

The title compound, 2-(4-nitrophenyl)-2-oxoethyl 2-chlorobenzoate, is relatively planar with the two aromatic rings being inclined to each other by 3.56 (11)°.
Keywords: crystal structure, supramolecular architecture, C—H⋯Cl hydrogen bonds, π–π interactions, Hirshfeld surface analysis
Abstract
The title compound, C15H10ClNO5, is relatively planar with the two aromatic rings being inclined to each other by 3.56 (11)°. The central —C(=O)—C–O—C(=O)— bridge is slightly twisted, with a C—C—O—C torsion angle of 164.95 (16)°. In the crystal, molecules are linked by C—H⋯O and C—H⋯Cl hydrogen bonds, forming layers parallel to the (101) plane. The layers are linked by a further C—H⋯O hydrogen bond, forming a three-dimensional supramolecular structure. There are a number of offset π–π interactions present between the layers [intercentroid distances vary from 3.8264 (15) to 3.9775 (14) Å]. Hirshfeld surface analyses, the d norm surfaces, electrostatic potential and two-dimensional fingerprint plots were examined to verify the contributions of the different intermolecular contacts within the supramolecular structure. The shape-index surface shows that two sides of the molecule are involved in the same contacts with neighbouring molecules, and the curvedness plot shows flat surface patches that are characteristic of planar stacking.
Chemical context
Due to their numerous applications in various fields of chemistry, phenacyl benzoates are of great importance (Rather & Reid, 1919 ▸; Literák et al., 2006 ▸; Sheehan & Umezawa, 1973 ▸; Huang et al., 1996 ▸; Gandhi et al., 1995 ▸; Zhang et al., 2009 ▸). In continuation of our work on such molecules (Kumar et al., 2014 ▸; Chidan Kumar et al., 2014 ▸), we report herein on the crystal and molecular structures of 2-(4-nitrophenyl)-2-oxoethyl chlorobenzoate (I). Its crystal and molecular structures are compared with those of 2-(4-nitrophenyl)-2-oxoethyl benzoate (II) (Sheshadri et al., 2019 ▸), published by us recently, and further details of uses and applications of such molecules are described therein.
Structural commentary
The molecular structure of the title compound, I, is shown in Fig. 1 ▸. The compound is composed of two aromatic rings (C1–C6 and C10–C15) linked by the –C7(=O2)—C8—O1—C9(=O3)– bridge. The bond lengths and angles in I are normal and similar to those reported for compound II. The two benzene rings are inclined to each other by 3.56 (11)°, indicating that they are almost coplanar, as in the structure of II. The nitro group (N1/O4/O5) lies almost in the plane of the benzene ring (C1–C6), with a dihedral angle between the two planes of 5.4 (4)°; the torsion angles C4—C3—N1—O4 and C2—C3—N1—O5 are −5.4 (3) and −5.1 (4)°, respectively. Atom Cl1 is displaced by 0.0749 (8) Å from the plane of benzene ring C10–C15.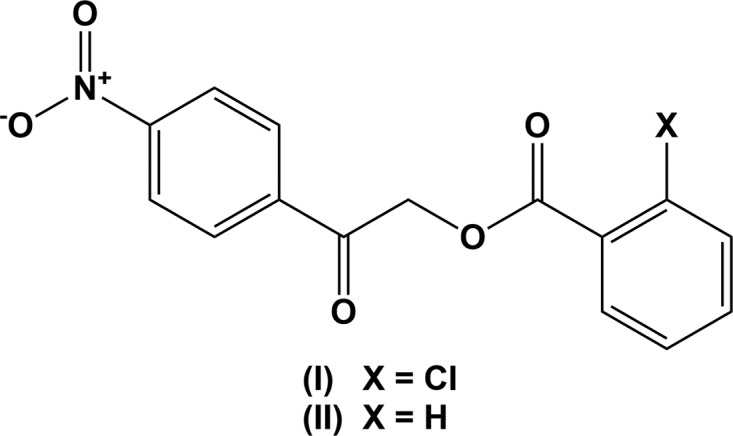
Figure 1.

The molecular structure of compound I, with the atom labelling. Displacement ellipsoids are drawn at the 50% probability level.
