

Abstract
Glucocorticoids in excess suppress osteoblast function and cause osteoporosis. We demonstrated that cortisol induces the expression of selected Notch receptors in osteoblasts, revealing a potential mechanism for the skeletal effects of glucocorticoids. However, it remains to be determined whether increased expression of Notch receptors results into enhanced signaling. Following activation of Notch, its intracellular domain (NICD) binds to the DNA-associated protein recombination signal binding protein for immunoglobulin kappa-J region (RBPJ) and induces the expression of target genes such as Hey1, Hey2 and HeyL. To determine whether glucocorticoids modulate Notch signaling in the skeleton, 1 month old wild-type mice were administered prednisolone or placebo and sacrificed after 72 h, and gene expression was analyzed in femoral bone. Prednisolone induced Tsc22d3, a glucocorticoid target gene, and suppressed Hey1 and HeyL expression, which is indicative of inhibited Notch receptor activity or direct Hey downregulation. To determine the mechanisms of Hey suppression, wild-type osteoblast-enriched cells were seeded on the Notch cognate ligand Delta-like (DLL)1 or transfected with constructs expressing the NOTCH1 NICD fragment and exposed to either cortisol or vehicle. Cortisol opposed the induction of mRNA and heterogeneous nuclear RNA for Hey1, Hey2 and HeyL by DLL1, but had no effect on mRNA stability, indicating that glucocorticoids inhibit Hey expression by transcriptional mechanisms. Transactivation studies and electrophoretic mobility shift assays revealed that cortisol did not oppose RBPJ-mediated transcription or RBPJ/DNA interactions, respectively. In conclusion, glucocorticoids suppress expression of Hey1, Hey2 and HeyL in osteoblasts by RBPJ-independent transcriptional mechanisms.
Keywords: Glucocorticoids, Hey, RBPJ, Osteoblast, Osteocyte
Osteoblasts derive from mesenchymal precursors and mature osteoblasts can differentiate further into bone lining cells or may become embedded in the newly mineralized bone matrix as osteocytes [Bianco and Gehron, 2000; Dallas et al., 2013]. Osteoblasts regulate bone formation, and in conjunction with osteocytes, govern osteoclast differentiation and bone resorption by secreting receptor activator of nuclear factor kappa-B ligand (RANKL) and its decoy receptor osteoprotegerin [Lacey et al., 1998]. Bone is continuously remodeled by the coordinated activities of osteoblasts and osteoclasts; this process ensures the renewal of bone tissue and allows the regulation of calcium metabolism [Canalis et al., 2007a]. The endocrine system is a central regulator of bone remodeling and multiple hormones influence osteoblast and osteoclast differentiation, function and lifespan.
Glucocorticoids are steroid hormones that bind to the glucocorticoid receptor and are necessary for osteoblastic function and bone homeostasis [Henneicke et al., 2014]. Glucocorticoids in excess, however, induce a transient increase in bone resorption caused by enhanced RANKL expression and by the suppression of osteoprotegerin. The bone resorptive response is followed by a decrease in bone formation due in part to a suppression of osteoblast differentiation and function, and the induction of osteoblast apoptosis. The effects of glucocorticoids on osteoblast differentiation and function and on the RANKL/osteoprotegerin axis are considered prominent mechanisms driving glucocorticoid-induced osteoporosis [Mazziotti et al., 2006]. Interactions of glucocorticoid signaling with the transcription factors peroxisome proliferator-activated receptor (PPAR)-γ and CCAAT-enhancer binding protein (CEBP)-β are believed to stimulate the differentiation of bone marrow precursor cells toward the adipocytic lineage [Rosen and Bouxsein, 2006]. As a consequence, osteoblastogenesis might be suppressed. The downregulation of insulin-like growth factor and Wnt/β-catenin signaling by glucocorticoids further contributes to the inhibitory effects of glucocorticoids on bone formation [Canalis et al., 2007b].
The Notch receptors and cognate ligands of the Delta-like (Dll) and Jagged (Jag) families are single-pass transmembrane proteins that relay signals between adjoining cells [Kopan and Ilagan, 2009]. Receptor-ligand interactions lead to the cleavage of Notch and the release of its intracellular domain (NICD). The latter translocates to the nucleus and associates with a DNA-associated protein termed recombination signal binding protein for immunoglobulin kappa J region (RBPJ), leading to the formation of an active transcriptional complex [Kovall, 2007]. Classical target genes induced by these events are Hey1, Hey2 and HeyL, which encode for closely related transcriptional repressors [Iso et al., 2003]. NOTCH1 and NOTCH2 regulate bone remodeling and their actions, like those of glucocorticoids, are determined by the degree of osteoblast maturity [Zanotti and Canalis, 2016]. Activation of NOTCH1 or NOTCH2 in osteoblasts suppresses cancellous bone formation, whereas activation of NOTCH1 in osteocytes causes osteopetrosis due to the induction of osteoprotegerin and the inhibition of osteoclastogenesis [Canalis et al., 2013a; Canalis et al., 2015; Canalis et al., 2013b; Canalis et al., 2016; Hilton et al., 2008; Yorgan et al., 2016; Zanotti et al., 2011; Zanotti et al., 2008; Zanotti et al., 2017].
Previous work demonstrated that cortisol induces Notch1 and Notch2 transcripts in the MC3T3 osteoblastic cell line, indicating that Notch signaling may contribute to the inhibitory effects of glucocorticoids on bone formation [Pereira et al., 2002]. In support of this possibility, overexpression of the NOTCH1 NICD in MC3T3 cells synergizes with cortisol to favor the acquisition of an adipocytic phenotype at the expense of osteoblastic differentiation by enhancing the expression of PPAR-γ and CEBP-β [Sciaudone et al., 2003]. However, it was not established whether the reported effects of cortisol translate into enhanced Notch receptor activation. In the present study, we sought to determine whether glucocorticoids regulate Notch signaling in osteocytes as well as in osteoblasts and explored the mechanisms involved.
