

Kumar et al. describe a detailed pathway for channeling fat from the liver into blood across fed/fasted cycles. Insulin, phosphatidic acid, and kinesin collaborate in hepatocytes to deliver lipid droplets to the smooth ER, where they are catabolized to supply fat for lipoprotein production and secretion.
Abstract
Triglyceride-rich lipid droplets (LDs) are catabolized with high efficiency in hepatocytes to supply fatty acids for producing lipoprotein particles. Fasting causes a massive influx of adipose-derived fatty acids into the liver. The liver in the fasted state is therefore bloated with LDs but, remarkably, still continues to secrete triglycerides at a constant rate. Here we show that insulin signaling elevates phosphatidic acid (PA) dramatically on LDs in the fed state. PA then signals to recruit kinesin-1 motors, which transport LDs to the peripherally located smooth ER inside hepatocytes, where LDs are catabolized to produce lipoproteins. This pathway is down-regulated homeostatically when fasting causes insulin levels to drop, thus preventing dangerous elevation of triglycerides in the blood. Further, we show that a specific peptide against kinesin-1 blocks triglyceride secretion without any apparent deleterious effects on cells. Our work therefore reveals fundamental mechanisms that maintain lipid homeostasis across metabolic states and leverages this knowledge to propose a molecular target against hyperlipidemia.
Introduction
Cytosolic lipid droplets (LDs) store triglycerides (TGs) that are substrates for energy and membrane synthesis in most tissues (Murphy, 2012; Thiam and Beller, 2017). LDs are catabolized to supply fatty acid for assembling VLDL (very low density lipoprotein) particles in the smooth ER (sER) of hepatocytes (Gibbons et al., 2004; Lehner et al., 2012; Rai et al., 2017). VLDL is secreted from the liver into blood, where it is detected as serum TG. Remarkably efficient mechanisms must exist to catabolize LDs for VLDL production, because the release rate/steady-state mass of TGs is 80-fold higher in liver than adipose tissue (Gibbons and Wiggins, 1995). We found that LDs purified from rat liver are transported vigorously by kinesin-1 on microtubules (Barak et al., 2013), and this transport delivers LDs to the sER inside hepatocytes, ensuring steady TG supply for VLDL production (Rai et al., 2017). The implications of these findings to liver biology were elaborated on in a commentary (Schulze and McNiven, 2019). Kinesin-1 knockdown in rat liver specifically inhibited TG secretion but had no effect on ApoB secretion, with ApoB appearing at higher density after knockdown because the secreted lipoprotein particles were TG deficient (Rai et al., 2017). Therefore, the molecular factors that sustain kinesin-driven LD transport in hepatocytes and TG secretion from liver are potential targets against hyperlipidemia.
To this end, here we elucidate a spatiotemporally defined sequence of molecular events that channels TG in cytosolic LDs toward production of VLDL particles in hepatocytes in the liver. We earlier reported (Rai et al., 2017) that the GTPase ADP-ribosylation factor 1 (ARF1), which generates “reactive” LDs (Thiam et al., 2013), appears abundantly on LDs in the liver in the fed state. Here we find that ARF1 also recruits phospholipase-D1 (PLD1) to LDs, which in turn generates phosphatidic acid (PA) on the LDs. PA signals on the LD membrane to recruit the microtubule plus end–directed motor kinesin-1, thus causing ARF1 and PA-rich reactive LDs to be transported to the peripherally located sER in hepatocytes. Most importantly, we show that this entire pathway operates downstream of insulin and is therefore toned down when insulin signaling is diminished in the fasted state. This allows the liver to protectively sequester away massive amounts of TG after fasting and, therefore, exert homeostatic control on circulating serum TG in the animal. Inhibiting any of the above molecules tempers VLDL-TG secretion, revealing a common pathway that can be targeted therapeutically at multiple levels. Indeed, overexpression of the kinesin-1 tail domain (KTD) blocks PA-dependent recruitment of kinesin-1 to LDs, and therefore the secretion of TG from hepatocytes. KTD shows no apparent deleterious effect on cells in our experiments and may therefore serve as a design template for interventions against hyperlipidemia.
Results
Insulin activates kinesin-driven transport of LDs in the liver
We have shown that the GTPase ARF1 and kinesin-1 appear abundantly on LDs in the fed state (insulin signaling high) but are both removed upon fasting (Rai et al., 2017). Cell culture studies suggest that insulin promotes binding of ARF1 to membranes (Shome et al., 1997), and ARF1 promotes VLDL secretion (Asp et al., 2000). We therefore asked if insulin controls ARF1 and kinesin-1 recruitment to LDs and, by virtue of this, also controls TG secretion from the liver. Primary hepatocytes were isolated from rat liver and cultured with oleic acid (OA) for 24 h to load LDs inside the cells. Cells were then treated with insulin (6 h), and TG secreted into culture medium was measured by quantitative lipidomic profiling (liquid chromatography-mass spectrometry [LC-MS]). Twofold more long-chain TGs were secreted from insulin-treated cells compared with control (Fig. 1 A). A cell-free assay has shown that ARF-1 influences VLDL production by activating PLD (Asp et al., 2000). We found that kinesin-1 recruitment to LDs requires PLD1 activity, which is significantly diminished in liver in the fasted state (Rai et al., 2017). In agreement with these results, a PLD inhibitor reversed the hypersecretion of TGs from OA-loaded and insulin-stimulated cells (Fig. 1 A). Reduced insulin signaling in the fasted state may therefore prevent hyper-secretion of TGs by inhibiting the ARF1-PLD1-kinesin pathway that channels TG efficiently toward VLDL production in the fed state.
Figure 1.

Physiological relevance of insulin-dependent kinesin-driven transport of LDs in the liver. (A) Primary hepatocytes isolated from rat liver were preloaded with OA to induce LD accumulation. Cells were then left untreated (control) or treated with insulin or insulin + PLD inhibitor (inhib) for 6 h. TG secretion into medium was measured by quantitative lipidomics. TG species of varying chain length are indicated. The data represent mean ± SEM. Data distribution was assumed to be normal, but this was not formally tested. ns, P > 0.1; *, P < 0.05; **, P < 0.01; ***, P < 0.001. Six biological replicates used, two-tailed t test for statistical significance. (B) Liver section of a fed rat stained with epoxide hydrolase (hydrol; sER marker). Central vein (CV) is marked. Part of image is magnified to show coimmunostaining with DAPI (nucleus), Na-K ATPase (plasma membrane marker), and epoxide hydrolase. The sER is localized at periphery of hepatocytes. (C) Liver sections of fed and 16-h fasted rats and 16-h fasted rats injected with insulin. All animals were sacrificed 4 h after insulin injection. The sER, labeled with epoxide hydrolase, is situated at the periphery of cells. LDs are immunostained with perilipin-2. Few LDs in the fed state colocalize with the sER. LD number increases after fasting, and these LDs are spread all over the cell. Insulin injection in the fasted state causes reduction in LD number and colocalization of LDs with the sER (similar to fed state). (D) Quantification of LD distribution inside rat liver hepatocytes. Significant peripheral distribution of LDs (DF close to 1) is observed in fed and fasted + insulin states, but not in the fasted state. Horizontal line represents mean of DF values; ***, P < 0.005. Two-tailed t test was used. (E) LDs are immunostained with perilipin-2 antibody (green) in heart tissue sections (cardiomyocytes) of rats. Relatively few LDs are seen in fed and fasted states. Insulin injection in the fasted state releases TGs from the liver, leading to massive increase of LDs in cardiac muscle (rightmost panel). (F) Same as E but in skeletal muscle (gracilis) from the leg of rat. The subsarcolemmar space is shown. (G) Quantification of LD numbers in cardiac and skeletal tissues across various conditions. The data represent mean ± SD; *, P < 0.05; ***, P < 0.005. Three biological replicates used; two-tailed t test for statistical significance.
Kinesin-1 knockdown in McA-RH7777 hepatoma cells disrupts peripheral localization of LDs near the sER (Rai et al., 2017). To examine if LDs localize to the cell periphery of hepatocytes in the liver, liver sections from fed rats were immunostained against the sER-marker epoxide hydrolase (Galteau et al., 1985). The sER indeed appeared localized toward the periphery of hepatocytes (Fig. 1 B, left). This was confirmed by double immunostaining of sER with plasma membrane (Fig. 1 B, right). We next asked if insulin induces LD localization to the sER in hepatocytes inside the liver. Liver sections were prepared from fed rats, fasted rats, and fasted rats that were injected with insulin after the fasting period. Significant reduction in blood glucose was observed within 1 h of insulin injection in the fasted animals (Fig. S1 A). Fig. 1 C shows a few LDs that largely localize to the periphery of hepatocytes in the fed state; however, fasting induced accumulation of LDs that are distributed all over the cells (also see Rai et al., 2017). Intracellular localization of LDs with respect to plasma membrane was quantified in terms of the fractional distance (DF; Fig. 1 D). A DF of 1 implies an LD at cell periphery. Injection of insulin in fasted rats caused a significant reduction in LD number (Fig. 1 C) and relocalization of these LDs to the peripherally located sER (Fig. 1 D). Therefore, insulin activated LD transport to the sER, which likely resulted in catabolism of LDs and TG secretion in the form of lipoproteins (Fig. 1 A). Lowered insulin signaling in the fasted state may therefore be required to prevent LD-sER interactions in a manner that tempers TG availability for lipoprotein production.
Elevated serum TG causes ectopic accumulation of LDs in tissues, leading to cardiac disease, diabetes, and other forms of lipotoxicity (Cohen et al., 2011; Cohen and Fisher, 2013). Because insulin is required for LD transport and VLDL-TG secretion from hepatocytes, lowered insulin signaling may homeostatically prevent hypersecretion of TG from the fasted liver, in turn protecting against the ectopic accumulation of LDs in peripheral tissues. To investigate, we imaged LDs in sections prepared from cardiac (Fig. 1 E) and skeletal (Fig. 1 F) tissue of fed, fasted, and fasted + insulin-treated rats. Unlike the liver (Fig. 1 C), only small differences in LD numbers were observed between fed and fasted peripheral tissues (Fig. 1 G), suggesting that the liver protects other tissues by sequestering away lipotoxic fatty acids into hepatic LDs in the fasted state. However, when this metabolic control was disrupted in fasted rats by injecting insulin, massive accumulation of LDs was obvious in heart and skeletal tissue (Fig. 1, E–G). Because experiments with primary hepatocytes showed that insulin enhances VLDL-TG secretion (Fig. 1 A), we believe that insulin administration to fasted rats also hypersecretes TGs from hepatocytes into blood. The externally supplied insulin concomitantly causes rapid uptake of glucose into peripheral tissues, thus reducing blood glucose within ∼1 h of insulin injection in fasted animals (Fig. S1 A). Once this happens, the glucose-starved peripheral tissues rapidly take up VLDL, thus reducing serum TG (Fig. S1 B) and causing ectopic LD accumulation in peripheral tissues (Fig. 1, E and F). Therefore, the effect of insulin shows up differently in primary hepatocytes (increased TG secretion into culture medium) compared with animals (ectopic LD accumulation in peripheral tissue leading to reduced serum TG). Taken together, in the absence of the insulin/kinesin control pathway, TG would be hypersecreted from the liver in the fasted state to accumulate as LDs in peripheral tissues (Fig. 1, E and F).
PA is enhanced dramatically on LDs in the liver in the fed state
We next focused on the molecular mechanism by which insulin may control kinesin driven transport of LDs to the sER. PLD1 activity is needed for TG secretion from primary hepatocytes (Fig. 1 A). PLD1 controls phospholipid metabolism, and because it is present on LDs (Nakamura et al., 2005), it could also modulate the phospholipid composition of LDs. We therefore performed LC-MS–based quantitative lipid profiling of LDs purified from liver of normally fed rats (NLDs) and 16-h-fasted rats (FLDs). The procedures for purifying LDs and LC-MS analysis have been described (Barak et al., 2014; Rai et al., 2017). Lipidomics data showed that multiple species of PA are elevated on NLDs compared with FLDs (Fig. 2 A). The percentage change in different lipids between NLDs and FLDs is shown in Fig. 2 B. Agreeing with earlier work (Chitraju et al., 2012; Rai et al., 2017; Sadh et al., 2017), TG content within LDs does not change (Figs. 2 B and S1 C), although the total number of LDs in liver increases after fasting (Fig. 1 C). Unlike LDs, the PA content was the same for microsomes prepared from liver of fed and fasted rats (Fig. S1 D; also see Fig. S1 E for purity of microsomes). This suggests that PA elevation on LDs in the fed state is not because of the transfer of PA from the ER membrane onto LDs.
Figure 2.