The overall molecular conformation of I is characterized by three torsion angles, viz. τ1 (C11—C10—C9—O3), τ2 (C7—C8—O1—C9) and τ3 (O2—C7—C6—C1). Torsion angle τ1 at −12.5 (3)° signifies a certain noncoplanarity between the benzene ring (C10–C15) and the adjacent carbonyl group (C9=O3) as a result of steric repulsion between the substituent Cl1 and the adjacent carbonyl group C9=O3. This is also reflected in the torsion angle τ2 of −164.95 (16)°, between the two carbonyl groups, C7=O2 and C9=O3, which have a –antiperiplanar conformation. Torsion angle τ3, involving the benzene ring (C1–C6) and the adjacent carbonyl group (C7=O2), is −3.6 (3)° and indicates a –synperiplanar conformation. A comparison of the torsion angles in I and II, indicates that the insertion of the Cl atom in I has the most significant influence on torsion τ2, which is −164.95 (16)° in I compared to 174.08 (9)° in II. Torsion angles τ1 of −12.5 (3)° and τ3 of −3.6 (3)° are slightly larger than the values observed in II, viz. 9.60 (16) and 1.88 (15)°, respectively. Hence, compound I has a less planar conformation than unsubstituted compound II.
Supramolecular features
The crystal structure of the title compound, is stabilized by intermolecular hydrogen bonds of the types C—H⋯O and C—H⋯Cl (Table 1 ▸). Molecules are linked by the C2—H2⋯O3i, C14—H14⋯O4i and C13—H13⋯Cl1iii hydrogen bonds to form layers lying parallel to the (101) plane; see Fig. 2 ▸ and Table 1 ▸. The layers are linked by C8—H8A⋯O3ii hydrogen bonds and offset π–π interactions (see Table 2 ▸), forming a supramolecular three-dimensional structure (Fig. 3 ▸).
Table 1. Hydrogen-bond geometry (Å, °).
| D—H⋯A | D—H | H⋯A | D⋯A | D—H⋯A |
|---|---|---|---|---|
| C2—H2⋯O3i | 0.93 | 2.54 | 3.258 (2) | 135 |
| C8—H8A⋯O3ii | 0.97 | 2.59 | 3.553 (3) | 171 |
| C13—H13⋯Cl1iii | 0.93 | 2.82 | 3.670 (3) | 153 |
| C14—H14⋯O4i | 0.93 | 2.50 | 3.211 (4) | 134 |
Symmetry codes: (i)  ; (ii)
; (ii)  ; (iii)
; (iii)  .
.
Figure 2.

A view normal to the (101) plane of the crystal packing of compound I. The hydrogen bonds are shown as dashed lines (Table 1 ▸; symmetry codes as in Table 1 ▸), and, for clarity, only the H atoms involved in hydrogen bonding have been included.
Table 2. π–π contacts (Å, °) in the crystal of compound I.
Cg1 and Cg2 are the centroids of the C1–C6 and C10–C15 rings, respectively.
| Cg(I) | Cg(J) | Cg(I)⋯Cg(J) (Å) | α (°) | β (°) | γ (°) | CgI_Perp (Å) | CgJ_Perp (Å) | Offset (Å) |
|---|---|---|---|---|---|---|---|---|
| Cg1 | Cg1iv | 3.9775 (14) | 0.02 (10) | 31.8 | 31.8 | 3.3791 (9) | 3.3791 (9) | 2.098 |
| Cg1 | Cg2v | 3.8801 (14) | 3.56 (11) | 30.1 | 29.1 | 3.3895 (9) | 3.3559 (10) | 1.948 |
| Cg2 | Cg2vi | 3.8264 (15) | 0.00 (11) | 24.8 | 24.8 | 3.4722 (10) | 3.4722 (10) | 1.608 |
Symmetry codes: (iv) −x + 1, −y + 1, −z; (v) −x + 1, −y + 1, −z + 1; (vi) −x, −y + 1, −z + 1.
Figure 3.

The crystal packing of compound I, viewed along the b axis, showing the layered stacking. For clarity, only the H atoms involved in hydrogen bonding have been included, and the hydrogen bonds are shown as dashed lines (Table 1 ▸).
