

Abstract
Despite the recent explosion in the use of monoclonal antibodies (mAbs) as drugs, it remains a significant challenge to generate antibodies with a combination of physicochemical properties that are optimal for therapeutic applications. We argue that one of the most important and underappreciated drug-like antibody properties is high specificity – defined here as low levels of antibody non-specific and self-interactions – which is linked to low off-target binding and slow antibody clearance in vivo and high solubility and low viscosity in vitro. Here we review the latest advances in characterizing antibody specificity and elucidating its molecular determinants as well as using these findings to improve the selection and engineering of antibodies with extremely high, drug-like specificity.
Keywords: mAb, solubility, aggregation, polyspecificity, pharmacokinetic
Graphical Abstract
Introduction
In hindsight, the success of antibodies as drugs seems obvious because of their many drug-like properties, including their high affinity, specificity and solubility as well as their slow clearance, low immunogenicity and low toxicity [1]. However, it is becoming increasingly clear that antibodies with similar IgG architectures and constant regions display highly variable and difficult-to-predict physicochemical properties that strongly influence their suitability for therapeutic applications. The variability in antibody physicochemical properties is mainly due to sequence and structural differences in their complementarity-determining regions (CDRs) and variable region frameworks [2–4], although differences outside the variable regions can also impact antibody performance [5,6].
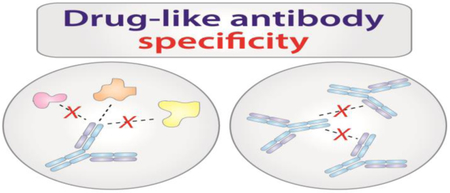
Specificity is one of the most complex antibody properties in general and one of the most important properties that impacts the efficacy of antibody drugs [7••,8••]. In this review, we define antibody specificity in terms of the relative propensity of antibodies to interact with molecules other than their antigens or receptors, which includes antibody non-specific (heterotypic) and self- (homotypic) interactions (Figure 1). Antibody specificity is poorly understood because it is a relative concept and dependent on the type of experimental measurements used to define it. Nevertheless, antibody specificity is critical to the success of antibody therapeutics because non-specific interactions can lead to off-target binding and result in fast antibody clearance in vivo [9,10], and self-interactions can lead to antibody aggregation and poor antibody solubility as well as abnormally high viscosity in vitro [11–14]
Figure 1. Drug-like antibodies have low levels of non-specific interactions.

Poor antibody specificity manifests itself in a number of observable phenomena both in vitro and in vivo. The impacts of high levels of non-specific (heterotypic) interactions are observed in vivo via fast antibody clearance, which has been linked to off-target binding and aberrant interactions with the neonatal Fc receptor (FcRn). Problems with high levels of antibody self- (homotypic) interactions arise during antibody formulation where it is often necessary to achieve exceedingly high protein concentrations. High levels of antibody self-interactions can lead to high solution viscosity, high aggregation, and/or low solubility. While heterotypic and homotypic interactions are often discussed independently, it is becoming increasingly clear that these undesirable behaviors often share similar molecular determinants, including excessively hydrophobic and/or charged interfaces.
The important role of antibody specificity in determining the relative likelihood of success of antibody drugs in the clinic was recently demonstrated in a comprehensive survey of the biophysical properties of 137 antibodies that are either approved drugs or in phase 2 or 3 clinical trials [7••]. The investigators evaluated twelve biophysical properties that are potentially important factors in determining the suitability of antibodies for therapeutic applications. These properties included antibody conformational (folding) stability, aggregation propensity, hydrophobicity, specificity (non-specific and self-interactions) and expression level in mammalian cells. Surprisingly, only specificity measurements (three non-specific interaction and two self-interaction assays) identified approved drugs as having superior biophysical properties relative to antibodies in clinical trials (Figure 2). Importantly, this finding suggests that approved antibody drugs are, on average, more specific than those in clinical trials and demonstrates that antibody specificity is a key property to optimize during the generation of antibody therapeutics.
Figure 2. Approved mAb drugs are more specific on average than mAbs in clinical trials.

Jain and colleagues performed a broad study of the biophysical properties of clinical-stage mAbs using twelve assays to identify the most important biophysical determinants of drug-like antibodies [7••]. Notably, measurements of antibody specificity (non-specific binding and self-association) were the only ones to statistically differentiate between approved antibody drugs and antibodies in clinical trials. This finding suggests that approved antibody drugs are, on average, more specific than those in clinical trials and highlights the importance of using specificity assays to guide the selection of antibodies with drug-like properties. The reported assays are: (A) baculovirus particle (BVP), (B) ELISA (panel of six biomolecules), (C) polyspecificity reagent (PSR), (D) clone self-interaction by biolayer interferometry (CSI-BLI), (E) affinity-capture self-interaction nanoparticle spectroscopy (AC-SINS), (F) salt gradient affinity-capture self-interaction nanoparticle spectroscopy (SGAC-SINS), (G) expression titer in HEK cells, (H) aggregation (accelerated stability), (I) folding stability (melting temperature of Fab), (J) cross-interaction chromatography (CIC), (K) hydrophobic interaction chromatography (HIC), and (L) standup monolayer adsorption chromatography (SMAC). It is important to note that poor performance in a single biophysical assay does not preclude the successful development of a therapeutic antibody.
