/z 454.1 [M+H]+.
4.3. General procedure for the synthesis of intermediate 29c. Method b.
A mixture of methyl 2-(4-iodobenzamido)benzoate (29c). (0.823 mg, 2.16 mmol) and NaH (121 mg, 3 mmol, 60% in oil) in anhydrous DMF (20 mL) was stirred at 0 °C for 15 minutes. Then, MeI (366 mg, 2.6 mmol) was added and the reaction was stirred at room temperature until completion. Then reaction was quenched by addition of MeOH and concentrated under reduced pressure. Desired intermediate was purified by flash column chromatography (0–50 % EtOAc in hexanes) to provide 551 mg (1.39 mmol, 65% yield) of methyl 2-(4-iodo-N-methylbenzamido)benzoate (29ć) as yellow oil.
4.3.1. methyl 2-(4-iodo-N-methylbenzamido)benzoate (29ć)
1H NMR (400 MHz, Methanol-d4) δ 7.79 (d, J = 7.9 Hz, 1H), 7.58 (t, J = 7.6 Hz, 1H), 7.55 – 7.49 (m, 2H), 7.45 (d, J = 8.0 Hz, 1H), 7.36 (t, J = 7.6 Hz, 1H), 7.01 – 6.93 (m, 2H), 3.85 (s, 3H), 3.40 (s, 3H). LC-MS (ESI) m/z 396.0 [M+H]+.
4.4. General procedure for the synthesis of intermediates 30a-u. Method c.
The syntheses of compounds 30a-u have been carried out according to general method c. To a solution of 29a-u (1 eq) in anhydrous DMF (11 mL/mmol) allyl alcohol (1.10 eq), NaHCO3 (2.5 eq), Pd(OAc)2 (0.06 eq) and tetrabutylammonium chloride (0.5 eq) were added and the mixture was stirred at 70 °C overnight. The reaction was cooled to room temperature and then ethyl acetate (50 mL/mmol) was added and the solution washed with saturated aqueous NH4Cl (3 × 100 mL/mmol). The organic phase was dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure. The desired product was obtained as a brown solid and was used in subsequent reaction without purification.[12]
4.4.1. methyl 1-(4-(3-oxopropyl)benzoyl)piperidine-2-carboxylate (30a)
(390 mg, 1.29 mmol, 91% yield). 1H NMR (400 MHz, CDCl3) δ 9.83 (t, J = 1.3 Hz, 1H), 7.40 – 7.35 (m, 2H), 7.26 – 7.21 (m, 2H), 5.54 – 5.45 (m, 1H), 3.78 (s, 3H), 3.70 – 3.62 (m, 1H), 3.27 – 3.18 (m, 1H), 2.98 (t, J = 7.2 Hz, 2H), 2.80 (t, J = 7.5 Hz, 3H), 2.38 – 2.30 (m, 1H), 1.82 – 1.73 (m, 3H), 1.48 – 1.37 (m, 2H). LC-MS (ESI) m/z 304.1 [M+H]+.
4.4.2. methyl 2-(4-(3-oxopropyl)benzamido)cyclohexane-1-carboxylate (30b)
(406 mg, 1.28 mmol, 91% yield). 1H NMR (400 MHz, CDCl3) δ 1H NMR (400 MHz, CDCl3) δ 1H NMR (400 MHz, Chloroform-d) δ 9.82 (t, J = 1.3 Hz, 1H), 7.73 – 7.68 (m, 2H), 7.27 – 7.23 (m, 3H), 4.32 (qd, J = 9.4, 4.5, 3.8 Hz, 2H), 3.71 (s, 3H), 2.99 (t, J = 7.4 Hz, 2H), 2.95 – 2.88 (m, 2H), 2.84 – 2.75 (m, 2H), 2.23 – 2.18 (m, 2H), 1.80 – 1.41 (m, 4H). LC-MS (ESI) m/z 318.1 [M+H]+.
4.4.3. methyl 2-(4-(3-oxopropyl)benzamido)benzoate (30c)
(338 mg, 1.09 mmol, 58% yield). 1H NMR (400 MHz, CDCl3) δ 12.02 (s, 1H),) , 9.85 (t, J = 1.2 Hz, 1H), 8.93 (d, J = 8.5 Hz, 1H), 8.12 – 8.07 (m, 1H), 8.01 – 7.96 (m, 2H), 7.61 (t, J = 8.3 Hz, 1H), 7.38 – 7.32 (m, 2H), 7.12 (t, J = 7.5 Hz, 1H), 3.97 (d, J = 1.4 Hz, 3H), 3.04 (t, J = 7.5 Hz, 2H), 2.84 (t, J = 7.5 Hz, 2H). LC-MS (ESI) m/z 312.1 [M+H]+.
4.4.4. methyl 2-(N-methyl-4-(3-oxopropyl)benzamido)benzoate (30ć)
(414 mg, 1.16 mmol, 53% yield). 1H NMR (400 MHz, CDCl3) δ 9.73 (s, 1H), 7.76 (d, J = 8.0 Hz, 1H), 7.45 (t, J = 7.8 Hz, 1H), 7.32 – 7.20 (m, 2H), 7.17 – 7.12 (m, 2H), 6.96 – 6.90 (m, 2H), 3.86 (s, 3H), 3.42 (s, 3H), 2.82 (t, J = 7.5 Hz, 2H), 2.66 (t, J = 7.5 Hz, 2H). LC-MS (ESI) m/z 326.1 [M+H]+.
4.4.5. methyl 3-(4-(3-oxopropyl)benzamido)benzoate (30d)
(404 mg, 1.29 mmol, 51% yield). 1H NMR (400 MHz, CDCl3) δ 9.84 (s, 1H), 8.13 (t, J = 2.0 Hz, 1H), 8.05 (dd, J = 8.0, 2.3 Hz, 1H), 7.92 (s, 1H), 7.83 – 7.80 (m, 3H), 7.46 (t, J = 7.9 Hz, 1H), 7.33 (d, J = 7.9 Hz, 2H), 3.92 (s, 3H), 3.03 (t, J = 7.4 Hz, 2H), 2.84 (t, J = 7.4 Hz, 2H). LC-MS (ESI) m/z 312.1 [M+H]+.
4.4.6. methyl 4-(4-(3-oxopropyl)benzamido)benzoate (30e)
(45 mg, 0.15 mmol, 39% yield). 1H NMR (500 MHz, CDCl3) δ 9.84 (t, J = 1.3 Hz, 1H), 8.15 (s, 1H), 7.97 – 7.94 (m, 2H), 7.72 – 7.68 (m, 2H), 7.47 – 7.44 (m, 2H), 7.35 – 7.31 (m, 2H), 3.93 (s, 3H), 3.04 (t, J = 7.5 Hz, 2H), 2.84 (t, J = 7.5 Hz, 2H). LC-MS (ESI) m/z 312.1 [M+H]+.
4.4.7. methyl 2-(2-(4-(3-oxopropyl)benzamido)phenyl)acetate (30f)
(0.78 g, 2.4 mmol, 83% yield). 1H NMR (400 MHz, CDCl3) δ 9.84 (t, J = 1.2 Hz, 1H), 9.68 (bs, 1H), 8.02 (d, J = 7.9 Hz, 1H), 7.99 – 7.95 (m, 2H), 7.39 – 7.32 (m, 3H), 7.25 – 7.22 (m, 1H), 7.14 (t, J = 7.7 Hz, 1H), 3.76 (s, 3H), 3.69 (s, 2H), 3.04 (t, J = 7.4 Hz, 2H), 2.87 – 2.81 (m, 2H). LC-MS (ESI) m/z 326.1[M+H]+.
4.4.8. methyl 3-(2-(4-(3-oxopropyl)benzamido)phenyl)propanoate (30g)
(3.93 g, 11.59 mmol, 72% yield). 1H NMR (400 MHz, CDCl3) δ 9.84 (t, J = 1.3 Hz, 1H), 9.57 (s, 1H), 8.05 – 8.01 (m, 2H), 7.83 (d, J = 8.1 Hz, 1H), 7.37 – 7.32 (m, 2H), 7.30 – 7.26 (m, 1H), 7.21 – 7.13 (m, 2H), 3.67 (s, 3H), 3.04 (t, J = 7.5 Hz, 2H), 2.94 – 2.89 (m, 2H), 2.87 – 2.81 (m, 2H), 2.80 – 2.75 (m, 2H). LC-MS (ESI) m/z 326.1[M+H]+.
4.4.9. N-(2-cyanophenyl)-4-(3-oxopropyl)benzamide (30h)
(1.09 g, 3.93 mmol, 83% yield). 1H NMR (400 MHz, CDCl3) δ 9.84 (t, J = 1.1 Hz, 1H), 8.59 (d, J = 8.5 Hz, 1H), 8.38 (s, 1H), 7.89 – 7.84 (m, 2H), 7.68 – 7.61 (m, 2H), 7.39 – 7.33 (m, 2H), 7.25 – 7.18 (m, 1H), 3.04 (t, J = 7.5 Hz, 2H), 2.84 (t, J = 7.5 Hz, 2H). LC-MS (ESI) m/z 279.1 [M+H]+.
4.4.10. methyl 2-((4-(3-oxopropyl)benzamido)methyl)benzoate (30i)
(0.44 g, 1.34 mmol, 99 % yield). 1H NMR (400 MHz, CDCl3) δ 9.80 (t, J = 1.3 Hz, 1H), 8.02 – 7.95 (m, 1H), 7.85 – 7.80 (m, 1H), 7.72 – 7.68 (m, 2H), 7.62 – 7.58 (m, 1H), 7.54 – 7.49 (m, 1H), 7.39 – 7.33 (m, 1H), 7.25 – 7.49 (m, 2H), 4.79 (d, J = 6.4 Hz, 2H), 3.95 (s, 3H), 3.01 – 2.95 (m, 2H), 2.78 (t, J = 7.5 Hz, 2H). LC-MS (ESI) m/z 326.1[M+H]+.
4.4.11. methyl 4-chloro-2-(4-(3-oxopropyl)benzamido)benzoate (30j)
(0.4 g, 1.16 mmol, 53% yield). 1H NMR (400 MHz, CDCl3) δ 12.06 (bs, 1H), 9.85 (t, J = 1.2 Hz, 1H), 9.05 (d, J = 2.0 Hz, 1H), 8.01 (d, J = 8.6 Hz, 1H), 7.99 – 7.94 (m, 2H), 7.39 – 7.34 (m, 2H), 7.10 (dd, J = 8.6, 2.1 Hz, 1H), 3.97 (s, 3H), 3.04 (t, J = 7.5 Hz, 2H), 2.87 – 2.81 (m, 2H). LC-MS (ESI) m/z 346.1 [M+H]+.
4.4.12. dimethyl 2-(4-(3-oxopropyl)benzamido)terephthalate (30k)
(326 mg, 0.88 mmol, 92% yield). 1H NMR (400 MHz, CDCl3) δ 11.98 (s, 1H), 9.85 (t, J = 1.2 Hz, 1H), 9.55 (d, J = 1.7 Hz, 1H), 8.15 (d, J = 8.3 Hz, 2H), 8.02 – 7.97 (m, 2H), 7.77 (dd, J = 8.3, 1.8 Hz, 2H), 7.40 – 7.34 (m, 2H), 4.00 (s, 3H), 3.96 (s, 3H), 3.04 (t, J = 7.5 Hz, 2H), 2.88 – 2.81 (m, 2H). LC-MS (ESI) m/z 370.1 [M+H]+.
4.4.13. methyl 4-methoxy-2-(4-(3-oxopropyl)benzamido)benzoate (30l)
(330 mg, 0.97 mmol, 79% yield). 1H NMR (400 MHz, DMSO-d6) δ 11.96 (s, 1H), 9.73 (t, J = 1.2 Hz, 1H), 8.38 (d, J = 2.7 Hz, 1H), 8.00 (d, J = 9.0 Hz, 1H), 7.89 – 7.86 (m, 2H), 7.50 – 7.44 (m, 2H), 6.80 (dd, J = 9.0, 2.6 Hz, 1H), 3.89 (s, 3H), 3.86 (s, 3H), 2.95 (t, J = 7.0 Hz, 2H), 2.88 – 2.82 (m, 2H). LC-MS (ESI) m/z 342.1 [M+H]+.
4.4.14. methyl 4-cyano-2-(4-(3-oxopropyl)benzamido)benzoate (30m)
(381 mg, 1.13 mmol, 92% yield). 1H NMR (400 MHz, DMSO-d6) δ 11.44 (s, 1H), 9.71 (t, J = 1.2 Hz, 1H), 8.79 (d, J = 1.6 Hz, 1H), 8.10 (d, J = 8.1 Hz, 1H), 7.88 – 7.84 (m, 2H), 7.68 (dd, J = 8.2, 1.7 Hz, 1H), 7.47 – 7.43 (m, 2H), 3.88 (s, 3H), 2.94 (t, J = 7.0 Hz, 2H), 2.83 (t, J = 7.2 Hz, 2H). LC-MS (ESI) m/z 337.1 [M+H]+.
4.4.15. methyl 4-(hydroxymethyl)-2-(4-(3-oxopropyl)benzamido)benzoate (30n)
(400 mg, 1.17 mmol, 96% yield). 1H NMR (400 MHz, CDCl3) δ 12.05 (s, 1H), 9.83 (t, J = 1.2 Hz, 1H), 9.06 (d, J = 1.6 Hz, 1H), 8.12 – 8.07 (m, 1H), 8.06 – 8.02 (m, 1H), 8.01 – 7.95 (m, 2H), 7.38 – 7.33 (m, 2H), 7.18 (dd, J = 8.4, 2.0 Hz, 1H), 5.41 (s, 2H), 3.97(s, 3H), 3.07 – 2.98 (m, 2H), 2.86 – 2.79 (m, 2H). LC-MS (ESI) m/z 342.1 [M+H]+.
4.4.16. methyl 4-(methoxymethyl)-2-(4-(3-oxopropyl)benzamido)benzoate (30o)
(150 mg, 0.422 mmol, 69% yield). 1H NMR (400 MHz, CDCl3) δ 12.05 (s, 1H), 9.85 (t, J = 1.0 Hz, 1H), 8.89 (s, 1H), 8.14 – 8.06 (m, 1H), 8.01 – 7.96 (m, 2H), 7.38 – 7.33 (m, 2H), 7.18 – 7.13 (m, 1H), 4.54 (s, 2H), 3.96 (s, 3H), 3.44 (s, 3H), 3.04 (t, J = 7.4 Hz, 2H), 2.84 (t, J = 7.5 Hz, 2H). LC-MS (ESI) m/z 356.1 [M+H]+.
4.4.17. methyl 4-carbamoyl-2-(4-(3-oxopropyl)benzamido)benzoate (30p)
(206 mg, 0.56 mmol, 96% yield). 1H NMR (400 MHz, CDCl3) δ 12.05 (s, 1H), 9.83 (t, J = 1.2 Hz, 1H), 9.06 (d, J = 1.6 Hz, 1H), 8.12 – 8.07 (m, 1H), 8.06 – 8.02 (m, 1H), 8.01 – 7.95 (m, 2H), 7.38 – 7.33 (m, 2H), 7.18 (dd, J = 8.4, 2.0 Hz, 1H), 5.41 (s, 2H), 3.97(s, 3H), 3.07 – 2.98 (m, 2H), 2.86 – 2.79 (m, 2H). LC-MS (ESI) m/z 342.1 [M+H]+.
