

Short abstract
In this study we investigated the association between intestinal dysbacteriosis with colorectal cancer progress and the underlying molecular mechanisms. Tumor progression was evaluated using xenograft mice model. The epithelial-mesenchymal transition (EMT) markers were quantified by both real-time PCR and immunoblotting. The serum content of IL-6 and TNF-α were measured with ELISA kits. Cell proliferation was determined by the Cell Counting Kit-8. Intestinal dysbacteriosis was successfully simulated by the administration of a large dose of antibiotics and was demonstrated to promote xenograft tumor growth and induce EMT. Accordingly, the serum concentrations of cytokines IL-6 and TNF-α were significantly increased. Furthermore, the production and secretion of IL-6 and TNF-α were remarkably elevated in macrophages isolated from intestinal dysbiotic mice in comparison with the normal counterparts, and conditioned medium from these was shown to significantly stimulate EMT process in HT29 cells in vitro. Macrophage depletion completely abrogated the pro-tumor effect of intestinal dysbacteriosis. Our results suggest that intestinal dysbacteriosis stimulates macrophage activation and subsequently induces EMT process via secreted pro-inflammatory cytokines IL-6 and TNF-α.
Keywords: Intestinal dysbacteriosis, macrophage, EMT, colorectal tumor, IL-6
Introduction
Colorectal cancer (CRC) is one of the most common malignances associated with the colon or rectum, and the fourth cause of cancer-related mortality.1 More than 1 million new cases are diagnosed per year and 715,000 deaths were reported in 2010. The incidence of CRC has been epidemiologically demonstrated to be intimately associated with old age and poor lifestyle, with hereditary genetic deficiency accounting for only a minority of cases.2 Well-recognized risk factors include diet, obesity, smoking and lack of exercise.3 Notably, chronic inflammatory bowel disease is increasingly understood to contribute to the tumorigenesis of CRC.4 The mainstream diagnostic standard for this disease is medical imaging, further confirmed with biopsy analysis of colon tissues acquired during sigmoidoscopy and colonoscopy. Clinical therapeutics mainly include surgical resection, radiation, chemotherapy, targeted therapy or combinations of these, based on empirical judgements with respect to individual health conditions and tumor stage.5
Evidence is accumulating to support the fundamental role of gut microbiome in the tumor biology of CRC.6 Although specific microbial infections are recognized as being associated with some human cancers, the scenario is quite distinct for the etiology of CRC, because of the rich diversity of microbes in the colorectum milieu. The emerging model suggests that intestinal dysbacteriosis stimulates mucosal immune response, which forges local chronic inflammatory reaction and consequently imposes detrimental effects on epithelial cells and contributes to tumorigenesis, along with other factors. Several hypotheses have been proposed to explain the intersections between colonic microbiota, immune response and malignant transformation. For instance, the alpha-bug hypothesis put forward by Sears and Pardoll suggests that CRC is initiated primarily by certain microbiota members with specific virulence determinants.7 The driver–passenger hypothesis proposed by Tjalsma suggests that the tumorigenesis of CRC initiated by the driver microbe may provoke the essential changes in the local milieu of infection, which in turn progresses to the loss of this initiator due to out-competition by other commensal bacteria (passengers).8
It is well recognized that chronic inflammation shapes a local milieu that is conducive to tumor initiation and progression. Tumor-associated macrophages (TAMs) are multifarious groups of cells originating from either peritumoral tissue or bone marrow and can be roughly categorized into two main subtypes, M1 and M2. The infiltration of M1 TAMs in the early stages of tumorigenesis inhibits tumor progression, whereas M2 predominantly promotes tumor growth.9,10 It has been reported that a high density of infiltrated macrophages is associated with advanced stages and poor prognosis.11,12 TAMs produce and secret a variety of cytokines and soluble factors involved in multiple biological processing during tumor initiation and progression, ranging from cell proliferation, cell survival, angiogenesis to epithelial-mesenchymal transition (EMT) and cancer stem cell etiology.13 EMT is one of the radical steps mediating intravasation and metastasis of localized tumor cells, which is frequently characterized in advanced human malignancies and is intimately associated with tumor progression and therapy resistance. Activation of EMT necessarily requires crosstalk between tumor cells and other components of the local microenvironment; therefore, acquiring a deeper insight into the signaling pathway interconnecting TAMs and EMT cascades may reveal opportunities to exploit promising therapeutics targeting tumor metastasis.
In this study, we systematically investigated the interactions between intestinal dysbacteriosis, macrophage activation and EMT in CRC using a xenograft mouse model. Intestinal dysbiosis was well-recapitulated by oral administration of a large dose of antibiotics.14 And the predominant role of macrophage in tumor progression in intestinal dysbiotic mice was further consolidated by clodronate-mediated depletion.15 Our data demonstrated, for the first time, that intestinal dysbacteriosis stimulated macrophages, while the resultant increase of circulating IL-6 and TNF-α consequently promoted EMT processing and tumor progression.