MATERIALS AND METHODS
Prednisolone Administration
To test the effects of glucocorticoids on Notch signaling in bone cells, pellets containing 5 mg of prednisolone (Innovative Research of America, Sarasota, FL), released over a 21-day period, were implanted subcutaneously in 1 month old wild-type C57BL/6J male mice (Jackson Laboratory, Bar Harbor, ME). Pellets containing 5 mg of placebo were implanted in littermates of the same sex and served as controls. Mice were sacrificed 72 h after implantation, which was equivalent to the administration of ~700 μg of prednisolone or placebo per mouse. Femurs were dissected, the epiphyses excised, the bone marrow removed by centrifugation and the remaining bone fragments frozen in liquid nitrogen and stored at −80°C.
Osteoblast-enriched Cultures
Parietal bones of 3 to 5 day old C57BL/6J wild-type mice were digested at 37ºC with either Liberase TL 1.2 U/ml (Sigma-Aldrich, St. Louis, MO) or type-II collagenase (Worthington Biochemical, Lake View, NJ) pretreated with N-α-tosyl-L-lysyl-chloromethylketone hydrochloride (Calbiochem, La Jolla, CA), for 20 min at 37°C and cells extracted in 5 sequential digestions [Yesil et al., 2009]. Cells extracted from the last 3 digestions were pooled and cultured in Dulbecco’s modified Eagle’s medium supplemented with non-essential amino acids (both from Thermo Fisher Scientific, Waltham, MA), 20 mM HEPES, 100 μg/ml ascorbic acid (both from Sigma-Aldrich) and heat-inactivated 10% fetal bovine serum (Atlanta Biologicals, Flowery Branch, GA) in a humidified 5% CO2 incubator at 37ºC. To activate Notch receptors, osteoblasts were trypsinized and seeded at a density of 22,000 cells/cm2 on a substrate coated with a DLL1-IgG2A fragment crystallizable recombinant fusion protein (referred to as DLL1, R&D Systems, Minneapolis, MN) at a concentration of 125 ng/cm2, as described [Nobta et al., 2005]. Cells seeded on an equimolar amount of bovine serum albumin (BSA, Sigma-Aldrich) served as controls [Hsu et al., 1999]. Cultures were allowed to recover for 24 h, serum-deprived for 24 h and subsequently exposed to cortisol 1 μM or ethanol, as control vehicle (both from Sigma-Aldrich), for 6 to 24 h. The effects of cortisol on mRNA decay were determined in selected cultures of osteoblasts seeded on DLL1 and exposed to either cortisol or control vehicle, and collected after different times of exposure to the transcriptional inhibitor actinomycin D (Sigma-Aldrich) at a concentration of 5 μg/ml [Chen et al., 2008; Rydziel et al., 2004]. To establish slopes of mRNA decay, percent of corrected gene expression before exposure to actinomycin D was transformed by a base 10 logarithmic function and fitted against time by linear regression. Experimental protocols were approved by the Institutional Animal Care and Use Committees of UConn Health.
RNA Integrity and Quantitative Reverse Transcription-polymerase Chain Reaction (qRT-PCR)
Femoral bone and osteocyte-enriched bone fragments were homogenized prior to chloroform/phenol extraction of total RNA with the micro RNeasy kit, whereas total RNA from osteoblast-enriched cells was extracted with the RNeasy kit (both from Qiagen, Valencia, CA). Integrity of the RNA was assessed by microfluidic electrophoresis on an Experion system (Bio-Rad, Hercules, CA), and gene expression measured by qRT-PCR in RNA of high quality, as defined by an RNA quality indicator number of 7.0 or higher [Nazarenko et al., 2002a; Nazarenko et al., 2002b]. Equal amounts of RNA were reverse-transcribed using the iScript RT-PCR kit (Bio-Rad) according to manufacturer’s instructions, and amplified in the presence of specific primers (all from Integrated DNA Technologies (IDT), Coralville, IA; Table 1) and iQ or Sso Advanced Universal SYBR Green Supermix (Bio-Rad) at 60°C for 35 cycles. Transcript copy number was estimated by comparison to a serial dilution of cDNA for Hey1 and Hey2 (T. Iso, Los Angeles, CA), HeyL (D. Srivastava, Dallas, TX), Rpl38 (American Type Tissue Culture Collection, Manassas, VA) and Tsc22d3 (GE Healthcare Dharmacon, Lafayette, CO) [Iso et al., 2001a; Nakagawa et al., 1999].
Table 1. Primers for qRT-PCR.
Forward (Fwd) and reverse (Rev) primers used for detection of mRNA and hnRNA by qRT-PCR. GenBank accession numbers indicate transcript variants with homologous sequences to primers.
| Gene | RNA species |
Strand | Primer Sequence 5’ to 3’ | GenBank Accession Numbers |
|---|---|---|---|---|
| Hey1 | mRNA | Fwd Rev |
ATCTCAACAACTACGCATCCCAGC GTGTGGGTGATGTCCGAAGG |
NM_010423 |
| hnRNA | Fwd Rev |
CCCAACGACATCGTCCCAGGTTT GGGTACTCACGCCTCTCCGTCTTT |
||
| Hey2 | mRNA | Fwd Rev |
AGCGAGAACAATTACCCTGGGCAC GGTAGTTGTCGGTGAATTGGACCT |
NM_013904 |
| hnRNA | Fwd Rev |
CGCCCTTGTGAGGAAACG CGCTCACCCAGGGTAATTG |
||
| HeyL | mRNA | Fwd Rev |
CAGTAGCCTTTCTGAATTGCGAC CCCAGCACAACTCCTCCCTA |
NM_013905 |
| hnRNA | Fwd Rev |
GAGAACGATCTTAGGTAAGACAGG AACTTGAGTGTGCTGGACTT |
||
| Rpl38 | mRNA | Fwd Rev |
AGAACAAGGATAATGTGAAGTTCAAGGTTC CTGCTTCAGCTTCTCTGCCCTTT |
NM_001048057 NM_023372 NM_001048058 |
| Tsc22d3 | mRNA | Fwd Rev |
GCGTCATCTCATGTGGAGAACTT CTCTTGGCACCAGCTATGTTAGG |
NM_010286 NM_001077364 |
To determine the effects of cortisol on Hey1, Hey2 and HeyL transcription, expression of the respective heterogeneous nuclear (hn)RNA was measured in reverse-transcribed RNA with specific primers (IDT; Table 1) and Sso Advanced Universal SYBR Green Supermix (Bio-Rad) at 60°C for 40 cycles. Synthetic DNA fragments (IDT) homologous to the ~100 bp spanning the exon2-intron2 junction of Hey1, or the exon1-intron1 junctions of Hey2 or HeyL were cloned into pcDNA3.1(−) (Thermo Fisher Scientific) by isothermal single reaction assembly (New England Biolabs, Ipswich, MA). Serial dilutions of these constructs were used to estimate copy number of the hnRNA [Gibson et al., 2009]. Reactions were conducted in a CFX96 qRT-PCR detection system (Bio-Rad), and fluorescence was monitored during every PCR cycle at the annealing step. Presence of a single PCR product was documented by melting curve analysis carried out after the final amplification cycle. Data are expressed as copy number corrected for Rpl38 [Tso et al., 1985].