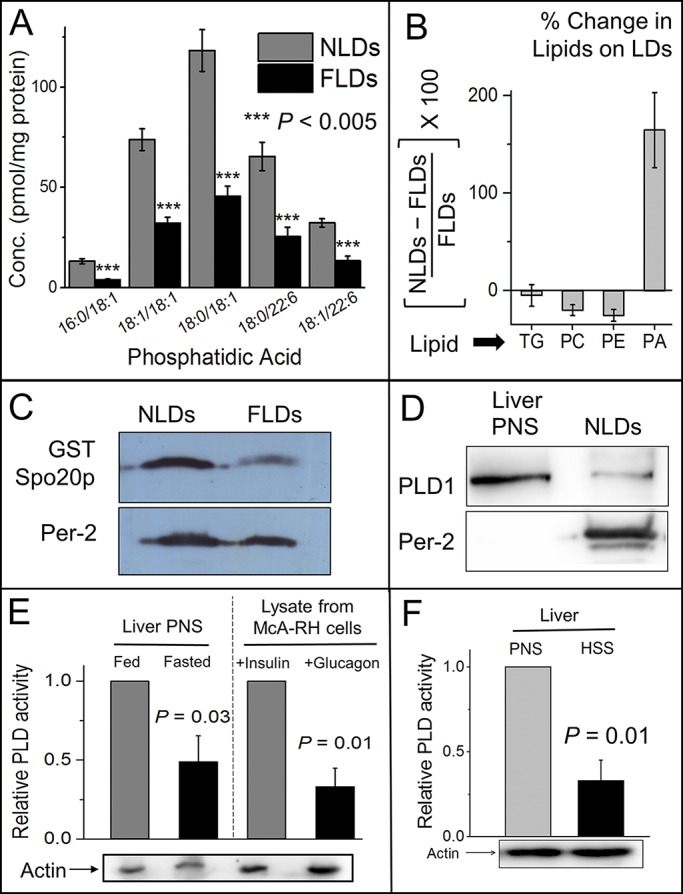
Insulin signaling activates PLD to generate PA on LDs. (A) Lipidomic analysis shows reduction of PA on FLDs. X axis denotes different species of PA. Experiment was performed with five independent pairs of animals (each pair consisting of a fed and a fasted rat). The data represent mean ± SEM across all pairs; ***, P < 0.005. Two-tailed t test used. Conc, concentration. (B) Percentage change of indicated lipids on LDs purified from fed and fasted rat livers, as obtained by quantitative lipidomics. Experiment was performed with five independent pairs of animals. Error bars are SD. (C) The PA sensor protein GST-Spo20p was incubated with NLDs and FLDs. The LDs were separated from unbound GST-Spo20p protein by flotation and immunoblotted with antibody against GST. Significantly more GST-Spo20p was detected on NLDs. Western blot with perilipin-2 shows equal loading of NLD and FLD samples. (D) LDs were isolated from liver of normally fed rat (NLDs). PNS was prepared from liver of fed rat. Immunoblotting was performed using antibodies against PLD1 and perilipin-2 (ADRP). (E) Relative activity of PLD measured in PNS (contains membranes) prepared from fed and fasted rat livers (left) and McA-RH7777 cells (right). Cells were treated with 100 nM insulin (in high-glucose medium) or 100 nM glucagon (in low-glucose medium supplemented with 200 µM OA). Western blot for actin was performed as loading control. The data represent mean ± SD; P value calculated using unpaired one-tailed t test. (F) Relative activity of PLD in PNS and HSS prepared from liver of fed rat. Western blot for actin was performed as loading control. The data represent mean ± SD; P value calculated using unpaired one-tailed t test.
We next expressed the PA binding sensor GST-Spo20p (see LD motility assay in Materials and methods; Kassas et al., 2012) and used its binding on LDs to infer the presence of PA on the LDs. Fig. S1 F shows Coomassie-stained SDS-PAGE gels for the bacterially expressed proteins used in this study. The specificity and graded response of Spo20p binding toward PA was verified using a lipid blot (Fig. S1 G). We found that Spo20p bound much more abundantly to NLDs than FLDs (Fig. 2 C). Along with the LC-MS evidence discussed above, this confirms that PA is elevated on LDs in the fed state compared with fasted. The precursors of PA (phosphatidylcholine [PC] and phosphatidylethanolamine [PE]) were lowered on NLDs compared with FLDs (Figs. 2 B and S2 A), whereas phosphatidylserine (PS) was unchanged (Fig. S2 A). This suggests PLD-catalyzed conversion of PC/PE to PA (Athenstaedt and Daum, 1999) in situ on LDs in liver in the fed state. The in situ generation of PA on LDs agrees with unchanged PA on microsomes across fed/fasted states (Fig. S1 D). Consistent with this, and agreeing with presence of PLD1 on LDs in cultured cells (Nakamura et al., 2005; Andersson et al., 2006), we detected PLD1 by Western blotting on NLDs (Fig. 2 D). PLD1 activity is enhanced in cultured hepatocytes after insulin stimulation (Donchenko et al., 1994). In agreement with this, a colorimetric assay showed that PLD activity is elevated in postnuclear supernatant (PNS; contains membranes) prepared from liver of fed rats (Fig. 2 E, left). Next, hepatoma-derived McA-RH7777 cells were treated with insulin to mimic the fed state or with glucagon + OA to mimic the fasted state. PLD activity was significantly higher in lysate from insulin-treated cells (Fig. 2 E, right). Based on this evidence, we hypothesize that elevated insulin signaling activates PLD1 in the fed state, which generates PA on LDs in the liver. Significant PLD activity was retained in a high-speed supernatant (HSS; devoid of membranes) prepared from rat liver (Fig. 2 F), although this activity was expectedly lower than a PNS fraction. This suggests that cytosolic PLD can also be recruited to the LD membrane in hepatocytes, where it generates PA.
PA-dependent recruitment of kinesin-1
PA is a bioactive signaling lipid known to recruit effector proteins on membranes. In the context of the above discussion, an effector protein of interest is the kinesin-1 motor. We therefore asked if PA exhibits specific affinity to kinesin-1. This was first investigated in lipid blots against an array of lipids. The membrane was incubated with PNS prepared from BRL3A rat liver fibroblast cells and then probed with antibody against kinesin-1. Kinesin-1 showed highly specific binding to PA on the membrane (Fig. 3 A). Soluble PA cannot bind to LDs, because it has truncated fatty acid chains, but the intact head-group may bind its protein partners. Therefore, soluble PA should bind kinesin-1 and sequester it away from LDs into the solution. Indeed, addition of soluble PA to the PNS reduced kinesin-1 binding to the PA-containing spot on the membrane (Figs. S2 B). We confirmed that unlike kinesin-1, other microtubule motors such as kinesin-2 and dynein do not show specific affinity to PA (Fig. 3 B).
Figure 3.

PA recruits kinesin-1 to LDs. (A) BRL3A hepatocyte PNS was overlaid on nitrocellulose membrane spotted with indicated lipids. The membrane was immunoblotted (IB) against kinesin-1 antibody. Kinesin-1 exhibited the highest affinity to PA. (B) PA and PC were spotted on nitrocellulose membrane and overlaid with PNS prepared from BRL3A hepatocytes. Binding of kinesin-1, kinesin-2, and dynein was probed using antibodies against the specific proteins. Only kinesin-1 shows specificity for PA. (C) Top: Schematic of experiment. ALDs were incubated with HSS prepared from liver of fed rat. HSS is devoid of membrane, organelles, and endogenous LDs. ALDs (PC only) were also incubated with HSS in presence of 50 µM PLD inhibitor. ALDs were removed by flotation after the incubation, and ALD-bound proteins were probed with kinesin-1 antibody. Bottom: Kinesin-1 binding to ALDs visualized by Western blot against kinesin-1 antibody. TLC for TG confirms equal loading of ALDs. The blot is representative of three independent experiments. (D) COS7 cells overexpressing PA sensor protein (GFP-Spo20p) were treated with DMSO (control) or Brefeldin-A (BFA). The cells were fixed, and LDs were stained with MDH immediately before imaging. Confocal images of cells show that BFA reduced GFP-Spo20p binding to LD membrane. Scale bar = 2 µm. (E) Same as D, but cells treated with DMSO (control) and PLD inhibitor. Scale bar = 5 µm.
We found that protein recruitment to hepatic LDs is sensitive to fed/fasted states of the animal (Sadh et al., 2017). Proteins can be recruited to LDs from the cytosol or from the ER via ER-LD bridges (Kory et al., 2016), possibly in a manner depending on phospholipid composition of the LDs. Analyzing proteins of low abundance (such as kinesin-1) on LDs becomes difficult, because endogenous LDs have low overall protein concentration (Rai et al., 2017). To overcome this problem, and to specifically test a role for PA on LDs, we prepared artificial lipid droplets (ALDs) with a phospholipid monolayer consisting of 100% PC, 95% PC+ 5% PA, or 95% PC + 5% PS. We verified the presence of a phospholipid coat on a TG core in these ALDs by fluorescence microscopy (Fig. S2 C). ALDs were incubated with HSS prepared from liver of fed or fasted rats. After the incubation, ALDs were separated by flotation into a buoyant top layer, and proteins recruited to ALDs from HSS were probed by Western blotting. The experimental procedure is shown as a schematic in Fig. 3 C (top). This ALD assay could be scaled up for addressing phospholipid-dependent mechanisms behind recruitment of kinesin-1 to LDs. Significantly more kinesin-1 was recruited to ALDs after PA addition (bottom panel, Fig. 3 C; lanes 1 and 2). This effect was not observed with PS, which also has a negatively charged head group similar to PA (Fig. 3 C; lane 3). We have reported higher levels of kinesin-1 on NLDs compared with FLDs (Rai et al., 2017). Because NLDs are also rich in PA (Fig. 2 A), the PA-dependent recruitment of kinesin-1 on ALDs (Fig. 3 C) recapitulates aspects of kinesin-1 recruitment to LDs in liver in the fed state (Rai et al., 2017). Note that the average size of ALDs (∼4 µm) was unchanged after PA addition (Fig. S2 D).
PLD1 activity generates PA on LDs, which recruits kinesin-1 to LDs
Very interestingly, kinesin-1 was also recruited to the PC-only ALDs (Fig. 3 C, lane 1). This suggests that PLD activity in liver-HSS converts PC to PA in situ on the ALDs, which then recruits kinesin-1 to LDs/ALDs. Indeed, addition of a PLD inhibitor reduced binding of kinesin-1 below the level seen for PC-only ALDs (Fig. 3 C, compare lane 4 with lane 1). We have found enhanced ARF1 activity on LDs in the fed state, this activity being required for kinesin-1 recruitment to NLDs (Rai et al., 2017). A direct interaction between ARF1 and PLD1 is known (Kim et al., 1998), and ARF1-dependent activation of PLD1 is specifically important for VLDL secretion (Asp et al., 2000). If ARF1 recruits PLD1 that generates PA on LDs, then the presence of PA on LDs must require upstream activity of ARF1 and PLD1. Agreeing with this, the PA sensor GFP-Spo20p localized to LDs in control cells but was reduced in cells treated with brefeldin-A (Fig. 3 D) or PLD inhibitor (Fig. 3 E). Brefeldin-A blocks ARF1 in the inactive GDP state. These results were replicated in vitro, as the PA sensor was recruited abundantly to PC-only ALDs incubated with liver HSS, but recruitment was reduced in presence of the PLD inhibitor (Fig. S2 E).
We have shown that LDs localize to the extreme periphery of McA-RH7777 hepatoma cells, and this localization is disrupted after kinesin-1 knockdown or brefeldin-A treatment (Rai et al., 2017). Treatment of McA-RH7777 cells with PLD inhibitor also disrupted the peripheral localization of LDs (Fig. 4 A). LD localization was again quantified in terms of the DF (Fig. 4 B; DF defined in Fig. 1 D). We next checked the relevance of PA for kinesin-1–driven in vitro motility of LDs. Individual NLDs were held in an optical trap and placed above a microtubule to score for motion (Barak et al., 2013, 2014; Rai et al., 2017). When added to the motility buffer, soluble PA or GST-Spo20p reduced NLD motion, but PE, PS, or GST had no effect (Fig. 4 C). This suggests that both soluble PA and GST-Spo20p competitively inhibit binding of kinesin-1 to PA on the LD surface. Indeed, incubation of ALDs with rat liver HSS in the presence of GST-Spo20p inhibited the recruitment of kinesin-1 to ALDs (Fig. 4 D). PA generated on LDs via PLD1 activity therefore recruits kinesin-1, which then transports LDs toward the cell periphery in hepatocytes (Barak et al., 2013; Rai et al., 2017).
Figure 4.