4.4.18. methyl 3-(methoxymethyl)-2-(4-(3-oxopropyl)benzamido)benzoate (30q)
(313 mg, 0.88 mmol, 75% yield). 1H NMR (400 MHz, CDCl3) δ 10.32 (s, 1H), 9.85 (t, J = 1.2 Hz, 1H), 7.97 – 7.90 (m, 3H), 7.78 – 7.74 (m, 1H), 7.37 – 7.33 (m, 2H), 7.30 (t, J = 7.8 Hz, 1H), 4.48 (s, 2H), 3.87 (s, 3H), 3.37 (s, 3H), 3.04 (t, J = 7.5 Hz, 2H), 2.87 – 2.81 (m, 2H). LC-MS (ESI) m/z 356.1 [M+H]+.
4.4.19. methyl 5-methoxy-2-(4-(3-oxopropyl)benzamido)benzoate (30r)
(835 mg, 2.45 mmol, 86% yield). 1H NMR (400 MHz, CDCl3) δ 11.75 (s, 1H), 9.84 (d, J = 1.3 Hz, 1H), 8.85 (d, J = 9.2 Hz, 1H), 7.99 – 7.94 (m, 2H), 7.57 (d, J = 3.1 Hz, 1H), 7.36 – 7.32 (m, 2H), 7.18 (dd, J = 9.3, 3.1 Hz, 1H), 3.97 (s, 3H), 3.84 (s, 3H), 3.03 (t, J = 7.5 Hz, 2H), 2.86 – 2.80 (m, 2H).. LC-MS (ESI) m/z 342.1 [M+H]+.
4.4.20. methyl 2-(4-methoxy-2-(4-(3-oxopropyl)benzamido)phenyl)acetate (30s)
(189 mg, 0.53 mmol, 88% yield). 1H NMR (400 MHz, DMSO-d6) δ 12.05 (s, 1H), 9.73 (t, J = 1.2 Hz, 1H), 8.18 (d, J = 2.7 Hz, 1H), 7.98 – 7.90 (m, 2H), 7.85 – 7.78 (m, 3H), 7.50 – 7.44 (m, 2H), 6.98 (dd, J = 8.7, 2.0 Hz, 1H), 3.89 (s, 3H), 3.86 (s, 3H), 2.98 (t, J = 7.0 Hz, 2H), 2.88 – 2.82 (m, 2H). LC-MS (ESI) m/z 356.1 [M+H]+.
4.4.21. methyl 2-(4-cyano-2-(4-(3-oxopropyl)benzamido)phenyl)acetate (30t)
(625 mg, 1.78 mmol, 45% yield). 1H NMR (400 MHz, CDCl3) δ 9.84 (t, J = 1.2 Hz, 1H), 8.45 (d, J = 1.7 Hz, 1H), 7.98 – 7.94 (m, 2H), 7.41 (dd, J = 7.9, 1.7 Hz, 1H), 7.38 – 7.33 (m, 3H), 3.79 (s, 3H), 3.74 (s, 2H), 3.05 (t, J = 7.4 Hz, 2H), 2.88 – 2.82 (m, 2H). LC-MS (ESI) m/z 351.1 [M+H]+.
4.4.22. methyl 4-(2-methoxy-2-oxoethyl)-3-(4-(3-oxopropyl)benzamido)benzoate (30u)
(2.05 g, 5.35 mmol, 94% yield). 1H NMR (400 MHz, CDCl3) δ 9.74 (t, J = 1.3 Hz, 1H), 9.09 (s, 1H), 8.21 (d, J = 1.9 Hz, 1H), 7.91 – 7.85 (m, 2H), 7.77 (dd, J = 8.0, 1.9 Hz, 1H), 7.48 – 7.41(m, 3H), 3.81 (s, 3H), 3.75 (s, 2H), 3.55 (s, 3H), 3.02 – 2.94 (m, 2H), 2.70 – 2.61 (m, 2H). LC-MS (ESI) m/z 384.1 [M+H]+.
4.5. General procedure for the synthesis of intermediates 31a-u. Method d.
Intermediates 31a-u have been obtained through general method d. To a solution of 3-oxopropyl derivates 30a-u (1 eq) in anhydrous THF (5mL/mmol) 5,5-dibromo-2,2-dimethyl-1,3-dioxane-4,6-dione (0.5 eq) and HCl (35%) (8.15 μL/mmol) were added. The mixture reaction was stirred at room temperature for 18 h. 5% NaHCO3 solution (20 mL/mmol) was added and the organic layer washed with H2O (3 × 20 mL/mmol) and dried over anhydrous Na2SO4. After filtration solvent was evaporated under reduced pressure and the desired intermediates, obtained as a orange solid, were used in subsequent reaction without purification.[12]
4.5.1. methyl 1-(4-(2-bromo-3-oxopropyl)benzoyl)piperidine-2-carboxylate (31a)
(275 mg, 0.72 mmol, 49% yield). 1H NMR (400 MHz, CDCl3) δ 9.53 (d, J = 2.1 Hz, 1H), 7.40 – 7.35 (m, 2H), 7.26 – 7.21 (m, 2H), 5.54 – 5.45 (m, 1H), 4.49 (ddd, J = 14.5, 6.4, 2.2 Hz, 1H), 3.78 (s, 3H), 3.70 – 3.62 (m, 1H), 3.58 (dd, J = 14.7, 6.3 Hz, 1H), 3.27 – 3.18 (m, 2H), 2.38 – 2.30 (m, 1H), 1.82 – 1.73 (m, 3H), 1.48 – 1.37 (m, 2H). LC-MS (ESI) m/z 383.1 [M+H]+.
4.5.2. methyl 2-(4-(2-bromo-3-oxopropyl)benzamido)cyclohexane-1-carboxylate (31b)
(362 mg, 0.91 mmol, 60% yield). 1H NMR (400 MHz, CDCl3) δ 9.50 (d, J = 2.2 Hz, 1H), 7.76 – 7.72 (m, 2H), 7.31 – 7.27 (m, 3H), 4.48 – 4.42 (m, 1H), 4.38 – 4.27 (m, 2H), 3.73 (s, 3H), 3.53 (dd, J = 14.5, 6.3 Hz, 1H), 3.23 – 3.15 (m, 1H), 2.95 – 2.89 (m, 2H), 2.23 – 2.14 (m, 2H), 1.85 – 1.63 (m, 3H), 1.52 – 1.42 (m, 1H). LC-MS (ESI) m/z 397.1 [M+H]+.
4.5.3. methyl 2-(4-(2-bromo-3-oxopropyl)benzamido)benzoate (31c)
(93 mg, 0.24 mmol, 31% yield).1H NMR (400 MHz, CDCl3) δ 1H NMR (400 MHz, CDCl3) δ 12.05 (s, 1H), 9.53 (d, J = 2.1 Hz, 1H), 8.93 (d, J = 7.5 Hz, 1H), 8.12 – 8.07 (m, 2H), 8.03 (d, J = 8.4 Hz, 1H), 7.62 (t, J = 8.2 Hz, 1H), 7.43 – 7.36 (m, 2H), 7.14 (t, J = 7.8 Hz, 1H), 4.53 – 4.46 (m, 1H), 3.98 (s, 3H), 3.58 (dd, J = 14.8, 6.3 Hz, 1H), 3.24 (dd, J = 14.7, 8.0 Hz, 1H). LC-MS (ESI) m/z 391.1 [M+H]+.
4.5.4. methyl 2-(4-(2-bromo-3-oxopropyl)-N-methylbenzamido)benzoate (31ć)
(313 mg, 0.78 mmol, 61% yield).1H NMR (400 MHz, CDCl3) δ 9.40 (s, 1H), 9.23 (s, 1H), 7.76 (d, J = 8.1 Hz, 1H), 7.45 (t, J = 7.7 Hz, 1H), 7.25 – 7.16 (m, 2H), 7.15 – 7.10 (m, 2H), 6.96 (d, J = 7.6 Hz, 1H), 4.35 – 4.28 (m, 1H), 3.86 (s, 3H), 3.43 (t, J = 4.9 Hz, 3H), 3.33 (dd, J = 14.8, 6.3 Hz, 1H), 3.03 (dd, J = 14.7, 8.0 Hz, 1H). LC-MS (ESI) m/z 406.0 [M+H]+.
4.5.5. methyl 3-(4-(2-bromo-3-oxopropyl)benzamido)benzoate (31d)
(450 mg, 1.15 mmol, 88% yield). 1H NMR (400 MHz, CDCl3) δ 9.53 (d, J = 2.1 Hz, 1H), 8.15 – 8.13 (m, 1H), 8.06 – 8.03 (m, 1H), 7.90 (s, 1H), 7.87 – 7.82 (m, 3H), 7.49 – 7.45 (m, 1H), 7.40 – 7.35 (m, 2H), 4.48 (ddd, J = 8.3, 6.3, 2.1 Hz, 1H), 3.93 (s, 3H), 3.57 (dd, J = 14.7, 6.3 Hz, 1H), 3.24 (dd, J = 14.7, 8.1 Hz, 1H). LC-MS (ESI) m/z 391.1 [M+H]+.
4.5.6. methyl 4-(4-(2-bromo-3-oxopropyl)benzamido)benzoate (31e)
(53 mg, 0.14 mmol, 91% yield). 1H NMR (400 MHz, CDCl3) δ 9.53 (d, J = 2.1 Hz, 1H), 8.15 (s, 1H), 7.97 – 7.94 (m, 2H), 7.72 – 7.68 (m, 2H), 7.47 – 7.44 (m, 2H), 7.35 – 7.31 (m, 2H), 4.48 (ddd, J = 8.3, 6.3, 2.1 Hz, 1H), 3.93 (s, 3H), 3.57 (dd, J = 14.7, 6.3 Hz, 1H), 3.24 (dd, J = 14.7, 8.1 Hz, 1H). LC-MS (ESI) m/z 391.1 [M+H]+.
4.5.7. methyl 2-(2-(4-(2-bromo-3-oxopropyl)benzamido)phenyl)acetate (31f)
(386.7 mg, 0.96 mmol, 40% yield). 1H NMR (400 MHz, CDCl3) δ 9.73 (bs, 1H), 9.53 (d, J = 2.2 Hz, 1H), 8.04 – 7.98 (m, 3H), 7.41 – 7.33 (m, 3H), 7.26 – 7.22 (m, 1H), 7.16 (t, J = 7.7 Hz, 1H), 4.49 (ddd, J = 8.3, 6.4, 2.2 Hz, 1H), 3.76 (s, 3H), 3.69 (s, 2H), 3.57 (dd, J = 14.6, 6.4 Hz, 1H), 3.24 (dd, J = 14.7, 8.1 Hz, 1H). LC-MS (ESI) m/z 406.0 [M+H]+.
4.5.8. methyl 3-(2-(4-(2-bromo-3-oxopropyl)benzamido)phenyl)propanoate (31g)
(2.23 g, 5.33 mmol, 46% yield). 1H NMR (400 MHz, CDCl3) δ 9.63 (s, 1H), 9.53 (d, J = 2.2 Hz, 1H), 8.11 – 8.07 (m, 2H), 7.84 (d, J = 8.0 Hz, 1H), 7.40 – 7.37 (m, 2H), 7.30 – 7.27 (m, 1H), 7.21 – 7.17 (m, 2H), 4.50 (ddd, J = 8.5, 6.6, 2.3 Hz, 1H), 3.97 (s, 3H), 3.58 (dd, J = 14.6, 6.5 Hz, 1H), 3.25 (dd, J = 14.7, 8.0 Hz, 1H), 2.95 – 2.92 (m, 2H), 2.81 – 2.79 (m, 2H). LC-MS (ESI) m/z 418.1 [M+H]+.
4.5.9. 4-(2-bromo-3-oxopropyl)-N-(2-cyanophenyl)benzamide (31h)
(830 mg, 2.32 mmol, 59% yield). 1H NMR (400 MHz, CDCl3) δ 9.53 (d, J = 2.1 Hz, 1H), 8.57 (d, J = 8.5 Hz, 1H), 8.41 (s, 1H), 7.90 – 7.85 (m, 2H), 7.69 – 7.62 (m, 2H), 7.40 – 7.34 (m, 2H), 7.24 – 7.17 (m, 1H), 4.53 – 4.46 (m, 1H), 3.58 (dd, J = 14.8, 6.3 Hz, 1H), 3.24 (dd, J = 14.7, 8.0 Hz, 1H). LC-MS (ESI) m/z 358.1 [M+H]+.
4.5.10. methyl 2-((4-(2-bromo-3-oxopropyl)benzamido)methyl)benzoate (31i)
(183 mg, 0.56 mmol, 42% yield). 1H NMR (500 MHz, CDCl3) δ 9.71 (s, 1H), 9.48 (d, J = 2.2 Hz, 1H), 8.04 – 7.97 (m, 3H), 7.76 – 7.71 (m, 2H), 7.68 – 7.64 (m, 1H), 7.55 – 7.51 (m, 1H), 7.39 – 7.36 (m, 1H), 4.82 (s, 2H), 4.43 (ddd, J = 8.4, 6.4, 2.3 Hz, 1H), 3.97 (s, 3H), 3.55 – 3.48 (m, 1H), 3.17 (dd, J = 14.7, 8.1 Hz, 1H). LC-MS (ESI) m/z 406.1 [M+H]+.
4.5.11. methyl 2-(4-(2-bromo-3-oxopropyl)benzamido)-4-chlorobenzoate (31j)
(234.2 mg, 0.554 mmol, 48% yield).1H NMR (500 MHz, CDCl3) δ 12.08 (s, 1H), 9.53 (d, J = 2.1 Hz, 1H), 9.04 (d, J = 2.2 Hz, 1H), 8.02 – 7.99 (m, 3H), 7.42 – 7.38 (m, 2H), 7.10 (dd, J = 8.5, 2.1 Hz, 1H), 4.49 (ddd, J = 8.3, 6.4, 2.1 Hz, 1H), 3.97 (s, 3H), 3.58 (dd, J = 14.6, 6.5 Hz, 1H), 3.24 (dd, J = 14.7, 8.1 Hz, 1H). LC-MS (ESI) m/z 424.1 [M+H]+.
4.5.12. dimethyl 2-(4-(2-bromo-3-oxopropyl)benzamido)terephthalate (31k)
(285 mg, 0.64 mmol, 73% yield). 1H NMR (400 MHz, CDCl3) δ 12.04 (s, 1H), 9.52 (d, J = 2.1 Hz, 1H), 8.18 – 8.14 (m, 1H), 8.07 – 8.03 (m, 1H), 7.81 – 7.76 (m, 2H), 7.59 – 7.55 (m, 2H), 7.40 (d, J = 7.9 Hz, 1H), 4.50 (ddd, J = 8.3, 6.3, 2.1 Hz, 1H), 4.01 (s, 3H), 3.97 (s, 3H), 3.58 (dd, J = 14.7, 6.2 Hz, 1H), 3.24 (dd, J = 14.7, 8.2 Hz, 1H). LC-MS (ESI) m/z 448.1 [M+H]+.