Material and methods
Intestinal dysbiosis mouse model
We obtained 8-wk old male C57BL/6 mice from Vital River Laboratory Animal Technology (Beijing, China) and then allowed 1 wk for acclimation. The animals were housed in a standard specific-pathogen-free (SPF) environment. The protocol for animal study was approved by the Committee of Animal Use and Care at Shanghai University of Traditional Chinese Medicine Affiliated PUTUO Hospital. To establish an intestinal dysbiosis model, mice were provided with ampicillin (1 g/l from Sigma, MO, USA), vancomycin (0.5 g/l from Abbott Labs), neomycin (1 g/l from Pharmacia), and metronidazole (1 g/l from Sidmack Labs) dissolved in drinking water for 4 wk. Given the relatively poor stability of antibiotics, fresh drinking water was replaced daily. A low concentration of sucrose was also supplemented for taste improvement purposes. A window of 4-wk antibiotics treatment referred to the previously established protocol to completely deplete the commensal as verified by bacteriologic analysis of colonic feces.
CRC cell lines
The human CRC cell line HT29 was obtained and authenticated by the American Type Culture Collection (ATCC, MA, USA). The cells were maintained in RPMI-1640 medium supplemented with 10% FBS and 1% penicillin-streptomycin-glutamine. Cells were cultured in a humidified incubator at 37°C with 5% CO2. The exponentially growing cells were harvested for further analysis.
CRC xenograft
HT29 cells at log phase were harvested by trypsin digestion and a single-cell suspension was prepared in HEPES buffer on ice. The cell suspension was mixed with an equal volume of Matrigel (BD BioSciences, CA, USA). The indicated mixture (6 × 106 cells/200 µl) was subcutaneously inoculated into the lower flank of mice with caution. The growth of xenograft tumor was monitored regular and the volume was estimated with the following formula: volume = (width)2 × length/2.
RT-PCR
RNA was extracted from indicated cells using TRIzol Reagent (ThermoFisher, MA, USA) in accordance with the manufacturer’s instruction. The quality and quantity of RNA was determined by BioAnalyzer 2100 (Agilent, CA, USA) prior to further processing. Reverse transcription was performed with the commercially available High-Capacity cDNA Reverse Transcription Kit (ThermoFisher, MA, USA) and the real-time PCR was performed with the PowerUp SYBR Green Master Mix (ThermoFisher, MA, USA), both in accordance with the manufacturer’s instructions. The relative expression was calculated using the 2-ΔΔCt method and normalized to GAPDH. The primers were listed as below:
human-E-cadherin-Forward: 5’-GTCGAGGGAAAAATAGGCTG-3’
human-E-cadherin-Reverse: 5’-GCCGAGAGCTACACGTTCAC-3’
human-N-cadherin-Forward: 5’-GGCATACACCATGCCATCTT-3’
human-N-cadherin-Reverse: 5’-GTGCATGAAGGACAGCCTCT-3’
human-Vimentin-Forward: 5’-GCAAAGATTCCACTTTGCGT-3’
human-Vimentin-Reverse: 5’-GAAATTGCAGGAGGAGATGC-3’
human-GAPDH-Forward: 5’-GGAGCGAGATCCCTCCAAAAT-3’
human-GAPDH-Reverse: 5’-GGCTGTTGTCATACTTCTCATGG-3’
Western blot
Cell lysate was prepared in radioimmunoprecipitation assay buffer lysis buffer and debris completely removed by refrigerated centrifugation. The protein concentration was determined by the bicinchoninic acid Protein Assay Kit (ThermoFisher, MA, USA). An equal amount of protein was resolved by SDS-PAGE and then transferred onto polyvinylidene difluoride (PVDF) membrane on ice. After brief blocking with 5% skim milk in Tris-buffered saline with 0.1% Tween-20 (TBST), the PVDF membrane was hybridized with indicated primary Abs (anti-human-E-cadherin, 1:1000, #14472; anti-human-N-cadherin, 1:1000, #14215; anti-human-vimentin, 1:1000, #3390; anti-mouse-IL-6, 1:1000, #12912; anti-mouse-TNF-α, 1:1000, #11948; anti-GAPDH, 1:1000, #2118; Cell Signaling Technology, MA, USA) at 4°C overnight. The unbound Abs were washed off with TBST next day and incubated with specific secondary Abs (anti-rabbit IgG, HRP-linked Ab, 1:5000, #7074; anti-mouse IgG, HRP-linked Ab, 1:5000, #7076; Cell Signaling Technology, MA, USA) at room temperature (25°C) for 1 h. The PVDF membrane was then rigorously washed with TBST for 30 min and target protein bands were visualized using the Enhanced Chemiluminescence Kit (Millipore, UT, USA) in accordance with the manufacturer’s instructions. The endogenous GAPDH was employed as loading control.