Hirshfeld surface analysis and two-dimensional fingerprint plots
The Hirshfeld surface analysis (Spackman & Jayatilaka, 2009 ▸) and the associated two-dimensional fingerprint plots (McKinnon et al., 2007 ▸) were performed with CrystalExplorer17 (Turner et al., 2017 ▸). Hirshfeld surface analysis enables the visualization of intermolecular interactions by different colours and colour intensity, representing short or long contacts and indicating the relative strength of the interactions. Fig. 4 ▸(a) shows the Hirshfeld surface mapped over d norm (−0.154 to 1.305) and for Fig. 4 ▸(b) the electrostatic potential. The Hirshfeld surface illustrated in Fig. 4 ▸(a) reflects the involvement of different atoms with the intermolecular interactions through the appearance of blue and red patches, which correspond to the regions of positive and negative electrostatic potential shown in Fig. 4 ▸(b). The shape-index surface (Fig. 5 ▸ a) clearly shows that the two sides of the molecule are involved in contacts with neighbouring molecules and the curvedness plot (Fig. 5 ▸ b) shows flat surface patches characteristic of planar stacking.
Figure 4.

A view of the Hirshfeld surface of compound I, mapped over d norm.
Figure 5.

Hirshfeld surface of compound I, mapped over (a) the shape-index and (b) the curvedness.
The overall two-dimensional fingerprint plot for the title compound and those delineated into O⋯H/H⋯O, H⋯H, C⋯H/H⋯C and Cl⋯H/H⋯Cl contacts are illustrated in Fig. 6 ▸. The percentage contributions from the different interatomic contacts to the Hirshfeld surfaces are as follows: O⋯H (34.8%), H⋯H (18.8%), C⋯H (14.7%) and Cl⋯H (10.1%), shown in the two-dimensional fingerprint plots, respectively, in Fig. 6 ▸. The percentage contributions for other intermolecular contacts are less than 5% in the Hirshfeld surface mapping.
Figure 6.

The two-dimensional fingerprint plots of compound I, showing the percentage contributions of all contacts and of individual atom–atom contacts.
Database survey
A search of the Cambridge Structural Database (CSD, Version 5.40, last update May 2019; Groom et al., 2016 ▸) using 2-oxo-2-phenylethyl benzoate as the main skeleton revealed the presence of 62 structures with different substituents on the terminal phenyl rings. In these structures, the two aromatic rings are inclined to each other by dihedral angles varying from ca 0 to 90°. There were seven structures with a nitro substituent on one of the aromatic rings. However, there is only one compound with the same skeleton as the title compound, i.e. 2-(biphenyl-4-yl)-2-oxoethyl 4-nitrobenzoate (CSD refcode CISSAB; Kwong et al., 2017 ▸). Here the two aromatic rings are inclined to each other by ca 70.96°, compared to only 3.56 (11)° in the title compound. In the crystal structure of the recently published compound 2-(4-nitrophenyl)-2-oxoethyl benzoate (II) (Sheshadri et al., 2019 ▸), this dihedral angle is 3.09 (5)°.
Synthesis and crystallization
The title compound, was synthesized as per the procedure reported earlier by Kumar et al. (2014 ▸). A mixture of 2-bromo-1-(4-nitrophenyl)ethanone (0.2 g, 0.5 mmol), potassium carbonate (0.087 g, 0.63 mmol) and 2-chlorobenzoic acid (0.156 g, 0.65 mmol) in dimethylformamide (5 ml) was stirred at room temperature for 2 h. After completion of the reaction, the reaction mixture was poured into ice-cold water. The solid product obtained was filtered, washed with water and recrystallized from ethanol to give colourless block-like crystals (m.p. 386–390 K).
Refinement
Crystal data, data collection and structure refinement details are summarized in Table 3 ▸. C-bound H atoms were positioned geometrically (C—H = 0.93–0.97 Å) and refined using a riding model, with U iso(H) = 1.2U eq(C).