4.5.13. methyl 2-(4-(2-bromo-3-oxopropyl)benzamido)-4-methoxybenzoate (31l)
(324 mg, 0.88 mmol, 91% yield). 1H NMR (400 MHz, DMSO-d6) δ 11.96 (s, 1H), 9.54 (d, J = 1.8 Hz, 1H), 8.36 (d, J = 2.8 Hz, 1H), 8.00 (d, J = 9.1 Hz, 1H), 7.95 – 7.88 (m, 2H), 7.62 – 7.58 (m, 2H), 6.80 (dd, J = 8.9, 2.7 Hz, 1H), 5.06 (ddd, J = 9.0, 5.7, 1.9 Hz, 1H), 3.89 (s, 3H), 3.86 (s, 3H), 3.60 (dd, J = 14.7, 5.7 Hz, 1H), 3.21 (dd, J = 14.8, 9.0 Hz, 1H). LC-MS (ESI) m/z 422.1 [M+H]+.
4.5.14. methyl 2-(4-(2-bromo-3-oxopropyl)benzamido)-4-cyanobenzoate (31m)
(357 mg, 0.98 mmol, 87% yield). 1H NMR (400 MHz, DMSO-d6) δ 11.50 (s, 1H), 9.54 (d, J = 1.9 Hz, 1H), 8.81 (d, J = 1.6 Hz, 1H), 8.13 – 8.09 (m, 1H), 8.03 – 7.99 (m, 2H), 7.74 – 7.71 (m, 2H), 7.54 (d, J = 8.1 Hz, 1H), 5.07 (ddd, J = 9.0, 5.6, 1.8 Hz, 1H), 3.89 (s, 3H), 3.61 (dd, J = 14.7, 5.6 Hz, 1H), 3.23 (dd, J = 14.6, 5.6 Hz, 1H). LC-MS (ESI) m/z 416.1 [M+H]+.
4.5.15. methyl 2-(4-(2-bromo-3-oxopropyl)benzamido)-4-(hydroxymethyl)benzoate (31n)
(77 mg, 0.17 mmol, 15% yield). 1H NMR (500 MHz, DMSO-d6) δ 11.62 (s, 1H), 9.57 (m, 1H), 8.70 (s, 1H), 8.04 – 7.87 (m, 6H), 7.51 (d, J = 8.0 Hz, 1H), 5.45 (s, 2H), 5.05 (ddd, J = 14.5, 6.4, 2.2 Hz, 1H), 3.90 (s, 3H), 3.63 – 3.56 (m, 1H), 3.25 – 3.19 (m, 1H). LC-MS (ESI) m/z 434.1 [M+H]+.
4.5.16. methyl 2-(4-(2-bromo-3-oxopropyl)benzamido)-4-(methoxymethyl)benzoate (31o)
(137 mg, 0.315 mmol, 77% yield). 1H NMR (400 MHz, CDCl3) δ 12.08 (s, 1H), 9.53 (d, J = 2.1 Hz, 1H), 8.89 (s, 1H), 8.10 – 8.00 (m, 3H), 7.57 – 7.54 (m, 1H), 7.39 (dd, J = 8.5, 2.1 Hz, 1H), 7.18 – 7.14 (m, 1H), 4.54 (s, 2H), 4.49 (ddd, J = 14.5, 6.4, 2.2 Hz, 1H), 3.97 (s, 3H), 3.58 (dd, J = 14.7, 6.3 Hz, 1H), 3.44 (s, 3H), 3.24 (dd, J = 14.7, 8.1 Hz, 1H). LC-MS (ESI) m/z 434.1 [M+H]+.
4.5.17. methyl 2-(4-(2-bromo-3-oxopropyl)benzamido)-4-carbamoylbenzoate (31p)
(167 mg, 0.39 mmol, 67% yield). 1H NMR (500 MHz, DMSO-d6) δ 11.45 (s, 1H), 9.53 (d, J = 2.1 Hz, 1H), 8.13 – 8.07 (m, 1H), 8.05 – 8.02 (m, 1H), 7.94 – 7.88 (m, 2H), 7.51 – 7.44 (m, 2H), 7.33 – 7.27 (m, Hz, 1H), 5.45 (s, 2H), 4.59 (ddd, J = 14.5, 6.4, 2.2 Hz, 1H), 3.89 (s, 3H), 3.62 – 3.55 (m, 1H), 3.25 – 3.19 (m, 1H). LC- MS (ESI) m/z 434.1 [M+H]+.
4.5.18. methyl 2-(4-(2-bromo-3-oxopropyl)benzamido)-3-(methoxymethyl)benzoate (31q)
(289 mg, 0.66 mmol, 75% yield). 1H NMR (500 MHz, DMSO-d6) δ 10.03 (s, 1H), 9.42 (d, J = 1.5 Hz, 1H), 7.93 – 7.91 (m, 2H), 7.75 (d, J = 8.1 Hz, 1H), 7.67 (d, J = 7.6 Hz, 1H), 7.46 – 7.39 (m, 3H), 4.62 – 4.56 (m, 1H), 4.42 (s, 2H), 3.96 (s, 3H), 3.70 (s, 3H), 3.57 (dd, J = 14.7, 5.9 Hz, 1H), 3.19 (dd, J = 14.7, 8.9 Hz, 1H). LC-MS (ESI) m/z 435.1 [M+H]+.
4.5.19. methyl 2-(4-(2-bromo-3-oxopropyl)benzamido)-5-methoxybenzoate (31r)
(888 mg, 2.11 mmol, 86% yield). 1H NMR (400 MHz, CDCl3) δ 11.78 (s, 1H), 9.52 (d, J = 2.2 Hz, 1H), 8.85 (d, J = 9.4 Hz, 1H), 8.01 – 7.98 (m, 2H), 7.57 (d, J = 3.1 Hz, 1H), 7.40 – 7.36 (m, 2H), 7.18 (dd, J = 9.2, 3.2 Hz, 1H), 4.53 – 4.44 (m, 1H), 3.97 (s, 3H), 3.84 (s, 3H), 3.57 (dd, J = 14.7, 6.4 Hz, 1H), 3.23 (dd, J = 14.7, 8.1 Hz, 1H). LC-MS (ESI) m/z 421.1 [M+H]+.
4.5.20. methyl 2-(2-(4-(2-bromo-3-oxopropyl)benzamido)-4-methoxyphenyl)acetate (31s)
(141 mg, 0.32 mmol, 63% yield). 1H NMR (400 MHz, CDCl3) δ 10.98 (s, 1H), 9.56 (d, J = 2.1 Hz, 1H), 8.16 (d, J = 2.7 Hz, 1H), 7.91 – 7.86 (m, 2H), 7.41 (dd, J = 7.9, 2.7 Hz, 1H), 7.40 – 7.35 (m, 2H), 7.09 – 7.05 (m, 1H), 4.49 (ddd, J = 14.5, 6.4, 2.2 Hz, 1H), 3.95 (s, 3H), 3.80 (s, 3H), 3.74 (s, 2H), 3.58 (dd, J = 14.5, 6.3 Hz, 1H), 3.27 – 3.19 (m, 1H). LC-MS (ESI) m/z 435.1 [M+H]+.
4.5.21. methyl 2-(2-(4-(2-bromo-3-oxopropyl)benzamido)-4-cyanophenyl)acetate (31t)
(688 mg, 1.60 mmol, 90% yield). 1H NMR (400 MHz, CDCl3) δ 9.90 (s, 1H), 9.53 (d, J = 2.1 Hz, 1H), 8.45 – 8.42 (m, 1H), 8.03 – 7.98 (m, 2H), 7.42 – 7.33 (m, 4H), 4.50 (ddd, J = 8.3, 6.4, 2.1 Hz, 1H), 3.79 (s, 3H), 3.74 (s, 2H), 3.55 (dd, J = 14.7, 6.4 Hz, 1H), 3.25 (dd, J = 14.7, 8.0 Hz, 1H). LC-MS (ESI) m/z 429.0 [M+H]+.
4.5.22. methyl 3-(4-(2-bromo-3-oxopropyl)benzamido)-4-(2-methoxy-2-oxoethyl)benzoate (31u)
(1.05 g, 2.27 mmol, 42% yield). 1H NMR (400 MHz, CDCl3) δ 9.89 (s, 1H), 9.72 (d, J = 1.9 Hz, 1H), 8.25 – 8.19 (m, 1H), 7.91 – 7.86 (m, 2H), 7.64 – 7.58 (m, 2H), 7.00 – 6.96 (m, 2H), 4.45 (ddd, J = 14.5, 6.4, 2.2 Hz, 1H), 3.80 (s, 3H), 3.74 (s, 2H), 3.65 (s, 3H), 3.55 (ddd, J = 14.5, 6.3 Hz, 1H), 3.23 (ddd, J = 14.5, 7.9, 1H). LC-MS (ESI) m/z 462.1 [M+H]+.
4.6. General procedure for the synthesis of final compound 7 and intermediates 32a-g, 32i-32u. Method e.
Intermediates 31a-u (1 eq) were added to solution of 2,6-diaminopyrimidin-4-ol (1.1 eq) and sodium acetate (2 eq) in water (3.13 mL/eq) and methanol (3.13 mL/eq). The reaction mixture was stirred at 45 °C for 2 hours. After solvent evaporation, intermediates were purified by flash column chromatography eluted initially with dichloromethane followed by 20% methanol in dichloromethane.[12]
4.6.1. 4-((2-amino-4-oxo-4,7-dihydro-3H-pyrrolo[2,3-d]pyrimidin-5-yl)methyl)-N-(2-cyanophenyl)benzamide (7)
(101 mg, 0.26 mmol, 11% yield). 1H NMR (400 MHz, DMSO-d6) δ 12.27 (s, 1H), 10.72 (s, 1H), 10.27 (s, 1H), 8.80 – 8.75 (m, 1H), 8.20 (dd, J = 7.8, 1.7 Hz, 1H), 8.05 – 8.01 (m, 2H), 7.52 – 7.46 (m, 2H), 7.30 – 7.25 (m, 1H), 7.14 (td, J = 7.5, 1.3 Hz, 1H), 6.35 (d, J = 1.8 Hz, 1H), 6.04 (s, 2H), 4.02 (s, 2H). 13C NMR (101 MHz, DMSO-d6) δ 166.4, 162.2, 157.8, 152.2, 147.3, 139.5, 134.5, 132.1, 128.1, 127.5, 127.1, 127.0, 126.6, 126.3, 123.5, 122.6, 122.1, 118.5, 48.2. HRMS (ESI): calc. for [M+H]+ C21H17N6O2+ 385.1408, found 385.1414.
4.6.2. methyl 1-(4-((2-amino-4-oxo-4,7-dihydro-3H-pyrrolo[2,3-d]pyrimidin-5-yl)methyl)benzoyl)piperidine-2-carboxylate (32a)
(60 mg, 0.147 mmol, yield 20% yield). 1H NMR (400 MHz, DMSO-d6) δ 10.72 (d, J = 2.1 Hz, 1H), 10.11 (s, 1H), 7.39 – 7.32 (m, 2H), 7.28 – 7.21 (m, 2H), 6.35 (d, J = 2.2 Hz, 1H), 5.99 (s, 2H), 4.47 – 4.36 (m, 1H), 3.96 (s, 2H), 3.71 (s, 3H), 3.59 – 3.50 (m, 1H), 3.13 – 3.02 (m, 1H), 2.20 – 2.12 (m, 1H), 1.71 – 1.63 (m, 1H), 1.58 – 1.48 (m, 1H), 1.44 – 1.36 (m, 1H), 1.30 – 1.23 (m, 1H), 1.20 – 1.13 (m, 1H). LC-MS (ESI) m/z 410.1 [M+H]+.
4.6.3. Methyl 2-(4-((2-amino-4-oxo-4,7-dihydro-3H-pyrrolo[2,3-d]pyrimidin-5-yl)methyl)benzamido) cyclohexane-1-carboxylate (32b)
(82.6 mg, 0.195 mmol, 20% yield). 1H NMR (400 MHz, DMSO-d6) δ 10.72 (s, 1H), 10.12 (s, 1H), 7.89 (d, J = 8.3 Hz, 1H), 7.65 – 7.59 (m, 2H), 7.37 – 7.31 (m, 2H), 6.32 (d, J = 2.1 Hz, 1H), 6.00 (s, 2H), 4.34 – 4.27 (m, 1H), 4.14 – 4.05 (m, 1H), 3.96 (s, 2H), 3.53 (s, 3H), 3.19 – 3.15 (m, 1H), 2.85 – 2.80 (m, 1H), 1.99 – 1.91 (m, 1H), 1.83 – 1.74 (m, 1H), 1.73 – 1.50 (m, 2H), 1.43 – 1.29 (m, 2H). LC-MS (ESI) m/z 424.1 [M+H]+.
4.6.4. methyl 2-(4-((2-amino-4-oxo-4,7-dihydro-3H-pyrrolo[2,3-d]pyrimidin-5-yl)methyl)benzamido)benzoate (32c)
(27 mg, 0.06 mmol, 25% yield). 1H NMR (400 MHz, DMSO-d6) δ 11.68 (s, 1H), 10.76 (d, J = 1.9 Hz, 1H), 10.14 (s, 1H), 8.72 (d, J = 2.1 Hz, 1H), 8.02 (d, J = 8.6 Hz, 1H), 7.85 – 7.80 (m, 2H), 7.53 – 7.49 (m, 2H), 7.30 (dd, J = 8.6, 2.1 Hz, 1H), 6.39 (d, J = 1.7 Hz, 1H), 6.02 (s, 2H), 4.03 (s, 2H), 3.90 (s, 3H). LC-MS (ESI) m/z 418.1 [M+H]+.
4.6.5. methyl 2-(4-((2-amino-4-oxo-4,7-dihydro-3H-pyrrolo[2,3-d]pyrimidin-5-yl)methyl)-N-methyl benzamido)benzoate (32ć)
(50 mg, 0.116 mmol, 15% yield). 1H NMR (400 MHz, DMSO-d6) δ 11.68 (s, 1H), 10.76 (d, J = 1.9 Hz, 1H), 10.14 (s, 1H), 8.72 (d, J = 2.1 Hz, 1H), 8.02 (d, J = 8.6 Hz, 1H), 7.85 – 7.80 (m, 2H), 7.53 – 7.49 (m, 2H), 7.30 (dd, J = 8.6, 2.1 Hz, 1H), 6.39 (d, J = 1.7 Hz, 1H), 6.02 (s, 2H), 4.03 (s, 2H), 3.90 (s, 3H). LC-MS (ESI) m/z 433.0 [M+H]+.
4.6.6. methyl 3-(4-((2-amino-4-oxo-4,7-dihydro-3H-pyrrolo[2,3-d]pyrimidin-5-yl)methyl)benzamido)benzoate (32d)
(57 mg, 0.13 mmol, 13 % yield). 1H NMR (400 MHz, DMSO-d6) δ 10.74 (d, J = 1.8 Hz, 1H), 10.33 (s, 1H), 10.14 (s, 1H), 8.45 (t, J = 2.0 Hz, 1H), 8.07 – 8.03 (m, 1H), 7.87 – 7.84 (m, 2H), 7.70 – 7.66 (m, 1H), 7.49 (t, J = 7.9 Hz, 1H), 7.46 – 7.43 (m, 2H), 6.36 (d, J = 2.2 Hz, 1H), 6.01 (s, 2H), 4.01 (s, 2H), 3.87 (s, 3H). LC-MS (ESI) m/z 418.1 [M+H]+.