ELISA
Blood samples from indicated mice were collected and sera were prepared in the serum separator tube. The serous content of IL-6 and TNF-α was determined with commercially available ELISA kits IL-6 High Sensitivity Human ELISA Kit ab46042 and Human TNF-α ELISA Kit ab181421 (Abcam, MA, USA) in according to the manufacturer’s instruction. Briefly, 100 µl sample and 50 µl biotinylated Ab solution was added into each well and allowed for 3-h incubation at room temperature. After wash, 100 µl of streptavidin-HRP solution was added and incubated at room temperature for 30 min. The chromogenic reaction was performed at room temperature with 100 µl of TMB solution in the dark for 15 min, and terminated with 100 µl of Stop Reagent. The absorption at 450 nm was recorded using the SpectraMax Plus 384 Microplate Reader (Molecular Devices, CA, USA) and concentration was calculated using purified recombinant mouse cytokine proteins as spiked standard.
Isolation of mouse macrophages
The C57BL6 mice were sacrificed by cervical dislocation and 7 ml of ice-cold sterile PBS was intraperitoneally perfused. After gentle massage on abdomen for 3 min, the ascites was collected by aspiration. The macrophages were recovered by centrifugation, washed with DMEM containing 10% FBS twice and subjected to standard cell culture. Each isolated batch was examined by flow cytometry to confirm that macrophages were the dominant population, before that batch was used for experiments.
Proliferation assay
Cell proliferation was determined by commercial Cell Counting Kit-8 (Dojindo, Dalian, China) in accordance with the manufacturer’s instruction. Briefly, the exponentially growing cells were seeded into 96-well plate in triplicate and cultured in a humidified CO2 incubator for 24 h. Ten µl of CCK-8 solution was added into each well and incubated at 37°C for 1 h. The absorption at 450 nm was measured by the microplate reader and relative cell viability was calculated.
Depletion of mouse macrophages
HT29 xenografts were established as described previously in both control and intestinal dysbiosis mice 15. After 2 wk, the tumor-bearing intestinal dysbiotic mice were randomly divided into two groups and subjected to either intravenous injection (tail vein) of 100 µl clodronate-liposomes solution (5 mg/10 g body mass, clodLIP B.V.) or saline every 3 d. The successful depletion of macrophages has been previously shown in liver, spleen, bone marrow and precursor blood monocytes. To monitor the tumor latency, mice were palpated and tumor size was measured with digital calliper every other day.
Statistical analysis
All data presented in this study were acquired from at least three independent experiments. Data analysis was performed with SPSS 23.0 software. The one-way ANOVA was employed for statistical comparison. The P value was calculated and P < 0.05 was considered as statistical significance.
Results
Intestinal dysbacteriosis promotes growth of xenograft tumor
To investigate the potential association of intestinal dysbacteriosis with tumor progression, here we first established intestinal dysbiosis model in HT29 xenograft tumor mice with large dose of antibiotics following the previously established protocol. The tumor growth was monitored up to 3 wk post-inoculation. As shown in Figure 1a, the xenograft tumor size in intestinal dysbiotic mice was significantly higher than control. Furthermore, the tumor mass was determined at the endpoint while experimental mice were sacrificed, which showed significant increase in intestinal dysbacteriosis group in comparison with control mice (Figure 1b). Our data demonstrated that intestinal dysbiosis promoted tumor progression in vivo.
Figure 1.

Intestinal dysbacteriosis promotes growth of xenograft tumor from inoculated colorectal cancer cells. (a) Colorectal cancer cell HT29 was inoculated into control and intestinal dysbacteriosis (ID) mice, respectively (n = 12), followed by measurements of tumor size on indicated days after cell inoculation. (b) On d 21, mice from both groups were sacrificed to extract the xenograft and weigh the tumor. Data were shown as mean ± SD (n = 12 each). *P < 0.05, **P < 0.01, compared with control.
Intestinal dysbacteriosis promotes EMT in xenograft tumor from inoculated CRC cells
Our above results suggested that intestinal dysbiosis significantly exacerbated xenograft tumor growth, next we sought to focus on the fundamental alterations at the molecular level. The tumor tissues were collected from both groups and the specific EMT markers were characterized. As shown in Figure 2a, the epithelial marker E-cadherin was significantly suppressed in intestinal dysbacteriosis mice in comparison with control (0.42 ± 0.09 vs. 1.0 ± 0.12). On the contrary, the mesenchymal markers N-cadherin and vimentin were markedly induced in intestinal dysbiotic animals (3.8 ± 0.4 vs. 1.0 ± 0.4 and 4.0 ± 0.4 vs. 1.0 ± 0.3, Figure 2b). We further confirmed our observations at protein levels via Western blotting. Correspondingly, the E-cadherin protein was decreased while N-cadherin and vimentin were increased compared with the control (Figure 2c). Our data demonstrated that EMT was significantly stimulated in intestinal dysbiosis mice.
Figure 2.