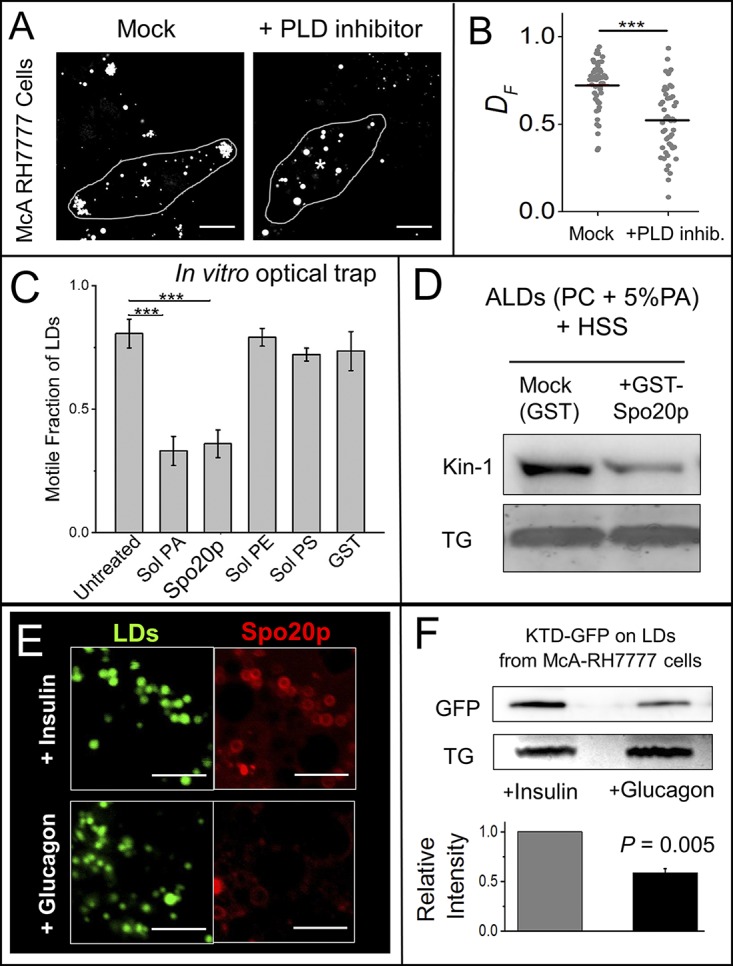
Kinesin-1 binding on LDs can be inhibited by PLD inhibitor, PA-binding agents, and glucagon. (A) McA-RH7777 cells were treated with DMSO (mock) or PLD inhibitor for 6 h, stained with MDH to label LDs, and imaged live on confocal microscope. Centroids of nuclei are indicated with asterisks. Scale bar = 10 µm. (B) Fractional distances (DF) of 50 randomly selected LDs from eight cells in each condition were calculated. Horizontal line indicates mean of DF values. ***, P < 0.001. Two-tailed t test used. (C) In vitro motility assay using LDs purified from liver of fed rats in presence of indicated reagents. Independent experiments were done using LDs isolated from three animals. The data represent mean ± SD; ***, P < 0.001. Two-tailed t test used. (D) ALDs were incubated with rat liver HSS in presence of GST (control) or GST-Spo20p. ALDs were separated by flotation and immunoblotted with kinesin-1 antibody. GST-Spo20p inhibits kinesin-1 binding to ALDs. TLC for TG was performed to confirm equal loading of ALDs. The Western blot is representative of three independent experiments. (E) PA-sensor (GFP-Spo20p)-overexpressing cells were treated with 100 nM insulin (in high-glucose medium) or 100 nM glucagon (in low-glucose medium). LDs were stained with MDH. Confocal microscopy was used to image LDs and PA-sensor. The PA-sensor was detected more abundantly on LDs in insulin-treated cells. Scale bar = 2 µm. (F) McA-RH7777 cells stably expressing KTD-GFP (does not have kinesin motor domain) were loaded with OA. Cells were treated with 100 nM insulin (in high-glucose medium) or 100 nM glucagon (in low-glucose medium with OA) for 24 h. LDs were isolated by flotation after cell lysis, and LD-bound proteins were immunoblotted with GFP antibody. TLC for TG shows equal loading of LDs. The data represent mean ± SEM. Two-tailed t test used.
Insulin signaling generates PA on LDs to recruit kinesin-1
Insulin activates ARF1 and localizes it to the plasma membrane in fibroblast-derived cells (Shome et al., 1997; Li et al., 2003). As discussed above, appearance of ARF1 on LDs is followed by PLD1 recruitment and PA generation on LDs. Because ARF1 is present on LDs (Wilfling et al., 2014), we hypothesized that insulin also activates ARF1 to localize it to the LD membrane in hepatocytes, and this localization is enhanced in the fed state (insulin signaling high). To verify that insulin initiates this sequence of events, cells expressing the PA sensor were treated with insulin or glucagon. We confirmed (Fig. S3 A) the activation of insulin and glucagon signaling in these cells (Dalle et al., 2004). Insulin significantly enhanced the presence of PA-sensor on LDs inside cells compared with glucagon (Fig. 4 E), suggesting that an insulin-ARF1-PLD1 pathway generates PA on LDs. To test if this pathway recruits kinesin-1 to LDs, we stably overexpressed the GFP-tagged KTD (KTD binds to cargo, as explained in next section) in McA-RH7777 cells, and then treated these cells with insulin or glucagon. Insulin-treated cells were cultured in high-glucose medium to mimic the fed state, while glucagon-treated cells were cultured in low-glucose medium supplemented with OA to mimic the fasted state (Shome et al., 1997; Nakamura et al., 2005). Cells were fractionated to purify endogenous LDs, and LD-associated proteins were subjected to Western blotting against anti-GFP antibody. Significantly more KTD-GFP was detected on LDs isolated from insulin-treated cells compared with glucagon (Fig. 4 F). We next used the ALD assay to verify that endogenous kinesin-1 (present in small amounts on LDs) is also recruited to LDs in an insulin-dependent manner. HSS (free of membranes and endogenous LDs) was prepared from insulin- and glucagon-treated McA-RH7777 cells. HSS was incubated with PC-ALDs, and the ALDs were separated by centrifugation and subjected to Western blotting. More kinesin-1 (endogenous) was recruited to ALDs incubated with HSS from insulin-treated cells (Fig. S3 B). These observations agree with the abundance of kinesin-1 on NLDs (fed state; insulin signaling and PA high) compared with FLDs (fasted state; insulin signaling and PA low).
KTD binds to PA on LDs
PA directly binds KTD to recruit kinesin-1 to matrix metalloprotease–containing vesicles in metastatic cells (Wang et al., 2017). Fig. 5 A is a cartoon showing the binding of kinesin-1 to cargo (e.g., an LD) via the KTD. Fig. 5 B shows the sequence of positively charged basic amino acids (aa 901–909, lysine and arginine rich) within KTD implicated in kinesin-1 binding, and the mutations therein that inhibit PA binding (Wang et al., 2017). Unlike vesicles with a bilayer membrane, LDs have a monolayer phospholipid membrane (Wilfling et al., 2014). Can PA on the monolayer membrane of LDs also recruit KTD? To test this, we expressed and purified WT GST-KTD and PA-binding–deficient GST-KTD-PA− from bacteria. Both these constructs lack the stalk and motor domains of kinesin (see Fig. S1 F for purity of proteins). We incubated the purified KTDs with ALDs prepared using PC or (PC + 5% PA). GST-KTD bound much more strongly to PA-containing ALDs than GST-KTD-PA− (Fig. 5 C). Negligible binding was observed for both constructs on PC-only ALDs. Next, we stably overexpressed GFP-tagged WT and PA-binding–deficient KTD in McA-RH7777 cells (Fig. S3 C). These KTD constructs were devoid of stalk and motor domains of kinesin-1. Immunoblotting of LD-associated proteins against GFP confirmed that KTD binds more strongly to LDs than KTD-PA– (Fig. 5 D). We also overexpressed full-length KIF5B-GFP and KIF5B-PA−-GFP in McA-RH7777 cells, purified endogenous LDs, and immunoblotted against GFP antibody. Full-length KIF5B-GFP also bound more strongly to LDs compared with KIF5B-PA−-GFP (Fig. S3 D). The purity of LDs prepared from cells was verified using perilipin-2 as a marker (Fig. S3 E).
Figure 5.
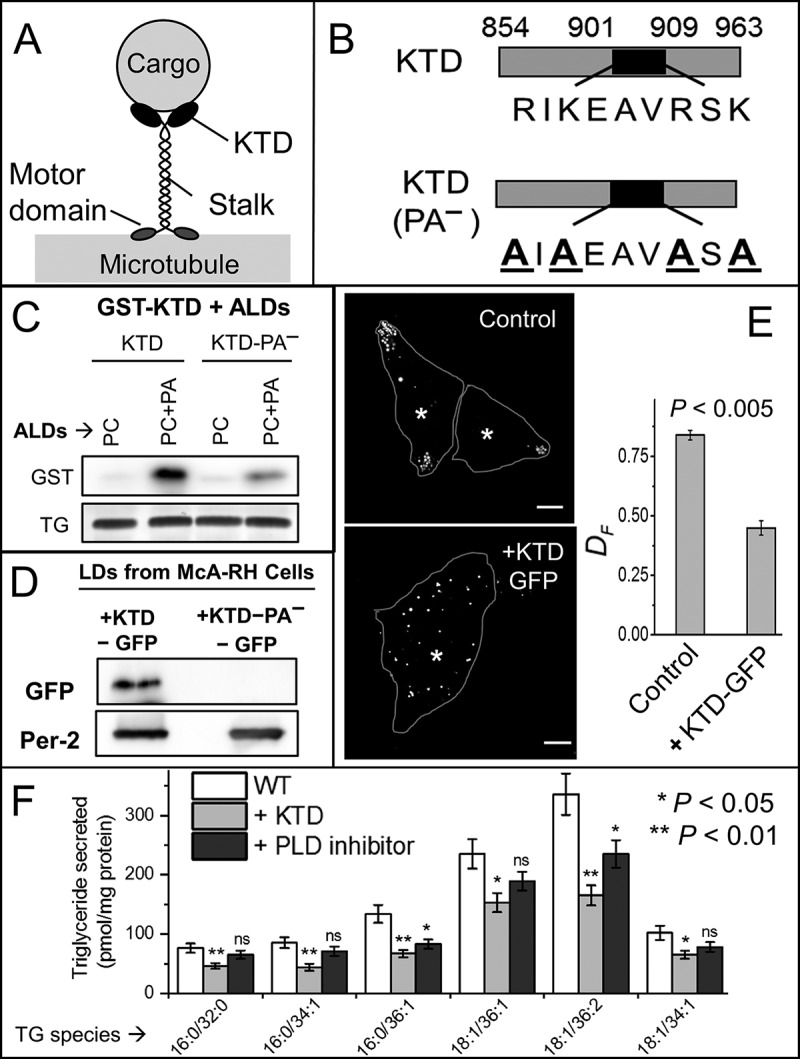
The KTD binds to PA on LDs. (A) Schematic of kinesin-1 bound to a cargo (e.g., LD). Different domains of kinesin-1 are shown. (B) Schematic of KTD (WT) and KTD-PA− (PA-binding mutant). In the mutant, lysine and arginine are replaced with alanine. (C) Purified GST-KTD or GST-KTD-PA− proteins were incubated with ALDs of indicated compositions; ALDs were separated by flotation, and LD-bound protein was probed with GST antibody. TLC for TG confirms equal loading of ALDs. The blot is representative of three independent experiments. (D) LDs were purified from McA-RH7777 cells stably expressing KTD-GFP or KTD-PA−−GFP. LD-bound proteins were probed with antibodies against GFP and perilipin-2 (loading control). The blot is representative of two independent experiments. (E) LDs were stained with MDH in naive and KTD-GFP–overexpressing McA-RH7777 cells, and imaged live. Cell boundaries (line) and centroid of nuclei (asterisks) are marked. Scale bar = 10 µM. DF is reduced upon overexpression of KTD (P < 0.005; averaged over 50 LDs from 15 cells of each type). Two-tailed t test used. (F) Untreated (WT), KTD-GFP–overexpressing, and PLD inhibitor–treated McA-RH7777 cells were loaded with 400 µM OA for 6 h. The cells were cultured in phenol red and serum-free medium for 4 h. The medium was collected, and TG secreted from the cells into medium was measured by quantitative lipidomics. Six independent replicates were used. The data represent mean ± SEM. Two-tailed t test used. Data distribution was assumed to be normal, but this was not formally tested. ns, not significant. P values corresponding to * and ** are indicated.
To test its effect in a functional assay, KTD-GFP (motorless) was stably overexpressed in McA-RH7777 cells, whereupon it disrupted the peripheral localization of LDs (Fig. 5 E). Accordingly, DF was also reduced for LDs in KTD-GFP–transfected cells (Fig. 5 E). A purpose of this study is to identify factors that can help lower serum TG. The KTD peptide is a candidate because it disrupts peripheral localization of LDs near the sER. To verify that KTD also removes kinesin-1 from LDs in the liver, we incubated PC-only ALDs with PNS from liver of fed rats, and then removed the ALDs by flotation. These ALDs were divided into three equal parts and treated with increasing concentrations of GST-KTD. Kinesin-1 was indeed replaced from ALDs by GST-KTD in a concentration-dependent manner (Fig. S3 F). Encouraged by these results, we performed quantitative lipidomics for the secreted TGs from KTD-GFP overexpressing McA-RH7777 cells. KTD-GFP overexpression reduced secretion of long-chain TGs significantly (Fig. 5 F). The PLD inhibitor also suppressed TG secretion from cells (Fig. 5 F), implicating kinesin-1 and LD-associated PA in a common pathway that facilitates VLDL-TG secretion.
We observed a similar number (∼35%) of BrdU-positive cells across control, KTD-GFP, or KTD-PA−-GFP overexpressing cells (Fig. 6 A). The cells therefore proliferate normally, without any apparent adverse effects of the KTD peptide. To better understand the effect of KTD on LDs versus other cellular organelles, we investigated the localization of other (bilayer) vesicles in McA-RH7777 cells. KTD consistently disrupted LD localization at the periphery to scatter LDs all over the cell (Fig. 6 B). However, KTD did not induce any obvious effect on the distribution of early endosomes, lysosomes, and mitochondria or on the organization of microtubules (Fig. 6 B). These results suggest that KTD-mediated inhibition of transport is more effective for (monolayer) LDs compared with other (bilayer) organelles, and therefore holds promise as a method to reduce TG secretion without disrupting other cellular processes in hepatocytes.
Figure 6.