4.6.7. methyl 4-(4-((2-amino-4-oxo-4,7-dihydro-3H-pyrrolo[2,3-d]pyrimidin-5-yl)methyl)benzamido)benzoate (32e)
(12 mg, 0.027 mmol, 20 % yield). 1H NMR (600 MHz, DMSO-d6) δ 10.74 (d, J = 1.8 Hz, 1H), 10.44 (s, 1H), 10.14 (s, 1H), 7.95 – 7.92 (m, 4H), 7.85 – 7.82 (m, 2H), 7.46 – 7.43 (m, 2H), 6.37 (d, J = 2.3 Hz, 1H), 6.01 (s, 2H), 4.01 (s, 2H), 3.83 (s, 3H). LC-MS (ESI) m/z 418.1 [M+H]+.
4.6.8. methyl 2-(2-(4-((2-amino-4-oxo-4,7-dihydro-3H-pyrrolo[2,3-d]pyrimidin-5-yl)methyl)benzamido)phenyl)acetate (32f)
(194 mg, 0.45 mmol, 47% yield). 1H NMR (400 MHz, DMSO-d6) δ 10.71 (d, J = 1.9 Hz, 1H), 9.87 (s, 1H), 7.81 – 7.77 (m, 2H), 7.44 – 7.40 (m, 2H), 7.39 – 7.36 (m, 1H), 7.34 – 7.29 (m, 2H), 7.21 (t, J = 7.4 Hz, 1H), 6.54 (s, 1H), 6.40 (s, 2H), 6.33 (d, J = 2.0 Hz, 1H), 4.00 (s, 2H), 3.72 (s, 3H), 3.50 (s, 2H). LC-MS (ESI) m/z 432.2 [M+H]+.
4.6.9. methyl 3-(2-(4-((2-amino-4-oxo-4,7-dihydro-3H-pyrrolo[2,3-d]pyrimidin-5-yl)methyl)benzamido) phenyl)propanoate (32g)
(300 mg, 0.67 mmol, 13% yield). 1H NMR (400 MHz, DMSO-d6) δ 10.76 (d, J = 1.6 Hz, 1H), 10.14 (s, 1H), 9.84 (s, 1H), 7.90 – 7.80 (m, 2H), 7.48 – 7.38 (m, 2H), 7.32 – 7.17 (m, 4H), 6.37 (d, J = 1.8 Hz, 1H), 6.02 (s, 2H), 4.01 (s, 2H), 3.54 (s, 3H), 2.85 (t, J = 7.7 Hz, 2H), 2.59 (t, J = 7.7 Hz, 2H). LC-MS (ESI) m/z 446.1 [M+H]+.
4.6.10. methyl 2-((4-((2-amino-4-oxo-4,7-dihydro-3H-pyrrolo[2,3-d]pyrimidin-5-yl)methyl)benzamido) methyl)benzoate (32i)
(91 mg, 0.21 mmol, 47% yield). 1H NMR (400 MHz, DMSO-d6) δ 10.74 (d, J = 2.1 Hz, 1H), 10.16 (s, 1H), 8.86 (t, J = 6.0 Hz, 1H), 7.86 (d, J = 7.7 Hz, 1H), 7.80 – 7.74 (m, 2H), 7.55 (t, J = 7.5 Hz, 1H), 7.42 – 7.33 (m, 4H), 6.35 (d, J = 2.1 Hz, 1H), 6.02 (s, 2H), 4.77 (d, J = 5.9 Hz, 2H), 3.98 (s, 2H), 3.85 (s, 3H). LC-MS (ESI) m/z 432.2 [M+H]+.
4.6.11. methyl 2-(4-((2-amino-4-oxo-4,7-dihydro-3H-pyrrolo[2,3-d]pyrimidin-5-yl)methyl)benzamido)-4-chlorobenzoate (32j)
(140 mg, 0.31 mmol, 44% yield). 1H NMR (400 MHz, DMSO-d6) δ 11.68 (s, 1H), 10.76 (d, J = 1.9 Hz, 1H), 10.14 (s, 1H), 8.72 (d, J = 2.1 Hz, 1H), 8.02 (d, J = 8.6 Hz, 1H), 7.85 – 7.80 (m, 2H), 7.53 – 7.49 (m, 2H), 7.30 (dd, J = 8.6, 2.1 Hz, 1H), 6.39 (d, J = 1.7 Hz, 1H), 6.02 (s, 2H), 4.03 (s, 2H), 3.90 (s, 3H). LC-MS (ESI) m/z 452.1 [M+H]+.
4.6.12. dimethyl 2-(4-((2-amino-4-oxo-4,7-dihydro-3H-pyrrolo[2,3-d]pyrimidin-5-yl)methyl)benzamido) terephthalate (32k)
(19 mg, 0.039 mmol, 6% yield). 1H NMR (400 MHz, DMSO-d6) δ 11.49 (s, 1H), 10.77 (d, J = 1.9 Hz, 1H), 10.15 (s, 1H), 9.11 (d, J = 1.7 Hz, 1H), 8.10 (d, J = 8.2 Hz, 1H), 7.88 – 7.83 (m, 2H), 7.77 (dd, J = 8.3, 1.7 Hz, 1H), 7.54 – 7.47 (m, 2H), 6.39 (d, J = 2.2 Hz, 1H), 6.03 (s, 2H), 4.02 (s, 2H), 3.91 (s, 6H). LC-MS (ESI) m/z 476.1 [M+H]+.
4.6.13. methyl 2-(4-((2-amino-4-oxo-4,7-dihydro-3H-pyrrolo[2,3-d]pyrimidin-5-yl)methyl)benzamido)-4-methoxybenzoate (32l)
(60 mg, 0.13 mmol, 15% yield). 1H NMR (400 MHz, DMSO-d6) δ 11.95 (s, 1H), 10.77 (d, J = 2.3 Hz, 1H), 10.14 (s, 1H), 8.38 (d, J = 2.6 Hz, 1H), 8.00 (d, J = 9.0 Hz, 1H), 7.86 – 7.81 (m, 2H), 7.54 – 7.48 (m, 2H), 6.79 (dd, J = 9.0, 2.6 Hz, 1H), 6.39 (d, J = 2.2 Hz, 1H), 6.02 (s, 2H), 4.02 (s, 2H), 3.88 (s, 3H), 3.86 (s, 3H). LC-MS (ESI) m/z 449.1 [M+H]+.
4.6.14. methyl 2-(4-((2-amino-4-oxo-4,7-dihydro-3H-pyrrolo[2,3-d]pyrimidin-5-yl)methyl)benzamido)-4-cyanobenzoate (32m)
(60 mg, 0.14 mmol, 14% yield). 1H NMR (400 MHz, DMSO-d6) δ 11.45 (s, 1H), 10.77 (d, J = 2.3 Hz, 1H), 10.15 (s, 1H), 8.82 (d, J = 1.7 Hz, 1H), 8.11 (d, J = 8.2 Hz, 1H), 7.86 – 7.82 (m, 2H), 7.70 (dd, J = 8.1, 1.7 Hz, 1H), 7.53 – 7.49 (m, 2H), 6.39 (d, J = 2.2 Hz, 1H), 6.03 (s, 2H), 4.03 (s, 2H), 3.90 (s, 3H). LC-MS (ESI) m/z 443.1 [M+H]+.
4.6.15. methyl 2-(4-((2-amino-4-oxo-4,7-dihydro-3H-pyrrolo[2,3-d]pyrimidin-5-yl)methyl)benzamido) -4- (hydroxymethyl)benzoate (32n)
(77 mg, 0.172 mmol, 19% yield). 1H NMR (400 MHz, DMSO-d6) δ 11.63 (s, 1H), 10.77 (d, J = 2.1 Hz, 1H), 10.15 (s, 1H), 8.70 (s, 1H), 8.08 – 7.80 (m, 5H), 7.51 – 7.44 (m, 2H), 6.37 (d, J = 2.1 Hz, 1H), 6.02 (s, 2H), 5.44 (s, 2H), 4.01 (s, 2H), 3.89 (s, 3H). LC-MS (ESI) m/z 448.1 [M+H]+.
4.6.16. methyl 2-(4-((2-amino-4-oxo-4,7-dihydro-3H-pyrrolo[2,3-d]pyrimidin-5-yl)methyl)benzamido)-4-(methoxymethyl)benzoate (32o)
(13 mg, 0.028 mmol, 8% yield). 1H NMR (400 MHz, DMSO-d6) δ 11.63 (s, 1H), 10.76 (d, J = 2.1 Hz, 1H), 10.13 (s, 1H), 8.61 (d, J = 1.5 Hz, 1H), 8.00 (d, J = 8.2 Hz, 1H), 7.86 – 7.82 (m, 2H), 7.52 – 7.48 (m, 2H), 7.16 (dd, J = 8.2, 1.6 Hz, 1H), 6.38 (d, J = 2.1 Hz, 1H), 6.02 (s, 2H), 4.51 (s, 2H), 4.02 (s, 2H), 3.90 (s, 3H), 3.35 (s, 3H). LC-MS (ESI) m/z 462.1 [M+H]+.
4.6.17. methyl 2-(4-((2-amino-4-oxo-4,7-dihydro-3H-pyrrolo[2,3-d]pyrimidin-5-yl)methyl)benzamido)-4-carbamoylbenzoate (32p)
(22.7 mg, 0.049 mmol, 13% yield). 1H NMR (400 MHz, DMSO-d6) δ 11.44 (s, 1H), 10.78 (d, J = 2.1 Hz, 1H), 10.15 (s, 1H), 8.90 (d, J = 2.3 Hz, 1H), 8.17 (s, 1H), 8.03 – 8.01 (m, 1H), 7.87 – 7.83 (m, 2H), 7.61 – 7.58 (m, 2H), 7.53 – 7.48 (m, 2H), 6.39 (d, J = 2.1 Hz, 1H), 6.04 (s, 2H), 4.02 (s, 2H), 3.89 (s, 3H). LC-MS (ESI) m/z 461.1 [M+H]+.
4.6.18. methyl 2-(4-((2-amino-4-oxo-4,7-dihydro-3H-pyrrolo[2,3-d]pyrimidin-5-yl)methyl)benzamido)-3-(methoxymethyl)benzoate (32q)
(81.9 mg, 0.177 mmol, 27% yield). (400 MHz, DMSO-d6) δ 10.77 (d, J = 2.2 Hz, 1H), 10.15 (s, 1H), 9.93 (s, 1H), 7.86 – 7.81 (m, 2H), 7.75 (dd, J = 7.6, 1.6 Hz, 1H), 7.68 (dd, J = 7.7, 1.6 Hz, 1H), 7.46 – 7.39 (m, 3H), 6.39 (d, J = 2.2 Hz, 1H), 6.03 (s, 2H), 4.45 (s, 2H), 4.01 (s, 2H), 3.69 (s, 3H), 3.29 (s, 3H). LC-MS (ESI) m/z 462.1 [M+H]+.
4.6.19. methyl 2-(4-((2-amino-4-oxo-4,7-dihydro-3H-pyrrolo[2,3-d]pyrimidin-5-yl)methyl)benzamido)-5-methoxybenzoate (32r)
(192.8 mg, 0.43 mmol, 20% yield). 1H NMR (400 MHz, DMSO-d6) δ 11.19 (s, 1H), 10.75 (d, J = 2.1 Hz, 1H), 10.13 (s, 1H), 8.39 (d, J = 9.1 Hz, 1H), 7.83 – 7.79 (m, 2H), 7.49 – 7.45 (m, 3H), 7.29 (dd, J = 9.2, 3.1 Hz, 1H), 6.37 (d, J = 2.1 Hz, 1H), 6.01 (s, 2H), 4.02 (s, 2H), 3.87 (s, 2H), 3.80 (s, 3H). LC-MS (ESI) m/z 448.1 [M+H]+.
4.6.20. methyl 2-(2-(4-((2-amino-4-oxo-4,7-dihydro-3H-pyrrolo[2,3-d]pyrimidin-5-yl)methyl) benzamido)-4-methoxyphenyl)acetate (32s)
(49 mg, 0.011 mmol, 34 % yield). 1H NMR (600 MHz, DMSO-d6) δ 10.74 (d, J = 1.9 Hz, 2H), 10.15 (s, 1H), 9.80 (s, 1H), 7.81 – 7.76 (m, 2H), 7.45 – 7.39 (m, 2H), 7.22 (d, J = 8.5 Hz, 1H), 7.02 (d, J = 2.6 Hz, 1H), 6.80 (dd, J = 8.4, 2.7 Hz, 1H), 6.36 (d, J = 2.1 Hz, 1H), 6.02 (s, 2H), 4.00 (s, 2H), 3.74 (s, 3H), 3.65 (s, 2H), 3.50 (s, 3H). LC-MS (ESI) m/z 462.1 [M+H]+.
4.6.21. methyl 2-(2-(4-((2-amino-4-oxo-4,7-dihydro-3H-pyrrolo[2,3-d]pyrimidin-5-yl)methyl)benzamido)-4-cyanophenyl)acetate (32t)
(316 mg, 0.691 mmol, 45% yield). 1H NMR (400 MHz, DMSO-d6) δ 10.74 (d, J = 2.2 Hz, 1H), 10.13 (s, 1H), 10.04 (s, 1H), 7.88 (d, J = 1.7 Hz, 1H), 7.82 – 7.77 (m, 2H), 7.69 (dd, J = 7.9, 1.8 Hz, 1H), 7.56 (d, J = 8.0 Hz, 1H), 7.46 – 7.42 (m, 2H), 6.37 (d, J = 2.1 Hz, 1H), 6.01 (s, 2H), 4.01 (s, 2H), 3.87 (s, 2H), 3.51 (s, 3H). LC-MS (ESI) m/z 457.1 [M+H]+.
4.6.22. methyl 3-(4-((2-amino-4-oxo-4,7-dihydro-3H-pyrrolo[2,3-d]pyrimidin-5-yl)methyl)benzamido)-4-(2-methoxy-2-oxoethyl)benzoate (32u)
(295 mg, 0.603 mmol, 47% yield). 1H NMR (400 MHz, CDCl3) δ 10.72 (d, J = 2.1 Hz, 1H), 10.13 (s, 1H), 10.05 (s, 1H), 8.07 (s, 1H), 7.82 – 7.77 (m, 2H), 7.78 (dd, J = 8.0, 1.7 Hz, 1H), 7.48 – 7.41 (m, 3H), 6.35 (d, J = 2.2 Hz, 1H), 6.01 (s, 2H), 4.02 (s, 2H), 3.86 (s, 3H), 3.74 (s, 2H), 3.60 (s, 3H). LC-MS (ESI) m/z 490.1 [M+H]+.