Intestinal dysbacteriosis promotes epithelial-mesenchymal transition in xenograft tumor from inoculated colorectal cancer cells. Colorectal cancer cell HT29 was inoculated into control and intestinal dysbacteriosis (ID) mice, respectively (n = 12). On d 21, mice from both groups were sacrificed to extract the xenograft tumor, followed by analyses of (a) E-cadherin mRNA, (b) N-cadherin and vimentin mRNA, and (c) their protein levels. Data were shown as mean ± SD (n = 12 each). *P < 0.05, **P < 0.01, compared with control.
Intestinal dysbacteriosis increases serum concentrations of cytokines IL-6 and TNF-α in experimental mice
We further evaluated the immune response in xenograft tumor mice with intestinal dysbiosis. Serum was collected from each group at the endpoint of experiments. The relative contents of inflammatory cytokines IL-6 and TNF-α were determined with commercial ELISA kits. Our results demonstrated that both serum IL-6 and TNF-α were significantly increased in intestinal dysbiotic mice in comparison with control ones (Figure 3a and b). The average level of IL-6 was about 11.8 ng/ml in ID mice while 4.2 ng/ml in control. Likewise, the concentration of TNF-α increased from 3 ng/ml in control mice to 14 ng/ml in intestinal dysbiotic mice. Our data indicated the notable inflammation in peripheral blood of intestinal dysbiotic mice with xenograft tumors.
Figure 3.
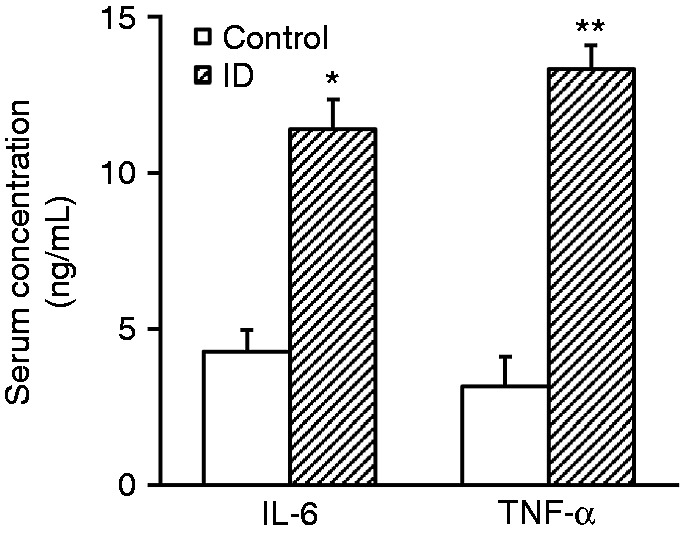
Intestinal dysbacteriosis increases serum concentrations of cytokines IL-6 and TNF-α in experimental mice. Serum was collected from both control and intestinal dysbacteriosis (ID) mice, respectively, followed by analyses of IL-6 and TNF-α concentrations by ELISA. Data were shown as mean ± SD (n = 12 each). *P < 0.05, **P < 0.01, compared with control.
Intestinal dysbacteriosis increases expression and secretion of cytokines IL-6 and TNF-α in the macrophages isolated from experimental mice
Next, we attempted to source the secretory IL-6 and TNF-α in intestinal dysbiotic mice. To this purpose, we first isolated macrophages from the experimental mice via peritoneal perfusion. The condition medium was collected from in vitro cultured macrophages and subjected to analysis of IL-6 and TNF-α contents. Consistent with our previous observations from peripheral blood, the concentrations of IL-6 and TNF-α were significantly higher in the conditioned medium from intestinal dysbiotic macrophages than control, both by Western blotting (Figure 4a) and ELISA analysis (Figure 4b, 25 ± 4 ng/ml vs. 5 ± 2 ng/ml and 22.5 ± 4.5 ng/ml vs. 6.1 ± 1.9 ng/ml). Our data suggested that macrophages from intestinal dysbacteriosis mice were more proficient in production and secretion of inflammatory cytokines.
Figure 4.
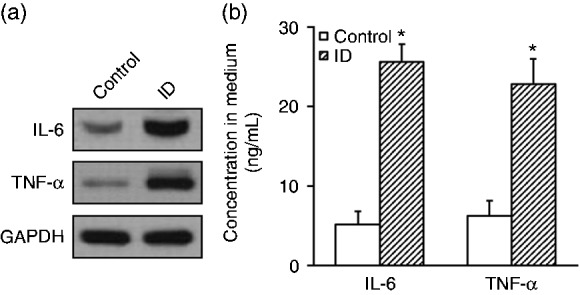
Intestinal dysbacteriosis increases expression and secretion of cytokines IL-6 and TNF-α in the macrophages isolated from experimental mice. Macrophages were isolated from both control and intestinal dysbacteriosis (ID) mice, respectively, followed by analyses of (a) IL-6 and TNF-α protein expressions by Western blot and (b) their secretions into the medium by ELISA. Data were shown as mean ± SD from at least three independent experiments. *P < 0.05, compared with control.
