

Abstract
Circulating tumor cells (CTCs) are broadly accepted as an indicator for early cancer diagnosis and disease severity. However, there is currently no reliable method available to capture and enumerate all CTCs as most systems require either an initial CTC isolation or antibody-based capture for CTC enumeration. Many size-based CTC detection and isolation microfluidic platforms have been presented in the past few years. Here we describe a new size-based, multiple-row cancer cell entrapment device that captured LNCaP-C4–2 prostate cancer cells with >95% efficiency when in spiked mouse whole blood at ~50 cells/mL. The capture ratio and capture limit on each row was optimized and it was determined that trapping chambers with five or six rows of micro constriction channels were needed to attain a capture ratio >95%. The device was operated under a constant pressure mode at the inlet for blood samples which created a uniform pressure differential across all the microchannels in this array. When the cancer cells deformed in the constriction channel, the blood flow temporarily slowed down. Once inside the trapping chamber, the cancer cells recovered their original shape after the deformation created by their passage through the constriction channel. The CTCs reached the cavity region of the trapping chamber, such that the blood flow in the constriction channel resumed. On the basis of this principle, the CTCs will be captured by this high-throughput entrapment chip (CTC-HTECH), thus confirming the potential for our CTC-HTECH to be used for early stage CTC enrichment and entrapment for clinical diagnosis using liquid biopsies.
Graphical Abstract
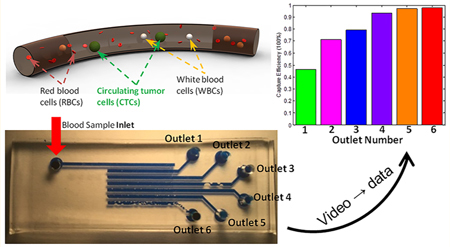
Measurement of circulating tumor cells (CTCs) represents a minimally invasive cancer screening method useful for initial staging of cancer patients and in monitoring recurrent or metastatic disease.1–4 The CTCs in peripheral blood play an important role in cancer metastasis. Considering the rare count of CTCs in blood with 1–100 cells/mL, the blood volume usually requires ~7.5 mL to achieve conclusive results for CTC detection and enrichment. The challenge of detecting and isolating the rare cells from peripheral blood encouraged scientists and engineers to develop many methods for CTC enrichment and enumeration. A limitation of the current CTC enumeration systems approved by the U.S. Food and Drug Administration (FDA), namely, CellSearch and AdnaTest, is their reliance upon expression of the epithelial cell adhesion molecule (EpCAM).5,6 Clinical studies show that CellSearch could detect CTCs in blood samples drawn from breast cancer, colorectal cancer, and prostate cancer patients.7–9 While EpCAM is expressed on the majority of cells within primary epithelial tumors, its expression is often lost during tumor progression.10–12 During epithelialmesenchymal transition (EMT), there is a loss of epithelial cell markers including EpCAM and an upregulation of mesenchymal surface proteins which corresponds to progression to more aggressive and metastatic cells.10–12 The level of EpCAM expression also varies for different cancer types and among patients. Furthermore, noncancerous EpCAM+ epithelial cells can exist in the blood.13 The same deficiency also applies to microfluidic chips that have been developed throughout the years that function based on such surface markers. A good example is the CTC chip containing over 10 000 microposts in an array that uses anti-EpCAM or aptamers for cell capture. The CTC detection and capture rate using CTC chip can reach 60% with approximately 50% purity.14,15 It is also notable that the surface markers used for CTC entrapment are fundamentally derived from cell lines and are based on primary tumor or metastatic tumor staining but very few have been validated on CTCs in patients.1,10,11
The ideal method for CTC isolation and enrichment should have minimal sample preprocessing to avoid CTC loss, high throughput, high efficiency, high sensitivity, high purity, and low cost. These criteria along with the limitations associated with surface-marker-based approaches have motivated the micro-fluidics community to develop new CTC technologies that rely on biophysical attributes of CTCs. This is primarily based on the assumption that these properties such as size, deformability, permittivity, and conductivity in CTCs differ from those of blood cells. Size-exclusion-based, deformability-based, and dielectrophoresis-based microchips have been explored and have shown promising results in CTC collection.16–18 Surface acoustic waves generating via tilted identical interdigital transducers on microfluidic channels also were used to collect CTCs from whole blood.19,20 The development of microfluidic chip for CTC enrichment is finding the balance between throughput, capture efficiency, and purity. Some existing technologies are listed in Table 1. In particular, separation of CTCs based on the hypothesis that they have a larger size compared to other cells in blood have been broadly investigated. Several review articles have been published summarizing these efforts.21–24 Some of the examples include using constriction channels,25 microfilter arrays,26 or fan-in-fan-out microcavity arrays27 to capture CTCs but these chips suffer from limitations in throughput and capture efficiency. Tai and Cote’s groups developed three-dimensional (3D) microfilters on a chip to select CTCs.28 The two-layers filter structure with micropores can capture ~85% of the spiked cancer cell lines LNCaP and MCF-7. The configuration and dimensions of the 3D microfilters need to be customized for specific cell lines, which increase the complexity of fabricating the parylene-C microfilters on polydimethylsiloxane (PDMS) devices. Toner’s group developed another type of microfilters with triangle barrier array to select single CTCs and CTC clusters.29 The trapping efficiency for larger CTC clusters reached 41% for clusters formed by two CTCs. However, single CTC cells could deform and escape from the triangle barrier without being captured. Ventana Medical Systems, Inc. also developed a microchannel-based filter for MCF-7 and MDA-MB-231 spiked blood sample with capture efficiency of ~75%.30 Because of the limited volume of capturing cavity before the microchannel filter, the maximum quantity of the CTC captured by this device is limited. The capture efficiency increased to 84% if more than 1000 of the cancer cells were spiked in 1 mL of blood sample, which indicated a limited application of capturing rare CTCs with only 1–100 cells/mL. Di Carlo’s group and the Vortex company demonstrated CTC isolation with generating rectangular reservoirs to form laminar vortices to isolate larger cancer cells from smaller blood cells.31–34 The dimensions of the rectangular reservoirs can be modified to make the device suitable for different size distributions of CTCs in either different cancer types or different cancer subpopulations.34 A recent report of using Vortex technique to isolate prostate CTCs with both cell line and patients reached an average of 1.88–93.75 CTCs/7.5 mL blood with purity of 1.74–37.59%,33 which had higher CTC detection than parallel CellSearch system comparison. The high-throughput Vortex (Vortex-HT) chip showed a capture efficiency of 51% and a purity of 55.2% after six cycles of sample screening with a high flow rate of 8 mL/min.33
Table 1.