Molecular determinants of antibody specificity
Non-specific antibody interactions are generally thought to be driven, in large part, by attractive electrostatic and hydrophobic interactions. This complicates the study of antibody specificity because some of the same residues that contribute to off-target binding also promote strong interactions with the target antigen, and it is often unclear what combination of residues are likely to cause reduced specificity. Nevertheless, focused studies on single or small sets of mAbs have advanced our understanding of the molecular determinants of specificity in recent years.
A major theme that has emerged in the study of antibody non-specific interactions is the risk conferred by excess positive charge. An early antibody engineering effort sought to increase cellular uptake and biodistribution of mAbs via cationization [15]. Despite successfully promoting cellular uptake in vitro, it was found that the positively charged mutations increased antibody clearance rates in mice by ninefold. Later, Igawa and colleagues demonstrated that the isoelectric point of mAbs is directly related to clearance rates for a small panel of antibodies [16]. They also showed that the antibody isoelectric point could be lowered by focused mutagenesis without significantly reducing affinity and that these mutations significantly reduced clearance rates in monkeys. A more complete review of the role of mAb charge on antibody pharmacokinetic behavior has been reported elsewhere [17].
Other major drivers of antibody non-specific interactions are solvent-exposed hydrophobic residues, especially when clustered in antibody CDRs. Analysis of the aggregation-prone mAb CNTO607 demonstrated that a hydrophobic hotspot (residues 99-FHW-100a) in heavy chain CDR3 was responsible for its aberrant behavior [18,19]. The investigators demonstrated that replacement of the hydrophobic residues with alanine eliminated aggregation but also severely compromised affinity for the target antigen, highlighting the importance of hydrophobicity in antigen recognition. Moreover, it is important to note that hydrophobic residues do not need to be consecutive in sequence to create hydrophobic clusters in the 3D antibody structure. Indeed, hydrogen-deuterium exchange mass spectrometry has been recently used to identify CDR residues in a mAb (MEDI1912) that become solvent-shielded due to self-association [20]. Unlike the consecutive residues identified in CNTO607, structural modeling was used to identify a discontinuous but spatially co-localized motif of hydrophobic residues in heavy chain CDR1 (W30 and F31) and CDR2 (L56). Mutation of all three hydrophobic residues to more hydrophilic ones (threonine or serine) reduced aggregation and improved pharmacokinetics without impacting antigen affinity. Finally, a recent study reaffirmed the detrimental nature of hydrophobic clusters by actively screening for antibodies with low specificity [21]. The investigators found that hydrophobic motifs such as three consecutive valines or tryptophans were enriched during positive selections for antibody non-specific interactions.
Two residues in particular, namely arginine and tyrosine, have proven to be particularly enigmatic in terms of their impact on antibody specificity. While the role of positive charge is often discussed in general terms, there are numerous reports demonstrating a prominent role for arginine in mediating non-specific interactions [21–24]. The distributed positive charge and relatively poor hydration of the guanidinium group as well as the long hydrophobic side chain are likely contributors to the ability of arginine to mediate antibody non-specific interactions. Nevertheless, arginine plays an important role in antigen recognition and it has been demonstrated that the contribution of arginine to antibody polyspecificity is context dependent [25]. Like arginine, the impact of tyrosine on antibody specificity is complex and context-dependent. Nevertheless, tyrosine has been primarily associated with specific interactions [21–23,26]. For example, highly specific antibodies have been isolated using antibody libraries with CDRs that are only diversified with a limited number of residues (including tyrosine), and these specific antibodies were generally enriched in tyrosine content [22,23,26]. However, there are examples of tyrosine contributing to polyspecificity in certain contexts including a recent report demonstrating that motifs such as two consecutive tyrosines in HCDR3 lead to increased non-specific antibody binding [21]. Interestingly, tyrosine is one of the most abundant CDR residues in naive and mature antibodies, which also highlights its important role in typically mediating specific antibody-antigen interactions [26–28].