Conditional medium from macrophages isolated from intestinal dysbacteriosis mice promotes proliferation and EMT of HT29 cells in vitro
Next, we further determined the potential effect of intestinal dysbacteriosis macrophages on the malignance behaviours of HT29. Conditioned medium from isolated macrophages was applied to HT29 culture in vitro, and cell proliferation and EMT transition were evaluated accordingly. As shown in Figure 5a, the relative cell viability was significantly stimulated by the conditioned medium from macrophages isolated from intestinal dysbiotic mice in comparison with the controls (1.42 ± 0.16 vs. 1.0 ± 0.15). In addition, the epithelial marker E-cadherin was greatly inhibited and mesenchymal marker N-cadherin and vimentin were induced in response to intestinal dysbacteriosis macrophage conditioned medium co-culture, which was consistent with our in vivo observation (Figure 5b). Our results suggested that the secretory cytokines from macrophages isolated from intestinal dysbiotic mice significantly contributed to both cell proliferation and EMT of CRC.
Figure 5.
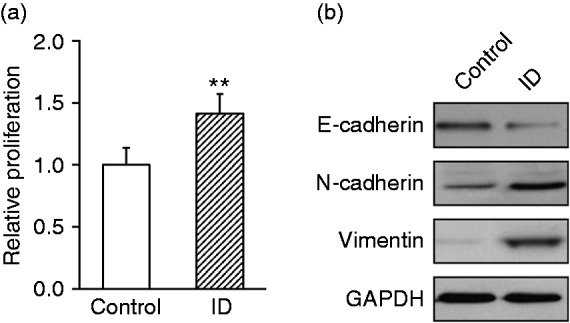
Conditional medium from macrophages isolated from intestinal dysbacteriosis mice promotes proliferation and epithelial-mesenchymal transition of HT29 cells in vitro. Macrophages were isolated from both control and intestinal dysbacteriosis (ID) mice, and cultured in vitro to collect their respective conditional medium. HT29 cells were then cultured with these two types of collected conditional medium, respectively, followed by assessments of (a) proliferation and (b) protein levels of E-cadherin, N-cadherin and vimentin. Data were shown as mean ± SD from at least three independent experiments. **P < 0.01, compared with control.
Growth promotion of xenograft tumor by intestinal dysbacteriosis requires macrophages
Next, we sought to assess the critical role of macrophages in the intestinal dysbacteriosis milieu on tumor growth in vivo. To this purpose, macrophages were completely depleted in the HT29 xenograft tumor-bearing mice via administration of a large dose of combinational antibiotics. The tumor growth was monitored and compared between control, intestinal dysbiotic mice and intestinal dysbiosis plus macrophage-depleted mice. As shown in Figure 6a, the xenograft tumor progression was significantly accelerated in the mice with intestinal dysbiosis in comparison with the controls, which was readily reversed by macrophage depletion. Similar results were observed with respect to tumor mass measured at the endpoint of experiments. The average mass of xenograft tumor increased from 302 ± 45 g in control to 408 ± 30 g in intestinal dysbacteriosis mice, whereas decreased to 275 ± 43 g in intestinal dysbacteriosis mice accompanied with macrophage depletion (Figure 6b). Our data consolidated the indispensable role of macrophages to promote xenograft growth in intestinal dysbiotic mice in vivo.
Figure 6.

Growth promotion of xenograft tumor by intestinal dysbacteriosis requires macrophages. (a) Colorectal cancer cell HT29 was inoculated into control + sham, intestinal dysbacteriosis (ID) + sham and ID-macrophage mice, respectively (n = 12), followed by measurements of tumor size on indicated days after cell inoculation. (b) On d 21, mice from all groups were sacrificed to extract the xenograft and weigh the tumor. Data were shown as mean ± SD (n = 12 each). *P < 0.05, **P < 0.01, compared with control + sham. #P < 0.05, compared with ID + sham.
EMT promotion of xenograft tumor by intestinal dysbacteriosis requires macrophages
Our previous data demonstrated that the pro-tumor effect of intestinal dysbacteriosis required macrophages; next, we sought to address its influence on the molecular events with respect to EMT, which has been fundamentally implicated in the malignancy progression. The relative expressions of E-cadherin, N-cadherin and vimentin were determined in the indicated mice at both transcript and protein levels. As shown in Figure 7a, E-cadherin transcript was remarkably inhibited in the intestinal dysbiotic mice in comparison with the controls, which was subsequently restored by macrophage depletion. Conversely, both N-cadherin and vimentin were induced in intestinal dysbacteriosis group whereas they were suppressed in response to macrophage deficiency (Figure 7b). The consistent changes were further confirmed by Western blotting (Figure 7c).
Figure 7.