Transient Transfections
To monitor RBPJ-mediated transcription, osteoblast-enriched cells were transfected with a plasmid containing 6 dimeric consensus sequences for RBPJ, previously referred to as core binding factor-1/suppressor of hairless/Lag-1 (CSL), cloned upstream of the β-globin basal promoter and Luciferase (12xCSL-Luc; L.J. Strobl, Munich, Germany) [Strobl et al., 1997]. The pGL2 plasmid alone was transfected to measure basal or non-specific expression of Luciferase. To induce Notch signaling, cells were either seeded on DLL1 or co-transfected with a 2.4 kilobase (kb) DNA fragment containing the coding sequence of the murine NOTCH1 NICD (J.S. Nye, New York, NY) cloned into pcDNA3.1(−) downstream of the cytomegalovirus (CMV) promoter (pcDNA-NICD) [Nye et al., 1994]. BSA or a pcDNA3.1(−) plasmid served as controls. Transfections were conducted in 70% confluent osteoblast-enriched cells with X-tremeGENE 9 (2 μl X-tremeGENE 9/1 μg DNA), according to manufacturer’s instructions (Roche, Indianapolis, IN). A plasmid where the CMV promoter directs the expression of β-galactosidase (Clontech, Mountain View, CA) was co-transfected to account for transfection efficiency. Cells were exposed to the X-tremeGENE 9-DNA mix for 16 h, transferred to DMEM without serum for 6 h, and exposed to cortisol 1 μM or vehicle for 24 h. Cells were harvested in reporter lysis buffer (Promega) and lysed by freezing and thawing. Luciferase and β-galactosidase activity were measured with a Luciferase Assay System kit (Promega) and with galacton plus (Life Technologies, Grand Island, NY), respectively, in accordance with manufacturers’ instructions on an Optocomp II luminometer (MGM Instruments, Hamden, CT).
Electrophoretic Mobility Shift Assay (EMSA)
Osteoblast-enriched cells were seeded on BSA or DLL1, allowed to achieve confluence, cultured for 24 h in the absence of serum, exposed to cortisol 1 μM or vehicle for 6 h, and nuclear extracts obtained as described [Schreiber et al., 1989]. A double-stranded DNA oligonucleotide biotinylated at the 5’-end and containing an RBPJ consensus sequence found in the EBNA2 promoter (forward strand: 5’-GGAAACACGCCGTGGGAAAAAATTTGGG-3’) was synthesized commercially (IDT) [Henkel et al., 1994]. Binding reactions of 1 μM biotinylated DNA with 5 μg of nuclear extracts were carried out with the LightShift Chemiluminescent EMSA Kit, as recommended by the manufacturer (Thermo Fisher Scientific). To determine the specificity of the binding reactions, unlabeled homologous DNA (IDT) was added in 200-fold excess. Nucleic acid/protein complexes were resolved on non-denaturing, non-reducing 4% polyacrylamide gels (Bio-Rad) for 45 min and then transferred to a nylon membrane with a 0.45 μm pore size (MP Biomedicals, Solon, OH) in 45 mM Tris, 45 mM boric acid (both from Sigma-Aldrich), 1 mM EDTA at pH 8.3. Transfer was conducted for 30 min at 4°C, and complexes were cross-linked to the membrane by 1 min exposure to 120 mJ/cm2 UV-light in a CL-1000 instrument (UVP, Upland, CA). The immobilized biotinylated DNA was detected with a streptavidin-horseradish peroxidase conjugate with the LightShift Chemiluminescent EMSA Kit detection module in accordance to manufacturers’ instructions and chemiluminescent images were acquired with a Chemidoc XSR molecular imager (Bio-Rad).
Statistical Analysis
Data are expressed as means ± standard deviation (SD). Statistical differences were determined by Student’s t-test or two-way analysis of variance (ANOVA) with Holm-Šídák post-hoc analysis for pairwise or multiple comparisons, respectively. Differences in the slopes of mRNA decay were tested by analysis of covariance (ANCOVA) [Sokal and Rohlf, 1981].
RESULTS
Prednisolone Suppresses the Expression of Notch Target Genes In Vivo
To establish whether glucocorticoids regulate Notch signaling in cells of the osteoblastic lineage in vivo, wild-type male mice were administered prednisolone pellets or placebo for 72 h, and the expression of Notch target genes was measured in total RNA from femoral bone fragments. Prednisolone induced the expression of Tsc22d3, a glucocorticoid target gene, and suppressed the transcript levels of Hey1 and HeyL, indicating that glucocorticoids suppress Notch target genes in osteoblastic cells in vivo (Figure 1) [D’Adamio et al., 1997]. A tendency toward reduced Hey2 expression also was noted in mice administered prednisolone, but the effect did not achieve statistical significance.
Figure 1. Prednisolone suppresses expression of Hey1 and HeyL in bone.
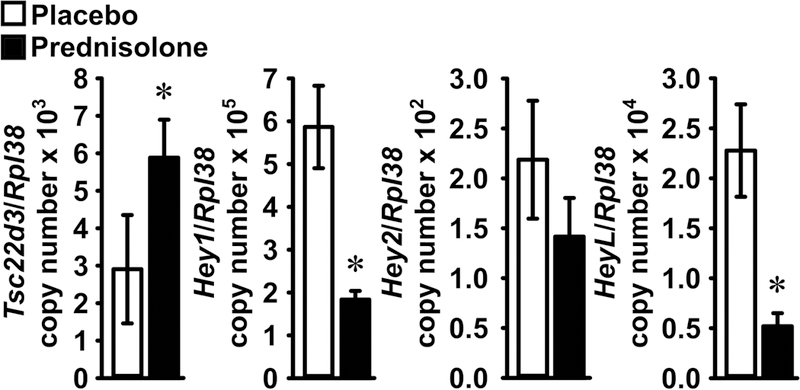
Pellets containing placebo (white bars) or prednisolone (black bars) were implanted in 1 month-old wild-type C57BL/6J male mice. Mice were sacrificed after 72 h and total RNA extracted from femoral bone fragment. Transcript levels were measured by qRT-PCR and data are presented as Tsc22d3, Hey1, Hey2 and HeyL copy number corrected for Rpl38 expression. Values are means ± SD, n = 4 for placebo; n = 3 for prednisolone. *Significant difference, p < 0.05 by unpaired Student’s t-test.