An often overlooked contributor to antibody specificity is glycosylation. Glycosylation generally reduces antibody non-specific interactions, likely by masking hydrophobic patches with hydrophilic and/or anionic carbohydrate moieties. In fact, several studies have demonstrated the effectiveness of incorporating N-linked, non-conventional glycosylation sites near residues that mediate antibody non-specific or self-interactions [19,29]. Further, the introduction of glycosylation sites in the Fab region of IgGs near predicted aggregation hotspots can also reduce aggregation propensity [30]. While glycosylation can be an effective approach for improving antibody specificity, its utility in therapeutic antibody development is limited because glycans at non-conventional sites increase antibody heterogeneity during production, and complicate analytical characterization and product validation.
Characterizing drug-like antibody specificity
Focused research studies on small numbers of antibodies have done much to expand our understanding of antibody specificity. However, there is a critical need for methodologies that can evaluate antibody specificity for large numbers of antibodies – which cannot be studied with the level of detail discussed above – during early discovery and lead optimization. Therefore, there has been intense interest in recent years in developing in vitro assays that can identify antibodies with poor physicochemical properties early in the development pipeline (Figure 3).
Figure 3. Preclinical assays for evaluating antibody specificity are important tools in drug development.

The rise in awareness of the importance of antibody specificity has prompted the development of several assays that report on different types of non-specific interactions. Antibody self-interactions can be probed using methods such as nanoparticle-based (e.g., AC-SINS) or biolayer interferometry (e.g., CSI-BLI) methods [41,53]. Non-specific interactions are often assessed using ELISA methods but now are also being evaluated using flow cytometry-based (PSR) methods [32,33•]. FcRn binding assays are also useful for identifying antibodies with increased risk of displaying poor pharmacokinetic properties due to abnormal interactions mediated via the antibody variable regions with FcRn [37,38]. Pharmacokinetic studies in model organisms (e.g., mouse) are a mainstay in drug development, but the creation of transgenic mice with humanized FcRn has been a major advance for the assessment of therapeutic antibodies [54]. Finally, cell binding assays provide a simple yet valuable assessment of potential antibody behavior in vivo [10]. These assays are most powerful when used in combination for preclinical antibody characterization and lead selection [8••].
Several studies have leveraged the versatility of ELISA methods to characterize antibody non-specific interactions. In a study of antibody maturation in vivo, Wardemann and colleagues discovered that immature B-cells often produce antibodies with clusters of positively charged amino acids in their CDRs and that these antibodies are highly polyspecific [31]. They used an ELISA method to demonstrate that these antibody variants were prone to interact non-specifically with diverse biomolecules (many of which were anionic), including lipopolysaccharides, DNA and proteins. Further, the prevalence of such clones is diminished during B-cell maturation in vivo, underscoring the risks of pursuing abnormally cationic antibodies for clinical development. More recently, ELISA methods have been developed to evaluate antibody non-specific binding to heparin [9,10]. In these studies, the investigators demonstrated that antibody charge distribution is a major contributor to non-specific interactions and that redistribution of positive charge may serve the same purpose as charge reduction. An unexpected development has been the use of virus particles (baculovirus particles, BVP) for the assessment of antibody non-specific interactions. Notably, measurements of non-specific binding to such viral particles – which present diverse types of lipids, glycans and proteins – were positively correlated with antibody clearance rates in humans and non-human primates [32].
It would be particularly significant if antibodies could be simultaneously selected for both strong antigen recognition and weak recognition of non-target molecules using in vitro antibody discovery methods such as phage and yeast surface display. Indeed, a powerful approach has emerged that leverages the high-throughput capacity of flow cytometry to profile antigen and non-specific binding in parallel [33•]. This approach employs a polyspecificity reagent (PSR) that is generated via biotinylation of soluble membrane proteins from eukaryotic cell lines (e.g., CHO). This strategy has been elegantly implemented using the yeast display platform [7••,33•,34]. Recently, efforts to reduce the potential variability of PSR preparations have demonstrated the use of well-characterized chaperones like Hsp90 as alternative and effective polyspecificity reagents [34].
It is also important to evaluate antibody non-specific interactions with natural antibody receptors. Of particular interest is the pH-dependent interaction of the antibody Fc region with its neonatal Fc receptor (FcRn), which promotes antibody recycling from cellular (endosomal) compartments [35]. Methods for assessing antibody-FcRn interactions include surface plasmon resonance (SPR), biolayer interferometry (BLI), and liquid chromatography [36–38]. A major finding in these studies is that antibodies with identical amino acid sequences in their Fc region and different sequences in their variable regions can have significantly different pH-dependent rates of association and dissociation with FcRn [36]. Although these methods have proven useful in studies of antibody clearance, no methodology to date has been reported for use in high-throughput screening. Assays that characterize aberrant FcRn interactions early in development are expected to be particularly useful in generating antibodies with drug-like specificity.