Kinesin tail domain specifically impairs intracellular transport of LDs to cell periphery. (A) McRH-7777 cells (naive, stably expressing KTD-GFP or KTD-PA−-GFP] were cultured for 4 h in presence of 10 µM BrdU. The cells were stained with BrdU antibody to assess proliferation. Nuclei were stained with DAPI. In the right panel, BrdU-positive cells are quantified as mean ± SEM. Scale bar = 25 µm. (B) McRH-7777 cells (naive or stably expressing KTD-GFP] were stained with MDH to label LDs. These cells were also immunostained with indicated antibodies against lysosomes, early endosomes, mitochondria, and microtubules. KTD specifically disrupts peripheral localization of LDs without any observable effect on localization of lysosomes, early endosomes, and mitochondria. Organization of microtubules is also unaffected after KTD treatment. Scale bars = 10 µm.
Possible role of PLD1 and PA in maintaining LD–ER contacts and protein transfer
LD–ER contacts are crucial for LD biogenesis (Joshi et al., 2018) and also for efficient transfer of lipids/proteins between these organelles (Prinz, 2014; Wilfling et al., 2014). Cideb, a protein present at sER–LD contacts in hepatocytes, is required for lipidation of VLDL particles (Ye et al., 2009). Cideb is more abundant on NLDs compared with FLDs, perhaps due to enhanced sER–LD interactions in the fed state (Rai et al., 2017). The data in Fig. 1 (A and C) suggest that kinesin-1–driven localization of LDs to cell periphery enhances LD–sER contacts to channel lipids into lipoproteins. However, just a physical localization of LDs at sER is not sufficient for lipid exchange, because the negatively charged phospholipids present an energy barrier to LD–ER exchanges. ARF1 activity may remove phospholipids from NLDs to lower this barrier. Phospholipids with intrinsic negative curvature (e.g., PA) can also stabilize the necklike region at LD–ER interfaces (Choudhary et al., 2018). Therefore, the abundance of PA on NLDs may facilitate LD–sER interactions for VLDL production. Because LD–sER contacts are difficult to resolve in cells, we addressed this question at the negative curvature region of LD–LD contacts. This is justified because PA also enhances LD–LD fusion (Fei et al., 2011). Immunofluorescence staining showed that PLD1 is indeed localized to the necklike region between LDs in cells (Fig. S4 A) and could therefore generate PA near these bridges. To test in vitro whether PA on LDs can enhance LD–ER interactions, we prepared ER-enriched microsomes from HEK293T cells expressing GFP-Rab18(Q67L), a constitutively active mimic of the Rab18 GTPase. Purity of microsomes was confirmed using specific markers (Fig. S4 B). It is known that Rab18 is enriched on LDs and also localizes to the ER, from where it engineers ER-LD contacts (Martin et al., 2005). Microsomes were incubated with ALDs containing PC or PC + 5% PA, following which ALDs were separated by flotation and imaged. GFP-Rab18–positive membranous structures (presumably ER microsomes) were more abundant on PA-containing ALDs (Fig. S4 C), suggesting that PA enhances ER–LD contacts. It therefore appears that PA on the LD membrane has multiple functions in VLDL secretion. First, it is an effector molecule that recruits kinesin-1 to initiate LD transport toward the sER. Second, the intrinsic negative curvature of PA may stabilize LD–sER bridges to facilitate lipid/protein exchanges.
Discussion
PA makes up only 1% of the total lipid content of cells (Kooijman et al., 2007), but this simplest phospholipid is the common biosynthetic precursor for membrane phospholipids as well as TG. This places PA at a crucial metabolic branch point between growth and storage (Barbosa et al., 2015), with the effect that de novo generated PA is channeled away for synthesizing phospholipids during growth or TG during starvation. The cellular mechanisms that flux lipids between these two pathways in response to metabolic demands must involve LDs, because LDs are the predominant lipid stores in cells (Murphy, 2012; Thiam and Beller, 2017). Such lipid fluxes occur daily on a massive scale when the body switches from the fed state (LD catabolism in liver) to the fasted state (LD storage in liver). PA acquires special relevance in hepatic fatty acid regulation, because lipins convert PA into diacylglycerol, which further yields TG (Khalil et al., 2010). Motivated by these observations, we show here how insulin, the key hormone that controls metabolism, engineers the generation of PA on hepatic LDs to control the storage and catabolism of fatty acids across fed-to-fasted transitions. Apart from its aforesaid role as a metabolic branch-point, PA is also a signaling lipid and a lipidic second messenger with established roles in vesicle trafficking (Shin and Loewen, 2011). This requires PA to recruit downstream effector proteins (e.g., kinesin) on membranes. PA is unique in that its phospho-monoester headgroup combines hydrogen bonding with electrostatics to recruit effectors with impressive specificity (Kooijman et al., 2007). Our present results reveal how the master hormone insulin uses this simplest phospholipid, PA, to control vesicular transport of LDs inside the liver in a manner that establishes lipid homeostasis in the animal.
Based on the present data and our earlier work (Rai et al., 2017), we propose a sequence of events that delivers TG efficiently for VLDL production and secretion in the liver in the fed state and also tempers this process to buffer away LDs in liver after fasting. This sequence is depicted schematically in Fig. 7 A, with specific steps detailed in Fig. 7 (B and C).
Figure 7.
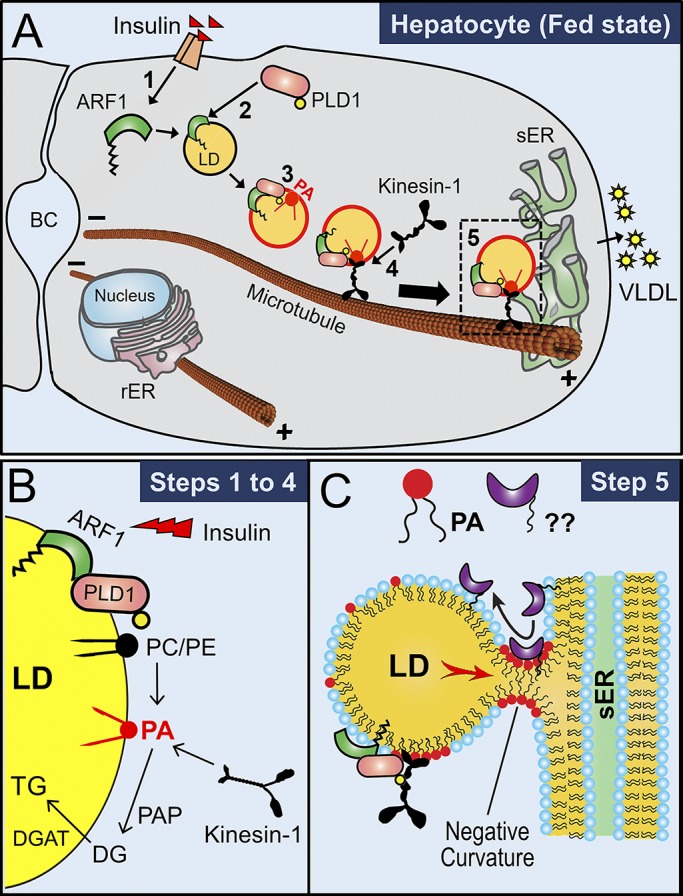
Mechanism for efficient VLDL-TG secretion from hepatocytes. (A) A hepatocyte in the fed state is depicted. (A1) Insulin binds to insulin receptors and activates the ARF1 GTPase. (A2) Active ARF1 binds to LDs and interacts with cytosolic PLD1 to recruit PLD1 on LDs. (A3) PLD1 converts PC into PA on the LDs. ARF1 also pinches off phospholipids to make these PA-containing LDs reactive (red outline). (A4) PA recruits kinesin-1 to LDs. Kinesin-1 then transports the PA-rich, reactive LDs along microtubules toward the sER located at the periphery of hepatocytes. Microtubules have minus ends near the apical bile canaliculi (BC) and plus ends near sER at the periphery of the cells. (A5) LDs interact with sER to supply TG for VLDL assembly in the sER lumen. (B) Suggested pathway for generation of PA on LD membrane (steps 1–4 of A). PA can also be converted into diacylglycerol (DG) by PA phosphohydrolases (PAPs; e.g., lipins) and then further into TG by diacylglycerol acyltransferase (DGAT). (C) This schematic is speculative. PA-induced negative membrane curvature promotes formation of lipidic bridges between LDs and sER. Specific proteins (shown with ??) may transfer to LDs across bridges to stabilize the LD-sER contacts. sER-resident lipases may also access the LD via this bridge (not shown) to hydrolyze LD-TG. TG is subsequently channeled into the sER lumen (red arrow; intermediate hydrolysis into DG and re-esterification into TG not shown). VLDL particles are lipidated with TG in sER and subsequently secreted into blood after further processing in Golgi (not shown). sER, smooth-ER.
Step 1: Insulin signaling activates ARF1, causing it to bind LDs. This is likely the first step because ARF1-GTP can bind PA-free LDs in vitro (Thiam et al., 2013; Wilfling et al., 2014). Step 2: LD-associated ARF1-GTP recruits and activates cytosolic PLD1 on LDs. ARF1 also pinches off phospholipids to make these LDs reactive, as shown by red color. Step 3: PLD1 activity generates PA on reactive LDs. In support of this, PA is elevated on NLDs, and PLD inhibition reduces PA on LDs in cells. Step 4: PA binds KTD to recruit kinesin-1 on LDs, causing reactive PA-rich LDs to be transported (black arrow, Fig. 7 A) to the sER at the periphery of hepatocytes. Step 5: Fatty acids are channeled from PA-rich, reactive LDs to the sER lumen via lipidic bridges that are possibly stabilized by negative curvature from PA (Fig. 7 C).
Note the involvement of PA at multiple steps (Fig. 7 B). Step 5, where LD–sER bridges are stabilized by the negative curvature from PA, remains speculative at present but is worth investigating further using advanced imaging techniques such as electron microscopy and superresolution microscopy. The physiologically relevant mechanisms that control VLDL secretion have been elusive because they cannot be studied entirely in animals or in cell culture. Our use of diverse in vivo and in vitro assays reveals fundamentally important biophysical and biochemical mechanisms inside the liver by which the master regulator (insulin) and the simplest phospholipid (PA) together harness the kinesin motor to channel LD toward VLDL production in a manner that establishes lipid homeostasis in the animal. These results elucidate not just interactions between lipids and proteins, but also reveal a precise spatiotemporal sequence of connected molecular events that operates to maintain physiologically relevant homeostasis of lipids in the animal (Fig. 7).
Materials and methods
Reagents
All reagents were purchased from Sigma-Aldrich unless otherwise mentioned. FBS (16000-044), trypsin-EDTA (15400054), Lipofectamine 2000 (11668-019), lipid-strips (P23751), BODIPY (D3922), and GFP (A11122) antibody were purchased from Invitrogen. Monodansylpentane/monodansylpentane (MDH; SM1000a) was purchased from Abgent. Thin layer chromatography (TLC) Silica gel 60 plates (1.05721.0001) were purchased from Merck Millipore. Protease inhibitor cocktail (13539320) was purchased from Roche Diagnostics. All phospholipids, Rhodamine-PE, and PLD inhibitor (VU0155056) were purchased from Avanti Polar Lipids. OA (O1383), PA (P9511), glyceryl trioleate (T7140), and α-tubulin antibody (T9026) were purchased from Sigma-Aldrich. PLD activity assay kit (ab183306), antibodies for epoxide hydrolase (ab96695), Na-K-ATPase (ab7671), kinesin-2/KIF3A (ab11259), KDEL (ab12223), LAMP1 (ab24170), and Hsp60 (ab46798) were purchased from Abcam. Antibodies for perilipin-2/ADRP (651102) were purchased from PROGEN Biotechnik; actin (AAN01) was purchased from Cytoskeleton; GST (TAX158Ge22) was purchased from Cloud-Clone Corp.; dynein intermediate chain (sc13524) and Rab18 (sc393168) were purchased from Santa Cruz Biotechnology; AKT (4691P), pAKT (4060S), CREB (9197S), pCREB (9198S), and EEA1 (2411S) antibodies were purchased from Cell Signaling Technology; PLD1 (A15081) was purchased from Abclonal; GM130 (610822) and PDI (612117) were purchased from BD Bioscience; and Golgin97 (A21270) was purchased from Life Technologies. An affinity-purified rabbit polyclonal antibody was raised against the peptide CDKNRVPYVKGCTER (rat KIF5c amino acids 159–172) by GenScript. This antibody was tested and reported by Hammond et al. (2008). Secondary antibodies were Alexa Fluor 488 donkey anti-mouse (A21202) and Alexa Fluor 555 donkey anti-rabbit (A31572) from Thermo Fisher Scientific; donkey anti-rabbit IgG-HRP (sc-2313) and donkey anti-mouse IgG-HRP (sc-2314) from Santa Cruz Biotechnology; GST-Spo20p(PABD), a generous gift from Nichole Vitale (Université de Strasbourg, Strasbourg, France; Kassas et al., 2012); GFP-Rab18(Q67L), a generous gift from Robert G. Parton (The University of Queensland, Brisbane, Australia; Ozeki et al., 2005). pCDH-KIF5B-GFP and pCDH-KIF5B(PA−)-GFP, kind gifts from Guangwei Du (University of Texas Health Science Center, Houston, TX; Wang et al., 2017); and psPAX2 and pMD2.G procured from Addgene. Restriction enzymes, Taq polymerase, and T4 DNA ligase were purchased either from Anza or Thermo Fisher Scientific. Collagenase type-4 (CLS-4, LS004188, 245 U/mg dry weight) was purchased from Worthington. Monocomponent Biosynthetic r-DNA insulin used for animal studies was procured from Novo Nordisk. Primers are listed in Table 1, and KTD amino acid sequences are listed in Table 2.