Selected Existing Technologies on CTC Enrichment
| devices | key observations (recovery rate, throughput, purity) | reference |
|---|---|---|
| micropillar chip; herringebone chip | 5–1281 CTCs/mL with 1 mL/h, detection rate 65%; 63 CTCs/mL with 1.2 mL/h, detection rate 91.8 ± 5% for PC3; 50.3 CTCs/mL with 8 mL/h, detection rate 98% SKBR3 | M. Toner15,35–37 |
| micropillar chip | 30 CTCs/mL with 1 mL/h, detection rate 97 ± 3% | B.J. Kirby38 |
| sinusoidal chip | 53 CTCs/mL with 15 mL/h, detection rates are 98% MCF-7 and 82% MDA-MB-231 | S. A Soper39–42 |
| microchannel array | 10 CTCs/mL with 36 μL/min, recover rate >95%; using additional grooved surface can achieve over 90% capture rate with >84% purity; throughput 3.6 mL/h | Z. Fan43,44 |
| μHall: antibody labeled magnetic nanoparticles | recovery rate of MDA-MB-468 99%; purity 100%; throughput 3.25 mL/h; analyzing ovarian 7.6 CTCs/mL | H. Lee45 |
| CellSearch system | positive selection to enrich tumor cells from whole blood; the enriched and stained cells are scanned and imaged on CellTrackersAnalyzer II | 8, 46–49 |
| microfilter | 0–12.5 CTCs/mL with 90 mL/h, detection rate 90% | M. Toner35,50 |
| portable filter | MCF-7, SK-BR-3:5 CTCs/mL at 0.5 psi constant pressure, detection rate >90% | R.J. Cote,51 |
| microfilters | 1–3 CTCs/mL with 2 mL/h, detection rate enhanced with antibody coating: 95% MCF-7; 97% MDA-MB-231 | C.T. Lim,52 |
| microsieve filter with uniform pore structure | undiluted whole blood with flow rate <2 mL/min; MCF-7 recover rate >80%; accurately detected CTC from eight patients | M. Li53 |
| 3D parylene-C micronlter | selection by both size and deformability; collect >85% viable spiked MCF-7 cells | Y. Tai28 |
| suspended microchannel resonator | flow rate 45 μL/h; analyze 500 cells/h to reach 73% unhealthy cells; healthy blood cells passage time <10 ms; CTC passage time >10 ms | S. Manalis54–56 |
| lab-on-a-disc: centrifugal microfluidic size selection | MCF-7 61% capture rate; have the potential for high throughput and easy detection by directly mL; throughput >3 mL/min; sensitivity 95.9 ± 3.1% recovery rate; selectivity > 2.5 log WBCs depletion | Y. Cho57,58 |
| vortex | capture CTC by fluidic vortex without having CTCs experience deformation; throughput 7.5 mL whole blood in 20 min; about 60–90 min on the system with no sample prep; no staining or any kind of affinity capture of the cells is required; efficiency of capture 60–80% | D. Di Carlo31,32,59–61 |
Here, we present our new size/deformability-based CTC high-throughput entrapment chip (CTC-HTECH) with multiple rows of microconstriction channels and trapping chambers that overcome the limitations of inefficient capture observed with some existing size-dependent CTC capture designs. The single CTC can be captured in trapping chambers, while blood cells can pass through the microchannels with blood flow. The localized entrapment allows determination of the number of CTCs by scanning the trapping chambers, a unique feature that is not available in other size-based CTC microfluidic chips.28,30,51 The following describes the chip design and fabrication along with the experimental results that are achieved by spiking blood samples with prostate cancer cells.
EXPERIMENTAL SECTION
Cell Line Selection and Sample Preparation.
Prostate cancer is the second leading cause of cancer-related death in men. Prostate cancer cells are good candidates to represent CTCs in peripheral blood.41,62 Prostate cancer cell line LNCaP-C4–2 (passage #7, expressing green fluorescence protein (GFP) by lentiviral transduction) was grown in RPMI with 10% fetal bovine serum and 1% PenStrep (100 U/mL penicillin and 100 μg/mL streptomycin). Cells were grown in T-25 cm2 culture flasks at 37 °C in a 5% CO2 in air atmosphere until cells were ready for subculture. The morphology of the prostate cancer cells was observed before trypsinization (Figure 1). The cells were then detached from the flask with trypsin-ethylenediaminetetraacetic acid solution (Sigma-Aldrich). The LNCaP-C4–2 cells were trypsinized at 37 °C for 5 min, respectively. LNCaP-C4–2 cells were mixed with murine whole blood at about ~50 cells/mL. The white blood cells (WBCs) count in the murine whole blood is ~2–3 × 106/mL; the lymphocytes count is ~1–2.5 × 106/mL; and the platelets count is ~109/mL.The size of blood cells including red blood cells (RBCs), WBCs, lymphocytes, and platelets are below 8–10 μm, while the prostate cancer cells are larger in size distributed mainly in the range of 10–15 μm according to previously published work.63
Figure 1.

Micrographs depicting the morphology of prostate cancer cell line LNCaP-C4–2.
Device Fabrication.