4.7. General procedure for the synthesis of final compounds 2–6, 9 and 11–25. Method f.
The synthesis of compounds 2–6, 9 and 11–25 has been carried out following general method f. The corresponding derivate 32a-g or 32h-u (1 eq) was dissolved in a mixture MeOH (33 mL/mmol) and 2N NaOH (33 mL/mmol) was added. The resulting mixture was stirred at 60 °C for 1h. The resulting solution was cooled in an ice bath, and the pH was adjusted to 3–4 using 1 N HCl. The resulting suspension was chilled in a dry ice/acetone bath and thawed to 4 °C overnight in a refrigerator. The precipitate was filtered, washed with cold water, and dried under reduced pressure.[12]
4.7.1. 2-(4-((2-amino-4-oxo-4,7-dihydro-3H-pyrrolo[2,3-d]pyrimidin-5-yl)methyl)benzamido)benzoic acid (2)
(4.4 mg, 0.011 mmol, 17% yield). 1H NMR (400 MHz, DMSO-d6) δ 12.18 (s, 1H), 10.75 (s, 1H), 10.13 (s, 1H), 8.71 (d, J = 8.3 Hz, 1H), 8.05 (d, J = 7.7 Hz, 1H), 7.86 – 7.77 (m, 2H), 7.64 (t, J = 7.8 Hz, 1H), 7.56 – 7.43 (m, 2H), 7.19 (t, J = 7.5 Hz, 1H), 6.37 (s, 1H), 6.01 (s, 2H), 4.02 (s, 2H). 13C NMR (101 MHz, DMSO-d6) δ 169.9, 164.7, 159.2, 152.2, 151.2, 147.0, 141.2, 134.2, 131.8, 131.3, 129.0, 126.9, 122.7, 119.7, 116.6, 116.5, 114.2, 98.6, 31.6. HRMS (ESI): calc. for [M+H]+ C21H18N5O4+ 404.1353, found 404.1359.
4.7.2. 3-(4-((2-amino-4-oxo-4,7-dihydro-3H-pyrrolo[2,3-d]pyrimidin-5-yl)methyl)benzamido)benzoic acid (3)
(13 mg, 0.032 mmol, 24% yield) as a green pale solid. 1H NMR (600 MHz, DMSO-d6) δ 12.94 (s, 1H), 11.05 (s, 1H), 10.71 (s, 1H), 10.32 (s, 1H), 8.40 (t, J = 1.9 Hz, 1H), 8.02 (ddd, J = 8.2, 2.2, 1.0 Hz, 1H), 7.88 – 7.84 (m, 2H), 7.66 (dt, J = 7.7, 1.2 Hz, 1H), 7.46 (t, J = 7.9 Hz, 1H), 7.44 – 7.42 (m, 2H), 6.69 (s, 2H), 6.47 – 6.44 (m, 1H), 4.02 (s, 2H). 13C NMR (151 MHz, DMSO-d6) δ 168.6, 166.0, 162.9, 152.6, 149.0, 134.9, 133.1, 131.4, 127.0, 126.9, 126.5, 126.1, 124.0, 119.1, 117.4, 116.8, 116.7, 111.3, 30.8. HRMS (ESI): calc. for [M+H]+ C21H18N5O4+ 404.1353, found 404.1341.
4.7.3. 4-(4-((2-amino-4-oxo-4,7-dihydro-3H-pyrrolo[2,3-d]pyrimidin-5-yl)methyl)benzamido)benzoic acid (4)
(1.6 mg, 0.004 mmol, 15% yield) as a yellow-green solid. 1H NMR (600 MHz, DMSO-d6) δ 12.73 (s, 1H), 10.78 (s, 1H), 10.41 (s, 1H), 10.23 (s, 1H), 7.97 – 7.86 (m, 4H), 7.83 (d, J = 8.1 Hz, 2H), 7.44 (d, J = 8.0 Hz, 2H), 6.38 (d, J = 2.2 Hz, 1H), 6.10 (s, 2H), 4.01 (s, 1H).13C NMR (151 MHz, DMSO-d6) δ 170.3, 166.3, 162.9, 152.6, 149.0, 138.9, 133.1, 131.4, 129.5, 126.7, 126.0, 122.6, 121.5, 119.7, 116.5, 111.3, 31.8. HRMS (ESI): calc. for [M+H]+ C21H18N5O4+ 404.1353, found 404.1348.
4.7.4. 2-(2-(4-((2-amino-4-oxo-4,7-dihydro-3H-pyrrolo[2,3-d]pyrimidin-5-yl)methyl)benzamido)phenyl) acetic acid (5)
(71.1 mg, 0.17 mmol, 73% yield) as a pink solid. 1H NMR (500 MHz, DMSO-d6) δ 12.34 (s, 1H), 10.75 (d, J = 1.9 Hz, 1H), 10.16 (s, 1H), 9.86 (s, 1H), 7.85 – 7.78 (m, 2H), 7.48 – 7.44 (m, 1H), 7.43 – 7.40 (m, 2H), 7.33 – 7.27 (m, 2H), 7.22 – 7.16 (m, 1H), 6.37 (d, J = 1.9 Hz, 1H), 6.04 (s, 2H), 4.00 (s, 2H), 3.65 (s, 2H). 13C NMR (101 MHz, DMSO-d6) δ 172.8, 165.2, 159.2, 152.2, 151.1, 146.3, 136.8, 131.8, 131.0, 130.5, 128.5, 127.5, 127.2, 126.2, 125.6, 116.9, 114.2, 98.6, 37.6, 31.6. HRMS (ESI): calc. for [M+H]+ C22H20N5O4+ 418.1510, found 418.1515.
4.7.5. 3-(2-(4-((2-amino-4-oxo-4,7-dihydro-3H-pyrrolo[2,3-d]pyrimidin-5-yl)methyl)benzamido)phenyl) propanoic acid (6)
(257 mg, 0.595 mmol, 89% yield) as a green pale solid. 1H NMR (400 MHz, DMSO-d6) δ 12.22 (s, 1H), 10.76 (d, J = 1.6 Hz, 1H), 10.15 (s, 1H), 9.89 (s, 1H), 7.91 – 7.78 (m, 2H), 7.48 – 7.39 (m, 2H), 7.34 (d, J = 7.2 Hz, 1H),7.30 (dd, J = 7.6, 1.7 Hz, 1H), 7.27 – 7.16 (m, 2H), 6.37 (d, J = 1.6 Hz, 1H), 6.03 (s, 2H), 4.01 (s, 2H), 2.82 (t, J = 7.4 Hz, 2H), 2.53 (d, J = 7.4 Hz, 2H). 13C NMR (151 MHz, DMSO-d6) δ 168.5, 166.4, 158.9, 152.0, 145.8, 140.7, 132.0, 131.6, 130.3, 129.3, 128.4, 127.1, 127.1, 127.0, 126.6, 117.2, 114.5, 98.7, 41.1, 31.5. HRMS (ESI): calc. for [M+H]+ C23H22N5O4+ 432.1666, found 432.1657.
4.7.6. 2-(4-((2-amino-4-oxo-4,7-dihydro-3H-pyrrolo[2,3-d]pyrimidin-5-yl)methyl)-N-methylbenzamido) benzoic acid (9)
(27 mg, 0.065 mmol, 56% yield). 1H NMR (400 MHz, DMSO-d6) δ 13.10 (s, 1H), 10.67 (s, 1H), 10.09 (s, 1H), 7.70 (d, J = 7.7 Hz, 1H), 7.50 (t, J = 7.5 Hz, 1H), 7.35 (d, J = 7.9 Hz, 1H), 7.29 (t, J = 7.6 Hz, 1H), 7.11 – 6.95 (m, 4H), 6.11 (s, 1H), 5.98 (s, 2H), 3.80 (s, 2H), 3.26 (s, 3H). 13C NMR (151 MHz, DMSO-d6) δ 169.2, 166.6, 159.2, 152.2, 151.1, 144.3, 143.3, 133.5, 133.0, 131.1, 130.2, 12.9, 127.8, 127.6, 127.4, 117.0, 114.0, 98.6, 37.8, 31.4. HRMS (ESI): calc. for [M+H]+ C22H20N5O4+ 418.1510, found 418.1522.
4.7.7. 2-((4-((2-amino-4-oxo-4,7-dihydro-3H-pyrrolo[2,3-d]pyrimidin-5-yl)methyl)benzamido)methyl) benzoic acid (11)
(39 mg, 0.093 mmol, 44% yield) as a yellow pale solid. 1H NMR (600 MHz, DMSO-d6) δ 13.00 (bs, 1H), 10.88 (d, J = 1.9 Hz, 1H), 10.40 (s, 1H), 8.85 (t, J = 6.0 Hz, 1H), 7.87 (dd, J = 7.7, 1.5 Hz, 1H), 7.81 – 7.77 (m, 2H), 7.51 (td, J = 7.6, 1.5 Hz, 1H), 7.40 – 7.32 (m, 4H), 6.39 (d, J = 2.2 Hz, 1H), 6.35 (s, 2H), 4.80 (d, J = 6.0 Hz, 2H), 3.99 (s, 2H). 13C NMR (151 MHz, DMSO-d6) δ 168.5, 166.4, 158.9, 152.0, 145.8, 140.7, 132.0, 131.6, 130.3, 129.3, 128.4, 127.1, 127.1, 127.0, 126.6, 117.2, 114.5, 98.7, 41.1, 31.5. HRMS (ESI): calc. for [M+H]+ C22H20N5O4+ 418.1510, found 418.1517.
4.7.8. 1-(4-((2-amino-4-oxo-4,7-dihydro-3H-pyrrolo[2,3-d]pyrimidin-5-yl)methyl)benzoyl) piperidine-2-carboxylic acid (12)
(60 mg, 0.147 mmol, 20% yield) as a white-yellow solid. 1H NMR (400 MHz, DMSO-d6) δ 11.89 (s, 1H), 10.72 (d, J = 2.1 Hz, 1H), 10.11 (s, 1H), 7.39 – 7.32 (m, 2H), 7.28 – 7.21 (m, 2H), 6.35 (d, J = 2.2 Hz, 1H), 5.99 (s, 2H), 4.47 – 4.36 (m, 1H), 3.96 (s, 2H), 3.59 – 3.50 (m, 1H), 3.13 – 3.02 (m, 1H), 2.20 – 2.12 (m, 1H), 1.71 – 1.63 (m, 1H), 1.58 – 1.48 (m, 1H), 1.44 – 1.36 (m, 1H), 1.30 – 1.23 (m, 1H), 1.20 – 1.13 (m, 1H). 13C NMR (101 MHz, DMSO-d6) δ 172.3, 170.4, 159.3, 152.2, 151.2, 144.0, 133.1, 128.5, 126.6, 117.0, 114.1, 98.7, 57.8, 51.7, 45.3, 31.5, 26.3, 24.8, 20.9. HRMS (ESI): calc. for [M+H]+ C20H22N5O4+ 396.1666, found 396.1675.
4.7.9. 2-(4-((2-amino-4-oxo-4,7-dihydro-3H-pyrrolo[2,3-d]pyrimidin-5-yl)methyl)benzamido)cyclohexan-1-carboxylic acid (13)
(4.1 mg, 0.010 mmol, 5% yield) as a white-green solid. 1H NMR (400 MHz, DMSO-d6) δ 12.17 (s, 1H), 10.73 (s, 1H), 10.16 (s, 1H), 7.85 (d, J = 8.3 Hz, 1H), 7.66 – 7.60 (m, 2H), 7.37 – 7.31 (m, 2H), 6.31 (s, 1H), 6.04 (s, 2H), 4.28 (s, 1H), 3.96 (s, 2H), 2.78 – 2.68 (m, 2H), 1.99 (s, 1H), 1.82 – 1.71 (m, 2H), 1.66 – 1.45 (m, 2H), 1.36 (s, 2H). 13C NMR (101 MHz, DMSO-d6) δ 173.6, 166.0, 162.9, 153.6, 150.5, 132.9, 132.7, 126.2, 125.9, 119.1, 116.7, 111.3, 53.5, 47.9, 31.8, 25.8, 25.0, 23.6, 23.5. HRMS (ESI): calc. for [M+H]+ C21H24N5O4+ 410.1823, found 410.1828.
4.7.10. 2-(4-((2-amino-4-oxo-4,7-dihydro-3H-pyrrolo[2,3-d]pyrimidin-5-yl)methyl)benzamido)-4-chloro benzoic acid (14)
(92.4 mg, 0.21 mmol, 68% yield) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 12.36 (s, 1H), 10.76 (s, 1H), 10.14 (s, 1H), 8.82 (d, J = 2.0 Hz, 1H), 8.05 (d, J = 8.5 Hz, 1H), 7.85 – 7.79 (m, 2H), 7.52 – 7.46 (m, 2H), 7.25 (dd, J = 8.7, 2.0 Hz, 1H), 6.38 (d, J = 1.4 Hz, 1H), 6.02 (s, 2H), 4.02 (s, 2H). 13C NMR (151 MHz, DMSO-d6) δ 169.3, 165.0, 159.3, 152.3, 151.3, 147.3, 142.3, 138.4, 133.0, 131.4, 130.1, 129.1, 127.0, 122.6, 119.0, 116.6, 114.3, 98.6, 31.7. HRMS (ESI): calc. for [M+H]+ C21H17ClN5O4+ 438.0964, found 438.0955.
4.7.11. 2-(4-((2-amino-4-oxo-4,7-dihydro-3H-pyrrolo[2,3-d]pyrimidin-5-yl)methyl)benzamido) terephthalic acid (15)
(10 mg, 0.022 mmol, 57% yield) as a white-green solid. 1H NMR (600 MHz, DMSO-d6) δ 13.25 (s, 2H), 10.76 (d, J = 2.3 Hz, 1H), 10.21 (s, 1H), 9.27 (d, J = 1.7 Hz, 1H), 8.12 (d, J = 8.2 Hz, 1H), 7.87 – 7.84 (m, 2H), 7.67 (d, J = 8.2 Hz, 1H), 7.50 – 7.47 (m, 2H), 6.37 (d, J = 2.1 Hz, 1H), 6.09 (s, 2H), 4.02 (s, 2H).13C NMR (151 MHz, DMSO-d6) δ 169.3, 166.7, 164.8, 159.3, 152.3, 151.3, 147.1, 141.0, 135.0, 131.7, 131.5, 131.4, 129.1, 127.0, 123.1, 120.6, 116.7, 114.2, 98.6, 31.7. HRMS (ESI): calc. for [M+H]+ C22H18N5O6+ 448.1252, found 448.1257.
4.7.12. 2-(4-((2-amino-4-oxo-4,7-dihydro-3H-pyrrolo[2,3-d]pyrimidin-5-yl)methyl)benzamido)-4-methoxy benzoic acid (16)
(40 mg, 0.092 mmol, 69% yield) as a white-yellow solid. 1H NMR (400 MHz, DMSO-d6) δ 13.48 (s, 1H), 12.38 (s, 1H), 10.77 (d, J = 1.9 Hz, 1H), 10.16 (s, 1H), 8.42 (d, J = 2.7 Hz, 1H), 8.00 (d, J = 8.9 Hz, 1H), 7.86 – 7.81 (m, 2H), 7.52 – 7.46 (m, 2H), 6.76 (dd, J = 8.9, 2.7 Hz, 1H), 6.38 (d, J = 2.2 Hz, 1H), 6.04 (s, 2H), 4.02 (s, 2H), 3.85 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 167.2, 166.0, 163.9, 158.3, 152.6, 159.0, 138.7, 133.1, 131.8, 128.7, 126.5, 127.1, 119.1, 116.9, 112.4, 111.3, 109.7, 101.2, 56.5, 31.8. HRMS (ESI): calc. for [M+H]+ C22H20N5O5+ 434.1459, found 434.1446.