Glucocorticoids Suppress the Expression of Notch Target Genes in Osteoblasts
To test whether the inhibition of Hey expression occurs in osteoblasts, cultures enriched in osteoblasts were obtained from C57BL/6J mice and carried out on a DLL1-coated substrate to activate the Notch receptors, or on BSA as a control. Cells were subsequently exposed to cortisol or vehicle. Cortisol increased the expression of its target Tsc22d3 either in the presence of DLL1 or under basal conditions, and DLL1 induced Hey1, Hey2 and HeyL, confirming activation of Notch signaling. Cortisol opposed the induction of Hey1, Hey2 and HeyL but had a non-significant effect on their basal expression (Figure 2).
Figure 2. Cortisol prevents the induction Hey1, Hey2 and HeyL by DLL1 in osteoblasts.
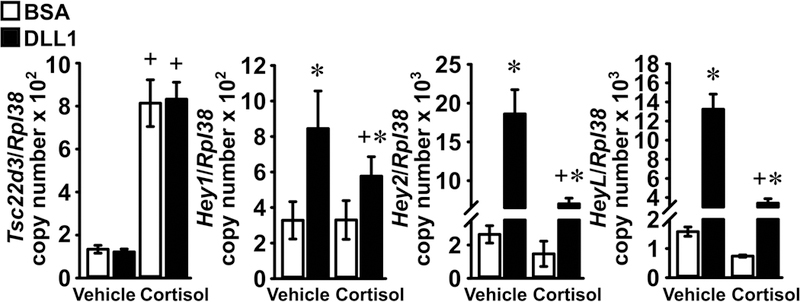
Primary osteoblast-enriched cells from the parietal bones of 3 to 5 day old wild-type C57BL/6J mice were seeded on BSA (white bars) or DLL1 (black bars). Cells were allowed to recover for 24 h, subsequently cultured for 24 h in the absence of serum and then exposed to vehicle or cortisol 1 μM for 6 h. Total RNA was extracted and mRNA levels quantified by qRT-PCR; data are expressed as Hey1, Hey2 and HeyL copy number corrected for Rpl38 expression. Values are means ± SD; n = 4 for all groups and genes with the exception of Hey1 expression in osteoblasts exposed to cortisol and BSA, n = 3. *Significantly different between Control and Notch, p < 0.05; +significantly different between vehicle and cortisol, p < 0.05; two-way ANOVA with Holm-Šídák post-hoc analysis.
Cortisol Represses Hey1, Hey2 and HeyL hnRNA in Osteoblasts
To determine whether glucocorticoids regulate the transcription of Hey1, Hey2 or HeyL, the expression of their respective hnRNA was measured in osteoblast-enriched cells cultured on a DLL1-coated substrate or on control BSA, either in the presence or absence of cortisol. As reported for the effects of DLL1 and cortisol on mRNA expression, DLL1 induced the hnRNA for Hey1, Hey2 and HeyL and cortisol opposed this effect, which is indicative of Notch signaling suppression through transcriptional mechanisms (Figure 3). To establish whether glucocorticoids regulate the stability of Hey1, Hey2 and HeyL transcripts, osteoblast-enriched cells cultured on DLL1-coated substrates were exposed to either cortisol or vehicle and then transcriptionally arrested with actinomycin D. Cortisol had no effect on the half-life of Hey1, Hey2 or HeyL, indicating that the inhibitory effects of cortisol on Notch signaling are primarily transcriptional (Figure 4).
Figure 3. Cortisol suppresses Hey1, Hey2 and HeyL expression in osteoblasts by transcriptional mechanisms.
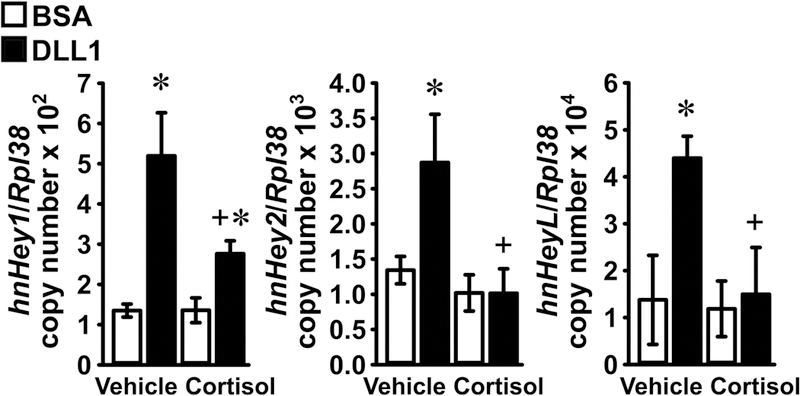
Primary osteoblast-enriched cells from the parietal bones of 3 to 5 day old wild-type C57BL/6J mice were seeded on BSA (white bars) or DLL1 (black bars). Cells were allowed to recover for 24 h, subsequently cultured for 24 h in the absence of serum and then exposed to vehicle or cortisol 1 μM for 6 h. Total RNA was extracted and hnRNA levels quantified by qRT-PCR; data are expressed as Hey1, Hey2 and HeyL hnRNA copy number corrected for Rpl38 expression. Values are means ± SD; n = 4. *Significantly different between Control and Notch, p < 0.05; +significantly different between vehicle and cortisol, p < 0.05; two-way ANOVA with Holm-Šídák post-hoc analysis.
Figure 4. Cortisol does not affect the stability of Hey1, Hey2 or HeyL mRNA.