Another important requirement for therapeutic antibodies is their ability to be formulated at concentrations that are several orders of magnitude higher than their final therapeutic levels in vivo. Antibodies with high propensity to self-associate can display aggregation and abnormally high viscosity at elevated antibody concentrations [39,40]. These characteristics are difficult to identify early in development because high antibody concentrations are typically required to detect them. However, a nanoparticle-based assay – namely affinity capture self-interaction nanoparticle spectroscopy (AC-SINS) – has been developed for high-throughput identification of antibodies with high levels of self-association [41,42], low solubility [41,43,44] and abnormally high viscosity [45]. Unlike traditional means of assessing these solution properties (e.g., light scattering), AC-SINS requires only very dilute (μg/mL) antibody concentrations. Additionally, AC-SINS measurements are correlated with various types of non-specific binding measurements [7••,8••,42,44] and, in some cases, with pharmacokinetic properties [8••,45].
Until recently, most studies of antibody specificity have focused on small sets of mAbs and have been unable to evaluate the broad applicability of novel screening methods to antibody development at large. Two recent studies have addressed this issue by analyzing relatively large panels of mAbs – including those that are approved drugs or in clinical development – using multiple specificity assays [7••,8••]. The first study by Jain and colleagues (discussed in the Introduction and Figure 2) applied twelve antibody characterization assays to 137 antibodies that were either approved drugs or in clinical trials [7••]. In addition to their notable finding that approved antibody drugs are, on average, more specific than antibodies in clinical trials, the investigators also found that antibodies developed or engineered using phage display had inferior biophysical properties relative to those generated using mammalian systems. The latter finding underscores the importance of implementing specificity assays during in vitro antibody discovery and optimization. A similar study by Avery and colleagues used specificity assays to evaluate a smaller set of clinical mAbs, although this study also included human and mouse pharmacokinetic data [8••]. They observed good correlations between specificity assays and clearance rates, but no single assay was predictive enough for robust identification of drug-like antibody candidates. Instead, the investigators elegantly demonstrated how a suite of specificity assays could be employed to identify antibodies with favorable pharmacokinetic properties.
Emerging computational methods for studying antibody specificity
Rapid advances in computational power and accumulation of publicly available experimental data have driven the emergence of in silico approaches in nearly every realm of biotechnology. The field of antibody discovery and engineering, including the study of antibody specificity, has not been exempt from this revolution. Indeed, a number of the studies discussed in this review integrated computational methods into their analysis of antibody non-specific interactions [10,19,20,29,30]. Recently, in an effort to engineer improved variants of an antibody that had unacceptably high viscosity at elevated concentrations, Nichols and colleagues made extensive use of homology modeling and molecular dynamics simulations to identify aggregation hot spots in the parent antibody, design antibody variants, and test their thermodynamic stability [48]. Importantly, the investigators experimentally evaluated the impact of their designed mutations on multiple antibody physicochemical properties (e.g. viscosity and solubility) and binding affinity in order to test the utility of their approach. One notable finding was the identification of a negatively charged patch in the variable regions that substantially contributed to antibody viscosity, an intriguing discovery given that several other studies highlight problematic electrostatic interactions associated with positively charged patches [13–15,48]. This suggests that excessive electrostatic charge leading to high viscosity can be either cationic or anionic in nature.
Homology modeling and molecular dynamics simulations have also been used to study how Fv properties impact FcRn binding for two antibodies with highly similar constant domains [51]. Through modeling, the investigators identified cationic domains that were predicted to be responsible for excessive FcRn binding and failure to dissociate efficiently from FcRn at neutral pH. They validated their computational results experimentally by creating chimeric variants in which they generated several combinations of heavy and light chain variable regions from the different antibodies and then experimentally probed their FcRn binding properties. Molecular dynamics simulations revealed that the mechanism for the Fv-mediated FcRn interaction is through the flexibility of the hinge region, which permitted the Fab regions to spatially co-localize with FcRn and these interactions were stabilized by the excess positive charge in the variable regions.
While the use of computational tools to study antibody structure are rapidly developing, many popular approaches are limited by the complexity of available modeling approaches. Thus, there is a critical need for methods that can predict antibody biophysical properties from sequence data alone. Such knowledge would be useful for guiding antibody library design and improving discovery programs. There are several excellent antibody databases that provide extensive sequence information for naturally produced antibodies, which can be used probe the sequence characteristics of antibodies generated by the immune system [27,52]. However, monoclonal antibody therapeutics require biophysical properties that are, in some cases, unique from those of naturally occurring antibodies. Thus, there is a need for predictive methods that relate antibody sequences to their biophysical properties. Sharma and colleagues demonstrated such an approach by developing a model to predict antibody clearance rates based on variable region charge and hydrophobicity in select CDRs [49•]. In particular, they discerned that antibodies with a combined hydrophobicity value of >4.0 (Eisenberg scale) in light chain CDRs 1 and 3 and heavy chain CDR 3, and/or an Fv charge of <0 or >6 (pH = 5.5) were at risk for fast clearance from cynomolgus monkeys.