Table 3. Experimental details.
| Crystal data | |
| Chemical formula | C15H10ClNO5 |
| M r | 319.69 |
| Crystal system, space group | Monoclinic, P21/c |
| Temperature (K) | 294 |
| a, b, c (Å) | 12.6646 (18), 12.4099 (18), 9.0902 (13) |
| β (°) | 99.947 (2) |
| V (Å3) | 1407.2 (3) |
| Z | 4 |
| Radiation type | Mo Kα |
| μ (mm−1) | 0.30 |
| Crystal size (mm) | 0.55 × 0.26 × 0.19 |
| Data collection | |
| Diffractometer | Bruker APEXII DUO CCD area-detector |
| Absorption correction | Multi-scan (SADABS; Bruker, 2012 ▸) |
| T min, T max | 0.796, 0.946 |
| No. of measured, independent and observed [I > 2σ(I)] reflections | 36449, 4122, 2586 |
| R int | 0.058 |
| (sin θ/λ)max (Å−1) | 0.705 |
| Refinement | |
| R[F 2 > 2σ(F 2)], wR(F 2), S | 0.054, 0.141, 1.06 |
| No. of reflections | 4122 |
| No. of parameters | 199 |
| H-atom treatment | H-atom parameters constrained |
| Δρmax, Δρmin (e Å−3) | 0.26, −0.44 |
Supplementary Material
Crystal structure: contains datablock(s) global, I. DOI: 10.1107/S2056989019014336/su5521sup1.cif
Structure factors: contains datablock(s) I. DOI: 10.1107/S2056989019014336/su5521Isup2.hkl
Additional supporting information: crystallographic information; 3D view; checkCIF report
Acknowledgments
CSCK extends his appreciation to Vidya Vikas Research & Development Centre for the facilities and encouragement. NS thanks Jain University for sanctioning research grants under minor project.
supplementary crystallographic information
Crystal data
| C15H10ClNO5 | F(000) = 656 |
| Mr = 319.69 | Dx = 1.509 Mg m−3 |
| Monoclinic, P21/c | Mo Kα radiation, λ = 0.71073 Å |
| Hall symbol: -P 2ybc | Cell parameters from 6264 reflections |
| a = 12.6646 (18) Å | θ = 2.3–27.5° |
| b = 12.4099 (18) Å | µ = 0.30 mm−1 |
| c = 9.0902 (13) Å | T = 294 K |
| β = 99.947 (2)° | Block, colourless |
| V = 1407.2 (3) Å3 | 0.55 × 0.26 × 0.19 mm |
| Z = 4 |
Data collection
| Bruker APEXII DUO CCD area-detector diffractometer | 4122 independent reflections |
| Radiation source: Rotating Anode | 2586 reflections with I > 2σ(I) |
| Graphite monochromator | Rint = 0.058 |
| Detector resolution: 18.4 pixels mm-1 | θmax = 30.1°, θmin = 1.6° |
| φ and ω scans | h = −17→17 |
| Absorption correction: multi-scan (SADABS; Bruker, 2012) | k = −17→17 |
| Tmin = 0.796, Tmax = 0.946 | l = −12→12 |
| 36449 measured reflections |
Refinement
| Refinement on F2 | 0 restraints |
| Least-squares matrix: full | Hydrogen site location: inferred from neighbouring sites |
| R[F2 > 2σ(F2)] = 0.054 | H-atom parameters constrained |
| wR(F2) = 0.141 | W = 1/[Σ2(FO2) + (0.0456P)2 + 0.8032P] WHERE P = (FO2 + 2FC2)/3 |
| S = 1.06 | (Δ/σ)max < 0.001 |
| 4122 reflections | Δρmax = 0.26 e Å−3 |
| 199 parameters | Δρmin = −0.44 e Å−3 |
Special details