EMT promotion of xenograft tumor by intestinal dysbacteriosis requires macrophages. Colorectal cancer cell HT29 was inoculated into control + sham, intestinal dysbacteriosis (ID) + sham and ID-macrophage mice, respectively (n = 12). On d 21, mice from all groups were sacrificed to extract the xenograft tumor, followed by analyses of (a) E-cadherin mRNA, (b) N-cadherin and vimentin mRNA, and (c) their protein levels. Data were shown as mean ± SD (n = 12 each). *P < 0.05, **P < 0.01, compared with control + sham. ##P < 0.01, #P < 0.05, compared with ID + sham.
Discussion
Intestinal dysbacteriosis is a risk factor linked to a range of human diseases such as periodontal disease, inflammatory bowel, chronic fatigue syndrome, obesity, cancer, bacterial vaginosis, and colitis.16 The dysbiosis is commonly caused by repeated and inappropriate antibiotic exposure, alcohol misuse and inappropriate diets.17 In this study, we concentrated on the etiology of CRC with regard to its complicated microenvironment and commensal microbiota. The dysbiotic conditions were well-recapitulated in mice with over-dosed antibiotics administrated in drinking water. The intestinal dysbacteriosis significantly promoted xenograft tumor progression in our system. In addition, we demonstrated that intestinal dysbacteriosis stimulated EMT. The peripheral inflammatory cytokines such as IL-6 and TNF-α were significantly increased in response to the intestinal dysbacteriosis occurrence. Consistently, the production and secretion of IL-6 and TNF-α in the isolated macrophages from dysbiotic mice were higher than those in normal controls. Furthermore, using a co-culture system, we demonstrated that conditioned medium from dysbiotic macrophages significantly stimulated the EMT process in HT29 cells. The exclusive role of macrophages in the pro-tumor effect of intestinal dysbacteriosis was highlighted in our macrophage-depletion experiment, wherein both the xenograft tumor growth and EMT were completely abolished.
EMT is the process by which the epithelial cells lose their cell polarity and cell–cell attachment, and subsequently gain migratory and invasive capacity and transform to mesenchymal stem cells.18 EMT is essential for a range of physiological processes including mesoderm formation and neural tube formation during development, and has been shown to be involved in wound healing, organ fibrosis and initiation of metastasis in tumor progression.19 Loss of E-cadherin is considered as the fundamental change during EMT, followed by increase in mesenchymal markers such as N-cadherin and vimentin. Many transcription factors have been shown to repress E-cadherin expression. For example, SNAI1, SNAI2, ZEB1, ZEB2, TCF3 and KLF8 can bind to specific motifs in E-cadherin promoter and inhibit its transcription.20 Signaling pathways including TGF-β, FGF, EGF, HGF, Wnt/β-catenin, Notch and hypoxia have been shown to be involved in EMT induction in addition.18 Furthermore, cancer-related inflammation was recently characterized in the stimulation of EMT. For instance, Wang et al. reported that TNF-α-induced EMT required AKT/GSK-3β-mediated stabilization of Snail in CRC.21 Huang et al. demonstrated that miR-19a was associated with lymph metastasis and mediated the TNF-α-induced EMT in CRC.22 Bhat et al. further showed that claudin-1 promoted TNF-α-induced EMT and migration in colorectal adenocarcinoma cells.23 Zhang et al. reported that TNF-α-induced EMT and increased stemness properties in renal cell carcinoma cells.24 With respect to IL-6, Rokavec et al. demonstrated that an IL-6R/STAT3/miR-34a feedback loop promoted EMT-mediated CRC invasion and metastasis.25 Castellana et al. proposed that interplay between YB-1 and IL-6 promoted the metastatic phenotype in breast cancer cells.26 Lee et al. further showed that IL-6 promoted growth and EMT of CD133+ cells in non-small cell lung cancer.27 Chen et al. reported that TNF-α-inducing protein of Helicobacter pylori induced EMT in gastric cancer cells through activation of the IL-6/STAT3 signaling pathway.28 In agreement with all the above mentioned observations, in our study we demonstrated that elevated serum content of both IL-6 and TNF-α in intestinal dysbacteriosis was positively correlated with EMT process in xenograft tumor, and conditioned medium containing both cytokines significantly stimulated EMT phenotype in HT29 cells in vitro. Of note is that the detailed molecular signaling pathway underlying the IL-6/TNF-α-regulated EMT markers is still to be defined in subsequent investigations.