In a follow-up study, these investigators used their empirical model to predict the pharmacokinetics of rationally engineered antibody variants and discovered that variants with poor in vivo pharmacokinetics could be predicted reasonably well using their computational framework [50]. Specifically, they selected one antibody known to have normal pharmacokinetics and a second one with fast clearance, and designed antibody mutants with increased or decreased Fv charge. They demonstrated that both cationized variants suffered from faster clearance than their parental mAbs while the more anionic variants corresponding to the fast-clearing parent showed improved pharmacokinetics and those variants corresponding to the slow clearing parent showed little change. In the future, the continued emergence and exploration of large data sets of biophysical properties for diverse antibody sequence variants will further the development of predictive methods.
Conclusions
Antibodies are regarded as the pinnacle of specific binding agents in the biological world and that characterization is accurate in many ways. However, as our interest in antibody versatility in the laboratory and clinic has grown, so has our awareness of antibody binding promiscuity. Important work in recent years has advanced our understanding of antibody specificity and spurred the development of assay platforms for the study and characterization of diverse types of antibody interactions. Continued assay development and optimization with a focus on high-throughput applications as well as predictive computational methods are areas where improvements could vastly change the pharmaceutical discovery landscape. In the future, it will be important to incorporate these concepts as early in the process as possible into antibody discovery and engineering programs.
Highlights.
Specificity is a critical attribute of drug-like monoclonal antibodies
Antibody specificity is compromised by excess localized charge and/or hydrophobicity
High-throughput detection of non-specific clones is enhancing development programs
Computational methods to predict antibody specificity are in high demand
Acknowledgements
We thank members of the Tessier lab for their helpful suggestions. This work was supported by the National Institutes of Health [R01GM104130 and R01AG050598 to P.M.T.], National Science Foundation [CBET 1813963 and 1605266 to P.M.T.], and the Albert M. Mattocks Chair (to P.M.T).
Footnotes
Conflict of interest
P.M.T. has received consulting fees and/or honorariums for presentations of this and/or related research findings at MedImmune, Eli Lilly, Bristol-Myers Squibb, Janssen, Merck, Genentech, Amgen, Pfizer, Adimab, Abbvie, Roche, Boehringer Ingelheim, Bayer, Abbott, DuPont, Schrodinger and Novo Nordisk.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
Papers of special interest (•)
Papers of outstanding (••) interest
- 1.Tiller KE, Tessier PM: Advances in Antibody Design. Annu Rev Biomed Eng 2015, 17:191–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rouet R, Lowe D, Christ D: Stability engineering of the human antibody repertoire. FEBS Left 2014, 588:269–277. [DOI] [PubMed] [Google Scholar]
- 3.Lowe D, Dudgeon K, Rouet R, Schofield P, Jermutus L, Christ D: Aggregation, stability, and formulation of human antibody therapeutics. Elsevier Inc.; 2011. [DOI] [PubMed] [Google Scholar]
- 4.Wang X, Das TK, Singh SK, Kumar S: Potential aggregation prone regions in biotherapeutics: A survey of commercial monoclonal antibodies. MAbs 2009, 1:254–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ito T, Tsumoto K: Effects of subclass change on the structural stability of chimeric, humanized, and human antibodies under thermal stress. Protein Sci 2013, 22:1542–1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chennamsetty N, Helk B, Voynov V, Kayser V, Trout BL: Aggregation-Prone Motifs in Human Immunoglobulin G. J Mol Biol 2009, 391:404–413. [DOI] [PubMed] [Google Scholar]
- 7. ••.Jain T, Sun T, Durand S, Hall A, Houston NR, Nett JH, Sharkey B, Bobrowicz B, Caffry I, Yu Y, et al. : Biophysical properties of the clinical-stage antibody landscape. Proc Natl Acad Sci 2017, 114:944–949.This study is the largest survey to date of the biophysical properties of therapeutic mAbs using a panel of twelve in vitro characterization assays, including five for specificity. Key findings include the ability of specificity assays to delineate between approved and clinical-stage antibodies, and the superior performance of antibodies discovered via immunization as opposed to those discovered via in vitro phage display. The authors have included in their report the sequences and biophysical measurements for the 137 mAbs.