| Geometry. Bond distances, angles etc. have been calculated using the rounded fractional coordinates. All su's are estimated from the variances of the (full) variance-covariance matrix. The cell esds are taken into account in the estimation of distances, angles and torsion angles |
| Refinement. Refinement on F2 for ALL reflections except those flagged by the user for potential systematic errors. Weighted R-factors wR and all goodnesses of fit S are based on F2, conventional R-factors R are based on F, with F set to zero for negative F2. The observed criterion of F2 > 2sigma(F2) is used only for calculating -R-factor-obs etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2 are statistically about twice as large as those based on F, and R-factors based on ALL data will be even larger. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
| x | y | z | Uiso*/Ueq | ||
| Cl1 | 0.13276 (6) | 0.19871 (4) | 0.64688 (9) | 0.0704 (3) | |
| O1 | 0.32241 (12) | 0.43016 (11) | 0.42430 (17) | 0.0451 (5) | |
| O2 | 0.44530 (13) | 0.57459 (11) | 0.32308 (19) | 0.0553 (6) | |
| O3 | 0.31616 (15) | 0.26717 (12) | 0.5238 (2) | 0.0662 (7) | |
| O4 | 0.8091 (3) | 0.28826 (18) | −0.0648 (4) | 0.1349 (15) | |
| O5 | 0.8233 (2) | 0.45298 (17) | −0.1127 (3) | 0.1031 (10) | |
| N1 | 0.78442 (18) | 0.38048 (17) | −0.0532 (3) | 0.0632 (8) | |
| C1 | 0.60280 (16) | 0.53537 (15) | 0.1467 (2) | 0.0378 (6) | |
| C2 | 0.68013 (17) | 0.51241 (15) | 0.0615 (2) | 0.0407 (6) | |
| C3 | 0.70239 (17) | 0.40590 (16) | 0.0379 (2) | 0.0432 (7) | |
| C4 | 0.65143 (19) | 0.32166 (16) | 0.0961 (3) | 0.0493 (7) | |
| C5 | 0.57408 (18) | 0.34589 (15) | 0.1802 (3) | 0.0458 (7) | |
| C6 | 0.54870 (15) | 0.45265 (14) | 0.2066 (2) | 0.0356 (6) | |
| C7 | 0.46407 (16) | 0.48220 (15) | 0.2957 (2) | 0.0367 (6) | |
| C8 | 0.40417 (18) | 0.38936 (16) | 0.3492 (2) | 0.0429 (6) | |
| C9 | 0.28099 (17) | 0.35666 (15) | 0.5069 (2) | 0.0394 (6) | |
| C10 | 0.19286 (15) | 0.40267 (15) | 0.5758 (2) | 0.0365 (6) | |
| C11 | 0.12673 (17) | 0.33809 (16) | 0.6478 (2) | 0.0414 (6) | |
| C12 | 0.05046 (19) | 0.3840 (2) | 0.7205 (3) | 0.0548 (8) | |
| C13 | 0.0388 (2) | 0.4942 (2) | 0.7222 (3) | 0.0604 (9) | |
| C14 | 0.10067 (19) | 0.55891 (18) | 0.6499 (3) | 0.0544 (8) | |
| C15 | 0.17715 (17) | 0.51397 (16) | 0.5769 (3) | 0.0443 (7) | |
| H1 | 0.58650 | 0.60680 | 0.16450 | 0.0450* | |
| H2 | 0.71620 | 0.56730 | 0.02110 | 0.0490* | |
| H4 | 0.66890 | 0.25050 | 0.07900 | 0.0590* | |
| H5 | 0.53830 | 0.29040 | 0.21980 | 0.0550* | |
| H8A | 0.37220 | 0.34550 | 0.26490 | 0.0510* | |
| H8B | 0.45330 | 0.34460 | 0.41680 | 0.0510* | |
| H12 | 0.00710 | 0.34040 | 0.76810 | 0.0660* | |
| H13 | −0.01150 | 0.52480 | 0.77300 | 0.0720* | |
| H14 | 0.09130 | 0.63320 | 0.64990 | 0.0650* | |