4.7.13. 2-(4-((2-amino-4-oxo-4,7-dihydro-3H-pyrrolo[2,3-d]pyrimidin-5-yl)methyl)benzamido)-4-cyanobenzoic acid (17)
(50 mg, 0.117 mmol, yield 86% yield) as a white-brown solid. 1H NMR (400 MHz, DMSO-d6) δ 12.09 (s, 1H), 10.77 (s, 1H), 10.16 (s, 1H), 9.09 (s, 1H), 8.16 – 8.05 (m, 3H), 7.87 – 7.82 (m, 2H), 7.65 – 7.54 (m, 1H), 7.49 (d, J = 8.0 Hz, 1H), 6.38 (s, 1H), 6.04 (s, 2H), 4.02 (s, 2H). 13C NMR (151 MHz, DMSO-d6) δ 168.2, 166.0, 163.9, 153.8, 150.5, 138.3, 134.1, 132.4, 130.5, 127.8, 127.3, 127.1, 121.3, 120.1, 119.7, 117.4, 112.6, 112.3, 111.8, 31.8. HRMS (ESI): calc. for [M+H]+ C22H17N6O4+ 429.1306, found 429.1302.
4.7.14. 2-(4-((2-amino-4-oxo-4,7-dihydro-3H-pyrrolo[2,3-d]pyrimidin-5-yl)methyl)benzamido)-4-(hydroxymethyl)benzoic acid (18)
(40 mg, 0.092 mmol, 54% yield) as a white-green solid. 1H NMR (400 MHz, DMSO-d6) δ 12.27 (s, 1H), 10.77 (s, 1H), 10.15 (s, 1H), 8.72 (s, 1H), 8.12 – 7.96 (m, 3H), 7.92 – 7.79 (m, 3H), 7.51 – 7.46 (m, 1H), 7.16 – 7.09 (m, 1H) 6.37 (d, J = 2.2 Hz, 1H), 6.03 (s, 2H), 4.57 (s, 2H), 4.02 (s, 2H). 13C NMR (151 MHz, DMSO-d6) δ 168.2, 166.0, 163.9, 153.6, 150.2, 141.8, 137.5, 134.3, 131.3, 128.8, 127.5, 127.1, 125.5, 123.7, 120.3, 118.6, 117.5, 111.3, 61.5, 30.9. HRMS (ESI): calc. for [M+H]+ C22H20N5O5+ 434.1459, found 434.1446.
4.7.15. 2-(4-((2-amino-4-oxo-4,7-dihydro-3H-pyrrolo[2,3-d]pyrimidin-5-yl)methyl)benzamido)-4-(methoxymethyl)benzoic acid (19)
(62 mg, 0.143 mmol, 83% yield) as a white-pink solid. 1H NMR (600 MHz, DMSO-d6) δ 13.74 (s, 1H), 12.19 (s, 1H), 10.76 (d, J = 2.3 Hz, 1H), 10.14 (s, 1H), 8.72 (d, J = 1.5 Hz, 1H), 8.02 (d, J = 8.1 Hz, 1H), 7.84 – 7.81 (m, 2H), 7.50 – 7.47 (m, 2H), 7.12 (dd, J = 8.1, 1.6 Hz, 1H), 6.38 (d, J = 2.1 Hz, 1H), 6.02 (s, 2H), 4.50 (s, 2H), 4.02 (s, 2H), 3.34 (s, 3H). 13C NMR (151 MHz, DMSO-d6) δ 169.9, 164.7, 159.3, 152.3, 151.3, 147.0, 145.2, 141.4, 131.8, 131.3, 129.1, 126.9, 121.2, 118.0, 116.6, 115.3, 114.2, 98.6, 73.1, 57.9, 31.6. HRMS (ESI): calc. for [M+H]+ C23H22N5O5+ 448.1615, found 448.1622.
4.7.16. 2-(4-((2-amino-4-oxo-4,7-dihydro-3H-pyrrolo[2,3-d]pyrimidin-5-yl)methyl)benzamido)-4-carbamoylbenzoic acid (20)
(10 mg, 0.022 mmol, 46% yield) as a white-brown solid. 1H NMR (400 MHz, DMSO-d6) δ 12.20 (bs, 1H), 10.73 (s, 1H), 10.12 (s, 1H), 9.06 (d, J = 1.7 Hz, 1H), 8.09 – 8.01 (m, 2H), 7.83 – 7.79 (m, 2H), 7.56 (d, J = 8.1 Hz, 2H), 7.50 (bs, 1H), 7.47 – 7.44 (m, 2H), 6.35 (d, J = 2.1 Hz, 1H), 5.99 (s, 2H), 3.99 (s, 2H). 13C NMR (101 MHz, DMSO-d6) δ 168.5, 167.8, 166.0, 165.9, 155.6, 152.0, 138.2, 134.9, 133.8, 132.6, 128.1, 127.5, 127.3, 127.1, 120.2, 117.5, 117.6, 115.8, 111.3, 31.8. HRMS (ESI): calc. for [M+H]+ C22H19N6O5+ 447.1411, found 447.1416.
4.7.17. 2-(4-((2-amino-4-oxo-4,7-dihydro-3H-pyrrolo[2,3-d]pyrimidin-5-yl)methyl)benzamido)-3-(methoxymethyl)benzoic acid (21)
(43 mg, 0.096 mmol, 56% yield) as a white-brown solid. 1H NMR (400 MHz, DMSO-d6) δ 12.88 (s, 1H), 10.76 (d, J = 2.0 Hz, 1H), 10.14 (s, 1H), 9.99 (s, 1H), 7.88 – 7.82 (m, 2H), 7.78 (dd, J = 7.8, 1.6 Hz, 1H), 7.66 (d, J = 7.6 Hz, 1H), 7.45 – 7.36 (m, 3H), 6.38 (d, J = 2.2 Hz, 1H), 6.03 (s, 2H), 4.41 (s, 2H), 4.00 (s, 2H), 3.28 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 168.4, 166.7, 162.9, 152.6, 151.4, 145.5, 137.1, 136.7, 135.4, 134.7, 132.5, 130.5, 130.1, 126.9, 123.1, 120.7, 111.6, 111.3, 68.6, 56.0, 31.8. HRMS (ESI): calc. for [M+H]+ C23H22N5O5+ 448.1615, found 448.1611.
4.7.18. 2-(4-((2-amino-4-oxo-4,7-dihydro-3H-pyrrolo[2,3-d]pyrimidin-5-yl)methyl)benzamido)-5-methoxybenzoic acid (22)
(121 mg, 0.279 mmol, 65% yield) as a white-green solid. 1H NMR (400 MHz, DMSO-d6) δ 13.77 (s, 1H), 11.80 (s, 1H), 10.76 (d, J = 2.3 Hz, 1H), 10.15 (s, 1H), 8.59 (d, J = 9.2 Hz, 1H), 7.83 – 7.79 (m, 2H), 7.51 (d, J = 3.2 Hz, 1H), 7.49 – 7.45 (m, 2H), 7.26 (dd, J = 9.2, 3.2 Hz, 1H), 6.37 (d, J = 2.2 Hz, 1H), 6.03 (s, 2H), 4.01 (s, 2H), 3.79 (s, 3H). 13C NMR (151 MHz, DMSO-d6) δ 169.5, 164.3, 159.3, 154.3, 152.3, 151.2, 146.8, 134.6, 132.0, 129.0, 126.8, 121.8, 120.3, 118.1, 116.7, 114.9, 114.2, 98.6, 55.4, 31.6. HRMS (ESI): calc. for [M+H]+ C22H20N5O5+ 434.1459, found 434.1465.
4.7.19. 2-(2-(4-((2-amino-4-oxo-4,7-dihydro-3H-pyrrolo[2,3-d]pyrimidin-5-yl)methyl)benzamido)-4-methoxyphenyl)acetic acid (23)
(27 mg, 0.061 mmol, 68% yield) as a white-pink solid. 1H NMR (400 MHz, DMSO-d6) δ 12.32 (s, 1H), 10.75 (s, 1H), 10.15 (s, 1H), 9.82 (s, 1H), 7.86 – 7.76 (m, 2H), 7.46 – 7.37 (m, 2H), 7.20 (d, J = 8.5 Hz, 1H), 7.14 (d, J = 2.2 Hz, 1H), 6.77 (dd, J = 8.5, 2.4 Hz, 1H), 6.36 (s, 1H), 6.03 (s, 2H), 4.00 (s, 2H), 3.74 (s, 3H), 3.57 (s, 2H). 13C NMR (101 MHz, DMSO-d6) δ 171.6, 168.0, 162.9, 158.7, 152.6, 151.3, 142.8, 137.1, 135.4, 132.3, 129.5, 128.7, 123.1, 119.7, 118.1, 115.3, 110.5, 104.9, 56.5, 39.9, 31.8. HRMS (ESI): calc. for [M+H]+ C23H22N5O5+ 448.1615, found 448.1624.
4.7.20. 2-(2-(4-((2-amino-4-oxo-4,7-dihydro-3H-pyrrolo[2,3-d]pyrimidin-5-yl)methyl)benzamido)-4-cyanophenyl)acetic acid (24)
(106 mg, 0.239 mmol, 38% yield) as a green pale solid. 1H NMR (600 MHz, DMSO-d6) δ 12.51 (bs, 1H), 10.82 (d, J = 2.3 Hz, 1H), 10.27 (s, 1H), 10.06 (s, 1H), 7.93 (d, J = 1.7 Hz, 1H), 7.84 – 7.79 (m, 2H), 7.67 (dd, J = 7.9, 1.8 Hz, 1H), 7.54 (d, J = 8.0 Hz, 1H), 7.46 – 7.40 (m, 2H), 6.39 (d, J = 2.1 Hz, 1H), 6.18 (s, 2H), 4.01 (s, 2H), 3.80 (s, 2H). 13C NMR (151 MHz, DMSO-d6) δ 171.8, 165.6, 159.1, 158.1, 152.1, 146.6, 137.8, 136.3, 132.5, 131.3, 129.4, 129.0, 128.5, 127.6, 118.5, 117.0, 114.3, 109.9, 98.6, 37.5, 31.6. HRMS (ESI): calc. for [M+H]+ C23H19N6O4+ 443.1462, found 443.1464.
4.7.21. 3-(4-((2-amino-4-oxo-4,7-dihydro-3H-pyrrolo[2,3-d]pyrimidin-5-yl)methyl)benzamido)-4-(carboxymethyl)benzoic acid (25)
(89 mg, 0.193 mmol, 38% yield) as a orange solid. 1H NMR (600 MHz, DMSO-d6) δ 12.85 (bs, 2H), 10.75 (d, J = 2.3 Hz, 1H), 10.15 (s, 1H), 10.03 (s, 1H), 8.05 (s, 1H), 7.84 – 7.81 (m, 2H), 7.76 (dd, J = 8.0, 1.7 Hz, 1H), 7.46 – 7.41 (m, 3H), 6.37 (d, J = 2.2 Hz, 1H), 6.03 (s, 2H), 4.01 (s, 2H), 3.74 (s, 2H). 13C NMR (151 MHz, DMSO-d6) δ 172.2, 166.9, 165.4, 159.3, 152.3, 151.2, 146.5, 137.0, 135.5, 131.6, 131.3, 129.8, 128.5, 127.5, 126.9, 126.3, 116.9, 114.1, 98.6, 37.7, 31.7. HRMS (ESI): calc. for [M+H]+ C23H20N5O6+ 462.1408, found 462.1414.
4.8. General procedure for the synthesis of final compound 8. Method g.
The synthesis of compound 8 has been carried out following for general method g. NaN3 (76 mg, 1.16 mmol) was added to a solution of 4-((2-amino-4-oxo-4,7-dihydro-3H-pyrrolo[2,3-d]pyrimidin-5-yl)methyl)-N-(2-cyanophenyl)benza-mide(7) (50 mg, 0.13 mmol) in DMSO (0.57 mL) and water (0.57 mL) and the reaction mixture stirred at 100 °C overnight. After solvent evaporation under reduced pressure the desired final compound was purified by flash column cromatography eluted initially with dichloromethane followed by 20% methanol in dichloromethane and finally 50% methanol in dichloromethane to give N-(2-(1H-tetrazol-5-yl)phenyl)-4-((2-amino-4-oxo-4,7-dihydro-3H-pyrrolo[2,3-d]pyrimidin-5-yl)methyl)benzamide (8) as a pale brown powder.
4.8.1. N-(2-(1H-tetrazol-5-yl)phenyl)-4-((2-amino-4-oxo-4,7-dihydro-3H-pyrrolo[2,3-d]pyrimidin-5-yl)methyl)benzamide (8)
(27 mg, 0.061 mmol, 49% yield). 1H NMR (400 MHz, DMSO-d6) δ 13.30 (s, 1H), 10.72 (s, 1H), 10.29 (s, 1H), 8.77 – 8.73 (m, 1H), 8.22 (dd, J = 7.8, 1.7 Hz, 1H), 8.07 – 8.02 (m, 2H), 7.50 – 7.44 (m, 2H), 7.29 – 7.25 (m, 1H), 7.10 (td, J = 7.5, 1.3 Hz, 1H), 6.35 (d, J = 2.1 Hz, 1H), 6.06 (s, 2H), 4.00 (s, 2H). 13C NMR (101 MHz, DMSO-d6) δ 164.4, 159.9, 159.1, 152.2, 150.3, 137.7, 136.5, 130.1, 129.1, 127.7, 127.3, 127.0, 126.8, 126.5, 123.9, 122.8, 122.7, 119.2, 48.6. HRMS (ESI): calc. for [M+H]+ C21H18N9O2+ 428.1578, found 428.1570.
4.9. Procedure for the synthesis of final compound 10.
4.9.1. Synthesis of intermediate 33
4,4,5,5-tetramethyl-2-vinyl-1,2,3-dioxoborolane (1.23 g, 8 mmol), K2CO3 (1.1 g, 8 mmol) and Pd(dppf)Cl2 (293 mg, 0.4 mmol) were added to a solution of methyl 2-bromobenzoate (0.86 g, 4 mmol) in dioxane/water (11 mL/0.5 mL) and the resulting mixture was stirred at 100 °C overnight. The mixture was diluted with water (15 mL) and extracted with ethyl acetate (3 × 25 mL). The combined organic layers were washed with brine (100 mL), dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure.[24] The residue was purified by flash column chromatography (hexanes to ethyl acetate) to provide methyl 2-vinylbenzoate (33) as transparent oil.
4.9.2. Synthesis of intermediate 37
4-iodophenyl trifluoromethanesulfonate (34) Trifluoromethanesulfonic anhydride (34 mL, 5.64 g, 20 mmol) was slowly added at 0 °C to a solution of 4-iodophenol (4 g, 18.17 mmol) in dry pyridine (9.3 mL). The resulting mixture was stirred at the same temperature for 5 minutes and then allowed to warm to 25 °C and stirred for overnight. The recation was poured into water and extracted with ethyl ether (50 mL). The organic layer was washed sequentially with water, 10% HCl (2x), water and brine, dried over anhydrous MgSO4 and concentrated under reduced pressure. The crude was purified by flash column chromatography (hexanes to ethyl acetate) to provide 4-iodophenyl trifluoromethanesulfonate (34) as transparent oil.
4.9.2.1. 4-(3-oxopropyl)phenyl trifluoromethanesulfonate (35).
Synthesized following Method c.
4.9.2.2. 4-(2-bromo-3-oxopropyl)phenyl trifluoromethanesulfonate (36).
Synthesized following Method d.