Table 1. Primers.
| Primer name | Nucleotide sequence (5′ to 3′) |
|---|---|
| KTD forward (1) | ATCGAATTCGCAGATCTCCGCTGTGAACT |
| KTD reverse (1′) | AAGCTCGAGTTACACTTGTTTGCCTCCTC |
| mKTD forward (2) | ATCTCTAGAGCAGATCTCCGCTGTGAACTTCC |
| mKTD-EGFP reverse (2′) | AAGGCGGCCGCTTACTTGTACAGCTCGTCCATGCC |
Table 2. WT and mutated amino acid sequence of KTD.
| Domain name | Amino acid sequence |
|---|---|
| KTD | …ADLRCELPKLEKRLRATAERVKALESALKEAKENASRDRKRYQQEVDRIKEAVRSKNMARRGHSAQIAKPIRPGQHPAASPTHPSAIRGGGAFVQNSQPVAVRGGGGKQV* |
| KTD(PA−) | …ADLRCELPKLEKRLRATAERVKALESALKEAKENASRDRKRYQQEVDAIAEAVASANMARRGHSAQIAKPIRPGQHPAASPTHPSAIRGGGAFVQNSQPVAVRGGGGKQV* |
Animal strains and procedures
Sprague-Dawley rats were bred and maintained by the animal house facility at Tata Institute of Fundamental Research, Mumbai, India. All animal protocols were approved by the Institutional Animal Ethics Committee formulated by the Committee for the Purpose of Control and Supervision of Experiments on Animals, India. Rats were maintained on a regular light (12-h)/dark (12-h) cycle and fed a standard laboratory diet. 12–16-wk-old male Sprague-Dawley rats were used for all the experiments. Rats from the same litter were used as a fed-fasted pair in the experiment. Animals in the fed group had ad libitum access to food and water. Animals in the fasted group were fasted for 16 h with ad libitum access to water.
Primary hepatocyte culture and TG secretion
Primary hepatocytes were isolated from rat liver and cultured as described previously (Klaunig et al., 1981; Shen et al., 2012). Sprague-Dawley rats 12–16 wk old were anesthetized using sodium thiopentone (50 mg/kg i.p.). The abdominal cavity was cut open, and the liver was perfused with 150 ml of 1× PBS via portal vein using a 20G cannula. Inferior vena cava was cut to drain the blood out of tissues. The liver was perfused with 150 ml 1× HBSS containing 25 mM Hepes and 0.5 mM EGTA, and then with 200 ml digestion medium (low-glucose DMEM with 1× penicillin-streptomycin, 15 mM Hepes, and 70 mg collagenase IV). All the buffers were used at 37°C, and collagenase was added to the digestion medium just before perfusion. Collagenase-perfused liver was dissected out and kept in a Petri plate with collagenase containing digestion medium, and the liver capsules were disrupted using a pair of fine forceps to release hepatocytes into the medium. The cell suspension was filtered through a 100-µm cell strainer and centrifuged at 50 g for 3 min at 4°C to sediment hepatocytes. The supernatant was discarded, and the pellet containing hepatocytes was resuspended in DMEM (without FBS). The washing steps were repeated three times. After the third centrifugation, the cells were resuspended in DMEM with 10% FBS and 1× penicillin-streptomycin. Viable cells were counted with Trypan blue (90–95% cells were viable) and plated on collagen-coated 60-mm Petri plates at 60–70% confluence. The cells were cultured overnight in standard culture conditions before use.
The adherent hepatocytes were loaded with 200 µM OA for 24 h. The cells were washed with 0.5 ml of phenol red–free DMEM (supplemented with 0.5% fatty acid–free BSA). The same medium (2 ml) was poured on the cells and treated with 100 nM insulin or 100 nM insulin + 20 µM PLD inhibitor (VU0155056) for 6 h. The medium was collected and analyzed for TG species as described earlier (Rai et al., 2017).
Immunohistochemistry with rat tissue samples
Normally fed or 16-h-fasted Sprague-Dawley rats, 12–16 wk old, were used for the experiment. A pair of fasted animals were injected with 0.75 ml of Tyloxapol (Triton WR-1339 lipoprotein lipase inhibitor; 10% vol/vol solution in 0.9% wt/vol sodium chloride) via tail vein. Immediately, 0.75 IU insulin was injected into one animal, while the other was injected with same volume of 0.9% sodium chloride. Blood samples were collected every 1 h for 4 h. The blood samples were kept at room conditions for 1 h to facilitate clot formation. The clotted blood was centrifuged at 500 g for 10 min at 4°C. After the final blood sample collection at hour 4, the animal was injected with sodium thiopentone (30 mg/kg i.p.) to anesthetize it. The abdominal cavity was cut open, and the diaphragm was dissected carefully to expose the heart. Whole-body perfusion-fixation was done via the left ventricle with 300 ml of 1× PBS and 4% PFA each. A small incision was made to the right atrium immediately after cannulation to drain the fixative out of the body. Perfusion was conducted until muscle twitching stopped and liver tissue was hardened. Liver (right medial lobe), heart (apical part), and skeleton muscle (gracilis from right leg) were collected and fixed overnight in 4% PFA at 4°C. The tissues were cut into ∼10 × 10 × 10-mm size and dipped into 40% sucrose (prepared in 1× PBS) until they sank to the bottom (generally 24–36 h). The tissues were embedded in OCT (optimal cutting temperature) compound, and sections of 20-µm thickness were prepared on a cryostat at −20°C. The sections were mounted on charged slides and dried at 37°C for 3 h. The dried sections were fixed again using 4% PFA at room conditions for 15 min. Antigen was retrieved at 95°C for 3–4 min using antigen retrieval buffer (10 mM sodium citrate and 0.05% Tween-20, pH 6.0). The sections were washed three times each for 10 min with 1× PBS and blocked with 3% BSA in 1× PBS. The blocked sections were washed once with 1× PBS for 10 min. Primary antibodies were diluted following manufacturer’s instructions and added to the sections. The tissue sections were incubated overnight at 4°C in a moist chamber. The sections were washed three times each for 10 min with 1× PBS to remove unbound antibodies. The secondary antibodies (dilution 1:500 in 1× PBS) were added to the sections and incubated for 1 h at room conditions. The sections were washed three times for 10 min each with 1× PBS to remove unbound secondary antibodies. The sections were mounted with DAPI containing mounting medium and imaged on an Olympus FV1000 confocal microscope using 20× dry or 63× oil-immersion objectives. All the postacquisition analysis was done using ImageJ software (National Institutes of Health).
Cell lines and growth conditions
BRL3A (ECACC, 85111503), HEK-293T (ATCC, CRL-11268), COS-7 (ATCC, CRL-1651), and McARH7777 (ATCC, CRL-1601) cells were used in this study. BRL3A cells were maintained in Coon’s medium supplemented with 10% FBS. HEK-293T and COS-7 cells were maintained in DMEM containing 10% FBS. McA-RH7777 cells were maintained in DMEM containing 20% FBS. All cells were maintained in a humidified atmosphere with 5% CO2 at 37°C. The cells were passaged at 80–90% confluence with trypsin-EDTA.
Microscopy
The microscope, detection system, and in vitro motility observation has been described (Soppina et al., 2009; Barak et al., 2013). Briefly, LD motility assay was performed at 37°C using a differential interference contrast microscope (Nikon TE2000-U) fitted with a 100× oil objective with numerical aperture 1.4. The microscope was placed in an acoustically protected room on a vibration isolated table (Newport). A Cohu 4910 series camera was used for imaging. Each pixel measured 98 × 98 nm. Image frames were digitized and saved as AVI files using an image acquisition card (National Instruments) for further analysis. The positions of LDs were tracked frame by frame using a custom-written software in LabVIEW (National Instruments), as described previously (Carter et al., 2005). Fluorescent images were acquired on Olympus confocal microscope (FV1000) equipped with a 63× oil objective. Sequential excitation of the fluorophores was achieved by 405-, 488-, and 561-nm lasers. Spectral band-pass emission filters were used for the acquisition of confocal images with high-sensitivity detectors. The images were saved as .oib files, and postacquisition processing was done with ImageJ or Fiji software.
Isolation of LDs from rat liver for in vitro motility and biochemical assays
Lipid droplets for in vitro motility and biochemical assays were purified from rat liver as previously reported (Barak et al., 2014; Rai et al., 2017). Briefly, 12–16-wk-old male Sprague-Dawley rats were anesthetized using sodium thiopentone (50 mg/kg i.p.). The abdominal cavity was cut open, and the liver was perfused through hepatic portal vein with 50 ml of ice-cold 1× PBS. The liver was dissected out, washed three times with 1× PBS, and weighed. The liver was chopped into small pieces and homogenized using a glass Dounce homogenizer with 1.5 volume of liver in 0.9 M MEPS buffer (M indicates molarity of sucrose in the solution; 35 mM Pipes, 5 mM EGTA, 5 mM MgSO4, and 1 M sucrose, pH 7.1) supplemented with 2× complete protease inhibitor cocktail, 4 mM PMSF, 2 µg/ml pepstatin A and 4 mM DTT. The homogenate was centrifuged at 1,800 g for 10 min at 4°C to obtain PNS.
Isolation of LDs for motility assays
The PNS was supplemented with all the protease inhibitors mentioned above except PMSF and loaded on a Sephacryl S-200 column (40-ml bed volume) to separate organelles from the cytosolic protein fraction. The first 4 ml of whitish eluate enriched with LDs was collected and supplemented with all protease inhibitors except PMSF. The elute was mixed with 1.5 volume of 2.5 M MEPS, and 6 ml of the content was loaded at the bottom of clear SW41 Beckman rotor tube. The bottom layer was overlaid with 2 ml each of 1.4, 1.2, and 0.5 M MEPS buffers. The gradient was centrifuged at 120,000 g at 4°C for 1 h. The topmost layer containing floating LD was collected with a 18G needle, flash frozen, and stored in liquid nitrogen. The aliquots were used for LD-motility experiments within 6 mo after preparation.
Isolation of LDs for biochemical assays
The PNS was supplemented with all the protease inhibitors above except PMSF and mixed with 1.5 volume of 2.5 M MEPS, and 15 ml of the content was loaded at the bottom of clear SW32 Beckman rotor tube. The bottom layer was overlaid with 5 ml each of 1.4, 1.2, 0.5, and 0 M MEPS buffers. The gradient was centrifuged at 120,000 g at 4°C for 1 h. The topmost whitish layer with floating LDs was collected with a 18G needle, flash frozen, and stored in liquid nitrogen. The LDs samples were used within a week after preparation.
Isolation of LDs from McA-RH7777 cells
LDs for biochemical assays were purified from adherent McA-RH777 cells using protocols previously reported (Barak et al., 2014; Rai et al., 2017), with minor modifications. Briefly, naive cells and KIF5B-GFP– and KIF5B(PA−)-GFP–overexpressing stable McA-RH7777 cells were cultured in ≥6 plates of 150 mm size each. The cells were loaded with 400 µM OA conjugated with fatty acid–free BSA (at a molar ratio of 6:1) for 12 h. The cells were harvested after trypsinization and washed once with 2 ml of 1× PBS. The cell pellet was suspended in 0.9 M MEPS buffer supplemented with 2× complete protease inhibitor cocktail, 4 mM PMSF, 2 µg/ml pepstatin A, and 4 mM DTT, and cells were lysed using 8–12 strokes of a cell cracker (Isobiotec) with 18-µm clearance. Up to 80% of cells were lysed. The homogenate was centrifuged at 1,800 g for 10 min at 4°C to obtain PNS. The PNS was mixed with two volumes of 2.5 M MEPS and loaded on the bottom of clear SW41 Beckman rotor tubes. The bottom layer was overlaid with 2 ml each of 1.2, 0.5, and 0.25 M MEPS buffer. The gradient was centrifuged at 174,000 g at 4°C for 1.5 h. The topmost whitish layer with floating LDs was collected with an 18G needle, flash frozen, and stored in liquid nitrogen. The LD samples were normalized at OD600, and proteins were precipitated using chloroform:acetone method as described below. The protein samples were solubilized in 2× SDS sample buffer and denatured at 95°C for 15 min for Western blotting.
Lipid extraction and targeted LC-MS performed for lipid profiling
NLDs and FLDs were purified from normally fed and 16-h-fasted rat livers, respectively, as mentioned above. The extraction of lipids and MS were performed as described (Rai et al., 2017; Pathak et al., 2018).
PLD activity assay
PLD activity of liver and cell lysate was measured using PLD activity assay kit as per the manufacturer’s instructions. Briefly, a master mix was prepared to the specified ratio. Rat liver was homogenized in PLD assay buffer (10 µg tissue per 100 µl buffer). McA-RH7777 lysate was prepared using cells collected from a 60-mm plate at 70–80% confluence in 100 µl PLD assay buffer. 50 µl of the liver lysate or cell homogenate was mixed with 50 µl of the master mix, and OD570 was measured in kinetic mode for ∼180 min at 37°C using a Tecan plate reader (Infinite M200 Pro). The ΔOD570 was calculated from a linear slope of the graph plotted with OD570 against time:
where A1 is the sample reading at time T1, A1BG is the background control sample at time T1, A2 is the sample reading at time T2, and A2BG is the background reading at time T2.
The ΔOD570 value was then used to calculate B (choline generated in nmol) by PLD during the reaction time (ΔT = T2 – T1). The PLD activity in the samples were calculated as
where B is the amount of choline in the sample, ΔT is the reaction time (min), V is the sample volume added into the reaction well (ml), and D is the sample dilution factor. The amount of choline generated per unit time was considered as the absolute PLD activity (nmol/min/ml = mU/ml). The absolute values of PLD activities were normalized for each independent experiment to calculate relative PLD activities in the samples.
