

Abstract
Inflammasome-mediated production of mature IL-1β and IL-18 cytokines represents an important innate immune response against infecting pathogens. Helicobacter pylori, one of the most successful and persistent human pathogens, induces severe inflammation leading to gastritis and more serious gastric diseases. H. pylori modulates different immune responses for its survival and inflammasome signaling is manipulated by the cag pathogenicity island (cagPAI), urease and VacA cytotoxin. Here we report that H. pylori regulates NLRP3 expression, an inflammasome forming regulator, in infected THP-1 monocytes. This response was independent of the major H. pylori pathogenicity-associated factors CagA, VacA, Cgt, FlaA and cagPAI. Two NLRP3 expression controlling factors, the NLRP3 mRNA targeting microRNA hsa-miR-223-3p and cytokine IL-10, were found to work in tandem for its regulation. H. pylori infection also induced copious amount of pro-IL-1β in THP-1 monocytes/macrophages but secreted a very low amount of mature IL-1β. Moreover, secreted IL-10 correlated with the down-regulation of nigericin-induced NLRP3 inflammasome activation of LPS-primed THP-1 monocytes and human PBMCs from volunteers. However, H. pylori-treated PBMCs secreted significantly more mature IL-1β throughout the infection period, which suggests a different mode of activation. Taken together, this study demonstrates targeting of inflammasome-forming NLRP3, an important innate immunity component, and crucial manipulation of pro- and anti-inflammatory cytokines in H. pylori infection.
Keywords: Inflammasome, NLRP3, H. pylori, miRNA, interleukin-1β, interleukin-10
Introduction
Helicobacter pylori is a Gram-negative bacterium colonizing half of the human world population, associated with different gastric pathologies such as gastritis, peptic ulcer, gastric cancer and mucosa associated lymphoid tissue lymphoma. The World Health Organization (WHO) classified H. pylori as a type-I carcinogen. The long history of coexistence with mankind had influenced the H. pylori genome to encode features for adaptation to multiple host characteristics.1 High recombination and mutation rates in the bacterium, along with natural transformation potency, influenced this adaptation process during evolution. A plethora of H. pylori and host factors orchestrate the survival at one of the most hostile environments for microbes, the gastric mucosa.2 Highly virulent H. pylori strains acquired a 40-kb pathogenicity island (cagPAI), which encodes a type IV secretion system to deliver the CagA effector protein into host cells for various signal transduction events.3 The urease enzyme activity helps in the survival of H. pylori by metabolizing urea to release ammonia for neutralizing the surrounding acidic environment. In addition, H. pylori produces the vacuolating toxin A (VacA), exerting different toxicity effects both in epithelial and immune cells. Moreover, H. pylori uses highly motile flagella for crossing the mucus layer during infection, and adhesion proteins such as BabA, SabA, AlpA/B, OipA, HopQ and others ensure adhering to the epithelial cell surface. With the help of these and other pathogenicity factors H. pylori colonizes for almost the entire life span of its human host, if not eradicated by antibiotic therapy.4–6 Most of the individuals harboring H. pylori produce asymptomatic gastritis, about 10–20% of infected people have a lifetime risk of developing peptic ulcer disease and 1–2% risk of developing distal gastric cancer.7 In contrast, persistent infection of H. pylori has also been found to be beneficial for the human host by suppressing other illnesses, including asthma, allergies and inflammatory bowel disease.8–11 Host gene polymorphisms play a crucial role in producing different associated pathologies of H. pylori infection. Among these, a specific IL-1β gene polymorphism has prominence and was directly related to gastric cancer development.12 IL-1β is well-known to act as an inhibitor of acid secretion and leads to gastric atrophy, which ultimately ends in metaplasia and gastric cancer.13 In addition, IL-1β was reported to directly induce proliferation of gastric carcinoma cells.14 Therefore, the production of IL-1β is implicated as one of the key host factors for H. pylori-associated gastric cancer progression.
Many studies proved that the production of mature forms of IL-1β and IL-18 is mediated through multimeric protein complexes called ‘inflammasomes’.15 Nod-like receptors (NLRs), including NLR family, pyrin domain containing 1 (NLRP1), NLRP3 and card domain containing 4 (NLRC4), form the well-studied group of canonical inflammasomes. Absent in melanoma 2 and pyrin/TRIM20 were also identified to assemble inflammasomes during microbial infections.16,17 NLR members like NLRP6, NLRP7 and NLRP12 have also been recently implicated in inflammasome formation.18 The NLRP3 inflammasome is the most studied complex and found to require two major signals for its activation. The first signal usually comes through the detection of PAMPs by toll-like receptors (TLRs) and NF-κB activation to induce the gene expression of NLRP3 and Pro-IL-1β.19 Activation occurs through a second signal induced by various microbial, crystalline, metabolic molecules (e.g. nigericin, urate, silica, ATP) or reactive oxygen species. However, mechanistically all seems to be converged on the effect of K+ efflux and decides the final activation step.20 Once activated, NLRP3 molecules form a ring-like structure, which allows the adaptor protein called apoptosis associated speck-like protein polymerization through their pyrin domain interaction and finally recruits zymogen pro-caspase-1 and undergoes auto-activation. Activated caspase-1 then cleaves the pro-forms of IL-1β and IL-18 to generate their mature forms. However, recent advances in this field showed that caspase-4/5 (caspase-11 in mouse) and caspase-8 were also capable of the cleavage in a NLRP3-dependent mechanism.21,22
Activated caspase-1 and caspase-4/5/11 cleave gasdermin-D, which leads to an inflammatory cell death called ‘pyroptosis’.23 Pyroptosis helps to restrict the growth of intracellular bacteria and is released prematurely, which exposes them to immune attack or naïve cells.24 Massive production of mature IL-1β at the site of acute infection will also lead to pro-inflammatory tissue damage and aggravated disease conditions. However, pathogens causing chronic infections should manage this activation in a specific way to ensure their survival. The regulation of NLRP3 inflammasome in microbial infections is an upcoming area and lacks major studies. Earlier studies using a mouse model of H. pylori infection reported the activation of NLRP3 inflammasome to secrete mature IL-1β and IL-18.10,25,26 However, a recent study using the mouse model of chronic H. pylori infection identified mucin-1 as a regulator of NLRP3 expression and mature IL-1β production.27 The above facts led us to study the NLRP3 expression regulation in H. pylori infected THP-1 monocytic leukemia cell lines, which is the most commonly used model cell line for inflammasome activation studies, as well as human PBMCs. Here we report a novel mechanism of a miRNA (hsa-miR-223-3p) and an anti-inflammatory cytokine (IL-10)-mediated regulation of inflammasome-forming NLRP3 expression and activation in cultured and primary human immune cells during H. pylori infection.
Materials and methods
Bacterial strains and cell line cultures
The H. pylori mutants ΔcagA, ΔvacA, Δcgt, ΔflaA and ΔcagPAI were generated from wild type (wt) strain P12 by insertion of a chloramphenicol or kanamycin resistance gene cassette, respectively.28,29 All H. pylori strains were grown on horse serum agar plates supplemented with vancomycin (10 µg/ml), nystatin (1 µg/ml) and trimethoprim (5 µg/ml), and, if required, with chloramphenicol (4 µg/ml) or kanamycin (8 µg/ml), at 37℃ for 2 d in an anaerobic jar containing a campygen gas mix (Oxoid, Wesel, Germany).30,31 H. pylori grown on agar plates was harvested and re-suspended in PBS (pH 7.4), using a sterile cotton swab (Carl Roth, Karlsruhe, Germany). The bacterial concentration was measured as OD at 550 nm using an Eppendorf spectrophotometer. The eukaryotic cells grown in medium without antibiotics and antimycotics were infected with H. pylori at a multiplicity of infection of 50 or 100.32,33 The mock control cells were incubated with an equal amount of PBS.
THP-1 (ATCC-TIB-202) monocytic leukemia cell line was cultured in RPMI-1640 medium supplemented with 10% (vol/vol) heat-inactivated FBS (Invitrogen, Darmstadt, Germany) and 1% antibiotic and antimycotic solution (Sigma-Aldrich, Hamburg, Germany) in a humidified incubator at 37℃ with 5% CO2.34,35 The cells were washed with PBS (pH 7.4), and the required number of cells re-suspended in antibiotic- and antimycotic-free medium before seeding in to the culture plates for infection or treating with Escherichia coli LPS (500 ng/ml) (Sigma-Aldrich) for 6 h or 24 h and nigericin (10 µM) (Invivogen, Toulouse, France) for the last 30 min or 1 h in a humidified incubator at 37℃ with 5% CO2.