Primary osteoblast-enriched cells from the parietal bones of 3 to 5 day old wild-type C57BL/6J mice were seeded on DLL1. Cells were allowed to recover for 24 h and subsequently cultured for 24 h in the absence of serum and then exposed to vehicle or cortisol 1 μM for 6 h. Cells were transcriptionally arrested with actinomycin D (Time 0) and harvested at the indicated times. Total RNA was extracted and mRNA levels quantified by qRT-PCR. Data are expressed as percent of Hey1, Hey2 and HeyL mRNA levels corrected for Rpl38 copy number prior to exposure to actinomycin D, plotted versus time. Values are means ± SD; n = 4. No differences between cortisol and control slopes of decay by ANCOVA.
Cortisol does not Inhibit RBPJ-mediated Notch Signaling
To establish whether the suppression of Hey1, Hey2 and HeyL mRNA and hnRNA by cortisol is due to an effect on NICD/RBPJ/DNA interactions, osteoblasts were transiently transfected with the 12xCSL-Luc reporter, which contains multiple consensus sequences for CSL, the human RBPJ ortholog. To activate Notch signaling, cells were seeded on DLL1 or control BSA, or co-transfected with pcDNA-NICD or a pcDNA3.1(−) control vector, and treated with cortisol or vehicle. Exposure to DLL1 or overexpression of the NOTCH1 NICD enhanced the transactivation of 12xCSL-Luc, and cortisol did not oppose these effects (Figure 5). These observations suggest that cortisol does not affect RBPJ-mediated Notch signaling.
Figure 5. Cortisol does not affect RBPJ-mediated Notch signaling in osteoblasts.
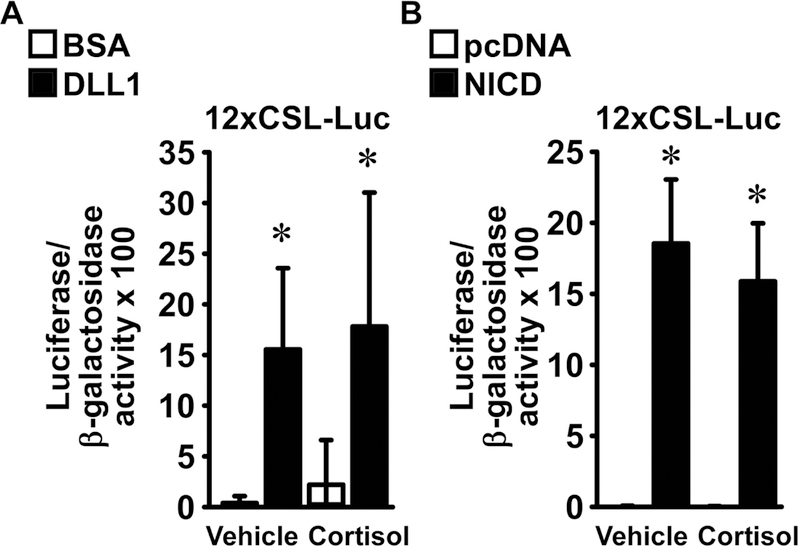
Primary osteoblast-enriched cells were harvested from the parietal bones of 3 to 5 day old wild-type C57BL/6J mice. In panel A, cells were seeded on BSA (white bars) or DLL1 (black bars) and transiently transfected with the 12xCSL-Luc reporter. In panel B, osteoblasts were transiently transfected with pcDNA3.1(−) (pcDNA) or with pcDNA-NICD (NICD) and co-transfected with the 12xCSL-Luc reporter. A CMV/β-galactosidase expression vector was co-transfected to test for transfection efficiency. Cells were allowed to recover for 16 h and subsequently cultured for 6 h in the absence of serum, exposed to vehicle or cortisol 1 μM, and harvested after 24 h. Data represent luciferase/β-galactosidase activity. Values are means ± SD, panel A n = 4; panel B n = 6. *Significantly different between BSA and DLL1 or pcDNA and NICD, p < 0.05; two-way ANOVA with Holm-Šídák post-hoc analysis.
To verify the findings from the transactivation experiments, EMSAs were carried out in osteoblast-enriched cells seeded on DLL1 or on BSA and exposed to cortisol or vehicle. A biotinylated oligonucleotide containing the consensus sequence for RBPJ was bound by nuclear protein extracts from cells cultured on BSA. The interaction was specific as it was competed away by an excess of unlabeled RBPJ oligonucleotides. Previous reports demonstrated that the binding of RBPJ to DNA decreases in the context of Notch activation; accordingly, the formation of a nuclear protein/RBPJ consensus DNA complex was reduced in osteoblasts seeded on DLL1 [Oswald et al., 2001]. Cortisol modestly enhanced the formation of a nuclear protein/DNA complex under basal conditions and had no effect on DNA binding in the context of DLL1 exposure (Figure 6). These results are congruent with observations in cells transfected with the 12xCSL-Luc reporter and demonstrate that glucocorticoids do not inhibit RBPJ/DNA interactions.
Figure 6. Cortisol has a modest impact on RBPJ/DNA interactions.
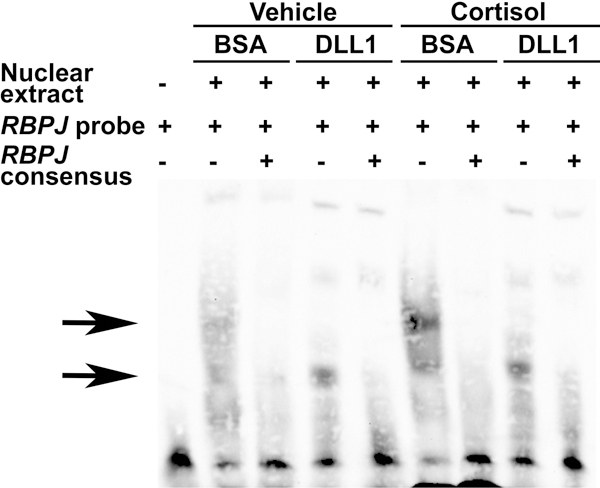
Primary osteoblast-enriched cells were seeded on BSA or DLL1, confluent cultures maintained for 24 h in the absence of serum and then exposed to vehicle or cortisol 1 μM for 6 h. Nuclear proteins were extracted and binding reactions carried out with a biotinylated oligonucleotide containing an RBPJ consensus sequence from the EBNA2 promoter. Competition of binding reactions was performed in the presence of unlabeled oligonucleotides containing homologous RBPJ consensus sequences in 200-fold excess. DNA-nuclear protein complexes were resolved by gel electrophoresis, transferred to a nylon membrane, cross-linked with UV-light, exposed to a streptavidin-horseradish peroxidase conjugate and visualized by chemiluminescence. The arrows indicate the position of the DNA/protein complexes.