- 8. ••.Avery LB, Wade J, Wang M, Tam A, King A, Piche-Nicholas N, Kavosi MS, Penn S, Cirelli D, Kurz JC, et al. : Establishing in vitro in vivo correlations to screen monoclonal antibodies for physicochemical properties related to favorable human pharmacokinetics. MAbs 2018, 10:244–255.This study correlated in vitro antibody characterization assay data with in vivo pharmacokinetic data in order to improve lead candidate selection based on multiple types of biophysical measurements. They found that most assays were partially predictive of in vivo pharmacokinetics, but none were sufficient as standalone determinants. Thus, they propose a compelling triage system in which results from a panel of assays are used to improve the selection of drug-like antibody candidates.
- 9.Datta-mannan A, Lu J, Witcher DR, Leung D, Tang Y, Wroblewski VJ: The interplay of non-specific binding, target-mediated clearance and FcRn interactions on the pharmacokinetics of humanized antibodies. MAbs 2015, 7:1084–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Datta-Mannan A, Thangaraju A, Leung D, Tang Y, Witcher DR, Lu J, Wroblewski VJ: Balancing charge in the complementarity-determining regions of humanized mAbs without affecting pl reduces non-specific binding and improves the pharmacokinetics. MAbs 2015, 7:483–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yadav S, Liu J, Shire SJ, Kalonia DS: Specific interactions in high concentration antibody solutions resulting in high viscosity. J Pharm Sci 2010, 99:1152–1168. [DOI] [PubMed] [Google Scholar]
- 12.Wang W, Singh S, Zeng DL, King K, Nema S: Antibody structure, instability, and formulation. J Pharm Sci 2007, 96:1–26. [DOI] [PubMed] [Google Scholar]
- 13.Connolly BD, Petry C, Yadav S, Demeule B, Ciaccio N, Moore JMR, Shire SJ, Gokarn YR: Weak interactions govern the viscosity of concentrated antibody solutions: High-throughput analysis using the diffusion interaction parameter. Biophys J 2012, 103:69–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Perchiacca JM, Tessier PM: Engineering Aggregation-Resistant Antibodies. Annu Rev Chem Biomol Eng 2012, 3:263–286. [DOI] [PubMed] [Google Scholar]
- 15.Pardridge WM, Kang Y-S, Yang J, Buciak JL: Enhanced cellular uptake and in vivo biodistribution of a monoclonal antibody following cationization. J Pharm Sci 1995, 84:943–948. [DOI] [PubMed] [Google Scholar]
- 16.Igawa T, Tsunoda H, Tachibana T, Maeda A, Mimoto F, Moriyama C, Nanami M, Sekimori Y, Nabuchi Y, Aso Y, et al. : Reduced elimination of IgG antibodies by engineering the variable region. 2010, 23:385–392. [DOI] [PubMed] [Google Scholar]
- 17.Boswell CA, Tesar DB, Mukhyala K, Theil FP, Fielder PJ, Khawli LA: Effects of charge on antibody tissue distribution and pharmacokinetics. Bioconjug Chem 2010, 21:2153–2163. [DOI] [PubMed] [Google Scholar]
- 18.Bethea D, Wu SJ, Luo J, Hyun L, Lacy ER, Teplyakov A, Jacobs SA, O’Neil KT, Gilliland GL, Feng Y: Mechanisms of self-association of a human monoclonal antibody CNTO607. Protein Eng Des Sel 2012, 25:531–537. [DOI] [PubMed] [Google Scholar]
- 19.Wu S, Luo J, Neil KTO, Kang J, Lacy ER, Canziani G, Baker A, Huang M, Tang QM, Raju TS, et al. : Structure-based engineering of a monoclonal antibody for improved solubility. 2010, 23:643–651. [DOI] [PubMed] [Google Scholar]
- 20.Dobson CL, Devine PWA, Phillips JJ, Higazi DR, Lloyd C, Popovic B, Arnold J, Buchanan A, Lewis A, Goodman J, et al. : Engineering the surface properties of a human monoclonal antibody prevents self-association and rapid clearance in vivo. Sci Rep 2016, 6:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kelly RL, Le D, Zhao J, Wittrup KD: Reduction of Nonspecificity Motifs in Synthetic Antibody Libraries. J Mol Biol 2018, 430:119–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Birtalan S, Fisher RD, Sidhu SS: The functional capacity of the natural amino acids for molecular recognition. Mol Biosyst 2010, 6:1186–1194. [DOI] [PubMed] [Google Scholar]
- 23.Birtalan S, Zhang Y, Fellouse FA, Shao L, Schaefer G, Sidhu SS: The Intrinsic Contributions of Tyrosine, Serine, Glycine and Arginine to the Affinity and Specificity of Antibodies. J Mol Biol 2008, 377:1518–1528. [DOI] [PubMed] [Google Scholar]