TAMs hold fundamental roles in tumor biology, including initiation, progression, metastasis and therapy resistance, and targeted depletion of TAMs represents a mainstream direction for therapeutic exploitation. For example, Wu et al. demonstrated that depletion of M2-like TAMs delayed cutaneous T-cell lymphoma development in vivo.29 Patwardhan et al. proposed that sustained inhibition of receptor tyrosine kinases and macrophage depletion by PLX3397 and rapamycin as a potential new approach for the treatment of malignant peripheral nerve sheath tumor.30 Tham et al. demonstrated that macrophage depletion reduced post-surgical tumor recurrence and metastatic growth in a spontaneous murine model of melanom.31 Zhang et al. showed that depletion of TAMs enhanced the effect of sorafenib in metastatic liver cancer models through anti-metastatic and anti-angiogenic effects.32 In addition, depletion of TAMs has been shown to enhance the anti-tumor immunity induced by a TLR agonist-conjugated peptide.33 In combination with anti-coagulant therapy, Shashkova et al. demonstrated macrophage depletion increased therapeutic window of systemic treatment with oncolytic adenovirus.34 Likewise, in our xenograft tumor model with intestinal dysbacteriosis, the clodronate liposomes-mediated macrophage depletion remarkably inhibited tumor progression and EMT. With regard to the aberrant activation of TAMs in dysbiosis animals, our data highlighted the potency of macrophage depletion-based immunotherapy against this disease. Notably, we would consolidate our major observations in the immunodeficient animal model in future work to exclude the potential involvement of host graft rejection response. In addition, metronidazole was reported to induce immunosuppression in Balb/c mice, causing a reduction in macrophages and TNF-α production,35 hinting that variations in the genetical background of mouse strains could contribute to different immune responses, as has been previously suggested.36 Therefore, further experiments involving a different mouse strain and different method to induce intestinal dysbacteriosis are needed to verify the generality of our results.
Conclusions
In summary, in this study we demonstrate that intestinal dysbacteriosis significantly stimulates macrophage activation, which in turn promotes production and secretion of inflammatory cytokines IL-6 and TNF-α. The elevated peripheral IL-6 and TNF-α subsequently promote the EMT process of CRC, and eventually contribute to tumor progression and metastasis. Our study highlights the fundamental role of macrophages and cytokines such as IL-6 and TNF-α in tumor progression in dysbiotic mice, indicating potential targets for intervention and therapeutic exploitation.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported by research projects at Shanghai University of Traditional Chinese Medicine Affiliated PUTUO Hospital (No:2016315A) and Shanghai University of Traditional Chinese Medicine Affiliated PUTUO Hospital (No: PTZP201616A).
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin 2016; 66: 7–30. [DOI] [PubMed] [Google Scholar]
- 2.Brenner H, Kloor M, Pox CP. Colorectal cancer. Lancet 2014; 383: 1490–1502. [DOI] [PubMed] [Google Scholar]
- 3.Movahedi M, Bishop DT, Macrae F, et al. Obesity, aspirin, and risk of colorectal cancer in carriers of hereditary colorectal cancer: a prospective investigation in the capp2 study. J Clin Oncol 2015; 33: 3591–3597. [DOI] [PubMed] [Google Scholar]
- 4.Grivennikov SI. Inflammation and colorectal cancer: colitis-associated neoplasia. Semin Immunopathol 2013; 35: 229–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.De Rosa M, Pace U, Rega D, et al. Genetics, diagnosis and management of colorectal cancer (Review). Oncol Rep 2015; 34: 1087–1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Van Raay T, Allen-Vercoe E. Microbial interactions and interventions in colorectal cancer. Microbiol Spectr 2017; 5. [DOI] [PubMed] [Google Scholar]
- 7.Sears CL, Pardoll DM. Perspective: alpha-bugs, their microbial partners, and the link to colon cancer. J Infect Dis 2011; 203: 306–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tjalsma H, Boleij A, Marchesi JR, et al. A bacterial driver-passenger model for colorectal cancer: beyond the usual suspects. Nat Rev Microbiol 2012; 10: 575–582. [DOI] [PubMed] [Google Scholar]
- 9.Noy R, Pollard JW. Tumor-associated macrophages: from mechanisms to therapy. Immunity 2014; 41: 49–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cassetta L, Kitamura T. Targeting tumor-associated macrophages as a potential strategy to enhance the response to immune checkpoint inhibitors. Front Cell Dev Biol 2018; 6: 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Biswas SK, Allavena P, Mantovani A. Tumor-associated macrophages: functional diversity, clinical significance, and open questions. Semin Immunopathol 2013; 35: 585–600. [DOI] [PubMed] [Google Scholar]
- 12.Zhong X, Chen B, Yang Z. The role of tumor-associated macrophages in colorectal carcinoma progression. Cell Physiol Biochem 2018; 45: 356–365. [DOI] [PubMed] [Google Scholar]
- 13.Zhang J, Yao H, Song G, et al. Regulation of epithelial-mesenchymal transition by tumor-associated macrophages in cancer. Am J Transl Res 2015; 7: 1699–1711. [PMC free article] [PubMed] [Google Scholar]