THP-1 monocytes were differentiated with 40 nM phorbol 12-myristate-13-acetate (PMA) in complete RPMI-1640 medium and cultured for 72 h with daily replenishment of fresh medium in a humidified incubator at 37℃ with 5% CO2. Next, the differentiated cells were rested in culture medium without PMA for 48 h to attain morphological and functional status of macrophage.36 These cells were washed with PBS, pH 7.4, and cultured in antibiotic- and antimycotic-free medium in a humidified incubator at 37℃ with 5% CO2 before H. pylori infection.
AGS (ATCC-CRL-1739) gastric adenocarcinoma cell line was cultured in RPMI-1640 medium as previously described.37,38 After adherent growth in culture flasks, the cells were washed with PBS (pH 7.4), and treated with trypsin-EDTA solution (Sigma-Aldrich) for 5 min and the reaction was stopped by adding fresh 10% FBS containing RPMI-1640 medium. The cell suspension prepared in 10% FBS containing RPMI-1640 medium was used for seeding cells in to culture plates and cultured in a humidified incubator at 37℃ with 5% CO2. The cells grown to 70–80% confluency in wells were used for transfection with plasmids and RNA, which is followed infection in antibiotic- and antimycotic-free medium.
Peripheral blood mononuclear cells (PBMC) isolation
PBMCs were isolated from venous blood of normal healthy and H. pylori-negative donors with written informed consent following approval by the local Friedrich Alexander University Erlangen-Nuremberg, Germany ethics committee (number: 36_12 B). Ficoll Paque (GE Life Sciences, Freiburg, Germany) density gradient centrifugation of venous blood was the method used (as per the protocol of the manufacturer) for isolation. The cells were gently treated with red blood cell lysis buffer (Sigma-Aldrich) for 1 min and washed with PBS (pH 7.4), and the required number of cells re-suspended in antibiotic- and antimycotic-free RPMI-1640 medium supplemented with 10% (vol/vol) heat-inactivated FBS before seeding in to culture plates for infection or treating with E. coli LPS (500 ng/ml) (Sigma-Aldrich) for 6 h or 24 h and nigericin (10 µM) (Invivogen) for the last 30 min and cultured in a humidified incubator at 37℃ with 5% CO2.19
mRNA, miRNA extraction and quantitative real-time PCR (RT-PCR)
H. pylori-infected and mock control THP-1 monocytes for the indicated time periods were harvested by centrifugation at 150 g for 5 min at 4℃. The supernatants were removed carefully and the cell pellet was washed with PBS before adding lysis buffer of the RNeasy Mini Kit (Qiagen, Hilden, Germany). Then, total RNA was extracted by the protocol provided by the manufacturer (Qiagen) and used to prepare cDNA by Reverse Transcriptase-PCR reagents (Invitrogen). The Taqman gene expression assay kit (Applied Biosystems, Foster City, CA, USA) was used for the analysis of mRNA expression and reactions were performed in Applied Biosystems sequence detection system 5700 as previously described.34 miRNA from infected and mock control THP-1 monocytes were extracted using the miRNeasy mini kit (Qiagen). The extracted RNAs were used for cDNA synthesis using miScript II RT kit (Qiagen). miScript Primer assay kit and Quantitect SYBR Green PCR master mix were used for setting up reactions in a Bio-Rad MyiQ Real Time PCR detection system for the analysis of miRNA expression by 2–ΔΔCt method.34
3’UTR Renilla luciferase assay
The AGS cells were grown to 70–80% confluency in 100 µl medium (96-well plate) and used for transfecting plasmids and/or miRNA followed by infection with H. pylori. One hundred ng pLigtswitch-3’UTR (expressing Renilla luciferase) control and pLigtswitch-NLRP3-3’UTR (Active Motif, Brabant Wallon, Belgium), alone or with 30 nM of anti-hsa-miR-223-3p RNA or with 30 nM of hsa-miR-223-3p RNA mimic (Qiagen) were transfected or co-transfected to the AGS cells using Lipofectamine 3000 transfection reagent in Opti-MEM serum-free media as per the manufacturers protocol (Thermo Fisher Scientific, Waltham, MA, USA). After transfection, cells were infected with H. pylori for 6 h in antibiotic- and antimycotic-free medium. Thereafter, the luciferase activity was measured in triplicate by adding 100 µl of assay buffer prepared as per the protocol of Lightswitch Luciferase assay kit (Active Motif) to the wells directly and kept at 25℃ for 30 min in dark before reading in an Orion Microplate Luminometer (Titertek Berthold, Pforzheim, Germany). The values were expressed as luminescence ratio of the relative light activity.
Recombinant human IL-10 activity assay
To test the inhibitory activity of IL-10 on NLRP3 and pro-IL-1β expression in PMA-differentiated THP-1 macrophages, the cells were induced with different concentrations (500–2000 pg/ml) of recombinant human (rh)-IL-10 (R&D Systems, Minneapolis, MN, USA) for 15 min before infecting with H. pylori or maintained as uninfected and incubated for 6 h in a humidified incubator at 37℃ with 5% CO2. After treatment, the cells were lysed in 2 × SDS buffer and boiled for 5 min and later analyzed by Western blotting, as described below.
Immunoprecipitation
The infected and mock control whole-cell extracts were prepared using lysis buffer containing pan-protease inhibitors (Roche, Mannheim, Germany). Then the whole-cell extracts were pre-cleared with Protein-G sepharose beads (Amersham Pharmacia, Munich, Germany) for 2 h at 4℃ and beads were removed by centrifugation at 2400 g for 5 min at 4℃. The Abs against human NLRP3 (Santa Cruz Biotech, Santa Cruz, CA, USA) were added to pre-cleared lysate and incubated for 16 h at 4℃ in a rotatory shaker at low speed. The immune complexes were precipitated using protein-G sepharose beads by incubating for 4 h at 4℃ and centrifugation at 2400 g for 5 min at 4℃.39,40 The protein-G sepharose bead-bound immune complexes were washed three times with PBS (pH 7.4) and then added 2 × SDS lysis buffer and boiled for 5 min before using for SDS-PAGE and Western blot analysis. A portion of the remaining lysate mixed with equal amount of 2 × SDS lysis buffer and boiled for 5 min, which is used for detection of loading control GAPDH protein.
Trichloroacetic acid (TCA) precipitation of culture supernatants
Culture supernatants of infected, LPS-primed, LPS-primed nigericin-induced and mock control in serum-free medium were used for TCA precipitation and subsequent analysis of the caspase-1 p10 fragment and mature IL-1β. Briefly, one-tenth the volume of 6.1 N trichloroacetic acid (Sigma-Aldrich) was added to each culture supernatants and incubated on ice for 1 h. After the incubation, the precipitated proteins were collected by centrifugation at 16,000 g for 30 min at 4℃. The supernatants were removed and the thin layer of pellet was washed with ice-cold acetone three times and finally dissolved in 2 × SDS-PAGE buffer and boiled for 5 min and later analyzed by Western blotting, as described below.
SDS-PAGE and immunoblotting
Infected and non-infected cells were harvested and mixed with equal amounts of 2 × SDS-PAGE buffer and boiled for 5 min. Proteins were separated by SDS-PAGE on 6–15% polyacrylamide gels and blotted onto PVDF membranes (Immobilon-P; Millipore, Billerica, MA, USA) and membranes were blocked in TBS-T buffer (140 mM NaCl, 25 mM Tris-HCl pH 7.4, 0.1% Tween-20) with 5% skimmed milk for 1 h at 25℃ as described.41,42 NLRP3,GAPDH and caspase-1 expression was monitored using Abs from Santa Cruz Biotech or Adipogen Life Sciences (San Diego, CA, USA). Ab against IL-1β was purchased from R&D systems. As secondary Abs, HRP-conjugated anti-rabbit, anti-mouse and anti-goat polyvalent pig and rabbit immunoglobulin, respectively, were used (Dako, Hamburg, Germany). Ab detection was performed with the ECL Plus chemiluminescence Western Blot system for immunostaining (Amersham Pharmacia Biotech).43,44
Quantification of cytokines
The supernatants of H. pylori infected and mock control cells were collected and centrifuged at maximum speed in a cold centrifuge at 4℃ to remove bacteria before storing at −80℃ until assayed. Human IL-1β and IL-10 concentration in the supernatant were determined by standard ELISA, with commercially available assay kits, described by the manufacturer (Becton Dickinson, Heidelberg, Germany).
Statistical analysis
All experiments were repeated at least three times with similar results. The data were evaluated using Student’s t-test with GraphPad statistical software online. P-Values ≤ 0.05 and ≤ 0.005 were considered as statistically significant.