The CTC-HTECH microfluidic channels were fabricated by polydimethylsiloxane (PDMS) soft lithography, followed by PDMS-glass bonding after plasma treatment (Figure 2). Forty microchannels with constriction channels (width, 8 μm; height, 8 μm; length, 100 μm/each) and trapping chambers (width, 30 μm; height, 30 μm; length, 40 μm/each) were connected in parallel in each row. The molds for micro-fluidic channels were fabricated on a silicon wafer with two layers of SU-8 (SU-8 3005 and SU-8 3025, MicroChem, Newton, MA) photolithography on a clean and dehydrated silicon prime wafer. Tridecafluoro-1,1,2,2-tetrahydrooctyl-1-trichlorosilane (Fisher Scientific) was coated on the surface of the molds for the easy release of PDMS.64,65 The PDMS channels were then bonded to a glass slide after air plasma treatment using plasma cleaner. Detailed fabrication procedures are available in the Supporting Information.
Figure 2.

Illustration of high-throughput entrapment chip for CTC (CTC-HTECH).
Experimental Setup.
The CTC-HTECH device is mounted on an inverted microscope (Zeiss Axio Observer LSM-510, Thornwood, NY). The blood sample is connected to the inlet and in all experiments with a constant pressure (500 mbar) applied to the sample reservoir by a pressure pump (Elveflow OB1, Paris). The resultant flow rate of the blood sample was ~2.4 mL/h. A smartphone with slow motion video function was positioned at the inlet and captured images at a rate of 240 frames/s (fps), which was sufficient to accurately count the number of cancer cells entering the chip. Only one inlet was open during each experimental run. As illustrated in Figure 2, cancer cells were deformed in multiple constriction channels arranged perpendicular to the inlet channel and cells recovered their shape within the cavities of the trapping chambers. RBCs, WBCs, platelets, and small lymphocytes passed through every constriction channel with the flow stream. The COMSOL simulation results of flow through the channels with and without cells in the constriction channels are presented in the Supporting Information (Figure S1). When the cancer cells enter and deform within the constriction channel, the blood flow temporarily slows down. Once inside the cavity of the trapping chamber, the cancer cells recover their original spherical shape after the deformation created by their passage through the constriction channel and the flow rate resumes. An individual CTC either continues through the series of constrictions and trapping cavities or remains in the cavity region of the trapping chamber. When the CTC remains in the trapping cavity, this defines an equilibrium point where the impetus of the blood flow stimulating cell progress through a subsequent constriction channel is offset by a CTC’s resistance to a subsequent deformation necessary to progress. On the basis of this principle, the CTCs are captured by this device. GFP prostate cancer cells diluted into whole mouse blood were visualized by their fluorescence. After 30 min the entire 1.2 mL sample passed through the device, and then camera images were taken of the device to ascertain the capture of cancer cells. The trapped CTCs were counted from camera images and the cancer cell trapping efficiency was determined by comparing the number of trapped GFP+ cancer cells to the number of GFP+ cancer cells entering the device at the inlet.
It is important to note that both the constant pressure and constant flow rate modes are used in microfluidic experiments.66,67 The constant flow rate mode using a syringe pump can guarantee the throughput for most microfluidic devices designed with a limited number of microchannels.67 However, when a constant flow rate is applied to a device with a large quantity of microchannels in an array, the pressure will be redistributed as cells are trapped in the microchannels. The pressure redistribution causes varying mechanical drag forces on the trapped cells within different channels. If three or more channels contain trapped cells under constant flow rate conditions, for example, the local pressure in the middle channel will increase and attempt to force the trapped cells through the constriction channel. The increased pressure will cause these cells to undergo greater deformation that could lead to changes in their cytoskeleton and possible cell damage, leading to poor recovery of viable cells.29,68,69 A sudden change in pressure will alter the cytoskeleton strength, membrane stiffness, and the biomechanical properties.70,71 In contrast, when a pressure pump is used in a constant pressure mode, the pressure supply is maintained constant by altering the back pressure. This results in a variation of the flow rate as the cells become trapped in the microchannels but avoids excessive pressure that imposes additional mechanical stress on the cells. In a microfluidic system with a low Reynolds number, the constant pressure mode will apply constant mechanical force on the cell membrane. Since the capturing and enrichment is related to the biomechanical properties and metastatic properties of CTCs, a gentle variation in both pressure and flow rate is needed. We used a programmable pressure pump to provide constant pressure to the blood samples at the inlet of the CTC-HTECH device. Consequently, an advance in our methodology based on the constant pressure inlet mode is the provision of a low stress environment for CTC trapping and enrichment.
RESULTS
Enrichment of Prostate Cancer Cells from Mouse Whole Blood.
Figure 3a shows the CTC-HTECH device is comprised of 6 rows, ①−⑥ where row ① is closest to the blood inlet port. Figure 3b shows a GFP+ cancer cell at the blood inlet. The outlets 1–6 were defined as waste collection site terminals from rows 1–6, respectively. Only one outlet was opened and connected to waste collection during each trial. For example, when outlet 1 is open and all other outlets are not punched open, outlet 1 collects the cells which pass through row ① (Figure 3). When outlet 2 is opened, cells pass through rows ① and ② and collect in outlet 2; when outlet 3 is open, cells pass through rows ①, ②, and ③ and collect in outlet 3, and so on.
Figure 3.

(a) Illustration of the configuration of the device (not to scale) with the inlet connected to programmable pressure pump; each row and each outlet was assigned and labeled individually; (b–g) the GFP+ LNCaP-C4–2 prostate cancer cells; (b) image of inlet with a GFP+ cell starting to enter row ①; (c) a GFP+ cell trapped in row ① after the blood flow ceased; (d) image of the waste collection at outlet 1; (e) a GFP+ cell deforming and passing through row ②; (f) two GFP+ cells in row ③ with one cell still passing and one cell exiting this row; (g) one GFP+ cell trapped in the trapping chamber of row ④; (h) the overall capture efficiency of each outlet. The data presented here is from three or four runs on CTC-HTECH device; (i) the percentage of the trapped GFP+ cells in every available row in the configuration where the indicated outlet was open (outlet 1 (green), outlet 2 (pink), outlet 3 (blue), outlet 4 (purple), outlet 5 (orange), and outlet 6 (red)).