4.9.2.3. 4-((2-amino-4-oxo-4,7-dihydro-3H-pyrrolo[2,3-d]pyrimidin-5-yl)methyl)phenyl trifluoromethane sulfonate (37).
Synthesized following Method e.
4.9.3. Synthesis of intermediate 38
Dry DMF (16 mL/eq), intermediates 37 (1eq) and 33 (1.25 eq), Pd(OAc)2 (0.01 eq), PPh3 (0.02 eq) and dry trimethylamine (4 eq) were added to a pressure vial. The reaction mixture was stirred at 100 °C for 16h. The crude reaction mixure was filtered through silica pad and washed with EtOAc.[25] The solvent was evaporated and the product was was purified by flash column cromatography eluted initially with dichloromethane followed by 20% methanol in dichloromethane to give (E)-2-(4-((2-amino-4-oxo-4,7-dihydro-3H-pyrrolo[2,3-d]pyrimidin-5-yl)methyl)styryl) benzoate (38).
4.9.4. Synthesis of final compound 10
Synthesized following Method f.
4.9.5. methyl 2-vinylbenzoate (33)
(286 mg, 1.76 mmol, 44% yield) 1H NMR (400 MHz, Methanol-d4) δ 7.87 (d, J = 7.6 Hz, 1H), 7.28–7.57 (m, 4H), 5.61 (d, J = 7.6 Hz, 1H), 5.35 (d, J = 1.2 Hz, 1H), 3.88 (s, 3H). LC-MS (ESI) m/z 163.1 [M+H]+.
4.9.6. 4-iodophenyl trifluoromethanesulfonate (34)
(2.47 g, 7.02 mmol, 39% yield) 1H NMR (400 MHz, CDCl3) δ 7.81 – 7.75 (m, 2H), 7.07 – 7.00 (m, 2H). LC- MS (ESI) m/z 353.1 [M+H]+.
4.9.7. 4-(3-oxopropyl)phenyl trifluoromethanesulfonate (35).
(1.86 g, 6.69 mmol, 55% yield). 1H NMR (400 MHz, CDCl3) δ 9.82 (t, J = 1.6 Hz, 1H), 7.30 – 7.24 (m, 2H), 7.21 – 7.17 (m, 2H), 3.02 – 2.94 (m, 2H), 2.84 – 2.77 (m, 2H). LC-MS (ESI) m/z 283.1 [M+H]+.
4.9.8. 4-(2-bromo-3-oxopropyl)phenyl trifluoromethanesulfonate (36).
(1.05 g, 4.02 mmol, 61% yield). 1H NMR (400 MHz, CDCl3) δ 9.51 (d, J = 1.7 Hz, 1H), 7.37 – 7.31 (m, 2H), 7.29 – 7.21 (m, 2H), 4.44 (ddt, J = 8.0, 6.4, 1.7 Hz, 1H), 3.56 – 3.50 (m, 1H), 3.18 (ddd, J = 14.8, 8.0, 1.5 Hz, 1H). LC-MS (ESI) m/z 262.1 [M+H]+.
4.9.9. 4-((2-amino-4-oxo-4,7-dihydro-3H-pyrrolo[2,3-d]pyrimidin-5-yl)methyl)phenyl trifluoromethane sulfonate (37).
(704 mg, 1.81 mmol, 45% yield). 1H NMR (400 MHz, DMSO-d6) δ 10.76 (d, J = 2.1 Hz, 1H), 10.13 (s, 1H), 7.48 – 7.42 (m, 2H), 7.37 – 7.31 (m, 2H), 6.39 (d, J = 2.1 Hz, 1H), 6.01 (s, 2H), 3.97 (s, 2H). LC-MS (ESI) m/z 389.1 [M+H]+.
4.9.10. (E)-2-(4-((2-amino-4-oxo-4,7-dihydro-3H-pyrrolo[2,3-d]pyrimidin-5-yl)methyl)styryl) benzoate (38)
(34.7 mg, 0.082 mmol, 62% yield). 1H NMR (500 MHz, DMSO-d6) δ 10.73(s, 1H), 10.13 (s, 1H), 7.98 – 7.64 (m, 3H), 7.62 – 7.00 (m, 6H), 6.36 – 6.23 (m, 2H), 6.03 (s, 2H), 3.97 (s, 2H), 3.88 (s, 3H). LC-MS (ESI) m/z 401.1 [M+H]+.
4.9.11. (E)-2-(4-((2-amino-4-oxo-4,7-dihydro-3H-pyrrolo[2,3-d]pyrimidin-5-yl)methyl)styryl)benzoic acid (10).
(13.6 mg, 0.035 mmol, 41% yield). 1H NMR (500 MHz, DMSO-d6) δ 12.96 (bs, 1H), 11.03 (s, 1H), 10.69 (s, 1H), 7.84 – 7.74 (m, 3H), 7.59 – 7.25 (m, 4H), 7.16 – 7.03 (m, 2H), 6.81 – 6.68 (m, 2H), 6.38 (s, 2H), 3.94 (s, 2H).13C NMR (151 MHz, DMSO-d6) δ 168.7, 158.5, 151.8, 149.0, 141.9, 137.9, 134.6, 131.9, 130.7, 130.3, 129.6, 129.0, 127.2, 126.5, 126.4, 126.3, 126.0, 114.6, 98.8, 31.3. HRMS (ESI): calc. for [M+H]+ C21H19N4O4+ 387.1452, found 387,1448.
4.10. General procedure for the synthesis of final compounds 26–27
4.10.1. Synthesis of intermediate 39
1-bromo-2-methoxyethane (205 μL, 2.21 mmol) was added to a solution of 3-bromo-5-methoxyphenol (373 mg, 1.84 mmol), K2CO3 (381 mg, 2.76 mmol) and anhydrous DMF (10 mL) and the reaction mixture was stirred overnight at 85 °C. The resulting mixyure was then diluted in EtOAc and washed with water (3 × 50 mL). The organic phase was dried over anhydrous Na2SO4, filtered and concentrated to give 1-bromo-3-methoxy-5-(2-methoxyethoxy)benzene (41) as a white powder. n-BuLi (0.63 mL, 1.58 mmol) was added under nitrogen atmosphere to a solution of 41 (411 mg, 1.58 mmol) in dry THF (5 ml) at −78° C. and the reaction stirred for 30 minutes. Trimethoxyborane (0.21 mL, 1.9 mmol) in a THF solution was slowly added dropwise at 70° C, and the mixture warmed to room temperature and stirred for 16 hours. After the completion of the reaction, 20% HCl (2.7 mL) was added and stirred for 3 hours. The mixture was extracted with ethyl acetate, washed with water and brine, and dried over anhydrous Na2SO4 to provide (3-methoxy-5-(2-methoxyethoxy)phenyl)boronic acid (39) as pale brown solid.
4.10.2. Synthesis of intermediate 40
PdCl2(dppf) (0.03eq), KOAc (2.9 eq) and bis(pinacolato)diboron (1.1 eq) were added to a solution of methyl 3-bromo-5-methoxybenzoate (1 eq) in dry dioxane (5.6 mL/mmol) under nitrogen. The solution was stirred at 80 °C for 2 hours and then was diluted with dichloromethane (25 mL) and washed with water (25 mL). The organic layer was dried over anhydrous Na2SO4, filtered and the solvent was eliminated under reduced pressure to give methyl 3-methoxy-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzoate (42). Crude arylboronic acid pinacol ester (1 eq) and sodium periodate (3 eq) were stirred in 8 mL/mmol of a 4:1 mixture of acetone and water for 30 min, at which time aqueous NH4OAc (1eq) was added to the suspension. The reaction mixture was stirred at room temperature for 17 h and then diluted with water (30 ml) and extracted with ethyl acetate (2 × 60 ml). The combined extracts were washed with water (2 × 30 ml) and brine (30 ml), dried over anhydrous Na2SO4, filtered, and concentrated to dryness by rotary evaporation to give a colourless solid, (3-methoxy-5-(methoxycarbonyl)phenyl)boronic acid (40).
4.10.3. Synthesis of final compounds 26 and 27
Compounds 26 and 27 have been synthesized through the reaction between trifluoromethane sulfonate derivate 37 (1 eq) and the corresponding boronic acid 39 or 40 following general method h or i, respectively. Method h: A mixture of 39 (1.5 eq), 37 (1.0 eq), Pd(PPh3)4 (0.01 eq) and TEA (2.6 eq) in dioxane (10 mL/mmol) was stirred overnight at 90 °C The solvent was evaporated under reduced pressure and the residue purified by flash column cromatography eluted initially with dichloromethane followed by 20% methanol in dichloromethane to give 2-amino-5-((3’-methoxy-5’-(2-methoxyethoxy)-[1,1’-biphenyl]-4-yl)methyl)-3,7-dihydro-4H-pyrrolo [2,3-d]pyrimidin-4-one (26). Pale pink powder. General method i: A solution of 40 (2 eq), 37 (1.0 eq), Pd(PPh3)2Cl2 (0.05 eq) and (2M) Na2CO3 (4 eq) in DMF (1 mL/mmol) was stirred for 2h at 100 °C. The solvent was evaporated under reduced pressure and the residue purified by flash column cromatography eluted initially with dichloromethane followed by 20% methanol in dichloromethane to give 4’-((2-amino-4-oxo-4,7-dihydro-3H-pyrrolo[2,3-d]pyrimidin-5-yl)methyl)-5-methoxy-[1,1’-biphenyl]-3-carboxylic acid (27). Pale green powder.
4.10.4. 1-bromo-3-methoxy-5-(2-methoxyethoxy)benzene (41)
(411 mg, 1.58 mmol, 86% yield) 1H NMR (400 MHz, CDCl3) δ 6.73 – 6.64 (m, 2H), 6.45 – 6.39 (m, 1H), 4.07 (t, J = 4.7 Hz, 2H), 3.76 (s, 3H), 3.73 (t, J = 4.6 Hz, 2H), 3.44 (s, 3H). LC-MS (ESI) m/z 262,1 [M+H]+.
4.10.5. (3-methoxy-5-(2-methoxyethoxy)phenyl)boronic acid (39)
(223 mg, 0.99 mmol, 63% yield). 1H NMR (400 MHz, CDCl3) δ 7.38 – 7.32 (m, 1H), 6.76 – 6.67 (m, 1H), 6.52 – 6.48 (m, 1H), 4.12 (t, J = 4.7 Hz, 2H), 3.78 (s, 3H), 3.75 (t, J = 4.6 Hz, 2H), 3.45 (s, 3H). LC-MS (ESI) m/z 227,0 [M+H]+.
4.10.6. methyl 3-methoxy-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzoate (42)
(651 mg, 2.23 mmol, 89% yield). 1H NMR (400 MHz, DMSO-d6) δ 7.86 – 7.84 (m, 1H), 7.54 (dd, J = 2.7, 1.5 Hz, 1H), 7.38 (dd, J = 2.7, 0.9 Hz, 1H), 3.86 (s, 3H), 3.83 (s, 3H), 1.31 (s, 12H). LC-MS (ESI) m/z 293,0 [M+H]+.
4.10.7. (3-methoxy-5-(methoxycarbonyl)phenyl)boronic acid (40)
(453 mg, 2.16 mmol, 97% yield). 1H NMR (400 MHz, DMSO-d6) δ 7.56 (t, J = 2.4, 1H), 7.51 (t, J = 2.4, 1H), 7.46 (t, J = 2.3 Hz, 1H), 3.89 (s, 3H), 3.88 (s, 3H). LC-MS (ESI) m/z 211,1 [M+H]+.
4.10.8. 2-amino-5-((3’-methoxy-5’-(2-methoxyethoxy)-[1,1’-biphenyl]-4-yl)methyl)-3,7-dihydro-4H-pyrrolo [2,3-d]pyrimidin-4-one (26).
(49 mg, 0.117 mmol, 39% yield). 1H NMR (600 MHz, Methanol-d4) δ 7.48 (d, J = 7.7 Hz, 2H), 7.34 (d, J = 7.7 Hz, 2H), 6.73 (d, J = 6.2 Hz, 2H), 6.47 (s, 1H), 6.31 (s, 1H), 4.14 (t, J = 4.5 Hz, 2H), 4.08 (s, 2H), 3.81 (s, 3H), 3.75 (t, J = 4.6 Hz, 2H), 3.43 (s, 3H). 13C NMR (151 MHz, DMSO-d6) δ 164.9, 161.7, 160.5, 153.7, 150.6, 142.1, 136.3, 134.3, 126.9, 126.3, 122.1, 117.9, 113.3, 109.1, 107.9, 102.5, 70.6, 67.5, 59.0, 55.8, 31.5. HRMS (ESI): calc. for [M+H]+ C23H25N4O4+ 421.1870, found 421.1886.
4.10.9. 4’-((2-amino-4-oxo-4,7-dihydro-3H-pyrrolo[2,3-d]pyrimidin-5-yl)methyl)-5-methoxy-[1,1’-biphenyl]-3-carboxylic acid (27).
(32 mg, 0.082 mmol, 96% yield) 1H NMR (600 MHz, DMSO-d6) δ 12.85 (bs, 1H), 10.64 (d, J = 2.3 Hz, 1H), 10.09 (s, 1H), 7.07 – 7.04 (m, 2H), 6.96 – 6.95 (m, 1H), 6.91 – 6.90 (m, 1H), 6.62 – 6.60 (m, 2H), 6.54 (t, J = 2.3 Hz, 1H), 6.20 (s, J = 2.1 Hz, 1H), 5.98 (s, 2H), 3.80 (s, 2H), 3.74 (s, 3H). 13C NMR (151 MHz, DMSO-d6) δ 167.6, 160.8, 158.9, 155.5, 152.6, 151.5, 132.8, 129.8, 122.2, 120.0, 119.0, 115.2, 114.0, 109.3, 106.1, 105.6, 99.1, 55.6, 31.3. HRMS (ESI): calc. for [M+H]+ C21H19N4O4+ 391.1401, found 391,1393.
4.11. Protein Preparation
ChTS-DHFR was expressed, purified and estimation of protein concentration was determined as reported in previous study.[13] His6-human TS was expressed, purified and estimation of protein concentration was determined as previously described.[26] Δ7–29 human TS deletion mutant (mhTS) was expressed, and an estimation of protein concentration was determined as previously reported.[19]
4.12. Enzyme assay, and data analysis
TS activity was determined by following the conversion of substrates dUMP and 5,10-methylenetetrahydrofolate to products dTMP and dihydrofolate at 340 nm (Δ ε = 6400 M−1 cm−1) at 25°C.[13] Reaction conditions used to determining IC50 values for TS are as described in a previous study.[12] Compound 1 was utilized as a positive antifolate control. Changes in absorption were determined on a SpectroMax M5 plate reader (Molecular Devices). Data were fitted to an [Inhibitor] vs normalized response with variable slope equation using GraphPad Prism software (version 7.01).
4.13. Protein crystallography
The purified ChTS-DHFR enzyme was co-crystalized with inhibitor compounds by hanging drop vaporization with a protein:well solution ratio of 2:1 to screen crystallization conditions. Reservoir conditions for crystallization included 16–22 % (v/v) PEG 6000, 200 mM ammonium sulfate, 60 mM lithium sulfate, and 100 mM Tris, pH 8.0. ChTS-DHFR enzyme (approximately 24 mg/ml) was incubated with 100 mM Tris, pH 8.0, 1.5 mM NADPH, 1.5 mM FdUMP, 0.5 mM methotrexate, and 5 mM inhibitor compounds on ice for 1 h before setting up drops. Crystals grew in approximately 2 days at 18°C. All crystals were frozen with a cryosolution containing 25 % (v/v) ethylene glycol and 5 mM of the appropriate inhibitor compounds and flash cooled with liquid nitrogen.