Results
Time-dependent NLRP3 expression in THP-1 monocytes infected by H. pylori
NLPR3 has been reported to produce six individual isoforms by alternative splicing.45,46 However, the exact role of these different isoforms is not yet clear. In the present study, we analyzed the NLRP3 expression in the THP-1 monocytes at two different time periods of H. pylori infection (early time point at 6 h and late time point at 24 h), which was normalized to GAPDH. We found that H. pylori infection regulated the expression of NLRP3 gene product. As expected, NLRP3 mRNA expression by Taqman quantitative real-time PCR (RT-PCR) was elevated at 6 h by wt H. pylori and isogenic mutants of major pathogenicity factors, such as ΔcagA, ΔvacA, Δcgt (cholesterol-α-glucosyltransferase), ΔflaA (flagellin A) and ΔcagPAI, when compared with mock control cells (Figure 1a). However, we were very surprised that the NLRP3 mRNA levels were significantly down-regulated at 24 h in all the infected (wt and mutants) cells (Figure 1a). In addition, we employed immunoprecipitation using an Ab raised against amino acids 25–90 of the N-terminal region of human NLRP3. This Ab immunoprecipitated known isoforms of the human NLRP3. Western blotting of the precipitated proteins from infected and mock control cells revealed bands of the different isoforms including isoform 1, 2 and 3 with predicted molecular masses of 106, 118 and 84 kDa, respectively (Figure 1b). All isoforms detected in the blot increased their intensity in the infected cells at 6 h (Figure 1b); however, the protein levels were found to be decreased at 24 h when compared with mock control cells (Figure 1b). The data demonstrate that the NLRP3 mRNA and protein expression are regulated in H. pylori infected THP-1 monocytes in a time-dependent fashion.
Figure 1.
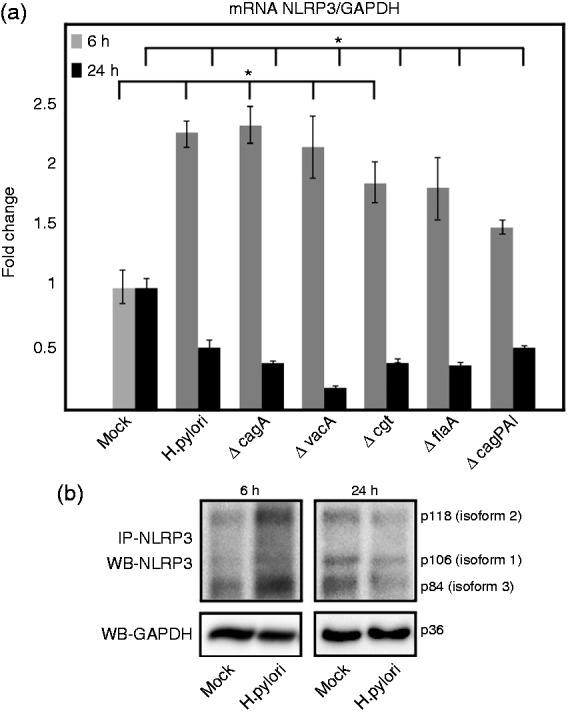
NLRP3 expression in THP-1 monocytes during H. pylori infection. Inflammasome-forming NLRP3 expression changes in THP-1 monocytes at early (6 h) and late (24 h) time points of H. pylori infection. (a) The mRNA level of expression was analyzed using quantitative Taqman RT-PCR of infected and mock control cells. Values are expressed as fold changes normalized to GAPDH expression derived from the 2–ΔΔCt method. (b) Immunoprecipitated NLRP3 proteins from infected and mock control cells were analyzed by Western blotting. The various NLRP3 isoforms are indicated. The GAPDH blot served as loading control. *P ≤ 0.05 and **P ≤ 0.005 were considered as statistically significant.
Up-regulated expression of NLRP3 targeting hsa-miR-223-3p (3’ arm miRNA) during H. pylori infection
miRNAs are short, endogenous, non-coding RNAs involved in the post-transcriptional regulation of gene expression.47 In general, seven conserved nucleotides (2–8) in a given miRNA determine its function through a base-pairing region at the 3’UTR of mRNA target. We screened two different miRNA databases, miRDB and TargetScan, and found that hsa-miR-223-3p has a predicted target at the 3’UTR of NLRP3 mRNA with a high target score (Figure 2; Supplementary Table S1). Mature miRNA is a double-stranded structure derived from both arms of the precursor and one arm of the mature miRNA preferentially accumulates with a small proportion of the opposite arm, commonly called miR*.48 In this case, hsa-miR-223-3p forms from the 3’ arm of the precursor accumulates over hsa-miR-223-5p forms from the 5’ arm. Using RT-PCR, we analyzed the differential expression of both mature miRNA levels normalized to RNU6-2 (RNA, U6 small nuclear 2) expression in H. pylori-infected THP-1 monocytes in comparison with the mock control cells. There was no change in the levels of hsa-miR-223-5p expression, even after 24 h of infection; however, the hsa-miR-223-3p levels were gradually increased from almost twofold at 6 h to over threefold at 24 h of infection period (Figure 3). To the best of our knowledge, this is the first report to show that H. pylori preferentially accumulates an NLRP3-targeting miRNA in a monocytic cell line until 24 h of infection. This data are well correlated with the decreased NLPR3 mRNA and protein level expression at 24 h of infection, as shown above.
Figure 2.

Prediction of hsa-miR-223-3p targeting of NLRP3 3’UTR with seed sequence base-pairing location and target score using two different prediction tools. miRDB database and TargetScan are two widely used miRNA target prediction tools. (a) The hsa-miR-223-3p has an 7mer target seed sequence base pairing region at 406 of the 613 nt length 3’UTR of the NLRP3 transcript variant 4 (NM_001127461.2) predicted by miRDB and a high target score. (b) TargetScan also predicted NLRP3 3’UTR targeting with similar score. 3’UTR region analysis of the other five reported transcript variants (NM_001243133.1, NM_001079821.2, NM_001127462.2, NM_004895.4, NM_183395.2) in curated non-redundant NCBI RefSeq database also showed hsa-miR-223-3p target in their respective 3’ UTR (Supplementary Table S1).
Figure 3.
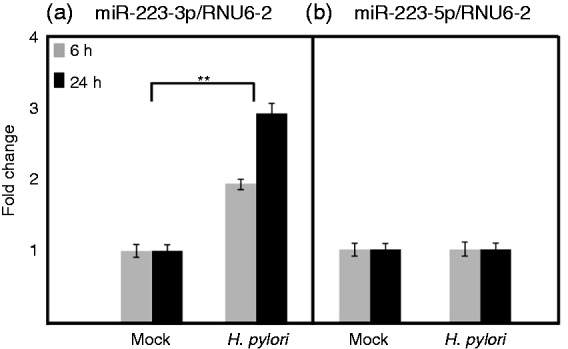
hsa-miR-223 expression during H. pylori infection of THP-1 monocytes. The NLRP3 targeting hsa-miR-223-3 p and its miR* partner hsa-miR-223-5p expression in H. pylori-infected and mock control cells were analyzed by quantitative real-time PCR using Qiagen miScript kits and SYBR Green PCR master mix. Values are expressed as fold changes of expression of (a) hsa-miR-223-3p and (b) hsa-miR-223-5 p normalized to the RNU6-2. *P ≤ 0.05 and **P ≤ 0.005 were considered as statistically significant.
Next, we analyzed the hsa-miR-223-3p expression in H. pylori-infected AGS gastric epithelial cells and found it to be gradually up-regulated after 6 h and 24 h (Figure 4a). This shows that H. pylori can up-regulate mature hsa-miR-223-3p expression in both epithelial and immune cells. To confirm the proposed NLRP3 3’UTR targeting, we transfected AGS cells with plasmid pLightswitch-3’UTR expressing a Renilla luciferase as control vector and NLRP3-3’UTR cloned (pLightswitch-NLRP3-3’UTR) at the 3’ end of the Renilla luciferase gene. We have also co-transfected 30 nM (best concentration selected) of anti-hsa-miR-223-3p (inhibitor) or hsa-miR-223-3p (mimic) along with control and NLRP3-3’UTR cloned vectors to validate the hsa-miR-223-3p targeting of NLRP3-3’UTR. The Renilla luciferase activity of infected and mock control cells was measured using Lightswitch luciferase assay kit (Active Motif) in a luminometer, as per the manufacturer’s protocol. The control vector transfected in mock control cells expressed high amounts of luciferase as expected; however, pLightswitch-NLRP3-3’UTR transfected mock control cells expressed significantly lower luciferase activity, which can be attributed to the increased NLRP3 3’UTR targeting in AGS cell line (data not shown). The luminescence ratio was calculated using relative light units from control and NLRP3 3’UTR vector transfected or co-transfected with inhibitor and mimic RNAs. The results from anti-hsa-miR-223-3p co-transfection experiments slightly increased the luminescence ratio. However, hsa-miR-223-3p miRNA mimetic reduced the ratio to almost half in comparison with vector alone transfected cells infected with H. pylori (Figure 4b; Supplementary Table S2). This confirmed that NLRP3-3’UTR targeting by hsa-miR-223-3p is increased during H. pylori infection. We therefore propose that these findings could have major implications on the functional steadiness, formation and activation of the NLRP3 inflammasome to produce mature forms of IL-1β and IL-18 during H. pylori infection.