To begin, GFP-labeled LNCaP-C4–2 cells were diluted into murine whole blood at a concentration of ~50 cells/mL. Outlet 1 was opened and the blood sample was video recorded at the inlet as cells entered row ①. Cells that do not exit row ① can be found in the constriction microchannels or in the trapping chambers. The blood flow passed through row ① and accumulated in the wider channel at outlet 1 waste collection. A GFP+ prostate cancer cell at the entrance of one constriction channel in row ① surrounded by unlabeled blood cells is shown in Figure 3c. Cancer cells that escaped from row ① were visible at the outlet 1 waste collection (Figure 3d). Blood did not flow into row ② when only outlet 1 was open because of the air remaining in the channels of rows ②–⑥. Figure 3e shows a GFP+ prostate cancer cell deformed in a constriction channel. Figure 3f shows two cells with one traveling in a constriction channel, and another escaped from the constriction channel and started to enter the next row of microchannels. Figure 3g shows a GFP+ prostate cancer cell stopped and trapped in a trapping chamber in row ④. Once the GFP+ prostate cancer cell recovers its original spherical shape, the cell occupies the upper space in the trapping chamber, as illustrated in Figure 2. This allows the blood flow to resume through the constriction channels.
In preliminary trials, we observed that some GFP+ cancer cells were still able to deform and pass through the second constriction channel; therefore, the CTC-HTECH design was adapted by duplicating the trapping chambers and increasing the quantity of microchannel rows to optimize trapping of the cancer cells. Figure 3h summarizes the results of sequentially selecting a different outlet (1–6) and monitoring the overall trapping efficiency of GFP+ cancer cells. When outlet 1 was open and only row 1 was used for capture, the trapping efficiency was 46.3% (Figure 3h green bar); when outlet 6 was open and rows 1–6 were used for capture, the trapping efficiency was 97.9% (Figure 3h red bar).
Figure 3i summarizes the GFP+ cell capture percentages for each outlet configuration. When only outlet 1 is open (green bar), cells transited only row ① and the capture efficiency of GFP+ prostate cancer cells was less than 50%. When outlet 2 was open (pink bars) cells transited both rows ① and ②. Here, the capture efficiency of row ① was ~55%; the number of captured GFP+ cells in row ② was somewhat less than 20%. We suggest that as the number of rows is increased, the overall fluidic resistance is also increased, and therefore, the trapping efficiency in each row varies. This said, the capture efficiency of row ① when either outlet 1, 2, or 3 was used was 45–55%. The capture efficiency of row ① when selecting either outlet 1, 2, or 3 was less than the capture efficiency of row ① when selecting either outlet 4, 5, or 6. However, the GFP+ cells that escape row ① can be captured by rows ② and ③ at a capturing ratio of ~20% and ~10%, respectively.
When the number of rows active in capture is ≤3, the flow rate at the outlet will be higher than when there are 6 active capture rows under the constant pressure mode. Therefore, a limiting factor for the capture efficiency in row ① appears to be the flow rate, with a slower flow rate resulting in more efficient cell capture in row ①. This could explain why the capture efficiency of row ① using outlets 1, 2, or 3 was 45–55% (Figure 3i, green bar row ①, pink bar row ①, and blue bar row ①). The configurations using either outlet 4 (Figure 3i, purple bars) or outlet 5 (Figure 3i, orange bars) had a higher capture efficiency of GFP+ cells in row ① of ~65%, while the capture efficiency in rows ② and ③ were similar to that of the configuration using outlet 3. The configuration using outlet 6 (Figure 3i, red bars) utilized all six rows and achieved the highest capture efficiency, ~70% in row ①. Row ② had a similar capture ratio of 15–20% when using outlets 2–6. The configuration using outlet 6 had more GFP+ cells captured in rows ② and ③ than rows ④, ⑤, and ⑥. The overall capture efficiency of all configurations is shown in Figure 3h. Use of outlets 5 and 6 allowed an overall capture ratio >95% to be reached.
As shown in the results from multiple devices in Figure 4, in each configuration (either open or closed) of outlets 1–6, we plotted the captured cell counts and the spiked cell counts. Each data point represented one experimental result from one device. Opening outlet 6 (red) included all 6 rows of channels, which had the best capture rate. As illustrated by red stars in Figure 4, the outlet 6 overall capture efficiency almost reaches 100% with a high linear correlation value of R2 = 0.9979. The outlet 5 and outlet 4 also achieved high capture efficiency by comparing the number of cells captured and the number of cells spiked in whole blood sample. The outlet 3 and outlet 2 capture efficiencies were reduced to 70–80%. The consistencies of outlets 2–6 were much better than outlet 1. Since outlet 1 contained only one row of micro-constriction channel and trapping chambers, the uncertainty of cancer cells deformation and trapping in row ① caused low capture rate. Even though we used constant pressure mode at the inlet, the flow rate will have higher variation if many cells are trapped in row ①. The local increasing flow rate dragged the trapped cancer cells through the trapping chambers. If outlet 6 is used, the overall pressure drop between the inlet and outlet will be smaller than that with use of outlet 1. The chance of driving trapped cancer cells from row ① using outlet 6 will be lower than that from using outlet 1.
Figure 4.

Number of cells captured compared to the number of cells spiked per 0.6−0.7 mL mouse whole blood in different outlet configurations. Results shown are the results testing n = 3−4 different devices at six different outlet configurations.
Control Group: Blood Cells Observation through CTCHTECH Outlet 6.
Mouse Whole Blood without Spiked LNCaPC4-2.
To estimate the trapping purities of the final CTC enrichment, we used mouse whole blood as a control group through the CTC-HTECH chip using outlet 6. The constant pressure was set up the same as the cancer cells spiked blood samples. As shown in Figure 5a, the RBCs and WBCs can pass through the micro-channels and the trapping chambers freely. After ~0.8 mL of mouse blood passing through the chip, to count the amount of blood cells trapped in the CTC-HTECH device, we switched the inlet to LNCaP-C4–2 culture medium to remove the blood cells that did not trap in the chip. The flow pressure of culture medium was kept constant. After 15 min of rinsing, blood cells were not observed in most of the microchannels (Figure 5b, Figure S2d). Only 17 out of 240 microchannels were found; WBCs or lymphocytes trapped in the microconstriction channels as some RBCs were also blocked in the trapping chambers. As shown in Figure S2f, the fluid coming to the reservoir of outlet 6 also contained little blood cells. Most of the RBCs were removed in the CTC-HTECH chip.