DISCUSSION
The studies presented in this manuscript demonstrate that selected glucocorticoids, namely prednisolone and cortisol, suppress the expression of the Notch target genes Hey1, Hey2 and HeyL in osteoblasts. Prednisolone suppressed Hey1 and HeyL expression in the context of basal Notch activation in vivo, whereas stimulation of Notch receptors with DLL1 was required to observe the inhibitory effect of cortisol on Hey expression in osteoblast-enriched cultures. The effects of glucocorticoids are of a transcriptional nature, as cortisol opposed the stimulatory effects of DLL1 on Hey1, Hey2 and HeyL hnRNA levels but did not affect the half-life of the respective mRNA. Transactivation experiments with the 12xCSL-Luc reporter, which contains multiple copies of the consensus sequence for the human ortholog of RBPJ, suggest that cortisol does not suppress Hey transcription by RBPJ-dependent mechanisms. This suggests that the expression of the Hey paralogs may be governed by glucocorticoid response elements (GRE) that oppose the transcriptional response to the Notch receptors. This possibility is substantiated by the observation that prednisolone-and cortisol-induced expression of Tsc22d3, a known target gene of the glucocorticoid receptor that harbors 4 GRE within the 2.5 kb upstream of the transcriptional start site [Wang et al., 2004]. Stimulation of Tsc22d3 is compatible with suppression of Hey1, Hey2 and HeyL as the sequence context of the GRE determines whether glucocorticoid receptors act as inducers or inhibitor of transcription [Wang et al., 2004].
EMSA confirmed that cortisol does not act by precluding RBPJ/DNA interactions. In fact, cortisol enhanced binding of nuclear proteins to the biotinylated RBPJ probes, possibly by increasing the expression of RBPJ or its affinity to cognate consensus sequences. The mechanisms of this effect remain to be determined. Enhanced binding of RBPJ to DNA does not necessarily imply an increase in Notch signaling, as RBPJ acts as a transcriptional inhibitor in the absence of the NICD [Kovall, 2008]. Accordingly, cortisol did not induce the activity of an acutely transfected 12xCSL-Luc reporter construct either in the context of DLL1 stimulation or overexpression of the NOTCH1 NICD [Strobl et al., 1997]. These observations demonstrate that despite the known stimulatory effects of cortisol on Notch1 and Notch2 expression in MC3T3 osteoblastic cell lines, glucocorticoids do not enhance the activity of the Notch receptors in osteoblasts. This is not necessarily unexpected, as cortisol did not appear to regulate the expression of Notch ligands in MC3T3 cells [Pereira et al., 2002].
Previous work in mice demonstrated that the misexpression of Hey1 or the global deletion of HeyL have a minimal impact on skeletal structure, whereas the conditional inactivation of Hey2 in osteoblasts leads to modest gains in bone mass [Canalis and Zanotti, 2017; Salie et al., 2010; Zanotti and Canalis, 2013]. The mild skeletal phenotypes of the individual Hey1, Hey2 or HeyL deletion can be explained by genetic redundancy that may compensate for the loss of a single gene within a group of closely related paralogs. This possibility is substantiated by the ability of Hey proteins to form homodimers as well as heterodimers and thereby determine the specificity of DNA binding [Fischer et al., 2007; Iso et al., 2001b]. Accordingly, global HeyL null mice harboring a heterozygous Hey1 deletion were reported to have increased bone mass, so that increased bone volume would be a plausible outcome of the Hey1, Hey2 and HeyL downregulation [Tu et al., 2012]. Although this postulated effect is difficult to reconcile with the skeletal impact of glucocorticoids in excess, suppression of Hey1, Hey2 and HeyL may contribute to the ability of glucocorticoids to maintain optimal osteoblastic function under physiological conditions [Rauch et al., 2010].
In conclusion, glucocorticoids suppress Hey1, Hey2 and HeyL expression in osteoblasts without inhibiting the activity of the Notch receptors or RBPJ/DNA interactions.
ACKNOWLEDGMENTS
The authors thank T. Iso for Hey1 and Hey2 cDNA, D. Srivastava for HeyL cDNA, L.J. Strobl for 12xCSL-Luc and J.S. Nye for pcDNA-NICD. The authors thank T.L. Eller for technical assistance and M. Yurczak for secretarial support.
Contract grant sponsor: National Institute of Diabetes and Digestive and Kidney Diseases; Contract grant number: DK045227
ABBREVIATIONS
- ANCOVA
analysis of covariance
- ANOVA
analysis of variance
- BSA
bovine serum albumin
- Cebp
CCAAT-enhancer binding protein
- CMV
cytomegalovirus
- CSL
core binding factor-1/suppressor of hairless/Lag-1
- Dll
delta-like
- EMSA
electrophoretic mobility shift assay
- Fwd
forward
- GRE
glucocorticoid response element
- hn
heterogeneous nuclear
- IDT
Integrated DNA Technologies
- Jag
Jagged
- kb
kilobase
- NICD
Notch intracellular domain
- PCR
polymerase chain reaction
- Ppar
peroxisome proliferator-activated receptor
- qRT-PCR
quantitative reverse transcription-PCR
- RANKL
receptor activator of nuclear factor kB ligand
- RBPJ
recombination signal binding protein Jk
- Rev
reverse
- SD
standard deviation
Footnotes
The authors have nothing to disclose.