Figure 4.
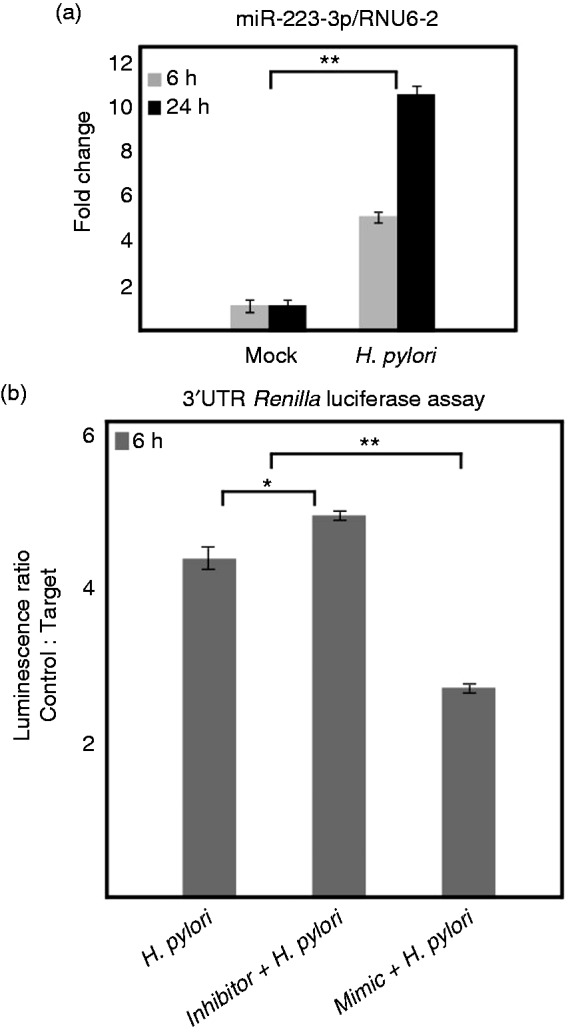
hsa-miR-223 expression and NLRP3 3’UTR targeting during H. pylori infection of AGS cells. (a) hsa-miR-223-3p expression in H. pylori-infected and mock-infected AGS cells was analyzed by quantitative real-time PCR using Qiagen miScript kits and SYBR Green PCR master mix. Values are expressed as fold changes of expression of hsa-miR-223-3p normalized to the RNU6-2. (b) pLightswitch-3’UTR and pLightswitch-NLRP3-3’UTR were transfected to AGS cells and Renilla luciferase activity analyzed after infection with H. pylori. In addition, anti-hsa-miR-223-3p (inhibitor) and hsa-miR-223-3p mimic were co-transfected to validate the effect. *P ≤ 0.05 and **P ≤ 0.005 were considered as statistically significant.
NLRP3 expression in THP-1 differentiated macrophages during H. pylori infection
Haneklaus et al. reported that miR-223 expression decreases as monocytes differentiate into macrophages and upon accumulation of the NLRP3 protein.49 To understand the effect on NLRP3 expression in macrophages during H. pylori infection, we differentiated THP-1 monocytes to macrophages by PMA pre-treatment followed by infection for two different time points. Western blot of the cell lysates from control and infected cells at different time points showed that NLRP3 isoform 3 (p84) was highly up-regulated at time points 6 h and 24 h when compared with the mock control (Figure 5a). However, isoform 1 concentration was not changed significantly after infection (Figures 5a and 6). The infected macrophages secreted significantly more mature IL-1β (Figure 5b); however, the pro-form was highly elevated until the late time point when compared with the mock control (Figure 5a).
Figure 5.
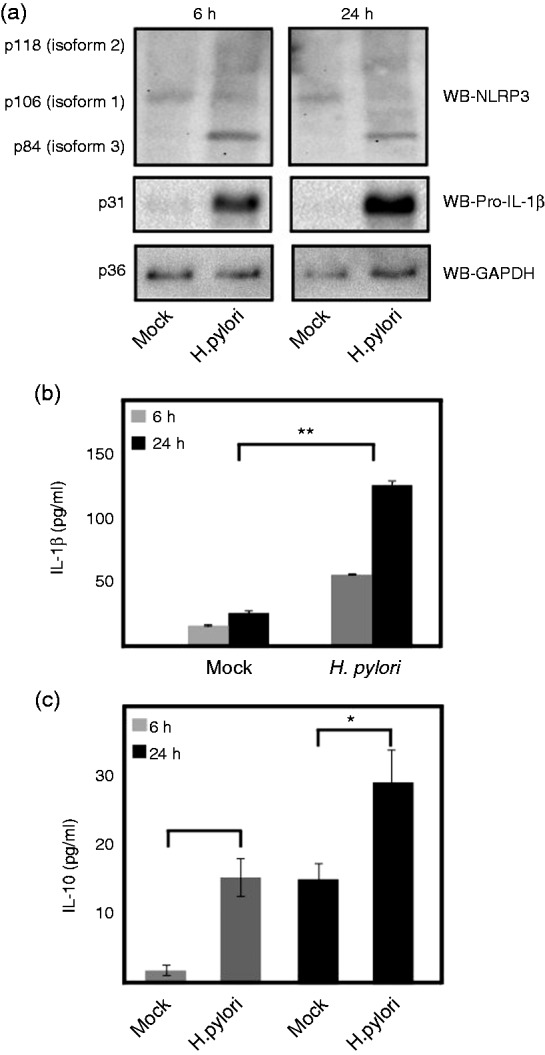
NLRP3 and pro-IL-1β expression and secretion of IL-1β and IL-10 in THP-1 macrophages in macrophages and monocytes. (a) NLRP3 and pro-IL-1β protein expression and (b) mature IL-1β secretion in PMA-differentiated THP-macrophages at early (6 h) and late (24 h) time points of H. pylori infection. (c) IL-10 secretion in PMA differentiated macrophages at early (6 h) and late (24 h) time points of H. pylori infection. *P ≤ 0.05 and **P ≤ 0.005 were considered as statistically significant.
Figure 6.
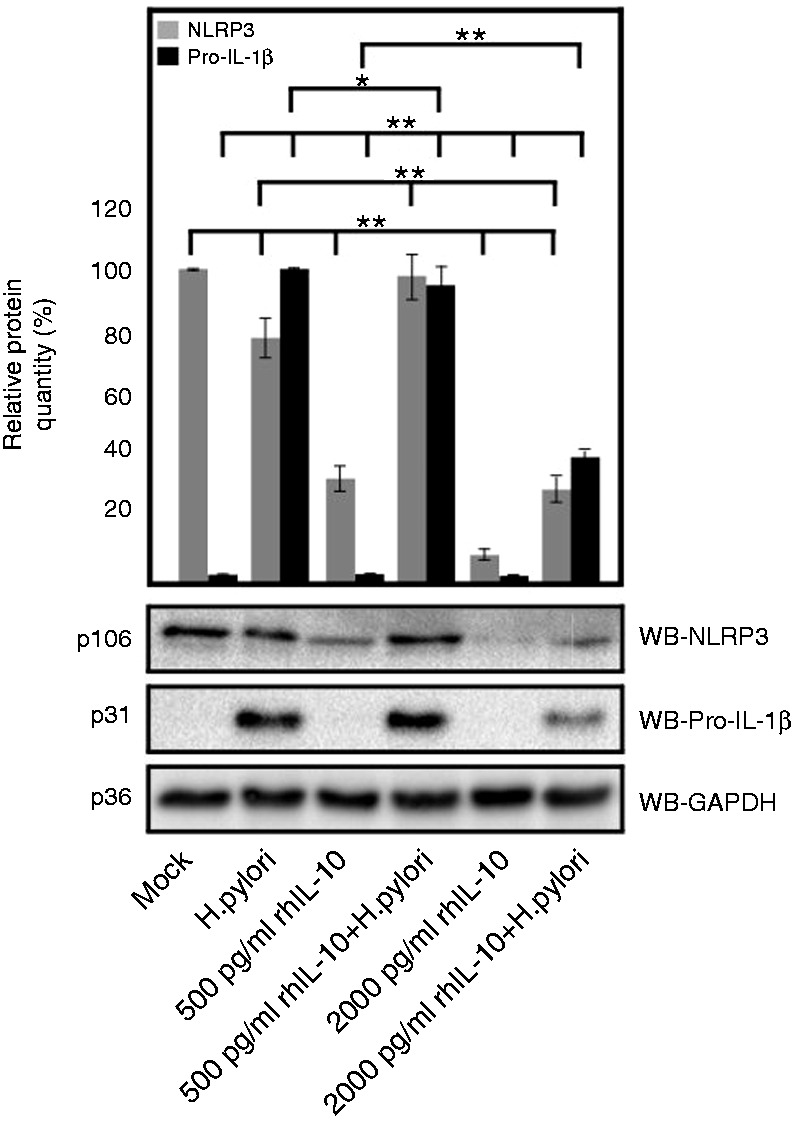
rhIL-10 mediated down-regulation of NLRP3 and pro-IL-1β expression in H. pylori-infected PMA-differentiated THP-1 macrophages in a dose-dependent manner at 6 h of treatment. The mean relative protein quantity was measured by multiple densitometric analysis of each protein bands by setting most intensity band as reference. *P ≤ 0.05 and **P ≤ 0.005 were considered as statistically significant.