Figure 5.

Image of the control group mouse whole blood without cancer cells spiked (a,b), and with cancer cells spiked (c,d) through the CTCHTECH: (a) whole blood passing through the microchannels; (b) the inlet switched to LNCaP-C4–2 culture medium to remove the blood cells;(c) image of a trapped cancer cell after 15 min rinsing by LNCaP-C4–2 medium; (d) image of a microchannel with WBCs/lymphocytes trapped after 15 min of rinsing by LNCaP-C4–2 medium.
Mouse Whole Blood with Spiked LNCaP-C4–2.
More control groups with mouse whole blood with spiked cancer cells were performed to estimate the amount of WBCs and lymphocytes remaining in the CTC-HTECH after rinsing with LNCaP-C4–2 culture medium. The LNCaP-C4–2 capture ratio again reached97.3 ± 2.5% (repeated three times on three devices) after 15 min rinsing. Most of the trapped cancer cells stayed in the trapping chambers (Figure 5c) while allowing the medium to freely flow through the channels. Similar to the control experiment, almost all blood cells passed through the microchannels. Only 19 out of 240 microchannels were observed with WBCs or lymphocytes trapped in the channel. Exact counting on these blood cells was difficult as they formed clusters. However, based on the size of the microchannels and trapping chambers, we estimated that no more than 100 WBCs or lymphocytes could trap in each micro-channel. Therefore, the total numbers of blood cells trapped in these chips were calculated to be less than 2000. This is similar to what we observed when only blood cells were drawn through the chip in our control experiment. Nevertheless, compared to the total number of blood cells in the original sample (~2.4−4.4 × 106 cells of WBCs and lymphocytes), CTC-HTECH has been able to remove millions of blood cells and increase the concentration of CTCs from <1:106 to ~1:50.
DISCUSSION
The CTC-HTECH provides a new size-based CTC entrapment microchip for low cost and high-throughput analysis of cancer cells in whole blood samples. The CTC-HTECH device with six rows of constriction channels and trapping chambers captured over 95% of GFP+ LNCaP-C4–2 metastatic human prostate cancer cells used as surrogates for human CTCs. The sample preparation was minimal as these cancer cells were captured from whole blood; the microchip run time was ~30 min for a 1.2 mL blood sample. Compared to magnetic particles with antibody methods, such as FDA approved CellSearch system, our CTCHTECH reduced the lengthy sample preprocessing and long analysis time of ~4–6 h. There is no requirement for antibodies, a high-speed camera, or sophisticated image analysis; the data can be collected using the video features of a smartphone.
Size is one of the major biophysical attributes of cells that have been utilized to separate CTCs from blood cells. Different micro-fluidic chips in PDMS, polycarbonate, parylene-C, and some other biocompatible polymers72 have been designed and fabricated to isolate larger epithelial tumor cells from smaller blood cells. The geometry and dimension selection of the size-based trapping methods are key to achieve desired CTC trapping efficiency and purity. In microfilters, for example, an array of small holes with a size around 8 μm can capture CTCs while allowing the majority of blood cells to pass through.73 Y. Chen’s group developed a conical-shaped hole to slightly increase the pressure once CTCs which are trapped in the small holes started to deform to enhance capture purity.74 From their simulation and experiment results on MCF-7 cells captured in cone-shaped hole, the blood flow pressure facilitated leukocytes to escape from the holes, which resulted in an increased capture efficiency and purity with 6.5–8.0 μm diameter cone-shaped hole.74 However, the results showed that the thickness of this microfilter also significantly affects the capture purity because more blood cells will accumulate and clog the cone-shaped holes if using a thicker microfilter. With this additional variable to the microfilter, finding the optimized parameters to achieve a balancing between capture efficiency and purity of the microfilter becomes challenging. A recent study from S. Lee’s group demonstrated a flow-restricted microfluidic channel with an array of trapping cavities that reached 97% trapping rate with single MDA-MB-231 cells spiked in mouse blood.75 The continuous blood flow in their delivery channel kept the spiked MDA-MB-231 cells staying in the trapping channel, which had less pressure to force the cancer cells passing through the constriction regions. However, the microchannel array still suffers from possible clogging when more CTCs are trapped in the same constriction. Therefore, a better CTC enrichment with microconstriction channels and trapping chambers requires proper selection size for CTCs with less chance of cells clogged in a single trapping cavity.
The CTC-HTECH device, presented here, operates on an enrichment principle whereupon large cancer cells are captured at “equilibrium point” cavities (trapping chambers) positioned between constriction microchannels, which allows the blood to continue flowing through either the same channel or neighboring ones. It is also important to note that all 40 parallel channels between two adjacent rows experience the same pressure drop. The CTC-HTECH system is operated under constant pressure for all scenarios described earlier. As a result, the overall flow rate decreases as the greater number of rows are utilized to trap CTCs. In each microchannel, the Reynolds number in constriction channel is around 2.77. On the basis of the dimension of each channel, including constriction channels and trapping chambers, the pressure drop across each channel is about 77 mbar. Assuming all channels are open and no large cell is trapped, the total fluid resistance across one row will be around 9 × 103 Pa·s/μL. In the case of using outlet 1, the low fluid resistance leads to higher flow rate which causes more cancer cells to escape from the chip. Therefore, using only a couple of rows will significantly decrease the number of trapped CTCs. Also, when only one row is used and cells continue to trap, the flow rate in neighboring trapping chambers that are still open will experience a higher increase in flow rate when we compare this case to a six-row configuration. In a six-row configuration, the change in the flow resistance due to cell trapping has less effect on the overall flow rate compared to the case where only one row is used. Furthermore, using a fewer number of rows reduces the probability of experiencing movement through multiple trapping chambers which in turn causes loss in enrichment efficiency. Since a greater number of cells are trapped in the first row, one alternative design that can be explored in future generations is to increase the number of parallel channels in the first row and decrease the number for the subsequent rows without changing the overall footprint of the chip.