REFERENCES
- Bianco P, Gehron RP. 2000. Marrow stromal stem cells. J. Clin. Invest. 105:1663–1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canalis E, Adams DJ, Boskey A, Parker K, Kranz L, Zanotti S. 2013a. Notch Signaling in Osteocytes Differentially Regulates Cancellous and Cortical Bone Remodeling. J. Biol. Chem. 288:25614–25625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canalis E, Bridgewater D, Schilling L, Zanotti S. 2015. Canonical Notch Activation in Osteocytes Causes Osteopetrosis. Am J Physiol Endocrinol Metab 310:E171–E182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canalis E, Giustina A, Bilezikian JP. 2007a. Mechanisms of anabolic therapies for osteoporosis. N Engl J Med 357:905–16. [DOI] [PubMed] [Google Scholar]
- Canalis E, Mazziotti G, Giustina A, Bilezikian JP. 2007b. Glucorticoid-Induced Osteoporosis: Pathophysiology and Therapy. Osteoporos. Int. 18:1319–1328. [DOI] [PubMed] [Google Scholar]
- Canalis E, Parker K, Feng JQ, Zanotti S. 2013b. Osteoblast Lineage-specific Effects of Notch Activation in the Skeleton. Endocrinology 154:623–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canalis E, Schilling L, Yee SP, Lee SK, Zanotti S. 2016. Hajdu Cheney Mouse Mutants Exhibit Osteopenia, Increased Osteoclastogenesis and Bone Resorption. J Biol Chem 291:1538–1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canalis E, Zanotti S. 2017. Hairy and Enhancer of Split-Related With YRPW Motif-Like (HeyL) Is Dispensable for Bone Remodeling in Mice. J Cell Biochem 118:1819–1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CY, Ezzeddine N, Shyu AB. 2008. Messenger RNA half-life measurements in mammalian cells. Methods Enzymol 448:335–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Adamio F, Zollo O, Moraca R, Ayroldi E, Bruscoli S, Bartoli A, Cannarile L, Migliorati G, Riccardi C. 1997. A new dexamethasone-induced gene of the leucine zipper family protects T lymphocytes from TCR/CD3-activated cell death. Immunity 7:803–12. [DOI] [PubMed] [Google Scholar]
- Dallas SL, Prideaux M, Bonewald LF. 2013. The osteocyte: an endocrine cell … and more. Endocr. Rev. 34:658–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer A, Steidl C, Wagner TU, Lang E, Jakob PM, Friedl P, Knobeloch KP, Gessler M. 2007. Combined loss of Hey1 and HeyL causes congenital heart defects because of impaired epithelial to mesenchymal transition. Circ. Res. 100:856–863. [DOI] [PubMed] [Google Scholar]
- Gibson DG, Young L, Chuang RY, Venter JC, Hutchison CA 3rd, Smith HO. 2009. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat Methods 6:343–5. [DOI] [PubMed] [Google Scholar]
- Henkel T, Ling PD, Hayward SD, Peterson MG. 1994. Mediation of Epstein-Barr virus EBNA2 transactivation by recombination signal-binding protein J kappa. Science 265:92–5. [DOI] [PubMed] [Google Scholar]
- Henneicke H, Gasparini SJ, Brennan-Speranza TC, Zhou H, Seibel MJ. 2014. Glucocorticoids and bone: local effects and systemic implications. Trends Endocrinol Metab 25:197–211. [DOI] [PubMed] [Google Scholar]
- Hilton MJ, Tu X, Wu X, Bai S, Zhao H, Kobayashi T, Kronenberg HM, Teitelbaum SL, Ross FP, Kopan R, Long F. 2008. Notch signaling maintains bone marrow mesenchymal progenitors by suppressing osteoblast differentiation. Nat Med 14:306–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu H, Lacey DL, Dunstan CR, Solovyev I, Colombero A, Timms E, Tan HL, Elliott G, Kelley MJ, Sarosi I, Wang L, Xia XZ, Elliott R, Chiu L, Black T, Scully S, Capparelli C, Morony S, Shimamoto G, Bass MB, Boyle WJ. 1999. Tumor necrosis factor receptor family member RANK mediates osteoclast differentiation and activation induced by osteoprotegerin ligand. Proc Natl Acad Sci U S A 96:3540–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iso T, Kedes L, Hamamori Y. 2003. HES and HERP families: multiple effectors of the Notch signaling pathway. J Cell Physiol 194:237–55. [DOI] [PubMed] [Google Scholar]
- Iso T, Sartorelli V, Chung G, Shichinohe T, Kedes L, Hamamori Y. 2001a. HERP, a new primary target of Notch regulated by ligand binding. Mol Cell Biol 21:6071–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iso T, Sartorelli V, Poizat C, Iezzi S, Wu HY, Chung G, Kedes L, Hamamori Y. 2001b. HERP, a novel heterodimer partner of HES/E(spl) in Notch signaling. Mol Cell Biol 21:6080–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopan R, Ilagan MX. 2009. The canonical Notch signaling pathway: unfolding the activation mechanism. Cell 137:216–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovall RA. 2007. Structures of CSL, Notch and Mastermind proteins: piecing together an active transcription complex. Curr. Opin. Struct. Biol. 17:117–127. [DOI] [PubMed] [Google Scholar]
- Kovall RA. 2008. More complicated than it looks: assembly of Notch pathway transcription complexes. Oncogene 27:5099–5109. [DOI] [PubMed] [Google Scholar]
- Lacey DL, Timms E, Tan HL, Kelley MJ, Dunstan CR, Burgess T, Elliott R, Colombero A, Elliott G, Scully S, Hsu H, Sullivan J, Hawkins N, Davy E, Capparelli C, Eli A, Qian YX, Kaufman S, Sarosi I, Shalhoub V, Senaldi G, Guo J, Delaney J, Boyle WJ. 1998. Osteoprotegerin ligand is a cytokine that regulates osteoclast differentiation and activation. Cell 93:165–176. [DOI] [PubMed] [Google Scholar]
- Mazziotti G, Angeli A, Bilezikian JP, Canalis E, Giustina A. 2006. Glucocorticoid-induced osteoporosis: an update. Trends Endocrinol. Metab. 17:144–149. [DOI] [PubMed] [Google Scholar]
- Nakagawa O, Nakagawa M, Richardson JA, Olson EN, Srivastava D. 1999. HRT1, HRT2, and HRT3: a new subclass of bHLH transcription factors marking specific cardiac, somitic, and pharyngeal arch segments. Dev. Biol. 216:72–84. [DOI] [PubMed] [Google Scholar]