| H15 | 0.21870 | 0.55860 | 0.52770 | 0.0530* |
Atomic displacement parameters (Å2)
| U11 | U22 | U33 | U12 | U13 | U23 | |
| Cl1 | 0.0741 (4) | 0.0363 (3) | 0.1119 (6) | −0.0032 (3) | 0.0471 (4) | 0.0109 (3) |
| O1 | 0.0529 (9) | 0.0353 (7) | 0.0540 (9) | 0.0031 (6) | 0.0285 (7) | 0.0035 (6) |
| O2 | 0.0648 (11) | 0.0323 (7) | 0.0754 (11) | −0.0003 (7) | 0.0311 (9) | −0.0077 (7) |
| O3 | 0.0755 (12) | 0.0382 (8) | 0.0983 (14) | 0.0158 (8) | 0.0524 (11) | 0.0190 (8) |
| O4 | 0.165 (3) | 0.0587 (13) | 0.222 (3) | 0.0172 (14) | 0.149 (3) | −0.0060 (16) |
| O5 | 0.1241 (19) | 0.0699 (13) | 0.144 (2) | −0.0058 (13) | 0.1036 (18) | 0.0043 (13) |
| N1 | 0.0670 (14) | 0.0509 (11) | 0.0825 (16) | −0.0031 (10) | 0.0433 (12) | −0.0073 (11) |
| C1 | 0.0436 (11) | 0.0268 (8) | 0.0437 (11) | −0.0021 (7) | 0.0095 (9) | −0.0019 (8) |
| C2 | 0.0446 (11) | 0.0342 (9) | 0.0456 (12) | −0.0058 (8) | 0.0141 (9) | 0.0031 (8) |
| C3 | 0.0444 (12) | 0.0394 (10) | 0.0504 (13) | −0.0019 (9) | 0.0214 (10) | −0.0037 (9) |
| C4 | 0.0588 (14) | 0.0296 (9) | 0.0659 (15) | −0.0004 (9) | 0.0284 (12) | −0.0048 (9) |
| C5 | 0.0554 (13) | 0.0288 (9) | 0.0591 (14) | −0.0051 (9) | 0.0268 (11) | −0.0009 (9) |
| C6 | 0.0393 (10) | 0.0295 (8) | 0.0392 (11) | −0.0031 (7) | 0.0106 (9) | −0.0026 (8) |
| C7 | 0.0412 (11) | 0.0320 (9) | 0.0383 (11) | −0.0015 (8) | 0.0105 (9) | −0.0013 (8) |
| C8 | 0.0517 (12) | 0.0341 (9) | 0.0486 (12) | 0.0005 (8) | 0.0250 (10) | −0.0021 (8) |
| C9 | 0.0428 (11) | 0.0322 (9) | 0.0458 (12) | −0.0011 (8) | 0.0149 (9) | 0.0010 (8) |
| C10 | 0.0355 (10) | 0.0339 (9) | 0.0410 (11) | 0.0012 (7) | 0.0089 (9) | 0.0032 (8) |
| C11 | 0.0409 (11) | 0.0368 (9) | 0.0491 (12) | −0.0001 (8) | 0.0149 (9) | 0.0046 (9) |
| C12 | 0.0500 (13) | 0.0566 (13) | 0.0644 (16) | 0.0006 (11) | 0.0288 (12) | 0.0066 (12) |
| C13 | 0.0540 (14) | 0.0560 (14) | 0.0790 (18) | 0.0104 (11) | 0.0336 (14) | −0.0026 (13) |
| C14 | 0.0537 (14) | 0.0369 (11) | 0.0762 (17) | 0.0096 (9) | 0.0214 (13) | −0.0022 (10) |
| C15 | 0.0456 (12) | 0.0343 (10) | 0.0553 (13) | 0.0018 (8) | 0.0150 (10) | 0.0030 (9) |
Geometric parameters (Å, º)
| Cl1—C11 | 1.732 (2) | C10—C11 | 1.401 (3) |
| O1—C8 | 1.428 (3) | C10—C15 | 1.396 (3) |
| O1—C9 | 1.344 (2) | C11—C12 | 1.384 (3) |
| O2—C7 | 1.206 (2) | C12—C13 | 1.376 (4) |
| O3—C9 | 1.197 (2) | C13—C14 | 1.367 (4) |
| O4—N1 | 1.196 (3) | C14—C15 | 1.383 (3) |
| O5—N1 | 1.199 (3) | C1—H1 | 0.9300 |
| N1—C3 | 1.470 (3) | C2—H2 | 0.9300 |
| C1—C2 | 1.379 (3) | C4—H4 | 0.9300 |
| C1—C6 | 1.396 (3) | C5—H5 | 0.9300 |
| C2—C3 | 1.376 (3) | C8—H8A | 0.9700 |
| C3—C4 | 1.381 (3) | C8—H8B | 0.9700 |
| C4—C5 | 1.376 (3) | C12—H12 | 0.9300 |
| C5—C6 | 1.394 (3) | C13—H13 | 0.9300 |
| C6—C7 | 1.496 (3) | C14—H14 | 0.9300 |
| C7—C8 | 1.506 (3) | C15—H15 | 0.9300 |