Preparation of HSS from rat liver tissue
HSS (free of membranes, organelles, and LDs) from liver tissue was prepared using a previously reported protocol (Adelman et al., 1973) with slight modification. Male Sprague-Dawley rats 12–14 wk old were anesthetized using sodium thiopentone (50 mg/kg i.p.). The perfused liver was dissected out and washed three times with 1× PBS. The liver was weighed and homogenized using a glass Dounce homogenizer in 1.5× (vol/wt) 1 M MEPS buffer supplemented with 2× complete protease inhibitor cocktail, 4 mM PMSF, 2 µg/ml of pepstatin A, and 4 mM DTT. The homogenate was centrifuged at 1,800 g for 10 min at 4°C to obtain PNS. The PNS was supplemented with the above protease inhibitors except PMSF and centrifuged again at 500,000 g for 1 h at 4°C. The uppermost white floating layer of LDs was discarded, and the middle clear layer (HSS) was collected in fresh tubes without disturbing the pellet consisting of membrane and organelles. HSS was supplemented with 1× Roche protease inhibitor cocktail, flash frozen, and stored in liquid nitrogen. The HSS was used within 3 d of preparation.
ALDs
ALDs were prepared by cyclic freeze–thaw or sonication method (Thiam et al., 2013; Prévost et al., 2018). For preparation of 1 ml ALDs, 70 µl glyceryl trioleate (TG) and 0.5 µmol egg PC were mixed together in acid-washed clean glass tubes. For preparation of PA- or PS-supplemented ALDs, 25 nmol of respective PLs were mixed with the above lipids. For imaging experiments, 1.6 nmol of Rhodamine-PE was mixed with the other lipids. The lipid mixture was dried in a nitrogen gas stream and kept in a vacuum for ≥4 h to remove residual organic solvents. 930 µl HKM buffer (50 mM Hepes, pH 7.4 with KOH, 120 mM potassium acetate, and 1 mM MgCl2) was added to the TG-PL mixture and vortexed continuously for 10 min. The whitish emulsion was poured into ultra-low temperature resistant tubes and flash frozen in liquid nitrogen. The frozen emulsion was immediately thawed in a water bath preset at 55°C. This freeze–thaw process was repeated for five cycles with intermittent vortexing before every freezing step. In an alternative sonication method, the emulsion was sonicated for six cycles of 10 s each. Examination of the suspension under a white light or confocal microscope displayed ALDs having a TG core with phospholipid coating. The ALDs were used within 3 h of preparation.
Flotation assay with ALDs
HSS and ALDs were prepared by the methods described in the previous sections. ALDs were mixed with freshly prepared HSS and GTP (100 µM) and incubated for 1.5 h at 37°C with intermittent mixing cycles of 5 s on and 5 s off. The ALD-HSS was mixed with 2.5 M MEPS buffer (1:1.5 vol/vol ratio) containing 1× complete protease inhibitor cocktail and poured to the bottom of a clear SW32 Beckman rotor tube. The bottom layer was overlaid with 5 ml each of 1.2, 1, 0.5, and 0 M MEPS buffers (molarity refers to sucrose) and centrifuged at 120,000 g for 1 h at 4°C. TG-containing buoyant ALDs along with recruited proteins floated to the top as a dense whitish layer and were collected using a 18G needle in a clean microfuge tube. The ALDs were centrifuged again on a table-top centrifuge at 20,000 g for 10 min at 4°C to separate ALDs from aqueous MEPS buffer. The MEPS buffer from the bottom was collected with a 20G needle and discarded to obtain a concentrated ALD sample.
For PLD inhibition assay, ALDs were incubated with rat liver HSS containing 50 µM PLD inhibitor (VU0155056) for 1.5 h at 37°C. After incubation, the reaction mix was subjected to sucrose gradient centrifugation to separate the ALDs. ALD samples were collected and normalized using OD600 as described for NLDs and FLDs (Rai et al., 2017). Equal volumes of concentrated ALD samples were taken, and proteins were precipitated from them using chloroform-acetone (1:1 vol/vol) method. The precipitated proteins were solubilized in 2× sample buffer and separated using 10% SDS-PAGE. Immunoblotting was performed with indicated primary and secondary antibodies following standard methods. Western blot quantification (densitometry) was performed using ImageJ.
Protein precipitation from LD sample and Western blotting
LDs were mixed with two volumes of acetone:chloroform (1:1) in acid-washed glass tubes, vortexed thoroughly, and stored overnight at −20°C. Precipitated proteins were collected by centrifugation at 20,000 g for 2 h. The pellet was solubilized in 2× sample buffer and kept at 95°C for 15 min. The proteins were separated on 10% SDS-PAGE and subjected to immunoblotting with indicated antibodies.
For immunoblotting, the proteins separated on SDS-PAGE were transferred to PVDF membrane. The membrane was blocked with 5% nonfat dry milk (NFDM) in 0.1% Tris-buffered saline with Tween-20 (TBST) for 1 h at room temperature. The membrane was incubated with primary antibody diluted in 5% NFDM in 0.1% TBST (as suggested by manufacturers) for 1 h at room temperature or overnight at 4°C. The membrane was washed with 0.1% TBST three times for 10 min each. The membrane was incubated with HRP-conjugated secondary antibody and diluted in 5% NFDM in 0.1% TBST (as suggested by manufacturers) for 45 min at room temperature. The membrane was washed with 0.1% TBST three times for 10 min each and developed using ECL kit (Millipore). The blots were imaged on an Amersham Imager 600, and band intensity was quantified using ImageJ.
Lipid/fat blot assay for lipid–protein interaction
The lipid blot assay to analyze lipid–protein interaction was done as described previously (Munnik and Wierzchowiecka, 2013). Briefly, indicated phospholipids dissolved in chloroform were spotted on a nitrocellulose membrane. Alternatively, phosphatidylinositol phosphate (PIP) Strips membranes spotted with a series of phospholipids were used. BRL3A cell lysate was prepared in buffer containing 25 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1% Triton X-100, 1 mM PMSF, 0.1% BSA, 5 mM EDTA, and 1× complete protease inhibitor cocktail (Roche). Lysate was centrifuged at 10,000 g for 10 min at 4°C to prepare PNS. The PNS was overlaid on preblocked NC membrane and incubated overnight at 4°C. The membranes were then immunoblotted with primary antibodies against kinesin-1, kinesin-2, and dynein. The bound primary antibodies were visualized using HRP-conjugated secondary antibodies.
TLC for TG quantification
Lipid droplets were purified from rat liver or synthesized using TG and PLs as described in previous sections. Lipids were extracted according to the standard method (Bligh and Dyer, 1959). Briefly, 5 µl ALDs/LDs of OD600 ≈0.2 (diluted 1:500 in Milli-Q water) was suspended in 500 µl water and mixed with 2 ml methanol and 1 ml chloroform. The mixture was briefly vortexed and kept overnight at 4°C for lipid extraction. The next day, 1 ml of chloroform and water each were added to the tube, vortexed well, and kept undisturbed to allow phase separation under gravity. Alternatively, they were centrifuged at 1,000 g for 5–10 min in a Pyrex conical centrifuge tube to speed up the phase separation process. The organic phase was transferred to a new glass tube, and solvent was evaporated under a nitrogen stream. The silica TLC plates were precleaned with chloroform followed by air drying and heating to 100°C for 15 min. The dried lipids at the bottom of the glass tube were solubilized in 35 µl chloroform and spotted onto the plates using glass capillaries. A two-solvent system was used to resolve TG from other PLs. The first solvent (n-hexane:diethyl ether:acetic acid, 60:40:4) was run half the length of the plate and air-dried. The plate was again run in second solvent (n-hexane:diethyl ether, 59:1) up to the full length of the plate. The plate was air-dried and visualized by spraying 10% CuSO4 in 8% H3PO4 followed by baking in the oven >180°C for 15–20 min. The plates were scanned, and the TG bands were quantified using ImageJ.
LD redistribution in McARH-7777 cells upon PLD inhibition
McA-RH7777 cells maintained in standard culture conditions were plated on custom-made glass-bottom Petri plates at ∼50–60% confluence. FBS containing high-glucose DMEM was replaced with lipoprotein-deficient serum-supplemented high-glucose DMEM. After 24 h, spent medium was replaced with FBS containing high-glucose DMEM in the presence of 20 µm PLD inhibitor or DMSO as control. The cells were cultured for 6 h in standard culture conditions. Intracellular LDs were stained for 10 min with 50 µM MDH dye. The plate with adherent cells was placed on an Olympus FV1000 confocal microscope fitted with a stage incubation chamber maintaining 5% CO2, 37°C temperature, and ambient humidity. MDH signal from stained LDs was acquired using a diode laser at 405 nm excitation, and emission was collected at 420–480 nm. The images were analyzed using ImageJ to calculate fractional distance of LDs as defined by Rai et al. (2017).
LD motility assay with soluble phospholipids and PA binding protein
LDs were purified from normally fed rat liver as described above. Soluble phospholipids (for a final concentration of 100 µM) dissolved in chloroform were dried in N2 stream to evaporate organic solvents. An aliquot of 40 µl LDs was added to the dried phospholipid, gently mixed with a pipette, and incubated for 20 min on ice. 2 µl of 20× ATP regenerating system (20 mM ATP, 20 mM MgCl2, 40 mM creatine phosphate, and 40 U/ml creatine kinase) was added to the LD-PL mixture. The motility mixture was then introduced in a flow chamber with microtubules sticking on poly-l-lysine–coated coverslip. A detailed method for LD motility has been published (Barak et al., 2013, 2014). The LDs were held in an optical trap and placed over a microtubule filament to score within a 30-s interval. The slides were used for a maximum of 20 min to quantitate motile fraction.
Spo20p is a yeast SNARE protein that preferentially binds to PA on membranes. Binding of Spo20 was assumed to represent the presence of PA on membrane of organelles (e.g., LDs). The plasmid for chimeric protein, GST-tagged PA-binding domain of Spo20p (denoted GST-Spo20p), was expressed in Escherichia coli BL21 (DE3), and protein was purified with glutathione-sepharose beads as described earlier (Vitale et al., 2001). An aliquot of 40 µl LDs was thawed on ice, and 20 µM GST-Spo20p protein was added to LDs and incubated on ice for 20 min. The LD-protein mix was introduced to a flow chamber with microtubule sticking on poly-l-lysine–coated coverslips (Barak et al., 2013, 2014). The motile fraction of the LDs was quantified as mentioned above. GST tag protein expressed and purified from bacteria was used as negative control.
Plasmid construction
pCDH-KIF5B-GFP and pCDH-KIF5B(PA−)-GFP plasmids, previously reported by Wang et al. (2017), were obtained as generous gift from Guangwei Du (University of Texas Health Science Center, Houston, TX). WT and PA binding mutant forms of KTDs were subcloned from PCDH-KIF5B-GFP and PCDH-KIF5B(PA−)-GFP into pGEX4T1 vector. The KIF5B nucleotide sequences from 2,560 to 2,892 were PCR amplified and ligated to pGEX4T1 vector using EcoR1 and Xho1 restriction sites to fuse with C-terminus of GST tag. Similarly, WT and PA binding mutant KTDs fused with GFP were subcloned from pCDH-KIF5B-GFP and pCDH-KIF5B(PA−)-GFP into pCDH-CMV-MCS-EF1-puro vector. The KTD-GFP nucleotide sequences were PCR amplified from pCDH-KIF5B-GFP and pCDH-KIF5B(PA−)-GFP and ligated using Xba1 and Not1 restriction sites.
Lentiviral particle production and stable overexpression in cultured mammalian cells
Lentiviral particles were produced in HEK-293T cells by transfecting three separate plasmids: pCDH-CMV-MCS-EF1-puro vector (containing desired inserts), psPAX2, and pMD2.G, using Lipofectamine 2000. The lentiviral particle containing medium was collected at 48, 60, and 72 h after transfection, and dead cells were removed by filtration with a 0.45-µm hydrophilic polyethersulfone filter (4614, Pall Life Sciences). The lentivirus-containing medium was aliquoted and stored at −80°C. For stable expression, McA-RH7777 cells (∼50% confluent) were infected with the lentivirus-containing medium supplemented with 8 µg/ml polybrene (hexadimethrine bromide) to increase the viral infection efficiency. Lentivirus particle containing medium was replaced with fresh medium after 12 h of infection. Cells overexpressing the desired proteins were further selected using 2 µg/ml puromycin 24 h after infection. Fresh medium supplemented with puromycin was changed every 3 d to remove dead cells.
Expression and purification of GST-tagged proteins from bacteria
GST-tagged fusion proteins were expressed in bacteria (E. coli, BL21) and purified using glutathione-conjugated sepharose beads (Qiagen, 27-4574-01) using the manufacturer’s protocol. In brief, bacterial cells (OD ≈0.6) expressing GST-tagged proteins were induced with 1 mM IPTG for 6 h. The cells were collected after centrifugation, resuspended in buffer A (50 mM Tris-HCl, 150 mM NaCl, 5 mM β-mercaptoethanol, 1 mg/ml lysozyme, 1% Triton X-100, and 1 mM PMSF) and lysed using probe sonicator. The lysate was centrifuged at 120,000 g for 45 min at 4°C to obtain a clarified lysate. The clarified cell lysate was incubated with prewashed glutathione-sepharose beads (1 ml bead/1 liter cultured bacterial cells) for 3 h at 4°C. The protein-bound beads were washed with buffer B (50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 5 mM β-mercaptoethanol, and 1 mM PMSF) and eluted with buffer C (50 mM Tris-HCl, pH 8.0, 150 mM NaCl, 5 mM β-mercaptoethanol, 20 mM reduced glutathione, and 1 mM PMSF). The purified proteins were dialyzed overnight against 1× PBS. The protein samples were run on 10% SDS-PAGE, and purity was checked by Coomassie Brilliant Blue staining.