- 24.Kelly RL, Zhao J, Le D, Wittrup KD: Nonspecificity in a nonimmune human scFv repertoire. MAbs 2017, 9:1029–1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tiller KE, Li L, Kumar S, Julian MC, Garde S, Tessier PM: Arginine mutations in antibody complementarity-determining regions display context-dependent affinity/specificity trade-offs. J Biol Chem 2017, 292:16638–16652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mahon CM, Lambert MA, Glanville J, Wade JM, Fennell BJ, Krebs MR, Armellino D, Yang S, Liu X, O’Sullivan CM, et al. : Comprehensive interrogation of a minimalist synthetic CDR-H3 library and its ability to generate antibodies with therapeutic potential. J Mol Biol 2013, 425:1712–1730. [DOI] [PubMed] [Google Scholar]
- 27.Swindells MB, Porter CT, Couch M, Hurst J, Abhinandan KR, Nielsen JH, Macindoe G, Hetherington J, Martin ACR: abYsis: Integrated Antibody Sequence and Structure–Management, Analysis, and Prediction. J Mol Biol 2017, 429:356–364. [DOI] [PubMed] [Google Scholar]
- 28.Clark LA, Ganesan S, Papp S, van Vlijmen HWT: Trends in Antibody Sequence Changes during the Somatic Hypermutation Process. J Immunol 2006, 177:333–340. [DOI] [PubMed] [Google Scholar]
- 29.Chuang GY, Zhang B, McKee K, O’Dell S, Kwon Y Do, Zhou T, Blinn J, Lloyd K, Parks R, Von Holle T, et al. : Eliminating antibody polyreactivity through addition of N-linked glycosylation. Protein Sci 2015, 24:1019–1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Courtois F, Agrawal NJ, Lauer TM, Trout BL: Rational design of therapeutic mAbs against aggregation through protein engineering and incorporation of glycosylation motifs applied to bevacizumab. MAbs 2016, 8:99–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wardemann H, Yurasov S, Nussenzweig MC, Young JW, Meffre E, Nussenzweig MC: Predominant Autoantibody Production by Early Human B Cell Precursors. Science (80-) 2003, 301:1374–1378. [DOI] [PubMed] [Google Scholar]
- 32.Hötzel I, Theil FP, Bernstein LJ, Prabhu S, Deng R, Quintana L, Lutman J, Sibia R, Chan P, Bumbaca D, et al. : A strategy for risk mitigation of antibodies with fast clearance. MAbs 2012. 4:753–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. •.Xu Y, Roach W, Sun T, Jain T, Prinz B, Yu TY, Torrey J, Thomas J, Bobrowicz P, Vasquez M, et al. : Addressing polyspecificity of antibodies selected from an in vitro yeast presentation system: A FACS-based, high-throughput selection and analytical tool. Protein Eng Des Sel 2013, 26:663–670.This study introduces the combination of a novel polyspecificity reagent (PSR) for studying non-specific interactions and its application to antibody discovery. Unlike many traditional assay platforms for evaluating off-target binding (e.g., ELISA), PSR permits screening for target antigen binding and off-target binding in parallel using yeast surface display. Although it has been developed for use with fluorescence-activated cell sorting (FACS), it could potentially be applied in other selection formats, including with magnetic-activated cell sorting (MACS).
- 34.Kelly RL, Geoghegan JC, Feldman J, Jain T, Kauke M, Le D, Zhao J, Wittrup KD: Chaperone proteins as single component reagents to assess antibody nonspecificity. MAbs 2017, 9:1036–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ward ES, Devanaboyina SC, Ober RJ: Targeting FcRn for the modulation of antibody dynamics. Mol Immunol 2015, 67:131–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang W, Lu P, Fang Y, Hamuro L, Pittman T, Carr B, Hochman J, Prueksaritanont T: Monoclonal antibodies with identical Fc sequences can bind to FcRn differentially with pharmacokinetic consequences. Drug Metab Dispos 2011, 39:1469–1477. [DOI] [PubMed] [Google Scholar]
- 37.Souders CA, Nelson SC, Wang Y, Crowley AR, Mark S, Jr WT, Souders CA, Nelson SC, Wang Y, Crowley AR, et al. : A novel in vitro assay to predict neonatal Fc receptor-mediated human IgG half-life. MAbs 2015, 0862:912–921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schlothauer T, Rueger P, Stracke JO, Hertenberger H, Fingas F, Kling L, Emrich T, Drabner G, Seeber S, Auer J, et al. : Analytical FcRn affinity chromatography for functional characterization of monoclonal antibodies. MAbs 2013, 5:576–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tomar DS, Kumar S, Singh SK, Goswami S, Li L: Molecular basis of high viscosity in concentrated antibody solutions: Strategies for high concentration drug product development. MAbs 2016, 8:216–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lee CC, Perchiacca JM, Tessier PM: Toward aggregation-resistant antibodies by design. Trends Biotechnol 2013, 31:612–620. [DOI] [PubMed] [Google Scholar]
