

Short abstract
To suppress virus multiplication, infected macrophages produce NO. However, it remains unclear how infecting viruses then overcome NO challenge. In the present study, we report the effects of accessory protein C from Sendai virus (SeV), a prototypical paramyxovirus, on NO output. We found that in RAW264.7 murine macrophages, a mutant SeV without C protein (4C(–)) significantly enhanced inducible NO synthase (iNOS) expression and subsequent NO production compared to wild type SeV (wtSeV). SeV 4C(-) infection caused marked production of IFN-β, which is involved in induction of iNOS expression via the JAK-STAT pathway. Addition of anti-IFN-β Ab, however, resulted in only marginal suppression of NO production. In contrast, NF-κB, a primarily important factor for transcription of the iNOS gene, was also activated by 4C(–) infection but not wtSeV infection. Induction of NO production and iNOS expression by 4C(–) was significantly suppressed in cells constitutively expressing influenza virus NS1 protein that can sequester double-stranded (ds)RNA, which triggers activation of signaling pathways leading to activation of NF-κB and IRF3. Therefore, C protein appears to suppress NF-κB activation to inhibit iNOS expression and subsequent NO production, possibly by limiting dsRNA generation in the context of viral infection.
Keywords: Accessory proteins, double-stranded RNA, macrophages, NO, paramyxovirus
Introduction
Airway epithelial cells are the primary targets of respiratory virus infection, although immune cells such as airway macrophages and dendritic cells are also susceptible.1,2 Macrophages are the critical first line of defense against respiratory pathogens.3 Thus, understanding how viruses evade or exploit macrophage function will provide greater insight into viral pathogenicity and antiviral responses. In response to viral infection, macrophages secrete NO as well as large amounts of cytokines. This in turn activates the expression of other immune genes, thereby controlling invading pathogens.1 Moreover, NO counters viral infectivity and replication, likely by reacting with viral or cellular factors.4,5 As a countermeasure, however, viral pathogens have evolved virulence factors that antagonize the NO pathway.6–8 We recently studied the ability of Sendai virus (SeV), as a representative of the Paramyxoviridae family, to inhibit innate immunity9–13 and found that the NO response to SeV without the C protein (4C(–)) was greater than that to wild type SeV (wtSeV) in infected RAW264.7 macrophages. This finding indicates that the C protein may inhibit NO activation and the inflammatory response. However, the underlying mechanism of how SeV regulates NO production in macrophages during viral infection remains unknown.
NO production is maximized by NF-κB and by IFN-stimulated gene factor 3 (ISGF3), a multiprotein complex consisting of STAT1, STAT2, and IRF9.14 The complex is formed after type I IFN triggers JAK-STAT signaling by binding to specific cell surface receptors. It has been shown that the SeV accessory proteins C and V overcome the type I IFN system by blocking the JAK-STAT pathway and/or limiting the production of IFN in HeLa cells.9,15–17 Therefore, we hypothesized that infection by SeV can regulate NO production in infected macrophages. Indeed, during our research on the effect of SeV on macrophages, we recently found that C protein inhibits TLR4-mediated NO production by blocking the ability of type I IFN to stimulate JAK-STAT.18 This finding indicates that the virus also regulates NO production by inhibiting JAK-STAT signaling in the context of virus infection in macrophages. However, unexpectedly, we found that 4C(–) activates production of NO in RAW264.7 cells, although IFN-β is only minimally involved in the associated increase in inducible NO synthetase (iNOS) and NO. Because this ability does not appear to be explained by the previously observed IFN antagonism, we here investigated the mechanism underlying this regulation. Our results show that the activation of NO is blocked by Bay-11-7082 (Bay), a chemical inhibitor of NF-κB, and is also inhibited by influenza virus nonstructural protein 1 (NS1), which sequesters double-stranded (ds) viral RNA. Thus, we report that the SeV C protein appears to limit the generation of dsRNA and thereby prevents NF-κB activation, iNOS expression, and NO output.