ALD binding assay with GST-KTD
ALDs were prepared using the method previously described (Prévost et al., 2018). In brief, phospholipids dissolved in chloroform were mixed with glyceryl trioleate at 0.5% molar concentration. The solvent was evaporated with nitrogen stream and kept in a vacuum desiccator for 4 h. Hydration buffer (20 mM Tris-HCl, pH 7.5, and 100 mM NaCl) was added to the lipid mix and vortexed for 10 min. The whitish solution was sonicated in a water bath sonicator for six cycles of 10 s each. The resultant oil-in-water emulsion was centrifuged at 20,000 g for 30 min to enrich and concentrate the buoyant ALDs. The floating whitish layer of ALDs was collected with an 18-G needle and incubated with 5 µg of purified GST-KTD or KTD(PA−) at 4°C for 30 min. The ALD–protein mix was supplemented with 2.5 M sucrose in MEPS buffer to adjust the final molarity to 1.5 M and loaded at the bottom of an MLS-50 clear ultracentrifuge tube. The bottom layer was overlaid with 1 ml each of 1.2 and 0.5 M sucrose in MEPS buffer. The topmost layer was 1.5 ml of MEPS buffer (without sucrose). The gradient was centrifuged at 175,000 g for 1 h at 4°C. The floating whitish layer of ALDs bound with GST-KTD was carefully collected without disturbing the underneath gradient. The ALDs were further concentrated by floating them up at 20,000 g at 4°C for 10 min to get a sample of desired concentration. The proteins bound to ALDs were precipitated using the acetone-chloroform method. The precipitated proteins were solubilized in 2× sample buffer and separated on 10% SDS-PAGE. The proteins were transferred to PVDF membrane and immunoblotted with GST antibody.
LD redistribution in McA-RH7777 cells upon KTD-GFP overexpression
McA-RH7777 cells stably expressing KTD-GFP or KTD(PA−)-GFP proteins were prepared using the protocol described above. The cells were plated on custom-made glass-bottom Petri dishes and cultured overnight. FBS containing DMEM was replaced with lipoprotein-deficient serum supplemented with high-glucose DMEM. After 24 h, 20% FBS containing high-glucose DMEM was added to the cells in the presence of 400 µM BSA-conjugated OA and cultured at standard conditions for 6 h. Intracellular LDs were stained with 50 µM MDH dye for 10 min and imaged live on a confocal microscope fitted with a stage incubator. The images were analyzed using ImageJ to calculate fractional distance of LDs as described (Rai et al., 2017).
Naive and KTD-WT–overexpressing McA-RH7777 cells were plated at ∼70% confluence on acid-washed glass coverslips. The cells were fixed with 4% PFA and 0.015% glutaraldehyde in 1× PBS for 15 min at room temperature. The cells were washed three times with 1× PBS. After washing, the cells were permeabilized with 0.1% Triton X-100 in PBS for 10 min. The cells were washed again with PBS, and nonspecific binding sites were blocked with 5% BSA in PBS. The blocked cells were incubated in a moist chamber with specified antibodies (dilution as instructed by manufacturer) for 16 h at 4°C. The cells were again washed and incubated with secondary antibody (dilution 1:500) at room temperature for 1 h. The unbound antibodies were removed by washing three times for 10 min with PBS and mounted in MDH (50 µM) containing mounting medium. The cells were imaged on an FV3000 confocal microscope using a 60× objective and 3× digital zoom.
Measurement of TG secreted from McA-RH7777 cells using targeted LC-MS
For measuring secreted TG, both naive and KTD-GFP–overexpressing stable McA-RH7777 cells were loaded with 0.4 mM OA conjugated with fatty acid–free BSA (at a molar ratio of 6:1) in incomplete medium for 6 h. At the end of incubation, cells were gently washed with phenol red–free incomplete medium containing 0.5% fatty acid–free BSA. The cells were further cultured in phenol red–free incomplete medium with 0.5% fatty acid–free BSA for 4 h. At the end of the chase period, the spent media were collected and used for LC-MS analysis as described (Rai et al., 2017).
Preparation of ER-enriched microsomes from rat liver and HEK293T cells
Microsomes were isolated from rat liver using established protocols (Wang et al., 2007; Rai et al., 2017), with slight modification. In brief, liver tissue was perfused with PBS, chopped into fine pieces, and homogenized using a loose glass Dounce homogenizer in homogenization buffer (20 mM Tris-HCl, pH 7.4, 250 mM sucrose, and 1 mM EDTA) by applying 10–12 gentle strokes. The homogenate was centrifuged at 500 g for 15 min to remove cell debris and unlysed tissue. The supernatant was transferred to a fresh tube and centrifuged again at 15,000 g for 10 min to pellet mitochondria down. This step was repeated three times. The supernatant was centrifuged again at 106,000 g for 1 h at 4°C to pellet the microsomes. The supernatant was discarded, while the microsome pellet was resuspended in 1 ml of buffer containing 10 mM Tris-HCl, pH 7.4, and 1 mM EDTA. The microsomes were stored at −80°C.
Microsomes were also prepared from GFP-Rab18(Q67L)–overexpressing HEK-293T cells. Two 100-mm plates of HEK293T cells at 60–70% confluence were transfected with GFP-Rab18(Q67L). The cells were collected 24 h after transfection using trypsin-EDTA. The spent medium was discarded and washed with 10 times the packed cell volume of 1× PBS. The cells were resuspended in three times the packed cell volume of hypotonic buffer (50 mM Hepes, pH 7.8, 5 mM EGTA, 125 mM KCl, and 1.25 M sucrose) and incubated at 4°C for 20 min. The cells were collected after centrifugation at 600 g for 5 min, and two volumes of isotonic buffer (100 mM Hepes, pH 7.8, 10 mM EGTA, and 250 mM KCl) was poured to the cells. The cells were lysed after 10–12 passages through an 18-µm clearance cell cracker (Isobiotec). The resultant homogenate was centrifuged at 1,000 g for 10 min at 4°C to pellet down unlysed cells and cellular debris. The supernatant was collected in a fresh tube and further centrifuged at 12,000 g for 15 min at 4°C to separate mitochondria. The last step was repeated twice to ensure complete removal of mitochondria. The supernatant was further centrifuged at 106,000 g for 60 min at 4°C. The pellet containing microsomes was resuspended in 100 µl of buffer containing 10 mM Tris-HCl, pH 7.4, and 1 mM EDTA, aliquoted, and stored at −80°C.
ALD-microsome fusion assay
ALDs and microsomes were prepared following the methods described above. 5 ml ALDs were mixed with 100 µl microsome (equivalent to 5 mg total microsomal protein) prepared from rat liver and 100 µM GTP. The reaction mixture was incubated at 37°C for 1 h with intermittent mixing cycle, set at 5 s on and off mode. ALDs were separated from unbound microsomes by step gradient ultracentrifugation. The proteins bound to ALDs were precipitated using the acetone-chloroform method. The proteins were transferred to PVDF membrane and immunoblotted with required antibodies.
For fusion assay, microsomes prepared from GFP-Rab(Q67L)-overexpressing HEK293T cells were incubated with ALDs (prepared with only PC or 0.5 μmol PC + 25 nmol PA + 1.6 nmol Rhodamine-PE) for 30 min at room temperature with intermittent mixing. ALDs were separated by flotation in a sucrose density gradient. The ALDs were introduced to a custom-made flow chamber, and the coverslip was kept upside down for 5–10 min in the dark to allow the sticking of ALDs to the glass slide. The ALDs were imaged on a confocal microscope.
BrdU cell proliferation assay
McA-RH7777 cells were plated at ∼70% confluence in custom-made glass-bottom culture plates. The cells were cultured with 10 µM BrdU for 4 h and fixed with 4% formaldehyde in PBS. After washing with PBS, cells were treated with prewarmed 2 M HCl for 20 min at room temperature. The cells were again washed with PBS and permeabilized with 0.1% Triton X-100 in PBS for 20 min. The nonspecific binding sites were blocked with 5% normal horse serum in 0.1% Triton X-100 in PBS and stained with anti-BrdU antibody. Finally, cells were stained with suitable secondary antibody and mounted in DAPI-containing mounting medium. The cells were imaged for DAPI and BrdU.
Estimation of PA on LDs by GFP-Spo20p (PA sensor protein) binding
COS-7 or McA-RH7777 cells were transiently transfected with 1 µg of GFP-Spo20p plasmid with Lipofectamine 2000 using the manufacturer’s protocol. The cells were induced with 200 µM BSA-complexed OA in complete medium for 12 h in standard culture conditions. The cells were treated with 100 nM insulin in high-glucose medium or 100 nM glucagon in low-glucose medium with 200 µM OA for 24 h. Pharmacological treatment with 20 µM PLD inhibitor (VU0155056) or 5 µg/ml Brefeldin-A was performed for 6 h. The cells were fixed with 4% PFA for 5 min at room temperature, washed twice with 1× PBS for 10 min each, and stained with MDH dye for 10 min before imaging on a confocal microscope.
Online supplemental material
Fig. S1 shows experiments related to Figs. 1 and 2, including the effect of injecting insulin on glucose and TG levels of fasted rats. Change in lipids (TG and PA) on LDs and microsomes across fed/fasted states, purity of microsomes and bacterial proteins, and specificity of GST-Spo20 for PA are shown. Fig. S2 shows change in phospholipid species (PC, PE, and PS) on LDs across fed/fasted states. Soluble PA is shown to remove kinesin in a lipid blot assay. Properties of the ALDs used in experiments is also shown, along with recruitment of GST-Spo20 to ALDs in the presence of PLD inhibitor. Fig. S3 shows the effect of insulin and glucagon on McA-RH7777 cells. Kinesin tail domain expression in cells, presence of KIF5B kinesin (normal and PA-binding mutant) on LDs purified from cells, and purity of LDs from McA-RH7777 cells are shown. GST-KTD is shown to remove kinesin-1 from ALDs in a dose-dependent manner. Fig. S4 shows the localization of PLD1 to LD-LD junctions. Purity of microsomes from HEK293T cells is shown, and GFP-Rab18 is shown to be recruited to ALDs from microsomes that were prepared from cells overexpressing GFP-Rab18.
Supplementary Material
Acknowledgments
We acknowledge N. Vitale for GFP-Spo20p and GST-Spo20p PA binding sensors; G. Du for the pCDH-KIF5B-GFP and pCDH-KIF5B-PA−-GFP constructs; R.G. Parton for GFP-Rab18(Q67L) construct; S. Nair (Tata Institute of Fundamental Research, Mumbai, India) for BrdU and antibodies; S.T. Suryavanshi and Tata Institute of Fundamental Research animal house facility; D.G. Mashek and R.J. Schulze for comments on the manuscript; and H. Bhonsle, J. Singh, P. Barak, D. Sengupta, and K. Sadh for suggestions and help with experiments.
This work was supported by funding from the Department of Atomic Energy, Government of India, the Wellcome Trust–DBT India Alliance (grants IA/S/11/2500255 to R. Mallik, IA/I/15/2/502058 to S.S. Kamat, and IA/E/11/1/500417 to P. Rai) and a Department of Science & Technology-Fund for Improvement of S&T Infrastructure in Universities & Higher Educational Institutions Infrastructure Development grant to Indian Institute of Science Education and Research Pune Biology Department.
The authors declare no competing financial interests.
Author Contributions: R. Mallik and M. Kumar wrote the manuscript with input from other authors. R. Mallik, S.S. Kamat, and M. Kumar designed the experiments. M. Kumar, S. Ojha, P. Rai, and A. Joshi did the experiments.