CONCLUSIONS
This low-cost, label-free, size-based CTC-HTECH device is able to effectively enrich the CTC sample in prostate cancer cell line spiked blood samples. The chip was able to achieve a trapping efficiency of 96%. From out of 42 cancer cells only 1 was not captured. This is significant as the number of CTCs has been shown to be a diagnostic or prognostic marker for tumor. The chip enables accurate enumeration of CTCs in blood and makes it a reliable tool for clinical settings for rapid CTC capture and subsequent counting. The difference between cancer cell line spiked in blood to patient CTC were discussed in previous publications.21,76–78 The blood cell size distribution, especially WBCs, lymphocytes, or other blood cells in peripheral blood from one patient or between different patients could vary from 8 to 20 μm.77,79 The deformability differences between cancer cells and blood cells can also be utilized in separating WBCs or lymphocytes.54 It is likely that the chip once using patient samples could require some adjustment in design to ensure highly efficient enrichment. However, out chip topography is very flexible and can be easily adopted to the clinical needs. We can envision new generations of this chip in which channels can be fabricated to have different sizes for both constriction regions and the trapping chambers. This may create a second degree of separation within the same chip. Also, a greater number of channels can be added to each row to make it more suitable for clinical samples where a greater number of CTCs may be present. We can also use CTC-HTECH chips in series whether having identical or different channel dimensions. CTC-HTECH has the potential to be connected in series as multistage multicycle CTC enrichment.
Supplementary Material
ACKNOWLEDGMENTS
The authors thank the National Institute of Health (NIH) National Cancer Institute (NCI) R21CA210216 and National Science Foundation (NSF) CBET-1403304 for supporting this research project. B.A.K. is supported by a NIH/NCI Pathway to Independence Award (R00CA175291). The authors acknowledge the Micro & Nano Fabrication Laboratory at Virginia Tech for the equipment support.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.analchem.8b01134.
Detailed fabrication procedure of the microchip CTCHTECH, including the protocols/equipment of each step; calculations of the microfluidic parameters in CTCHTECH, and COMSOL simulation of the CTCHTECH pressure distributions; additional images of the control group experiment using mouse whole blood (PDF)
The authors declare no competing financial interest.
REFERENCES
- (1).Kerr BA; Miocinovic R; Smith AK; West XZ; Watts KE; Alzayed AW; Klink JC; Mir MC; Sturey T; Hansel DE; et al. Oncotarget 2015, 6, 1889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Harris KS; Kerr BA Stem Cells Int 2017, 2017, 8629234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Hegemann M; Stenzl A; Bedke J; Chi KN; Black PC; Todenhöfer T BJUInt 2016, 118, 855–863. [DOI] [PubMed] [Google Scholar]
- (4).Siravegna G; Marsoni S; Siena S; Bardelli A Nat. Rev. Clin. Oncol 2017, 14, 531. [DOI] [PubMed] [Google Scholar]
- (5).Thalgott M; Rack B; Maurer T; Souvatzoglou M; Eiber M; Kreß V; Heck MM; Andergassen U; Nawroth R; Gschwend JE; et al. J. Cancer Res. Clin. Oncol 2013, 139, 755–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Josefsson A; Linder A; Flondell Site D; Canesin G; Stiehm A; Anand A; Bjartell A; Damber JE; Welén K Prostate 2017, 77, 849–858. [DOI] [PubMed] [Google Scholar]
- (7).De Bono JS; Scher HI; Montgomery RB; Parker C; Miller MC; Tissing H; Doyle GV; Terstappen LW; Pienta KJ; Raghavan D Clin. Cancer Res 2008, 14, 6302–6309. [DOI] [PubMed] [Google Scholar]
- (8).Riethdorf S; Fritsche H; Müller V; Rau T; Schindlbeck C; Rack B; Janni W; Coith C; Beck K; Jänicke F; et al. Clin. Cancer Res 2007, 13, 920–928. [DOI] [PubMed] [Google Scholar]
- (9).Cohen SJ; Punt CJ; Iannotti N; Saidman BH; Sabbath KD; Gabrail NY; Picus J; Morse M; Mitchell E; Miller MC J. Clin. Oncol 2008, 26, 3213–3221. [DOI] [PubMed] [Google Scholar]
- (10).Schilling D; Todenhöfer T; Hennenlotter J; Schwentner C; Fehm T; Stenzl A Nat. Rev. Urol 2012, 9, 448–463. [DOI] [PubMed] [Google Scholar]
- (11).Lowes LE; Goodale D; Xia Y; Postenka C; Piaseczny MM; Paczkowski F; Allan AL Oncotarget 2016, 7, 76125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Rycaj K; Tang DG In Cancer Stem Cells; Lathia JD, Ed.; Academic Press: Boston, 2016; pp 317–340. [Google Scholar]