Very recent studies have identified cytokine IL-10 as another regulator of NLRP3 and pro-IL1β expression in human monocytes and mouse bone marrow-derived macrophages.50–53 H. pylori-infected macrophages secreted significantly more IL-10 than mock control; however, it was < 30 pg/ml, even after 24 h of infection. This low level of IL-10 was not enough to control NLRP3 expression in macrophages (Figure 5a, c). To test whether higher concentration of IL-10 can down-regulate the expression of NLRP3 in macrophages, we designed an activity assay using different concentrations of exogenously added rhIL-10 for 6 h. The results showed that the addition of 500 pg/ml rhIL-10 significantly decreased the NLRP3 (isoform 1, p106) concentration in uninfected control cells; however, H. pylori-infected cells maintained NLRP3 expression. Moreover, exogenously added rhIL-10 at 2000 pg/ml was able to down-regulate NLRP3 expression significantly in both infected and uninfected cells (Figure 6). In addition, the pro-IL-1β expression in macrophages induced by H. pylori was also down-regulated significantly at 2000 pg/ml of exogenously added rhIL-10 (Figure 6).
Inflammasome-mediated caspase-1 activation and mature IL-1β production in THP-1 monocytes
Inflammasome activation and production of mature IL-1β requires the optimal expression of NLR proteins like NLRP3, and pro-IL-1β.16 The expressional difference in any one of the component can affect the typical activation of caspase-1 and mature IL-1β production. H. pylori infection highly induced the pro-IL-1β production in THP-1 monocytes that was gradually increased until the late time point (Figure 7a). As outlined above, we have shown that NLRP3 targeting hsa-miR-223-3p concentration was elevated in H. pylori-infected monocytes and that significantly reduced the NLRP3 expression in the late time point. Therefore, we have compared the caspase-1 activation in H. pylori-infected and LPS-primed and nigericin-induced THP-1 monocytes at early time point where NLRP3 concentration was high. As shown in Figure 7, caspase-1 activation and subsequent pyroptotic cell death, analyzed through GAPDH calibration, was only observed in LPS-primed and nigericin-induced cells, but not in H. pylori infected, LPS-primed or LPS-primed and H. pylori-infected cells. H. pylori infected THP-1 monocytes secreted very low levels of mature IL-1β secretion during the infection period (Figure 7b). However, LPS-primed and nigericin-induced cells sufficiently cleaved pro-caspase-1 and pro-IL-1β and secreted copious amounts of caspase-1 p10 fragment and mature IL-1β in the supernatant (Figure 7b, c).
Figure 7.
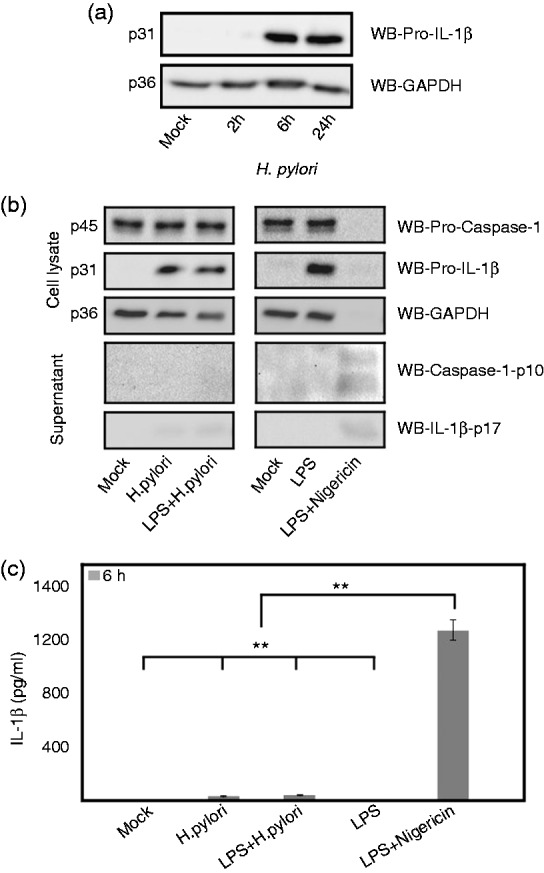
Analysis of NLRP3 inflammasome activation in E. coli LPS-primed and nigericin-induced (1 h) and H. pylori-infected THP-1 monocytes. (a) Time-dependent expression of pro-IL-1β in THP-1 monocytes during H. pylori infection. (b) The expression and cleavage of pro-caspase-1 and pro-IL-1β in E. coli LPS-primed nigericin-induced, E. coli LPS-primed, H. pylori-infected and E. coli LPS-primed H. pylori-infected THP-1 monocytes at 6 h. (c) The secreted mature IL-1β from the above treated and infected THP-1 monocytes at 6 h were measured by ELISA. *P ≤ 0.05 and **P ≤ 0.005 were considered as statistically significant.
IL-10 levels and inflammasome activation in THP-1 monocytes and human PBMCs
As described above, and reported by others, cytokine IL-10 is another regulator of NLRP3 expression in monocytes and macrophages.50–53 We analyzed this effect by measuring the levels of IL-10 secretion in LPS-primed and nigericin-induced THP-1 monocytes and human PBMCs in relation to secreted mature IL-1β. As expected, the results showed that IL-10 levels were gradually increased from 6 h of treatment to 24 h, and the secreted levels of mature IL-1β were significantly reduced (Figure 8a, b), which could be correlated with the abovementioned recent findings of the effect of IL-10 on NLRP3 expression and mature IL-1β secretion. We noted the correlation of increased IL-10 levels on regulating the classical activation of NLRP3-mediated mature IL-1β secretion. To investigate whether H. pylori can induce IL-10 in infected THP-1 monocytes and PBMCs, we analyzed the secretion of IL-10 by ELISA. Both cell systems produced copious amount of IL-10 secretion at early and late stages of infection (Figure 8a). Gurung et al. reported that reduced mature IL-1β secretion during classical NLRP3 inflammasome activation was more pronounced in the late time point, owing to the decreased NLRP3 expression.52 Our findings of increased hsa-miR-223-3p and gradually increasing IL-10 in these infected cell systems could be correlated with decreased expression of NLRP3 at the late time point of H. pylori infection (24 h). We also noted that H. pylori-infected THP-1 monocytes secreted very low levels of mature IL-1β secretion during the entire infection period (Figure 8b). This was also analyzed in various H. pylori wt strain-infected THP-1 monocytes and induced similar amounts (data not shown). In addition, pro-IL-18 was pre-formed in the THP-1 monocytes and found to be decreased during the time course of infection with no detectable secreted IL-18 in the supernatant (data not shown). However, infected human PBMCs secreted a moderately high amount of mature IL-1β at 6 h and was found to be significantly increased after 24 h of infection (Figure 8b).
Figure 8.