Materials and methods
Cells and viruses
Murine RAW264.7 macrophages were obtained from Riken BioResource Center (Tokyo, Japan) and maintained in Roswell Park Memorial Institute 1640 medium (Sigma Chemicals, St. Louis, MO, USA) containing 5% heat-inactivated FCS (Gibco-Invitrogen, Carlsbad, CA, USA). Vero cells were maintained in DMEM (Sigma Chemicals, St. Louis, MO, USA) containing 10% heat-inactivated FCS. A RAW264.7 cell clone stably expressing Flag-tagged NS1 from influenza A virus PR8 was isolated from neomycin-resistant colonies after transfection with pCXN2-Flag-NS1. This clone was provided by Y. Nakatsu, along with a control clone isolated in a similar manner after transfection with empty pCXN2.19 wtSeV, a cDNA-derived Z strain, was propagated in Vero cells in the presence of 3 μg/ml trypsin,9 along with the mutant strain 4C(–), from which C protein had been knocked out.20,21
Reagents
LPS from Escherichia coli O55:B5 and S-nitroso-N-acetyl-DL-penicillamine (SNAP) were purchased from Sigma Chemicals (St. Louis, MO, USA) and Dojindo (Kumamoto, Japan), respectively. Abs to p65 (#8242), p65 phosphorylated at Ser276 (#3037), STAT1 (#9172), STAT1 phosphorylated at Y701 (#9171), and STAT2 (#4597) were purchased from Cell Signaling Technology (Beverly, MA, USA). Abs to iNOS (#06-573) and STAT2 phosphorylated at Y689 (#07-224), as well as Bay 11-7082, a chemical inhibitor of NF-κB, were obtained from Millipore (Temecula, CA, USA). Abs to cyclooxygenase-2 (COX-2) (#610203) were obtained from BD Transduction Laboratories (San Jose, CA, USA), whereas neutralizing Abs to mouse IFN-β (#32401-1) were obtained from R&D Systems (Minneapolis, MN, USA). Sera against SeV and its C protein were produced in rabbits as described previously.22,23
Quantification of NO and cytokines
Nitrite, a stable degradation product of NO, was quantified in culture media by the Griess reaction, using a microplate reader as described previously.24 Potassium nitrite diluted in complete culture medium was used as a standard. TNF-α and IL-6 were quantified by ELISA kits from R&D Systems (Minneapolis, MN, USA), whereas IFN-β was assayed by a PBL InterferonSource ELISA kit (Piscataway, NJ, USA). Data were analyzed by Student t test, with P < 0.01 considered statistically significant, and are reported as mean ± SD from triplicates. All experiments were performed independently at least three times.
Immunoblotting
Cells were lysed in 50 mM Tris-HCl pH 7.4, 150 mM NaCl, 1% Triton X-100, 1 mM EDTA, 10 mM sodium fluoride, 1 mM sodium orthovanadate, and protease inhibitor cocktail (Sigma). Total proteins (20 μg) were separated by electrophoresis on 10% to 15% SDS-polyacrylamide gels, and electroblotted onto membranes (Durapore, Merck Millipore, Billerica, MA, USA).18 The membranes were then blocked for one h in PBS containing 5% skim milk and 0.05% Tween 20, probed for one h at room temperature with primary Abs, labeled for one h with anti-mouse or anti-rabbit IgG conjugated to HRP (Thermo Fisher Scientific, Waltham, MA, USA) and visualized by chemiluminescence using SuperSignal West Dura (Thermo Fisher Scientific) and an AE6955 light capture system with CS software (Atto, Tokyo, Japan).
Luciferase reporter assay
Using FuGENE HD transfection reagent, RAW264.7 cells in 24-well plates were transfected in triplicate with 200 ng/well pCA7HA vector with or without the C gene, along with 200 ng/well of piNOS-Luc or pISRE-Luc and 5 ng/well pRL-TK-Luc, which express luciferase under the control of the iNOS or IFN-stimulated response element (ISRE) and thymidine kinase (TK) promoter, respectively. At 24 h posttransfection, cells were mock-infected or infected with SeV 4C(–) at MOI 5. Relative luciferase activity was determined after eight h by a dual-luciferase reporter assay system (Promega, Madison, WI, USA). Activation of the promoter of iNOS or ISRE was assessed based on the mean relative luciferase activity from three independent experiments.18
Statistical analysis
All graphs were generated using the GraphPad Prism software (GraphPad Software Inc, San Diego, CA, USA). Data are presented as the means ± SD, and P values were calculated using an unpaired Student t test with two-tailed analysis. P < 0.01 was considered statistically significant.
Results
NO suppresses the infectivity of SeV
In vitro, NO was reported to have no apparent antiviral effect on SeV but to have potent mutagenic activity postinfection.25 To determine other effects of NO on SeV infection, we examined the effect of NO exposure preinfection. The addition of SNAP, an NO source that is often used to investigate the physiological actions of NO,1,5,6,26 to the culture medium increased the concentration of NO in a concentration-dependent manner (Figure 1a). Accordingly, Vero cells were pretreated with various concentrations of SNAP and then infected with wtSeV at MOI 0.01. The cells have defective type I IFN genes and do not mount an antiviral response via such genes. After incubation for 72 h in trypsin, the surviving cells were stained with amido black. As shown in Figure 1b, pretreatment with more than 10 μM SNAP inhibited the cytopathic effects of SeV, which are pronounced in the absence of the NO donor. In addition, SNAP significantly suppressed the expression of the viral proteins P, N, and M (Figure 1c). However, in agreement with a previous report,25 posttreatment with SNAP did not inhibit the cytopathic effects of SeV (Figure 1b, lower panel). Collectively, these results imply that NO suppresses the infectivity, but not the replication, of SeV.
Figure 1.