- (13).Punnoose EA; Atwal SK; Spoerke JM; Savage H; Pandita A; Yeh R-F; Pirzkall A; Fine BM; Amler LC; Chen DS; et al. PLoS One 2010, 5, No. e12517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Maheswaran S; Sequist LV; Nagrath S; Ulkus L; Brannigan B; Collura CV; Inserra E; Diederichs S; Iafrate AJ; Bell DW; et al. N. Engl. J. Med 2008, 359, 366–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Nagrath S; Sequist LV; Maheswaran S; Bell DW; Irimia D; Ulkus L; Smith MR; Kwak EL; Digumarthy S; Muzikansky A; et al. Nature 2007, 450, 1235–1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Shim S; Stemke-Hale K; Noshari J; Becker FF; Gascoyne PR Biomicrofluidics 2013, 7, 011808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Shim S; Stemke-Hale K; Tsimberidou AM; Noshari J; Anderson TE; Gascoyne PR Biomicrofluidics 2013, 7, 011807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Moon H-S; Kwon K; Kim S-I; Han H; Sohn J; Lee S; Jung H-I Lab Chip 2011, 11, 1118–1125. [DOI] [PubMed] [Google Scholar]
- (19).Li P; Mao Z; Peng Z; Zhou L; Chen Y; Huang P-H; Truica CI; Drabick JJ; El-Deiry WS; Dao M; et al. Proc. Natl. Acad. Sci. U.S. A 2015, 112, 4970–4975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Chen Y; Li P; Huang P-H; Xie Y; Mai JD; Wang L; Nguyen N-T; Huang TJ Lab Chip 2014, 14, 626–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Qian W; Zhang Y; Chen W Small 2015, 11, 3850–3872. [DOI] [PubMed] [Google Scholar]
- (22).Wu J; Chen Q; Lin J-M Analyst 2017, 142, 421–441. [DOI] [PubMed] [Google Scholar]
- (23).Guo Q; Duffy SP; Ma H In Microtechnology for Cell Manipulation and Sorting; Springer, 2017; pp 225–254. [Google Scholar]
- (24).Huang L; Bian S; Cheng Y; Shi G; Liu P; Ye X; Wang W Biomicrofluidics 2017, 11, 011501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Nora Dickson M; Tsinberg P; Tang Z; Bischoff FZ; Wilson T; Leonard EF Biomicrofluidics 2011, 5, 034119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Fan X; Jia C; Yang J; Li G; Mao H; Jin Q; Zhao J Biosens. Bioelectron 2015, 71, 380–386. [DOI] [PubMed] [Google Scholar]
- (27).Shen F-M; Zhu L; Ye H; Yang Y-J; Pang D-W; Zhang Z-L Sci. Rep 2015, 5, 11937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Zheng S; Lin HK; Lu B; Williams A; Datar R; Cote RJ; Tai Y-C Biomed. Microdevices 2011, 13, 203–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Sarioglu AF; Aceto N; Kojic N; Donaldson MC; Zeinali M; Hamza B; Engstrom A; Zhu H; Sundaresan TK; Miyamoto DT; et al. Nat. Methods 2015, 12, 685–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Riahi R; Gogoi P; Sepehri S; Zhou Y; Handique K; Godsey J; Wang Y Int. J. Oncol 2014, 44, 1870–1878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Hur SC; Mach AJ; Di Carlo D Biomicrofluidics 2011, 5, 022206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Lee W; Tseng P; Di Carlo D In Microtechnology for Cell Manipulation and Sorting, Lee W, Tseng P, Di Carlo D, Eds.; Springer International Publishing, 2017; pp 1–14. [Google Scholar]
- (33).Renier C; Pao E; Che J; Liu HE; Lemaire CA; Matsumoto M; Triboulet M; Srivinas S; Jeffrey SS; Rettig M; et al. NPJ. Precis. Oncol 2017, 1, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Haddadi H; Naghsh-Nilchi H; Di Carlo D Biomicrofluidics 2018, 12, 014112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Stott SL; Hsu C-H; Tsukrov DI; Yu M; Miyamoto DT; Waltman BA; Rothenberg SM; Shah AM; Smas ME; Korir GK; et al. Proc. Natl. Acad. Sci. U. S. A 2010, 107, 18392–18397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Yu M; Ting DT; Stott SL; Wittner BS; Ozsolak F; Paul S; Ciciliano JC; Smas ME; Winokur D; Gilman AJ; et al. Nature 2012, 487, 510–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Ozkumur E; Shah AM; Ciciliano JC; Emmink BL; Miyamoto DT; Brachtel E; Yu M; Chen P.-i.; Morgan B; Trautwein J; et al. Sci. Transl. Med 2013, 5, 179ra47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Gleghorn JP; Pratt ED; Denning D; Liu H; Bander NH; Tagawa ST; Nanus DM; Giannakakou PA; Kirby BJ Lab Chip 2010, 10, 27–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Dharmasiri U; Njoroge SK; Witek MA; Adebiyi MG; Kamande JW; Hupert ML; Barany F; Soper SA Anal. Chem 2011, 83, 2301–2309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Jackson JM; Witek MA; Hupert ML; Brady C; Pullagurla S; Kamande J; Aufforth RD; Tignanelli CJ; Torphy RJ; Yeh JJ; et al. Lab Chip 2014, 14, 106–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Dharmasiri U; Balamurugan S; Adams AA; Okagbare PI; Obubuafo A; Soper SA Electrophoresis 2009, 30, 3289–3300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Adams AA; Okagbare PI; Feng J; Hupert ML; Patterson D; Göttert J; McCarley RL; Nikitopoulos D; Murphy MC; Soper SA J. Am. Chem. Soc 2008, 130, 8633–8641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Sheng W; Chen T; Kamath R; Xiong X; Tan W; Fan ZH Anal. Chem 2012, 84, 4199–4206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Sheng W; Ogunwobi OO; Chen T; Zhang J; George TJ; Liu C; Fan ZH Lab Chip 2014, 14, 89–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Issadore D; Chung J; Shao H; Liong M; Ghazani AA; Castro CM; Weissleder R; Lee H Sci. Transl. Med 2012, 4, 141ra92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Van der Auwera I; Peeters D; Benoy I; Elst H; Van Laere S; Prove A; Maes H; Huget P; Van Dam P; Vermeulen P; et al. Br. J. Cancer 2010, 102, 276–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Peeters D; De Laere