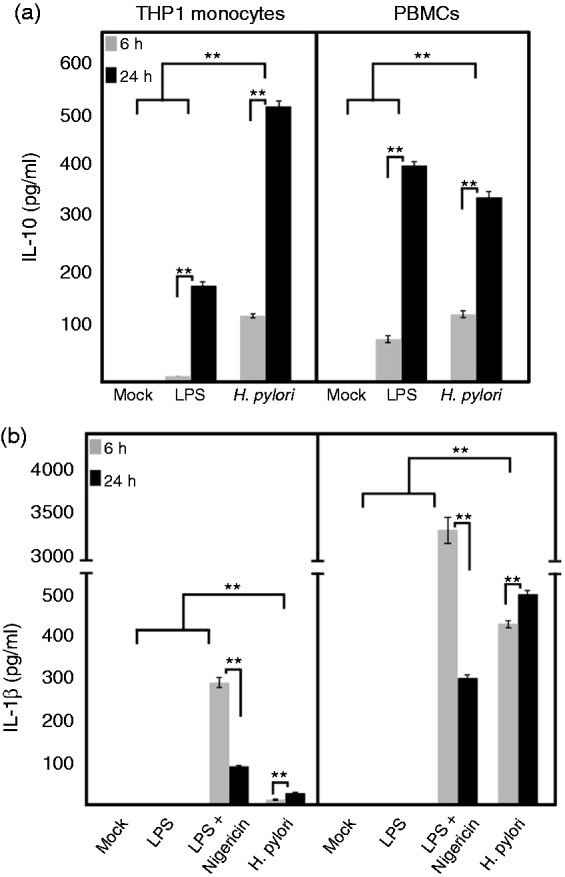
IL-10 secretion and inflammasome activation in THP-1 monocytes and PBMCs during H. pylori infection. (a) H. pylori-infected and E. coli LPS-primed THP-1 monocytes and PBMCs were analyzed for IL-10 secretion at early and late time points when compared with the mock control. (b) NLRP3 inflammasome activation by treating with nigericin for the last 30 min of E. coli LPS-primed THP-1 monocytes and PBMCs secreted very significant amount of mature IL-1β at the early time point (6 h); however, it was significantly down-regulated at late time point (24 h). H. pylori-infected THP-1 monocytes secreted very low amount of mature IL-1β at both time points, whereas infected PBMCs secreted significant amount at early and late time points. *P ≤ 0.05 and **P ≤ 0.005 were considered statistically significant.
Discussion
Inflammasome signaling and activation are important and crucial events during microbial infections and other inflammatory processes.54 The activated inflammasome commonly induces the production of mature caspase-1 and cleavage of pro-IL-1β and pro-IL-18, respectively. The mature IL-1β and IL-18 forms produced from their pro-forms play crucial roles in the innate and adaptive immunity responses. IL-1β is a known pyrogen involved in the recruitment of leukocytes and the production of mediators of inflammation and related immuno-pathologies.13,55 IL-18 does not have pyrogenic activity; however, it induces the production of IFN-γ and has a role in deciding the fate of activated T-cell populations depending on the cytokine milieu in this microenvironment.55 Fever is one of the important symptoms of enteropathogenic bacteria like Salmonella, Shigella, Yersinia and cholera infection, and is known to activate the inflammasome, producing large amounts of IL-1β. However, lifelong colonization of H. pylori does not generally produce fever-like symptoms. We propose that this observation could be related to the controlled production of pyrogenic IL-1β. Previous studies reported the activation of inflammasome and production of mature IL-1β and IL-18 in a mouse model of H. pylori infection.10,25,26 Another recent study showed that mucin-1 expression tightly regulated the NLRP3 inflammasome activation in a long-term mouse model of H. pylori infection.27 This finding revealed that regulation of inflammasome activation and pyroptosis is an important strategy of the gastric pathogen H. pylori that may help the survival in its niche without much destruction of the infection site. The reported studies on inflammasome activation during H. pylori infection were mainly dependent on NLRP3.10,25,26 Early studies using human gastric biopsies and recent in vitro studies using primary cells and cultured cell lines have also reported the expression and secretion of mature IL-1β during H. pylori infection.56–58 However, the regulation of NLRP3 expression during H. pylori infection in cells of human origin has not been described before. The results of our study showed that NLRP3 is constitutively expressed in non-infected THP-1 monocytes. During infection, H. pylori first up-regulated the expression of NLRP3 at early time point (6 h) and found to be down-regulated at later time point (24 h). The isogenic mutants of major pathogenicity-associated factors in H. pylori such as ΔcagA, ΔvacA, Δcgt, ΔflaA and ΔcagPAI exhibited a similar pattern of expression in infected cells. This demonstrates that these important H. pylori factors are not involved in this expressional variation of NLPR3. Previous studies described the cagPAI-dependent activation of NLRP3 inflammasome in infected mouse immune cells.10,25,26 Moreover, there are contradictory reports on the involvement of VacA on activation of NLRP3 inflammasome in mouse immune cells and human PBMCs.25,26 In our study, ΔvacA mutant-infected THP-1 monocytes showed the maximum suppression in the NLRP3 mRNA expression. In addition, infection of THP-1 monocytes induced high levels of pro-IL-1β production and continued until the late time point but secreted low amounts of mature IL-1β in the supernatant. Moreover, pro-IL-18 appears to be constitutively present in monocytes;59 however, H. pylori-infected THP-1 monocytes did not secrete mature IL-18 in the supernatant (unpublished data).
Our search for the factors regulating the NLRP3 expression have identified a miRNA, hsa-miR-223-3p. Analysis of hsa-miR-223-3p expression in THP-1 monocytes showed that hsa-miR-223-3p expression level was significantly increased after H. pylori infection in the whole period tested without any change in the miR* partner, hsa-miR-223-5p. Thus, this is the first study to show the up-regulation of a miRNA targeting NLRP3 in monocytes infected with H. pylori. This increased expression of hsa-miR-223-3p correlates with decreased NLRP3 expression in H. pylori-infected THP-1 monocytes. This effect was pronounced at the late time point of the infection, which was reflected in the corresponding mRNA and protein levels. This demonstrates that H. pylori induced a miRNA, which targets a major inflammasome forming protein in monocytes. Moreover, a very recent study reported that up-regulated expression of miR-223 in monocytes controls intestinal inflammation in inflammatory bowel disease (IBD) through repression of NLRP3 expression.60 To confirm this finding, we used a Renilla luciferase-expressing vector with the 3’ UTR sequence of human NLRP3. AGS gastric epithelial cells, which also exhibited up-regulated hsa-miR-223-3p expression in H. pylori infection, were transfected with the control and NLRP3 3’ UTR vectors and luciferase activity was measured after infection. Co-transfection of anti-hsa-miR-223-3p RNA and hsa-miR-223-3p mimic RNA with luciferase vector was applied to further validate NLRP3 3’ UTR targeting. The luminescence ratio of control vs. target vector-transfected cells have confirmed the NLRP3 targeting by hsa-miR-223-3p during H. pylori infection. In addition, Ma et al. reported similar increased hsa-miR-223 expression in H. pylori-infected gastric cancer patients, which was associated with increased cancer cell proliferation and migration.61 This emphasizes an important role of hsa-miR-223-3p in regulating the expression of inflammasome forming NLRP3 and production of mature IL-1β and IL-18 in H. pylori infection, inflammation resolution or persistence.
As reported by Haneklaus et al.,49 miR-223 expression decreased in macrophages and led to accumulation of NLRP3 protein. THP-1 differentiated macrophage infection with H. pylori specifically up-regulated the NLRP3 isoform 3 (p84) instead of other isoforms (the implications of this isoform switching is not clear) and also maintained the isoform 1 (p106) expression. However, the result confirms the previous study showing that monocyte-differentiated macrophages reduced the miR-223 and thereby accumulated the NLRP3 expression. The infection also induced elevated levels of pro-IL-1β in macrophages; however, that the amount of secreted mature IL-1β was not correspondingly elevated in the whole period of infection emphasizes insufficient or lack of inflammasome activation.
In addition, we tested the inflammasome activation and subsequent pro-caspase-1 and pro-IL-1β cleavage in LPS-primed and nigericin-induced THP-1 monocytes and compared this with H. pylori-infected cells at 6 h, where NLRP3 concentration was up-regulated. Classically activated (LPS–nigericin) cells have shown sufficient cleavage of pro-caspase-1 and pro-IL-1β and the cleaved fragments, caspase-1 p10 and mature IL-1β, respectively, were secreted in the supernatants in very high amounts when compared with H. pylori-infected and mock control cells. Moreover, the loading control GAPDH was not visible in the Western blot in LPS–nigericin treated cells and it is reported to be a target of activated caspase-1 during pyroptotic cell death in inflammasome-activated cells.62 These facts, discussed above, clearly point out the lack of inflammasome activation by H. pylori in THP-1 monocytes, even at high NLRP3 concentration, after 6 h of infection. Therefore, this study reveals two important aspects of H. pylori infection: inflammasome activation is compromised at early phase of infection and that it was intensified and assured through the down-regulation of NLRP3 expression in the late phase of infection.