- 14.Kanwal S, Joseph TP, Owusu L, et al. A polysaccharide isolated from Dictyophora indusiata promotes recovery from antibiotic-driven intestinal dysbiosis and improves gut epithelial barrier function in a mouse model. Nutrients 2018; 10: e1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Carron EC, Homra S, Rosenberg J, et al. Macrophages promote the progression of premalignant mammary lesions to invasive cancer. Oncotarget 2017; 8: 50731–50746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Petersen C, Round JL. Defining dysbiosis and its influence on host immunity and disease. Cell Microbiol 2014; 16: 1024–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Butto LF, Haller D. Dysbiosis in intestinal inflammation: cause or consequence. Int J Med Microbiol 2016; 306: 302–309. [DOI] [PubMed] [Google Scholar]
- 18.Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest 2009; 119: 1420–1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Micalizzi DS, Farabaugh SM, Ford HL. Epithelial-mesenchymal transition in cancer: parallels between normal development and tumor progression. J Mammary Gland Biol Neoplasia 2010; 15: 117–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Peinado H, Olmeda D, Cano A. Snail, Zeb and bHLH factors in tumour progression: an alliance against the epithelial phenotype? Nat Rev Cancer 2007; 7: 415–428. [DOI] [PubMed] [Google Scholar]
- 21.Wang H, Wang HS, Zhou BH, et al. Epithelial-mesenchymal transition (EMT) induced by TNF-alpha requires AKT/GSK-3beta-mediated stabilization of snail in colorectal cancer. PLoS One 2013; 8: e56664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huang L, Wang X, Wen C, et al. Hsa-miR-19a is associated with lymph metastasis and mediates the TNF-alpha induced epithelial-to-mesenchymal transition in colorectal cancer. Sci Rep 2015; 5: 13350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bhat AA, Ahmad R, Uppada SB, et al. Claudin-1 promotes TNF-alpha-induced epithelial-mesenchymal transition and migration in colorectal adenocarcinoma cells. Exp Cell Res 2016; 349: 119–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang L, Jiao M, Wu K, et al. TNF-alpha induced epithelial mesenchymal transition increases stemness properties in renal cell carcinoma cells. Int J Clin Exp Med 2014; 7: 4951–4958. [PMC free article] [PubMed] [Google Scholar]
- 25.Rokavec M, Oner MG, Li H, et al. IL-6R/STAT3/miR-34a feedback loop promotes EMT-mediated colorectal cancer invasion and metastasis. J Clin Invest 2014; 124: 1853–1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Castellana B, Aasen T, Moreno-Bueno G, et al. Interplay between YB-1 and IL-6 promotes the metastatic phenotype in breast cancer cells. Oncotarget 2015; 6: 38239–38256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lee SO, Yang X, Duan S, et al. IL-6 promotes growth and epithelial-mesenchymal transition of CD133+ cells of non-small cell lung cancer. Oncotarget 2016; 7: 6626–6638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen G, Tang N, Wang C, et al. TNF-alpha-inducing protein of Helicobacter pylori induces epithelial-mesenchymal transition (EMT) in gastric cancer cells through activation of IL-6/STAT3 signaling pathway. Biochem Biophys Res Commun 2017; 484: 311–317. [DOI] [PubMed] [Google Scholar]
- 29.Wu X, Schulte BC, Zhou Y, et al. Depletion of M2-like tumor-associated macrophages delays cutaneous T-cell lymphoma development in vivo. J Invest Dermatol 2014; 134: 2814–2822. [DOI] [PubMed] [Google Scholar]
- 30.Patwardhan PP, Surriga O, Beckman MJ, et al. Sustained inhibition of receptor tyrosine kinases and macrophage depletion by PLX3397 and rapamycin as a potential new approach for the treatment of MPNSTs. Clin Cancer Res 2014; 20: 3146–3158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tham M, Khoo K, Yeo KP, et al. Macrophage depletion reduces postsurgical tumor recurrence and metastatic growth in a spontaneous murine model of melanoma. Oncotarget 2015; 6: 22857–22868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang W, Zhu XD, Sun HC, et al. Depletion of tumor-associated macrophages enhances the effect of sorafenib in metastatic liver cancer models by antimetastatic and antiangiogenic effects. Clin Cancer Res 2010; 16: 3420–3430. [DOI] [PubMed] [Google Scholar]
- 33.Shen KY, Song YC, Chen IH, et al. Depletion of tumor-associated macrophages enhances the anti-tumor immunity induced by a Toll-like receptor agonist-conjugated peptide. Hum Vaccin Immunother 2014; 10: 3241–3250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shashkova EV, Doronin K, Senac JS, et al. Macrophage depletion combined with anticoagulant therapy increases therapeutic window of systemic treatment with oncolytic adenovirus. Cancer Res 2008; 68: 5896–5904. [DOI] [PubMed] [Google Scholar]
- 35.Fararjeh M, Mohammad MK, Bustanji Y, et al. Evaluation of immunosuppression induced by metronidazole in Balb/c mice and human peripheral blood lymphocytes. Int Immunopharmacol 2008; 8: 341–350. [DOI] [PubMed] [Google Scholar]
- 36.Sellers RS, Clifford CB, Treuting PM, et al. Immunological variation between inbred laboratory mouse strains:points to consider in phenotyping genetically immunomodified mice. Vet Pathol 2012; 49: 32–43. [DOI] [PubMed] [Google Scholar]