- Nazarenko I, Lowe B, Darfler M, Ikonomi P, Schuster D, Rashtchian A. 2002a. Multiplex quantitative PCR using self-quenched primers labeled with a single fluorophore. Nucleic Acids Res. 30:e37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nazarenko I, Pires R, Lowe B, Obaidy M, Rashtchian A. 2002b. Effect of primary and secondary structure of oligodeoxyribonucleotides on the fluorescent properties of conjugated dyes. Nucleic Acids Res 30:2089–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nobta M, Tsukazaki T, Shibata Y, Xin C, Moriishi T, Sakano S, Shindo H, Yamaguchi A. 2005. Critical regulation of bone morphogenetic protein-induced osteoblastic differentiation by Delta1/Jagged1-activated Notch1 signaling. J. Cell Biochem. 280:15842–15848. [DOI] [PubMed] [Google Scholar]
- Nye JS, Kopan R, Axel R. 1994. An activated Notch suppresses neurogenesis and myogenesis but not gliogenesis in mammalian cells. Development 120:2421–2430. [DOI] [PubMed] [Google Scholar]
- Oswald F, Tauber B, Dobner T, Bourteele S, Kostezka U, Adler G, Liptay S, Schmid RM. 2001. p300 acts as a transcriptional coactivator for mammalian Notch-1. Mol. Cell Biol. 21:7761–7774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira RM, Delany AM, Durant D, Canalis E. 2002. Cortisol regulates the expression of Notch in osteoblasts. J. Cell Biochem. 85:252–258. [DOI] [PubMed] [Google Scholar]
- Rauch A, Seitz S, Baschant U, Schilling AF, Illing A, Stride B, Kirilov M, Mandic V, Takacz A, Schmidt-Ullrich R, Ostermay S, Schinke T, Spanbroek R, Zaiss MM, Angel PE, Lerner UH, David JP, Reichardt HM, Amling M, Schutz G, Tuckermann JP. 2010. Glucocorticoids suppress bone formation by attenuating osteoblast differentiation via the monomeric glucocorticoid receptor. Cell Metab 11:517–31. [DOI] [PubMed] [Google Scholar]
- Rosen CJ, Bouxsein ML. 2006. Mechanisms of disease: is osteoporosis the obesity of bone? Nat. Clin. Pract. Rheumatol. 2:35–43. [DOI] [PubMed] [Google Scholar]
- Rydziel S, Delany AM, Canalis E. 2004. AU-rich elements in the collagenase 3 mRNA mediate stabilization of the transcript by cortisol in osteoblasts. J. Biol. Chem. 279:5397–5404. [DOI] [PubMed] [Google Scholar]
- Salie R, Kneissel M, Vukevic M, Zamurovic N, Kramer I, Evans G, Gerwin N, Mueller M, Kinzel B, Susa M. 2010. Ubiquitous overexpression of Hey1 transcription factor leads to osteopenia and chondrocyte hypertrophy in bone. Bone 46:680–694. [DOI] [PubMed] [Google Scholar]
- Schreiber E, Matthias P, Muller MM, Schaffner W. 1989. Rapid detection of octamer binding proteins with ‘mini-extracts’, prepared from a small number of cells. Nucleic Acids Res. 17:6419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sciaudone M, Gazzerro E, Priest L, Delany AM, Canalis E. 2003. Notch 1 impairs osteoblastic cell differentiation. Endocrinology 144:5631–5639. [DOI] [PubMed] [Google Scholar]
- Sokal RR, Rohlf FJ. 1981. Biometry, 2nd Editioneditorêditors Biometry, 2nd Edition San Francisco, CA: W. H. Freeman. [Google Scholar]
- Strobl LJ, Hofelmayr H, Stein C, Marschall G, Brielmeier M, Laux G, Bornkamm GW, Zimber-Strobl U. 1997. Both Epstein-Barr viral nuclear antigen 2 (EBNA2) and activated Notch1 transactivate genes by interacting with the cellular protein RBP-J kappa. Immunobiology 198:299–306. [DOI] [PubMed] [Google Scholar]
- Tso JY, Sun XH, Kao TH, Reece KS, Wu R. 1985. Isolation and characterization of rat and human glyceraldehyde-3-phosphate dehydrogenase cDNAs: genomic complexity and molecular evolution of the gene. Nucleic Acids Res. 13:2485–2502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu X, Chen J, Lim J, Karner CM, Lee SY, Heisig J, Wiese C, Surendran K, Kopan R, Gessler M, Long F. 2012. Physiological notch signaling maintains bone homeostasis via RBPjk and Hey upstream of NFATc1. PLoS Genet. 8:e1002577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JC, Derynck MK, Nonaka DF, Khodabakhsh DB, Haqq C, Yamamoto KR. 2004. Chromatin immunoprecipitation (ChIP) scanning identifies primary glucocorticoid receptor target genes. Proc Natl Acad Sci U S A 101:15603–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yesil P, Michel M, Chwalek K, Pedack S, Jany C, Ludwig B, Bornstein SR, Lammert E. 2009. A new collagenase blend increases the number of islets isolated from mouse pancreas. Islets 1:185–90. [DOI] [PubMed] [Google Scholar]
- Yorgan T, Vollersen N, Riedel C, Jeschke A, Peters S, Busse B, Amling M, Schinke T. 2016. Osteoblast-specific Notch2 inactivation causes increased trabecular bone mass at specific sites of the appendicular skeleton. Bone 87:136–146. [DOI] [PubMed] [Google Scholar]
- Zanotti S, Canalis E. 2013. Hairy and Enhancer of Split-related with YRPW Motif (HEY)2 Regulates Bone Remodeling in Mice. J. Biol. Chem. 288:21547–21557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanotti S, Canalis E. 2016. Notch Signaling and the Skeleton. Endocr. Rev. 37:223–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanotti S, Smerdel-Ramoya A, Canalis E. 2011. Reciprocal regulation of Notch and nuclear factor of activated T-cells (NFAT) c1 transactivation in osteoblasts. J Biol Chem 286:4576–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanotti S, Smerdel-Ramoya A, Stadmeyer L, Durant D, Radtke F, Canalis E. 2008. Notch inhibits osteoblast differentiation and causes osteopenia. Endocrinology 149:3890–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanotti S, Yu J, Sanjay A, Schilling L, Schoenherr C, Economides AN, Canalis E. 2017. Sustained Notch2 signaling in osteoblasts, but not in osteoclasts, is linked to osteopenia in a mouse model of Hajdu-Cheney syndrome. J Biol Chem 292:12232–12244. [DOI] [PMC free article] [PubMed] [Google Scholar]