IL10 polymorphisms were also reported to be involved in the H. pylori-associated gastric tumorigenesis.63 In addition, IL-10 was ascertained in the production of regulatory T cells and beneficial effects in suppressing allergy and asthma in H. pylori-colonized individuals.8,11,64 IL-10 is a major anti-inflammatory cytokine known to suppress antigen presentation, cytokine gene expression and mast cell degranulation. All these effects of IL-10 could be a determining factor for its known role on controlling allergy, autoimmunity and inflammation.65 In addition to miRNA, anti-inflammatory cytokines, like IL-10, were also reported to regulate NLRP3 expression in human and mouse monocytes and dendritic cells.51–53 In this study, we used THP-1 macrophages as a model with which to analyze the reported effect of IL-10 on NLRP3 expression. H. pylori-infected THP-1 macrophages secreted very small amounts of IL-10, which reflected the increased expression of NLRP3 in both infected and mock control cells. This could also be due to the lack of NLRP3 targeting of has-miR-223-3p in this system. This prompted us to analyze the effect of exogenously added rhIL-10 on NLRP3 protein expression. As expected, the levels of NLRP3 were decreased significantly in a dose-dependent manner in mock control and H. pylori-infected cells. However, H. pylori infected cells required a high amount of rhIL-10 to decrease the NLRP3 expression vs. mock control cells. This shows that H. pylori can maintain NLRP3 expression in THP-1 macrophages at moderately high levels of rhIL-10. A lesser amount may be required for an autocrine effect of endogenously produced IL-10, which may be the case in THP-1 monocytes and PBMCs, as described below. H. pylori-infected and LPS-treated THP1 monocytes and human PBMCs secreted gradually elevated amounts of IL-10 in the whole period of exposure or infection. The effect of secreted IL-10 on expression of NLRP3 and inflammasome activation was reflected in the low level of mature IL-1β secretion seen at the late time point of LPS–nigericin-treated THP1 monocytes and PBMCs. However, H. pylori-infected PBMCs secreted significantly elevated levels of mature IL-1β throughout the infection period, which suggests a different mode of activation in mature IL-1β secretion. Taken together, the above data show suppressive effects of IL-10 and hsa-miR-223-3p on expression of NLRP3, inflammasome activation and secretion of mature IL-1β in cells of human origin. In addition, one critical finding of this study is the disparity in mature IL-1β secretion by THP1 monocytes/macrophages and PBMCs during H. pylori infection. Further studies are underway in our laboratory to disover the disparity in secretion of mature IL-1β in infected primary and cultured cell lines.
Supplementary Material
Acknowledgements
We thank Christoph Holzberger for technical support in the experiments, and Dr. Heiko Bruns, University Hospital Erlangen, for providing human blood samples.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the German Science Foundation (project A04 in CRC-1181 to S.B.).
References
- 1.Moodley Y, Linz B. Helicobacter pylori sequences reflect past human migrations. Genome Dyn 2009; 6: 62–74. [DOI] [PubMed] [Google Scholar]
- 2.Salama NR, Hartung ML, Müller A. Life in the human stomach: persistence strategies of the bacterial pathogen Helicobacter pylori. Nat Rev Microbiol 2013; 11: 385–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Backert S, Tegtmeyer N, Fischer W. Composition, structure and function of the Helicobacter pylori cag pathogenicity island encoded type IV secretion system. Future Microbiol 2015; 10: 955–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Amieva MR, El-Omar EM. Host-bacterial interactions in Helicobacter pylori infection. Gastroenterology 2008; 134: 306–323. [DOI] [PubMed] [Google Scholar]
- 5.Atherton JC, Blaser MJ. Coadaptation of Helicobacter pylori and humans: ancient history, modern implications. J Clin Invest 2009; 119: 2475–2487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pachathundikandi SK, Tegtmeyer N, Backert S. Signal transduction of Helicobacter pylori during interaction with host cell protein receptors of epithelial and immune cells. Gut Microbes 2013; 4: 454–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kusters JG, van Vliet AH, Kuipers EJ. Pathogenesis of Helicobacter pylori infection. Clin Microbiol Rev 2006; 19: 449–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Oertli M, Sundquist M, Hitzler I, et al. DC-derived 1L-18 drives Treg differentiation, murine Helicobacter pylori-specific immune tolerance, and asthma protection. J Clin Invest 2012; 122: 1082–1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Engler DB, Leonardi I, Hartung ML, et al. Helicobacter pylori-specific protection against inflammatory bowel disease requires the NLRP3 inflammasome and 1L-18. Inflamm Bowel Dis 2015; 21: 854–861. [DOI] [PubMed] [Google Scholar]
- 10.Koch KN, Hartung ML, Urban S, et al. Helicobacter urease-induced activation of the TLR2/NLRP3/IL-18 axis protects against asthma. J Clin Invest 2015; 125: 3297–3302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hussain K, Letley DP, Greenaway AB, et al. Helicobacter pylori-mediated protection from allergy is associated with IL-10-secreting peripheral blood regulatory T cells. Front Immunol 2016; 7: 71–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.El-Omar EM, Carrington M, Chow WH, et al. Interleukin-1 polymorphisms associated with increased risk of gastric cancer. Nature 2000; 404: 398–402. [DOI] [PubMed] [Google Scholar]
- 13.Apte RN, Dotan S, Elkabets M, et al. The involvement of IL-1 in tumorigenesis, tumor invasiveness, metastasis and tumor-host interactions. Cancer Metastasis Rev 2006; 25: 387–408. [DOI] [PubMed] [Google Scholar]
- 14.Resende C, Regalo G, Durães C, et al. Interleukin-1B signaling leads to increased survival of gastric carcinoma cells through a CREB-C/EBPβ-associated mechanism. Gastric Cancer 2016; 19: 74–84. [DOI] [PubMed] [Google Scholar]
- 15.Backert, S. Inflammasome signaling and bacterial infections. 1st ed. Heidelberg: Springer, 2016.
- 16.Lamkanfi M, Dixit VM. Mechanisms and functions of inflammasomes. Cell 2014; 157: 1013–1022. [DOI] [PubMed] [Google Scholar]
- 17.Gavrilin MA, Abdelaziz DH, Mostafa M, et al. Activation of the pyrin inflammasome by intracellular Burkholderia cenocepacia. J Immunol 2012; 188: 3469–3477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Broz P, Monack DM. Newly described pattern recognition receptors team up against intracellular pathogens. Nat Rev Immunol 2013; 13: 551–565. [DOI] [PubMed] [Google Scholar]
- 19.Bauernfeind FG, Horvath G, Stutz A, et al. Cutting edge: NF-kappaB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J Immunol 2009; 183: 787–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.He Y, Zeng MY, Yang D, et al. NEK7 is an essential mediator of NLRP3 activation downstream of potassium efflux. Nature 2016; 530: 354–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Baker PJ, Boucher D, Bierschenk D, et al. NLRP3 inflammasome activation downstream of cytoplasmic LPS recognition by both caspase-4 and caspase-5. Eur J Immunol 2015; 45: 2918–2926. [DOI] [PubMed] [Google Scholar]
- 22.Gurung P, Burton A, Kanneganti TD. NLRP3 inflammasome plays a redundant role with caspase 8 to promote IL-1β-mediated osteomyelitis. Proc Natl Acad Sci U S A 2016; 113: 4452–4457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shi J, Zhao Y, Wang K, et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 2015; 526: 660–665. [DOI] [PubMed] [Google Scholar]
- 24.Storek KM, Monack DM. Bacterial recognition pathways that lead to inflammasome activation. Immunol Rev 2015; 265: 112–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kim DJ, Park JH, Franchi L, et al. The Cag pathogenicity island and interaction between TLR2/NOD2 and NLRP3 regulate 1L-1β production in Helicobacter pylori infected dendritic cells. Eur J Immunol 2013; 43: 2650–2658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Semper RP, Mejías-Luque R, Groß C, et al. Helicobacter pylori-induced 1L-1β secretion in innate immune cells is regulated by the NLRP3 inflammasome and requires the cag pathogenicity island. J Immunol 2014; 193: 3566–3576. [DOI] [PubMed] [Google Scholar]
- 27.Ng GZ, Menheniott TR, Every AL, et al. The MUC1 mucin protects against Helicobacter pylori pathogenesis in mice by regulation of the NLRP3 inflammasome. Gut 2016; 65: 1087–1099. [DOI] [PubMed] [Google Scholar]
- 28.Wiedemann T, Hofbaur S, Tegtmeyer N, et al. Helicobacter pylori CagL dependent induction of gastrin expression via a novel αvβ5-integrin-integrin linked kinase signaling complex. Gut 2012; 61: 986–996. [DOI] [PubMed] [Google Scholar]