| C9—C10 | 1.485 (3) | ||
| C8—O1—C9 | 114.31 (15) | C10—C11—C12 | 120.71 (19) |
| O4—N1—O5 | 123.0 (3) | C11—C12—C13 | 120.0 (2) |
| O4—N1—C3 | 118.4 (3) | C12—C13—C14 | 120.5 (2) |
| O5—N1—C3 | 118.6 (2) | C13—C14—C15 | 120.1 (2) |
| C2—C1—C6 | 120.74 (17) | C10—C15—C14 | 121.0 (2) |
| C1—C2—C3 | 118.07 (18) | C2—C1—H1 | 120.00 |
| N1—C3—C2 | 118.53 (18) | C6—C1—H1 | 120.00 |
| N1—C3—C4 | 118.41 (19) | C1—C2—H2 | 121.00 |
| C2—C3—C4 | 123.1 (2) | C3—C2—H2 | 121.00 |
| C3—C4—C5 | 118.17 (19) | C3—C4—H4 | 121.00 |
| C4—C5—C6 | 120.72 (19) | C5—C4—H4 | 121.00 |
| C1—C6—C5 | 119.24 (18) | C4—C5—H5 | 120.00 |
| C1—C6—C7 | 118.47 (16) | C6—C5—H5 | 120.00 |
| C5—C6—C7 | 122.29 (18) | O1—C8—H8A | 110.00 |
| O2—C7—C6 | 122.01 (18) | O1—C8—H8B | 110.00 |
| O2—C7—C8 | 122.19 (19) | C7—C8—H8A | 110.00 |
| C6—C7—C8 | 115.80 (16) | C7—C8—H8B | 110.00 |
| O1—C8—C7 | 109.30 (16) | H8A—C8—H8B | 108.00 |
| O1—C9—O3 | 121.9 (2) | C11—C12—H12 | 120.00 |
| O1—C9—C10 | 111.65 (16) | C13—C12—H12 | 120.00 |
| O3—C9—C10 | 126.36 (19) | C12—C13—H13 | 120.00 |
| C9—C10—C11 | 122.06 (17) | C14—C13—H13 | 120.00 |
| C9—C10—C15 | 120.13 (18) | C13—C14—H14 | 120.00 |
| C11—C10—C15 | 117.74 (18) | C15—C14—H14 | 120.00 |
| Cl1—C11—C10 | 122.61 (16) | C10—C15—H15 | 120.00 |
| Cl1—C11—C12 | 116.66 (17) | C14—C15—H15 | 119.00 |
| C9—O1—C8—C7 | −164.95 (16) | C5—C6—C7—O2 | 177.1 (2) |
| C8—O1—C9—O3 | 5.3 (3) | C5—C6—C7—C8 | −2.8 (3) |
| C8—O1—C9—C10 | −177.15 (15) | O2—C7—C8—O1 | 3.6 (3) |
| O4—N1—C3—C2 | 174.8 (3) | C6—C7—C8—O1 | −176.59 (15) |
| O4—N1—C3—C4 | −5.1 (4) | O1—C9—C10—C11 | 170.13 (17) |
| O5—N1—C3—C2 | −5.4 (3) | O1—C9—C10—C15 | −13.2 (3) |
| O5—N1—C3—C4 | 174.7 (3) | O3—C9—C10—C11 | −12.5 (3) |
| C6—C1—C2—C3 | −0.2 (3) | O3—C9—C10—C15 | 164.3 (2) |
| C2—C1—C6—C5 | 0.4 (3) | C9—C10—C11—Cl1 | −6.6 (3) |
| C2—C1—C6—C7 | −178.83 (17) | C9—C10—C11—C12 | 175.2 (2) |
| C1—C2—C3—N1 | 179.79 (19) | C15—C10—C11—Cl1 | 176.60 (17) |
| C1—C2—C3—C4 | −0.4 (3) | C15—C10—C11—C12 | −1.6 (3) |
| N1—C3—C4—C5 | −179.5 (2) | C9—C10—C15—C14 | −175.3 (2) |
| C2—C3—C4—C5 | 0.7 (4) | C11—C10—C15—C14 | 1.6 (3) |
| C3—C4—C5—C6 | −0.5 (4) | Cl1—C11—C12—C13 | −178.1 (2) |
| C4—C5—C6—C1 | 0.0 (3) | C10—C11—C12—C13 | 0.2 (3) |
| C4—C5—C6—C7 | 179.2 (2) | C11—C12—C13—C14 | 1.3 (4) |
| C1—C6—C7—O2 | −3.7 (3) | C12—C13—C14—C15 | −1.3 (4) |
| C1—C6—C7—C8 | 176.42 (17) | C13—C14—C15—C10 | −0.2 (4) |
Hydrogen-bond geometry (Å, º)
| D—H···A | D—H | H···A | D···A | D—H···A |
| C2—H2···O3i | 0.93 | 2.54 | 3.258 (2) | 135 |
| C8—H8A···O3ii | 0.97 | 2.59 | 3.553 (3) | 171 |
| C13—H13···Cl1iii | 0.93 | 2.82 | 3.670 (3) | 153 |
| C14—H14···O4i | 0.93 | 2.50 | 3.211 (4) | 134 |
Symmetry codes: (i) −x+1, y+1/2, −z+1/2; (ii) x, −y+1/2, z−1/2; (iii) −x, y+1/2, −z+3/2.