The purified mhTS enzyme was co-crystalized with 14 or 23 by hanging drop vaporization with a protein:well solution ratio of 1:1 to screen crystallization conditions. Reservoir conditions included 16–18 % (v/v) PEG monomethyl ether 5000, and 100 mM Bis-Tris, pH 7.0. mhTS enzyme was co-crystalized with 5 by hanging drop vaporization with a protein:well solution ratio of 1:1 to screen crystallization conditions. Reservoir conditions included 28 % (v/v) PEG 4000, 200 mM lithium sulfate, and 100 mM Tris, pH 7.5. mhTS enzyme was co-crystalized with 15 by hanging drop vaporization with a protein:well solution ratio of 1:1 to screen crystallization conditions. Reservoir conditions included 40 % (v/v) PEG 400, and 100 mM imidazole, pH 8.0. mhTS enzyme (approximately 13 mg/ml) was incubated with 20 mM Bis-Tris, pH 7.0, 2 mM DTT, 2 mM dUMP, and 5 mM of inhibitor compounds on ice for 1 h before setting up drops. Crystals grew in approximately 1 week at 22 °C. All crystals were frozen with a cryosolution containing 25 % (v/v) ethylene glycol and 5 mM of the appropriate inhibitor compound and flash cooled with liquid nitrogen.
Diffraction data for ChTS-DHFR with 2, 5, 11, 14, 15, 17, 20, and 23 and mhTS with 5, 14, 15, and 23 were collected on beam line 24-ID-E, while diffraction data for ChTS-DHFR with 6, 16, and 25 was collected on beam line 24-ID-C through NE-CAT, using wavelength 0.979 Å. Diffraction data for ChTS-DHFR with 24 was collected on beam line 17-ID-FMX at NSLSII, using wavelength 0.979 Å. Data sets for best diffracting crystals were processed by XDS.[27] Phases were solved via molecular replacement using the program phaser.[28, 29] For ChTS-DHFR:compound a ChTS-DHFR structure in complex with four ligands (PDB ID 4Q0E) was used as the search model for molecular replacement. For mhTS:compound an apo mhTS structure (PDB ID 1HZW) was used as the search model for molecular replacement. The program COOT[30] was used for model building of the ligands into the electron density as well as residues 307–313 of mhTS. Structures were refined using Phenix[31] until acceptable R factors, geometry statistics (RMSDs for ideal bond lengths and angles), and Ramachandran statistics were achieved for the respective structures. Iterative build omit σA− weighted 2mFo-Fc electron density maps were generated using Phenix Autobuild.[32] Refinement statistics and PDB codes for structures are listed in Supplementary Table S2.
4.14. Molecular modeling
The computational procedures that were utilized are exactly the same as in earlier studies.[33–35] Starting from a crystal structure of the protein, the BOMB program[36] was used to build initial structures for the desired protein-ligand complex. Conjugate-gradient energy minimizations were performed to relax the structure, and Monte Carlo free-energy perturbation (MC/FEP) was carried out to compute relative free energies of binding. The calculations were performed with the MCPRO program[37] using the OPLS-AA force field for the protein,[38] OPLS/CM1A for the ligands,[39] and the TIP4P water model.[40]
Supplementary Material
Scheme 2.

General scheme for the synthesis of compound 10.
Highlights.
Twenty-six novel antifolate analogues were synthesized
Biochemical and X-ray studies were performed to determine ChTS inhibition
2-phenylacetate methylene linker important for optimal position in ChTS active site
Active site rigidity driving force of inhibitor selectivity
Acknowledgments
This work us supported by the National Institutes of Health under Award Numbers (AI083146) to K.S.A., and (GM32136) to W.L.J. and Training Grant (5T32AI007404–23) to D.J.C. This work is based upon research conducted at the Northeastern Collaborative Access Team beamlines, which are funded by the National Institute of General Medical Sciences (NIGMS) from the NIH [P41 GM103403]. Crystals screening was conducted with supports in the Yale Macromolecular X-ray Core Facility [1S10OD018007–01]. This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02–06CH11357. This research used FMX beamline of the National Synchrotron Light Source II, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Brookhaven National Laboratory under Contract No. DE-SC0012704. The Life Science Biomedical Technology Research resource is primarily supported by the NIH, NIGMS through a Biomedical Technology Research Resource P41 grant (P41 GM111244), and by the DOE Office of Biological and Environmental Research (KP1605010).
References
- [1].Checkley W, White AC Jr., Jaganath D, Arrowood MJ, Chalmers RM, Chen XM, Fayer R, Griffiths JK, Guerrant RL, Hedstrom L, Huston CD, Kotloff KL, Kang G, Mead JR, Miller M, Petri WA Jr., Priest JW, Roos DS, Striepen B, Thompson RC, Ward HD, Van Voorhis WA, Xiao L, Zhu G, Houpt ER, A review of the global burden, novel diagnostics, therapeutics, and vaccine targets for cryptosporidium, Lancet Infect Dis, 15 (2015) 85–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Kotloff KL, Nataro JP, Blackwelder WC, Nasrin D, Farag TH, Panchalingam S, Wu Y, Sow SO, Sur D, Breiman RF, Faruque AS, Zaidi AK, Saha D, Alonso PL, Tamboura B, Sanogo D, Onwuchekwa U, Manna B, Ramamurthy T, Kanungo S, Ochieng JB, Omore R, Oundo JO, Hossain A, Das SK, Ahmed S, Qureshi S, Quadri F, Adegbola RA, Antonio M, Hossain MJ, Akinsola A, Mandomando I, Nhampossa T, Acacio S, Biswas K, O’Reilly CE, Mintz ED, Berkeley LY, Muhsen K, Sommerfelt H, Robins-Browne RM, Levine MM, Burden and aetiology of diarrhoeal disease in infants and young children in developing countries (the Global Enteric Multicenter Study, GEMS): a prospective, case-control study, Lancet, 382 (2013) 209–222. [DOI] [PubMed] [Google Scholar]
- [3].Miller CN, Panagos CG, Mosedale WRT, Kvac M, Howard MJ, Tsaousis AD, NMR metabolomics reveals effects of Cryptosporidium infections on host cell metabolome, Gut Pathog, 11 (2019) 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Kissinger JC, Evolution of Cryptosporidium, Nat Microbiol, 4 (2019) 730–731. [DOI] [PubMed] [Google Scholar]
- [5].Martucci WE, Udier-Blagovic M, Atreya C, Babatunde O, Vargo MA, Jorgensen WL, Anderson KS, Novel non-active site inhibitor of Cryptosporidium hominis TS-DHFR identified by a virtual screen, Bioorg Med Chem Lett, 19 (2009) 418–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Mazurie AJ, Alves JM, Ozaki LS, Zhou S, Schwartz DC, Buck GA, Comparative genomics of cryptosporidium, Int J Genomics, 2013 (2013) 832756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Laurent F, Lacroix-Lamande S, Innate immune responses play a key role in controlling infection of the intestinal epithelium by Cryptosporidium, Int J Parasitol, 47 (2017) 711–721. [DOI] [PubMed] [Google Scholar]
- [8].Lee S, Ginese M, Girouard D, Beamer G, Huston CD, Osbourn D, Griggs DW, Tzipori S, Piperazine-Derivative MMV665917: an effective drug in the diarrheic piglet model of Cryptosporidium hominis, The Journal of infectious diseases, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Ruiz V, Czyzyk DJ, Valhondo M, Jorgensen WL, Anderson KS, Novel allosteric covalent inhibitors of bifunctional Cryptosporidium hominis TS-DHFR from parasitic protozoa identified by virtual screening, Bioorg Med Chem Lett, 29 (2019) 1413–1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Borad A, Ward H, Human immune responses in cryptosporidiosis, Future Microbiol, 5 (2010) 507–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Martucci WE, Vargo MA, Anderson KS, Explaining an unusually fast parasitic enzyme: folate tail-binding residues dictate substrate positioning and catalysis in Cryptosporidium hominis thymidylate synthase, Biochemistry, 47 (2008) 8902–8911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Kumar VP, Cisneros JA, Frey KM, Castellanos-Gonzalez A, Wang Y, Gangjee A, White AC Jr., Jorgensen WL, Anderson KS, Structural studies provide clues for analog design of specific inhibitors of Cryptosporidium hominis thymidylate synthase-dihydrofolate reductase, Bioorg Med Chem Lett, 24 (2014) 4158–4161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Atreya CE, Anderson KS, Kinetic characterization of bifunctional thymidylate synthase-dihydrofolate reductase (TS-DHFR) from Cryptosporidium hominis: a paradigm shift for ts activity and channeling behavior, J Biol Chem, 279 (2004) 18314–18322. [DOI] [PubMed] [Google Scholar]
- [14].Martucci WE, Rodriguez JM, Vargo MA, Marr M, Hamilton AD, Anderson KS, Exploring novel strategies for AIDS protozoal pathogens: alpha-helix mimetics targeting a key allosteric protein-protein interaction in C. hominis TS-DHFR, Medchemcomm, 4 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Yuthavong Y, Kamchonwongpaisan S, Leartsakulpanich U, Chitnumsub P, Folate metabolism as a source of molecular targets for antimalarials, Future Microbiology, 1 (2006) 113–125. [DOI] [PubMed] [Google Scholar]
- [16].Kumar VP, Frey KM, Wang Y, Jain HK, Gangjee A, Anderson KS, Substituted pyrrolo[2,3-d]pyrimidines as Cryptosporidium hominis thymidylate synthase inhibitors, Bioorg Med Chem Lett, 23 (2013) 5426–5428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Carreras CW, Santi DV, The catalytic mechanism and structure of thymidylate synthase, Annu Rev Biochem, 64 (1995) 721–762. [DOI] [PubMed] [Google Scholar]
- [18].Benkovic SJ, Hammes-Schiffer S, A perspective on enzyme catalysis, Science, 301 (2003) 1196–1202. [DOI] [PubMed] [Google Scholar]
- [19].Czyzyk DJ, Valhondo M, Jorgensen WL, Anderson KS, Understanding the Structural Basis of Species Selective, Stereospecific Inhibition for Cryptosporidium and Human Thymidylate Synthase, FEBS Lett, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Doan LT, Martucci WE, Vargo MA, Atreya CE, Anderson KS, Nonconserved residues Ala287 and Ser290 of the Cryptosporidium hominis thymidylate synthase domain facilitate its rapid rate of catalysis, Biochemistry, 46 (2007) 8379–8391. [DOI] [PubMed] [Google Scholar]
- [21].Aso K, Imai Y, Yukishige K, Ootsu K, Akimoto H, Pyrrolo[2,3-d]pyrimidine thymidylate synthase inhibitors: design and synthesis of one-carbon bridge derivatives, Chem Pharm Bull (Tokyo), 49 (2001) 1280–1287. [DOI] [PubMed] [Google Scholar]
- [22].Dahlgren MK, Schyman P, Tirado-Rives J, Jorgensen WL, Characterization of biaryl torsional energetics and its treatment in OPLS all-atom force fields, J Chem Inf Model, 53 (2013) 1191–1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Zhang G, Liu C, Yi H, Meng Q, Bian C, Chen H, Jian JX, Wu LZ, Lei A, External Oxidant-Free Oxidative Cross-Coupling: A Photoredox Cobalt-Catalyzed Aromatic C-H Thiolation for Constructing C-S Bonds, J Am Chem Soc, 137 (2015) 9273–9280. [DOI] [PubMed] [Google Scholar]
- [24].Kim R, Kuduk SD, Liverton N, Zhuo G, N-Heteroarylethyldiamines as orexin receptor antagonists and their preparation, in, Merck Sharp & Dohme Corp., USA. 2016, pp. 116pp. [Google Scholar]
- [25].Demmer CS, Rombach D, Liu N, Nielsen B, Pickering DS, Bunch L, Revisiting the Quinoxalinedione Scaffold in the Construction of New Ligands for the Ionotropic Glutamate Receptors, ACS Chem. Neurosci, 8 (2017) 2477–2495. [DOI] [PubMed] [Google Scholar]
- [26].Pedersen-Lane J, Maley GF, Chu E, Maley F, High-level expression of human thymidylate synthase, Protein Expr Purif, 10 (1997) 256–262. [DOI] [PubMed] [Google Scholar]
- [27].Kabsch W, Xds, Acta Crystallogr D Biol Crystallogr, 66 (2010) 125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].McCoy AJ, Solving structures of protein complexes by molecular replacement with Phaser, Acta Crystallogr D Biol Crystallogr, 63 (2007) 32–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ, Phaser crystallographic software J Appl Crystallogr, 40 (2007) 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Emsley P, Cowtan K, Coot: model-building tools for molecular graphics, Acta Crystallogr D Biol Crystallogr, 60 (2004) 2126–2132. [DOI] [PubMed] [Google Scholar]
- [31].Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH, PHENIX: a comprehensive Python-based system for macromolecular structure solution, Acta Crystallogr D Biol Crystallogr, 66 (2010) 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Terwilliger TC, Grosse-Kunstleve RW, Afonine PV, Moriarty NW, Adams PD, Read RJ, Zwart PH, Hung LW, Iterative-build OMIT maps: map improvement by iterative model building and refinement without model bias, Acta Crystallogr D Biol Crystallogr, 64 (2008) 515–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Lee WG, Gallardo-Macias R, Frey KM, Spasov KA, Bollini M, Anderson KS, Jorgensen WL, Picomolar inhibitors of HIV reverse transcriptase featuring bicyclic replacement of a cyanovinylphenyl group, J Am Chem Soc, 135 (2013) 16705–16713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Bollini M, Domaoal RA, Thakur VV, Gallardo-Macias R, Spasov KA, Anderson KS, Jorgensen WL, Computationally-guided optimization of a docking hit to yield catechol diethers as potent anti-HIV agents, J Med Chem, 54 (2011) 8582–8591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Jorgensen WL, Bollini M, Thakur VV, Domaoal RA, Spasov KA, Anderson KS, Efficient discovery of potent anti-HIV agents targeting the Tyr181Cys variant of HIV reverse transcriptase, J Am Chem Soc, 133 (2011) 15686–15696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Jorgensen WL, Efficient drug lead discovery and optimization, Acc Chem Res, 42 (2009) 724–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Jorgensen WL, Tirado-Rives J, Molecular modeling of organic and biomolecular systems using BOSS and MCPRO, J Comput Chem, 26 (2005) 1689–1700. [DOI] [PubMed] [Google Scholar]
- [38].Jorgensen WL, Maxwell DS, TiradoRives J, Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids, Journal of the American Chemical Society, 118 (1996) 11225–11236. [Google Scholar]
- [39].Jorgensen WL, Tirado-Rives J, Potential energy functions for atomic-level simulations of water and organic and biomolecular systems, Proc Natl Acad Sci U S A, 102 (2005) 6665–6670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML, Comparison of Simple Potential Functions for Simulating Liquid Water, J Chem Phys, 79 (1983) 926–935. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.