- 29.Roure S, Bonis M, Chaput C, et al. Peptidoglycan maturation enzymes affect flagellar functionality in bacteria. Mol Microbiol 2012; 86: 845–856. [DOI] [PubMed] [Google Scholar]
- 30.Brisslert M, Enarsson K, Lundin S, et al. Helicobacter pylori induce neutrophil transendothelial migration: role of the bacterial HP-NAP. FEMS Microbiol Lett 2005; 249: 95–103. [DOI] [PubMed] [Google Scholar]
- 31.Tegtmeyer N, Wittelsberger R, Hartig R, et al. Serine phosphorylation of cortactin controls focal adhesion kinase activity and cell scattering induced by Helicobacter pylori. Cell Host Microbe 2011; 9: 520–531. [DOI] [PubMed] [Google Scholar]
- 32.Conradi J, Tegtmeyer N, Woźna M, et al. An RGD helper sequence in CagL of Helicobacter pylori assists in interactions with integrins and injection of CagA. Front Cell Infect Microbiol 2012; 2: 70–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lind J, Backert S, Pfleiderer K, et al. Systematic analysis of phosphotyrosine Abs recognizing single phosphorylated EPIYA-motifs in CagA of Western-type Helicobacter pylori strains. PLOS ONE 2014; 9: e96488–e96488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pachathundikandi SK, Brandt S, Madassery J, et al. Induction of TLR-2 and TLR-5 expression by Helicobacter pylori switches cagPAI-dependent signaling leading to the secretion of IL-8 and TNFα. PLOS ONE 2011; 6: e19614–e19614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Moese S, Selbach M, Zimny-Arndt U, et al. Identification of a tyrosine-phosphorylated 35 kDa carboxy-terminal fragment (p35CagA) of the Helicobacter pylori CagA protein in phagocytic cells: processing or breakage? Proteomics 2001; 1: 618–629. [DOI] [PubMed] [Google Scholar]
- 36.Daigneault M, Preston JA, Marriott HM, et al. The identification of markers of macrophage differentiation in PMA-stimulated THP-1 cells and monocyte-derived macrophages. PLOS ONE 2010; 5: e8668–e8668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mueller D, Tegtmeyer N, Brandt S, et al. c-Src and c-Abl kinases control hierarchic phosphorylation and function of the CagA effector protein in Western and East Asian Helicobacter pylori strains. J Clin Invest 2012; 122: 1553–1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tegtmeyer N, Lind J, Schmid B, et al. Helicobacter pylori CagL Y58/E59 mutation turns-off type IV secretion-dependent delivery of CagA into host cells. PLOS ONE 2014; 9: e97782–e97782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Saha A, Backert S, Hammond CE, et al. Helicobacter pylori CagL activates ADAM17 to induce repression of the gastric H, K-ATPase alpha subunit. Gastroenterology 2010; 139: 239–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tegtmeyer N, Hartig R, Delahay RM, et al. A small fibronectin-mimicking protein from bacteria induces cell spreading and focal adhesion formation. J Biol Chem 2010; 285: 23515–23526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Boehm M, Hoy B, Rohde M, et al. Rapid paracellular transmigration of Campylobacter jejuni across polarized epithelial cells without affecting TER: role of proteolytic-active HtrA cleaving E-cadherin but not fibronectin. Gut Pathog 2012; 4: 3–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hoy B, Geppert T, Boehm M, et al. Distinct roles of secreted HtrA proteases from gram-negative pathogens in cleaving the junctional protein and tumor suppressor E-cadherin. J Biol Chem 2012; 287: 10115–10120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tegtmeyer N, Rivas Traverso F, Rohde M, et al. Electron microscopic, genetic and protein expression analyses of Helicobacter acinonychis strains from a Bengal tiger. PLOS ONE 2013; 8: e71220–e71220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hirsch C, Tegtmeyer N, Rohde M, et al. Live Helicobacter pylori in the root canal of endodontic-infected deciduous teeth. J Gastroenterol 2012; 47: 936–940. [DOI] [PubMed] [Google Scholar]
- 45.Aganna E, Martinon F, Hawkins PN, et al. Association of mutations in the NALP3/CIAS1/PYPAF1 gene with a broad phenotype including recurrent fever, cold sensitivity, sensorineural deafness, and AA amyloidosis. Arthritis Rheum 2002; 46: 2445–2452. [DOI] [PubMed] [Google Scholar]
- 46.Gerhard DS, Wagner L, Feingold EA, et al. The status, quality, and expansion of the NIH full-length cDNA project: the Mammalian Gene Collection (MGC). Genome Res 2004; 14: 2121–2127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Winter J, Jung S, Keller S, et al. Many roads to maturity: microRNA biogenesis pathways and their regulation. Nat Cell Biol 2009; 11: 228–234. [DOI] [PubMed] [Google Scholar]
- 48.Griffiths-Jones S, Hui JH, Marco A, et al. MicroRNA evolution by arm switching. EMBO Rep 2011; 12: 172–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Haneklaus M, Gerlic M, Kurowska-Stolarska M, et al. Cutting edge: miR-223 and EBV miR-BART15 regulate the NLRP3 inflammasome and IL-1β production. J Immunol 2012; 189: 3795–3799. [DOI] [PubMed] [Google Scholar]
- 50.Guarda G, Braun M, Staehli F, et al. Type I interferon inhibits interleukin-1 production and inflammasome activation. Immunity 2011; 34: 213–223. [DOI] [PubMed] [Google Scholar]
- 51.Greenhill CJ, Jones GW, Nowell MA, et al. Interleukin-10 regulates the inflammasome-driven augmentation of inflammatory arthritis and joint destruction. Arthritis Res Ther 2014; 16: 419–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gurung P, Li B, Subbarao Malireddi RK, et al. Chronic TLR stimulation controls NLRP3 inflammasome activation through IL-10 mediated regulation of NLRP3 expression and caspase-8 activation. Sci Rep 2015; 5: 14488–14488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hofmann SR, Kubasch AS, Ioannidis C, et al. Altered expression of IL-10 family cytokines in monocytes from CRMO patients result in enhanced IL-1β expression and release. Clin Immunol 2015; 161: 300–307. [DOI] [PubMed] [Google Scholar]
- 54.von Moltke J, Ayres JS, Kofoed EM, et al. Recognition of bacteria by inflammasomes. Annu Rev Immunol 2013; 31: 73–106. [DOI] [PubMed] [Google Scholar]
- 55.Dinarello CA. Immunological and inflammatory functions of the interleukin-1 family. Annu Rev Immunol 2009; 27: 519–550. [DOI] [PubMed] [Google Scholar]
- 56.Jung HC, Kim JM, Song IS, et al. Helicobacter pylori induces an array of pro-inflammatory cytokines in human gastric epithelial cells: quantification of mRNA for interleukin-8; -1 alpha/beta, granulocyte-macrophage colony-stimulating factor, monocytechemoattractant protein-1 and tumour necrosis factor-alpha. J Gastroenterol Hepatol 1997; 12: 473–480. [DOI] [PubMed] [Google Scholar]
- 57.Kameoka S, Kameyama T, Hayashi T, et al. Helicobacter pylori induces IL-1b protein through the inflammasome activation in differentiated macrophagic cells. Biomed Res 2016; 37: 21–27. [DOI] [PubMed] [Google Scholar]
- 58.Pérez-Figueroa E, Torres J, Sánchez-Zauco N, et al. Activation of NLRP3 inflammasome in human neutrophils by Helicobacter pylori infection. Innate Immun 2016; 22: 103–112. [DOI] [PubMed] [Google Scholar]
- 59.Hentze H, Lin XY, Choi MS, et al. Critical role for cathepsin B in mediating caspase-1-dependent interleukin-18 maturation and caspase-1-independent necrosis triggered by the microbial toxin nigericin. Cell Death Differ 2003; 10: 956–968. [DOI] [PubMed] [Google Scholar]
- 60.Neudecker V, Haneklaus M, Jensen O, et al. Myeloid-derived miR-223 regulates intestinal inflammation via repression of the NLRP3 inflammasome. J Exp Med 2017; 214: 1737–1752. [DOI] [PMC free article] [PubMed]
- 61.Ma L, Chen Y, Zhang B, et al. Increased microRNA-223 in Helicobacter pylori-associated gastric cancer contributed to cancer cell proliferation and migration. Biosci Biotechnol Biochem 2014; 78: 602–608. [DOI] [PubMed]
- 62.Shao W, Yeretssian G, Doiron K, et al. The caspase-1 digestome identifies the glycolysis pathway as a target during infection and septic shock. J Biol Chem 2007; 282: 36321–36329. [DOI] [PubMed] [Google Scholar]
- 63.Kim J, Cho YA, Choi IJ, et al. Effects of interleukin-10 polymorphisms, Helicobacter pylori infection, and smoking on the risk of noncardia gastric cancer. PLOS ONE 2012; 7: e29643–e29643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Assis S, Marques CR, Silva TM, et al. IL-10 single nucleotide polymorphisms are related to upregulation of constitutive IL-10 production and susceptibility to Helicobacter pylori infection. Helicobacter 2014; 19: 168–173. [DOI] [PubMed] [Google Scholar]
- 65.Hawrylowicz CM, O'Garra A. Potential role of interleukin-10-secreting regulatory T cells in allergy and asthma. Nat Rev Immunol 2005; 5: 271–283. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.