- 41.Sule SV, Dickinson CD, Lu J, Chow CK, Tessier PM: Rapid analysis of antibody self-association in complex mixtures using immunogold conjugates. Mol Pharm 2013, 10:1322–1331. [DOI] [PubMed] [Google Scholar]
- 42.Liu Y, Caffry I, Wu J, Geng SB, Jain T, Sun T, Reid F, Cao Y, Estep P, Yu Y, et al. : High-throughput screening for developability during early-stage antibody discovery using self-interaction nanoparticle spectroscopy. MAbs 2014, 6:483–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wu J, Schultz JS, Weldon CL, Sule SV, Chai Q, Geng SB, Dickinson CD, Tessier PM: Discovery of highly soluble antibodies prior to purification using affinity-capture self-interaction nanoparticle spectroscopy. Protein Eng Des Sel 2015, 28:403–414. [DOI] [PubMed] [Google Scholar]
- 44.Geng SB, Wittekind M, Vigil A, Tessier PM: Measurements of Monoclonal Antibody Self-Association Are Correlated with Complex Biophysical Properties. Mol Pharm 2016, 13:1636–1645. [DOI] [PubMed] [Google Scholar]
- 45.Geoghegan JC, Fleming R, Damschroder M, Bishop SM, Sathish HA, Esfandiary R: Mitigation of reversible self-association and viscosity in a human IgG1 monoclonal antibody by rational, structure-guided Fv engineering. MAbs 2016, 8:941–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Alam ME, Geng SB, Bender C, Ludwig SD, Linden L, Hoet R, Tessier PM: Biophysical and Sequence-Based Methods for Identifying Monovalent and Bivalent Antibodies with High Colloidal Stability. Mol Pharm 2018, 15:150–163. [DOI] [PubMed] [Google Scholar]
- 47.Betts A, Keunecke A, van Steeg TJ, van der Graaf PH, Avery LB, Jones H, Berkhout J: Linear pharmacokinetic parameters for monoclonal antibodies are similar within a species and across different pharmacological targets: A comparison between human, cynomolgus monkey and hFcRn Tg32 transgenic mouse using a population-modeling approach. MAbs 2018, 10:751–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nichols P, Li L, Kumar S, Buck PM, Singh SK, Goswami S, Balthazor B, Conley TR, Sek D, Allen MJ: Rational design of viscosity reducing mutants of a monoclonal antibody: Hydrophobic versus electrostatic inter-molecular interactions. MAbs 2015, 7:212–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. •.Sharma VK, Patapoff TW, Kabakoff B, Pai S, Hilario E, Zhang B, Li C, Borisov O, Kelley RF, Chorny I, et al. : In silico selection of therapeutic antibodies for development: Viscosity, clearance, and chemical stability. Proc Natl Acad Sci 2014, 111:18601–18606.This study presents a series of computational approaches for predicting antibody physicochemical properties based on sequence and structural information. Of particular interest are methods for predicting antibody viscosity based on amino acid sequences and an empirical framework for identifying antibodies with rapid clearance profiles based on Fv charge and CDR hydrophobicity.
- 50.Yadav DB, Sharma VK, Boswell CA, Hotzel I, Tesar D, Shang Y, Ying Y, Fischer SK, Grogan JL, Chiang EY, et al. : Evaluating the use of antibody variable region (Fv) charge as a risk assessment tool for predicting typical cynomolgus monkey pharmacokinetics. J Biol Chem 2015, 290:29732–29741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schoch A, Kettenberger H, Mundigl O, Winter G, Engert J, Heinrich J, Emrich T: Charge-mediated influence of the antibody variable domain on FcRn-dependent pharmacokinetics. Proc Natl Acad Sci 2015, 112:5997–6002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lefranc MP, Giudicelli V, Duroux P, Jabado-Michaloud J, Folch G, Aouinti S, Carillon E, Duvergey H, Houles A, Paysan-Lafosse T, et al. : IMGT R, the international ImMunoGeneTics information system R 25 years on. Nucleic Acids Res 2015, 43:D413–D422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sun T, Reid F, Liu Y, Cao Y, Estep P, Nauman C, Xu Y: High throughput detection of antibody self-interaction by bio-layer interferometry. MAbs 2013, 5:838–841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Avery LB, Wang M, Kavosi MS, Joyce A, Kurz JC, Fan Y: Utility of a human FcRn transgenic mouse model in drug discovery for early assessment and prediction of human pharmacokinetics of monoclonal antibodies. MAbs 2016, 8:1064–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]