References
- Adelman M.R., Blobel G., and Sabatini D.D.. 1973. An improved cell fractionation procedure for the preparation of rat liver membrane-bound ribosomes. J. Cell Biol. 56:191–205. 10.1083/jcb.56.1.191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson L., Boström P., Ericson J., Rutberg M., Magnusson B., Marchesan D., Ruiz M., Asp L., Huang P., Frohman M.A., et al. 2006. PLD1 and ERK2 regulate cytosolic lipid droplet formation. J. Cell Sci. 119:2246–2257. 10.1242/jcs.02941 [DOI] [PubMed] [Google Scholar]
- Asp L., Claesson C., Boren J., and Olofsson S.O.. 2000. ADP-ribosylation factor 1 and its activation of phospholipase D are important for the assembly of very low density lipoproteins. J. Biol. Chem. 275:26285–26292. 10.1074/jbc.M003520200 [DOI] [PubMed] [Google Scholar]
- Athenstaedt K., and Daum G.. 1999. Phosphatidic acid, a key intermediate in lipid metabolism. Eur. J. Biochem. 266:1–16. 10.1046/j.1432-1327.1999.00822.x [DOI] [PubMed] [Google Scholar]
- Barak P., Rai A., Rai P., and Mallik R.. 2013. Quantitative optical trapping on single organelles in cell extract. Nat. Methods. 10:68–70. 10.1038/nmeth.2287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barak P., Rai A., Dubey A.K., Rai P., and Mallik R.. 2014. Reconstitution of microtubule-dependent organelle transport. Methods Enzymol. 540:231–248. 10.1016/B978-0-12-397924-7.00013-3 [DOI] [PubMed] [Google Scholar]
- Barbosa A.D., Sembongi H., Su W.-M., Abreu S., Reggiori F., Carman G.M., and Siniossoglou S.. 2015. Lipid partitioning at the nuclear envelope controls membrane biogenesis. Mol. Biol. Cell. 26:3641–3657. 10.1091/mbc.E15-03-0173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bligh E.G., and Dyer W.J.. 1959. A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol. 37:911–917. 10.1139/y59-099 [DOI] [PubMed] [Google Scholar]
- Carter B.C., Shubeita G.T., and Gross S.P.. 2005. Tracking single particles: a user-friendly quantitative evaluation. Phys. Biol. 2:60–72. 10.1088/1478-3967/2/1/008 [DOI] [PubMed] [Google Scholar]
- Chitraju C., Trötzmüller M., Hartler J., Wolinski H., Thallinger G.G., Lass A., Zechner R., Zimmermann R., Köfeler H.C., and Spener F.. 2012. Lipidomic analysis of lipid droplets from murine hepatocytes reveals distinct signatures for nutritional stress. J. Lipid Res. 53:2141–2152. 10.1194/jlr.M028902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choudhary V., Golani G., Joshi A.S., Cottier S., Schneiter R., Prinz W.A., and Kozlov M.M.. 2018. Architecture of Lipid Droplets in Endoplasmic Reticulum Is Determined by Phospholipid Intrinsic Curvature. Curr. Biol. 28:915–926.e9. 10.1016/j.cub.2018.02.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen D.E., and Fisher E.A.. 2013. Lipoprotein metabolism, dyslipidemia, and nonalcoholic fatty liver disease. Semin. Liver Dis. 33:380–388. 10.1055/s-0033-1358519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen J.C., Horton J.D., and Hobbs H.H.. 2011. Human fatty liver disease: old questions and new insights. Science. 332:1519–1523. 10.1126/science.1204265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalle S., Longuet C., Costes S., Broca C., Faruque O., Fontés G., Hani E.H., and Bataille D.. 2004. Glucagon promotes cAMP-response element-binding protein phosphorylation via activation of ERK1/2 in MIN6 cell line and isolated islets of Langerhans. J. Biol. Chem. 279:20345–20355. 10.1074/jbc.M312483200 [DOI] [PubMed] [Google Scholar]
- Donchenko V., Zannetti A., and Baldini P.M.. 1994. Insulin-stimulated hydrolysis of phosphatidylcholine by phospholipase C and phospholipase D in cultured rat hepatocytes. Biochim. Biophys. Acta. 1222:492–500. 10.1016/0167-4889(94)90059-0 [DOI] [PubMed] [Google Scholar]
- Fei W., Shui G., Zhang Y., Krahmer N., Ferguson C., Kapterian T.S., Lin R.C., Dawes I.W., Brown A.J., Li P., et al. 2011. A role for phosphatidic acid in the formation of “supersized” lipid droplets. PLoS Genet. 7:e1002201 10.1371/journal.pgen.1002201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galteau M.M., Antoine B., and Reggio H.. 1985. Epoxide hydrolase is a marker for the smooth endoplasmic reticulum in rat liver. EMBO J. 4:2793–2800. 10.1002/j.1460-2075.1985.tb04005.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbons G.F., and Wiggins D.. 1995. Intracellular triacylglycerol lipase: its role in the assembly of hepatic very-low-density lipoprotein (VLDL). Adv. Enzyme Regul. 35:179–198. 10.1016/0065-2571(94)00006-O [DOI] [PubMed] [Google Scholar]
- Gibbons G.F., Wiggins D., Brown A.M., and Hebbachi A.M.. 2004. Synthesis and function of hepatic very-low-density lipoprotein. Biochem. Soc. Trans. 32:59–64. 10.1042/bst0320059 [DOI] [PubMed] [Google Scholar]
- Hammond J.W., Griffin K., Jih G.T., Stuckey J., and Verhey K.J.. 2008. Co-operative versus independent transport of different cargoes by Kinesin-1. Traffic. 9:725–741. 10.1111/j.1600-0854.2008.00722.x [DOI] [PubMed] [Google Scholar]
- Joshi A.S., Nebenfuehr B., Choudhary V., Satpute-Krishnan P., Levine T.P., Golden A., and Prinz W.A.. 2018. Lipid droplet and peroxisome biogenesis occur at the same ER subdomains. Nat. Commun. 9:2940 10.1038/s41467-018-05277-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kassas N., Tryoen-Tóth P., Corrotte M., Thahouly T., Bader M.F., Grant N.J., and Vitale N.. 2012. Genetically encoded probes for phosphatidic acid. Methods Cell Biol. 108:445–459. 10.1016/B978-0-12-386487-1.00020-1 [DOI] [PubMed] [Google Scholar]
- Khalil M.B., Blais A., Figeys D., and Yao Z.. 2010. Lipin - The bridge between hepatic glycerolipid biosynthesis and lipoprotein metabolism. Biochim. Biophys. Acta. 1801:1249–1259. [DOI] [PubMed] [Google Scholar]
- Kim J.H., Lee S.D., Han J.M., Lee T.G., Kim Y., Park J.B., Lambeth J.D., Suh P.G., and Ryu S.H.. 1998. Activation of phospholipase D1 by direct interaction with ADP-ribosylation factor 1 and RalA. FEBS Lett. 430:231–235. 10.1016/S0014-5793(98)00661-9 [DOI] [PubMed] [Google Scholar]
- Klaunig J.E., Goldblatt P.J., Hinton D.E., Lipsky M.M., Chacko J., and Trump B.F.. 1981. Mouse liver cell culture. I. Hepatocyte isolation. In Vitro. 17:913–925. 10.1007/BF02618288 [DOI] [PubMed] [Google Scholar]
- Kooijman E.E., Tieleman D.P., Testerink C., Munnik T., Rijkers D.T.S., Burger K.N.J., and de Kruijff B.. 2007. An electrostatic/hydrogen bond switch as the basis for the specific interaction of phosphatidic acid with proteins. J. Biol. Chem. 282:11356–11364. 10.1074/jbc.M609737200 [DOI] [PubMed] [Google Scholar]
- Kory N., Farese R.V. Jr., and Walther T.C.. 2016. Targeting Fat: Mechanisms of Protein Localization to Lipid Droplets. Trends Cell Biol. 26:535–546. 10.1016/j.tcb.2016.02.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehner R., Lian J., and Quiroga A.D.. 2012. Lumenal lipid metabolism: implications for lipoprotein assembly. Arterioscler. Thromb. Vasc. Biol. 32:1087–1093. 10.1161/ATVBAHA.111.241497 [DOI] [PubMed] [Google Scholar]
- Li H.-S., Shome K., Rojas R., Rizzo M.A., Vasudevan C., Fluharty E., Santy L.C., Casanova J.E., and Romero G.. 2003. The guanine nucleotide exchange factor ARNO mediates the activation of ARF and phospholipase D by insulin. BMC Cell Biol. 4:13 10.1186/1471-2121-4-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin S., Driessen K., Nixon S.J., Zerial M., and Parton R.G.. 2005. Regulated localization of Rab18 to lipid droplets: effects of lipolytic stimulation and inhibition of lipid droplet catabolism. J. Biol. Chem. 280:42325–42335. 10.1074/jbc.M506651200 [DOI] [PubMed] [Google Scholar]
- Munnik T., and Wierzchowiecka M.. 2013. Lipid-binding analysis using a fat blot assay. Methods Mol. Biol. 1009:253–259. 10.1007/978-1-62703-401-2_23 [DOI] [PubMed] [Google Scholar]
- Murphy D.J. 2012. The dynamic roles of intracellular lipid droplets: from archaea to mammals. Protoplasma. 249:541–585. 10.1007/s00709-011-0329-7 [DOI] [PubMed] [Google Scholar]
- Nakamura N., Banno Y., and Tamiya-Koizumi K.. 2005. Arf1-dependent PLD1 is localized to oleic acid-induced lipid droplets in NIH3T3 cells. Biochem. Biophys. Res. Commun. 335:117–123. 10.1016/j.bbrc.2005.07.050 [DOI] [PubMed] [Google Scholar]
- Ozeki S., Cheng J., Tauchi-Sato K., Hatano N., Taniguchi H., and Fujimoto T.. 2005. Rab18 localizes to lipid droplets and induces their close apposition to the endoplasmic reticulum-derived membrane. J. Cell Sci. 118:2601–2611. 10.1242/jcs.02401 [DOI] [PubMed] [Google Scholar]
- Pathak D., Mehendale N., Singh S., Mallik R., and Kamat S.S.. 2018. Lipidomics Suggests a New Role for Ceramide Synthase in Phagocytosis. ACS Chem. Biol. 13:2280–2287. 10.1021/acschembio.8b00438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prévost C., Sharp M.E., Kory N., Lin Q., Voth G.A., Farese R.V. Jr., and Walther T.C.. 2018. Mechanism and Determinants of Amphipathic Helix-Containing Protein Targeting to Lipid Droplets. Dev. Cell. 44:73–86.e4. 10.1016/j.devcel.2017.12.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prinz W.A. 2014. Bridging the gap: membrane contact sites in signaling, metabolism, and organelle dynamics. J. Cell Biol. 205:759–769. 10.1083/jcb.201401126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rai P., Kumar M., Sharma G., Barak P., Das S., Kamat S.S., and Mallik R.. 2017. Kinesin-dependent mechanism for controlling triglyceride secretion from the liver. Proc. Natl. Acad. Sci. USA. 114:12958–12963. 10.1073/pnas.1713292114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadh K., Rai P., and Mallik R.. 2017. Feeding-fasting dependent recruitment of membrane microdomain proteins to lipid droplets purified from the liver. PLoS One. 12:e0183022 10.1371/journal.pone.0183022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulze R.J., and McNiven M.A.. 2019. Fasting Inhibits the Recruitment of Kinesin-1 to Lipid Droplets and Stalls Hepatic Triglyceride Secretion. Hepatology. 69:444–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen L., Hillebrand A., Wang D.Q.-H., and Liu M.. 2012. Isolation and primary culture of rat hepatic cells. J. Vis. Exp. 64:e3917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin J.J.H., and Loewen C.J.R.. 2011. Putting the pH into phosphatidic acid signaling. BMC Biol. 9:85 10.1186/1741-7007-9-85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shome K., Vasudevan C., and Romero G.. 1997. ARF proteins mediate insulin-dependent activation of phospholipase D. Curr. Biol. 7:387–396. 10.1016/S0960-9822(06)00186-2 [DOI] [PubMed] [Google Scholar]
- Soppina V., Rai A.K., Ramaiya A.J., Barak P., and Mallik R.. 2009. Tug-of-war between dissimilar teams of microtubule motors regulates transport and fission of endosomes. Proc. Natl. Acad. Sci. USA. 106:19381–19386. 10.1073/pnas.0906524106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiam A.R., and Beller M.. 2017. The why, when and how of lipid droplet diversity. J. Cell Sci. 130:315–324. 10.1242/jcs.192021 [DOI] [PubMed] [Google Scholar]
- Thiam A.R., Antonny B., Wang J., Delacotte J., Wilfling F., Walther T.C., Beck R., Rothman J.E., and Pincet F.. 2013. COPI buds 60-nm lipid droplets from reconstituted water-phospholipid-triacylglyceride interfaces, suggesting a tension clamp function. Proc. Natl. Acad. Sci. USA. 110:13244–13249. 10.1073/pnas.1307685110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitale N., Moss J., and Vaughan M.. 2001. Purification and properties of ARD1, an ADP-ribosylation factor (ARF)-related protein with GTPase-activating domain. Methods Enzymol. 329:324–334. 10.1016/S0076-6879(01)29094-6 [DOI] [PubMed] [Google Scholar]
- Wang H., Gilham D., and Lehner R.. 2007. Proteomic and lipid characterization of apolipoprotein B-free luminal lipid droplets from mouse liver microsomes: implications for very low density lipoprotein assembly. J. Biol. Chem. 282:33218–33226. 10.1074/jbc.M706841200 [DOI] [PubMed] [Google Scholar]
- Wang Z., Zhang F., He J., Wu P., Tay L.W.R., Cai M., Nian W., Weng Y., Qin L., Chang J.T., et al. 2017. Binding of PLD2-Generated Phosphatidic Acid to KIF5B Promotes MT1-MMP Surface Trafficking and Lung Metastasis of Mouse Breast Cancer Cells. Dev. Cell. 43:186–197.e7. 10.1016/j.devcel.2017.09.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilfling F., Thiam A.R., Olarte M.J., Wang J., Beck R., Gould T.J., Allgeyer E.S., Pincet F., Bewersdorf J., Farese R.V. Jr, and Walther T.C.. 2014. Arf1/COPI machinery acts directly on lipid droplets and enables their connection to the ER for protein targeting. eLife. 3:. 10.7554/eLife.01607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye J., Li J.Z., Liu Y., Li X., Yang T., Ma X., Li Q., Yao Z., and Li P.. 2009. Cideb, an ER- and lipid droplet-associated protein, mediates VLDL lipidation and maturation by interacting with apolipoprotein B. Cell Metab. 9:177–190. 10.1016/j.cmet.2008.12.013 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.