B; Van den Eynden G; Van Laere S; Rothé F; Ignatiadis M; Sieuwerts A; Lambrechts D; Rutten A; Van Dam P; et al. Br. J. Cancer 2013, 108, 1358–1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Miller MC; Doyle GV; Terstappen LW J. Oncol 2010, 2010, 617421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Schwed Lustgarten DE; Thompson J; Yu G; Vachani A; Vaidya B; Rao C; Connelly M; Udine M; Tan KS; Heitjan DF; et al. Ann. Am. Thorac. Soc 2013, 10, 582–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Au SH; Storey BD; Moore JC; Tang Q; Chen Y-L; Javaid S; Sarioglu AF; Sullivan R; Madden MW; O’Keefe R; et al. Proc. Natl. Acad. Sci. U. S. A 2016, 113, 4947–4952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Lin HK; Zheng S; Williams AJ; Balic M; Groshen S; Scher HI; Fleisher M; Stadler W; Datar RH; Tai Y-C; et al. Clin. Cancer Res 2010, 16, 5011–5018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Tan SJ; Yobas L; Lee GYH; Ong CN; Lim CT Biomed. Microdevices 2009, 11, 883–892. [DOI] [PubMed] [Google Scholar]
- (53).Lim LS; Hu M; Huang MC; Cheong WC; Gan ATL; Looi XL; Leong SM; Koay ES-C; Li M-H Lab Chip 2012, 12, 4388–4396. [DOI] [PubMed] [Google Scholar]
- (54).Bagnall JS; Byun S; Begum S; Miyamoto DT; Hecht VC; Maheswaran S; Stott SL; Toner M; Hynes RO; Manalis SR Sci. Rep 2016, 5, 18542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Bagnall JS; Byun S; Miyamoto DT; Kang JH; Maheswaran S; Stott SL; Toner M; Manalis SR Integr. Biol 2016, 8, 654–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Grover WH; Bryan AK; Diez-Silva M; Suresh S; Higgins JM; Manalis SR Proc. Natl. Acad. Sci. U. S. A 2011, 108, 10992–10996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Lee A; Park J; Lim M; Sunkara V; Kim SY; Kim GH; Kim M-H; Cho Y-K Anal. Chem 2014, 86, 11349–11356. [DOI] [PubMed] [Google Scholar]
- (58).Kim T-H; Lim M; Park J; Oh JM; Kim H; Jeong H; Lee SJ; Park HC; Jung S; Kim BC; et al. Anal. Chem 2017, 89, 1155–1162. [DOI] [PubMed] [Google Scholar]
- (59).Cruz I; Ciudad J; Ramos M; Gómez-Alonso A; Adansa JC; Rodríguez C; Orfao A; Cruz JJ Am. J. Clin. Pathol 2005, 123, 66–74. [DOI] [PubMed] [Google Scholar]
- (60).Di Carlo D J. Lab. Autom 2012, 17, 32–42. [DOI] [PubMed] [Google Scholar]
- (61).Sollier E; Go DE; Che J; Gossett DR; O’Byrne S; Weaver WM; Kummer N; Rettig M; Goldman J; Nickols N; et al. Lab Chip 2014, 14, 63–77. [DOI] [PubMed] [Google Scholar]
- (62).Paris PL; Kobayashi Y; Zhao Q; Zeng W; Sridharan S; Fan T; Adler HL; Yera ER; Zarrabi M; Zucker S; et al. Cancer Lett 2009, 277, 164–173. [DOI] [PubMed] [Google Scholar]
- (63).Park S; Ang RR; Duffy SP; Bazov J; Chi KN; Black PC; Ma H PLoS One 2014, 9, No. e85264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Ren X; Ghassemi P; Babahosseini H; Strobl J; Agah M ACS Sens 2017, 2, 290–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Ren X; Kumbur EC; Zhou JG; Noh M; Chong PL-G J. Membr. Sci 2017, 540, 27–34. [Google Scholar]
- (66).Babahosseini H; Srinivasaraghavan V; Zhao Z; Gillam F; Childress E; Strobl JS; Santos WL; Zhang C; Agah M Lab Chip 2016, 16, 188–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Ren X; Lu H; Zhou JG; Chong PL-G; Yuan W; Noh MJ Microelectromech. Syst 2017, 26, 120–126. [Google Scholar]
- (68).Mohamed H; Murray M; Turner JN; Caggana MJ Chromatogr. A 2009, 1216, 8289–8295. [DOI] [PubMed] [Google Scholar]
- (69).Mohamed H; McCurdy LD; Szarowski DH; Duva S; Turner JN; Caggana M IEEE Trans.Nanobioscience 2004, 3, 251–256. [DOI] [PubMed] [Google Scholar]
- (70).Babahosseini H; Ketene AN; Schmelz EM; Roberts PC; Agah M Nanomedicine 2014, 10, e1013–e1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (71).Babahosseini H; Strobl JS; Agah M Nanotechnology 2015, 26, 354004. [DOI] [PubMed] [Google Scholar]
- (72).Zheng S; Lin H; Liu J-Q; Balic M; Datar R; Cote RJ; Tai Y-CJ Chromatogr. A 2007, 1162, 154–161. [DOI] [PubMed] [Google Scholar]
- (73).Vona G; Sabile A; Louha M; Sitruk V; Romana S; Schütze K; Capron F; Franco D; Pazzagli M; Vekemans M; et al. Am. J. Pathol 2000, 156, 57–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (74).Tang Y; Shi J; Li S; Wang L; Cayre YE; Chen Y Sci. Rep 2015, 4, 06052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (75).Yoon Y; Lee J; Yoo K-C; Sul O; Lee S-J; Lee S-B Micromachines 2018, 9, 106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (76).Dong Y; Skelley AM; Merdek KD; Sprott KM; Jiang C; Pierceall WE; Lin J; Stocum M; Carney WP; Smirnov DA J. Mol. Diagn 2013, 15, 149–157. [DOI] [PubMed] [Google Scholar]
- (77).Allard WJ; Matera J; Miller MC; Repollet M; Connelly MC; Rao C; Tibbe AG; Uhr JW; Terstappen LW Clin. Cancer Res 2004, 10, 6897–6904. [DOI] [PubMed] [Google Scholar]
- (78).Wan Y; Tan J; Asghar W; Kim Y -t.; Liu, Y.; Iqbal, S. M. J. Phys. Chem. B 2011, 115, 13891–13896. [DOI] [PubMed] [Google Scholar]
- (79).Krombach F; Münzing S; Allmeling A-M; Gerlach JT; Behr J; Dörger M Environ. Health Perspect 1997, 105, 1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.