References
- Bruker (2012). APEX2, SAINT and SADABS. Bruker AXS Inc., Madison, Wisconsin, USA.
- Chidan Kumar, C. S., Fun, H.-K., Tursun, M., Ooi, C. W., Chandraju, S., Quah, C. K. & Parlak, C. (2014). Spectrochim. Acta A Mol. Biomol. Spectrosc. 124, 595–602. [DOI] [PubMed]
- Gandhi, S. S., Bell, K. L. & Gibson, M. S. (1995). Tetrahedron, 51, 13301–13308.
- Groom, C. R., Bruno, I. J., Lightfoot, M. P. & Ward, S. C. (2016). Acta Cryst. B72, 171–179. [DOI] [PMC free article] [PubMed]
- Huang, W., Pei, J., Chen, B., Pei, W. & Ye, X. (1996). Tetrahedron, 52, 10131–10136.
- Kumar, C. S. C., Chia, T. S., Ooi, C. W., Quah, C. K., Chandraju, S. & Fun, H. K. (2014). Z. Kristallogr. Cryst. Mater. 229, 328–342.
- Kwong, H. C., Chidan Kumar, C. S., Mah, S. H., Chia, T. S., Quah, C. K., Loh, Z. H., Chandraju, S. & Lim, G. K. (2017). PLoS One, 12, e0170117. [DOI] [PMC free article] [PubMed]
- Literák, J., Dostálová, A. & Klán, P. (2006). J. Org. Chem. 71, 713–723. [DOI] [PubMed]
- Macrae, C. F., Bruno, I. J., Chisholm, J. A., Edgington, P. R., McCabe, P., Pidcock, E., Rodriguez-Monge, L., Taylor, R., van de Streek, J. & Wood, P. A. (2008). J. Appl. Cryst. 41, 466–470.
- McKinnon, J. J., Jayatilaka, D. & Spackman, M. A. (2007). Chem. Commun. pp. 3814. [DOI] [PubMed]
- Rather, J. B. & Reid, E. (1919). J. Am. Chem. Soc. 41, 75–83.
- Sheehan, J. C. & Umezawa, K. (1973). J. Org. Chem. 38, 3771–3774.
- Sheldrick, G. M. (2008). Acta Cryst. A64, 112–122. [DOI] [PubMed]
- Sheshadri, S. N., Chidan Kumar, C. S., Naveen, S., Veeraiah, M. K., Raghava Reddy, K. & Warad, I. (2019). Acta Cryst. E75, 1719–1723. [DOI] [PMC free article] [PubMed]
- Spackman, M. A. & Jayatilaka, D. (2009). CrystEngComm, 11, 19–32.
- Spek, A. L. (2009). Acta Cryst. D65, 148–155. [DOI] [PMC free article] [PubMed]
- Turner, M. J., McKinnon, J. J., Wolff, S. K., Grimwood, D. J., Spackman, P. R., Jayatilaka, D. & Spackman, M. A. (2017). CrystalExplorer17. University of Western Australia. http://hirshfeldsurface.net.
- Westrip, S. P. (2010). J. Appl. Cryst. 43, 920–925.
- Zhang, L., Shen, Y., Zhu, H. J., Wang, F., Leng, Y. & Liu, J. K. (2009). J. Antibiot. 62, 239–242. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Crystal structure: contains datablock(s) global, I. DOI: 10.1107/S2056989019014336/su5521sup1.cif
Structure factors: contains datablock(s) I. DOI: 10.1107/S2056989019014336/su5521Isup2.hkl
Additional supporting information: crystallographic information; 3D view; checkCIF report