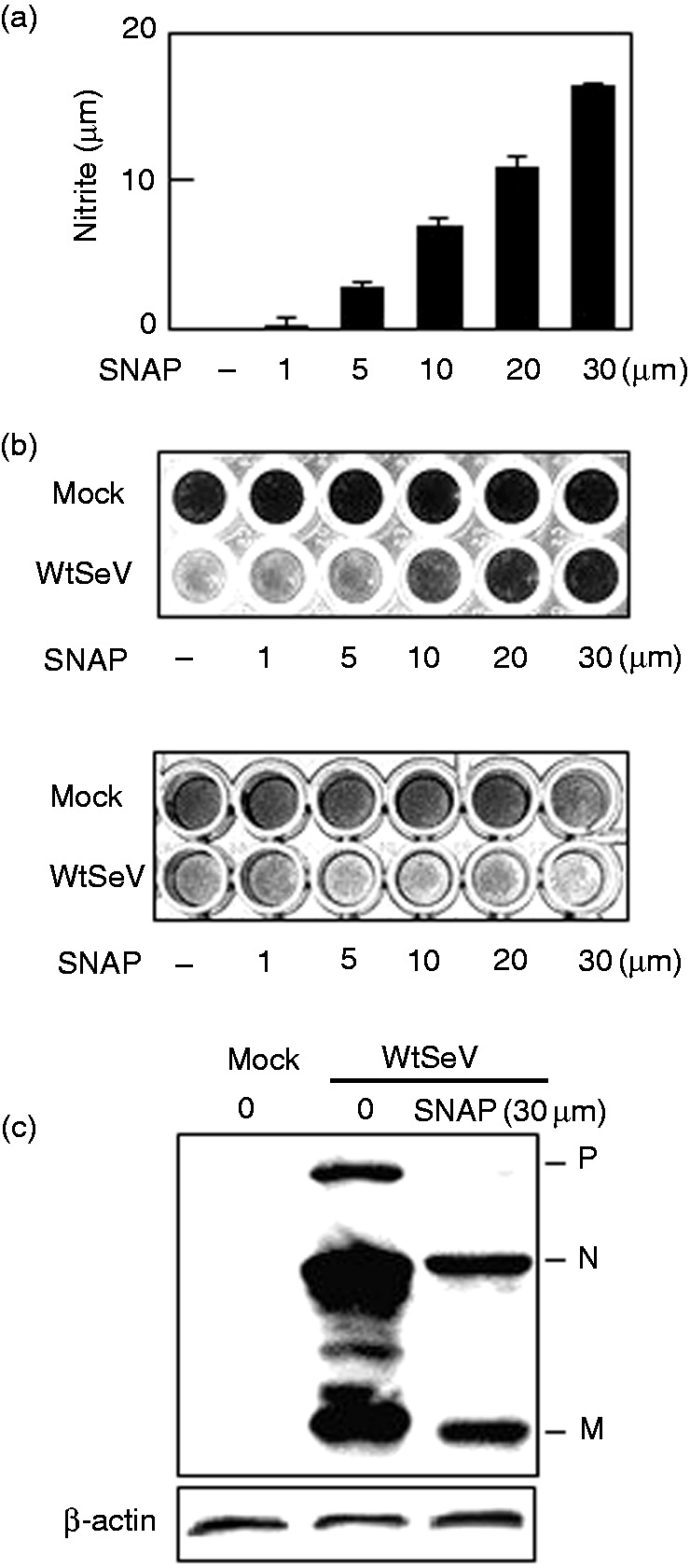
Effect of NO on SeV multiplication. Following treatment with the indicated concentrations (a, b) or 30 μM (c) SNAP, one h before infection (b, upper panel, and c) or one h after infection (b, lower panel), Vero cells in a 96-well plate were mock-infected or infected with wtSeV at MOI 0.001 and incubated in 3 μg/ml trypsin. Culture media were collected 24 h postinfection and assayed for nitrite (a). Cells were also stained 72 h postinfection with 0.5% w/v amido black 10B dissolved in 20% ethanol and 10% acetic acid (b) or immunoblotted with rabbit serum against SeV (c). Viral proteins P, N, and M are marked in (c). SeV: Sendai virus; SNAP: S-nitroso-N-acetyl-DL-penicillamine; wt: wild type.
SeV C protein suppresses NO production in infected RAW264.7 macrophages
The SeV accessory protein C overcomes the type I IFN system by blocking the JAK-STAT pathway and/or limiting the production of IFN.9,15–17 Therefore, we surmised that NO output in infected cells is also regulated by these proteins. Thus, we tested the effects of this accessory protein on host NO production in murine RAW264.7 macrophages, which are often used as a model to investigate NO activity against many viruses. In response to respiratory infection, NO is abundantly produced by various cells, including alveolar macrophages and airway epithelial cells,1 via induction of iNOS, one of the key enzymes that generate NO from l-arginine.27
NO was quantified in culture media from RAW264.7 cells infected with SeV 4C(–) (Figure 2a). Whereas wtSeV elicited minimal NO production, infection with SeV 4C(–) caused significant NO output in a concentration- and time-dependent fashion (Figure 2b, c). iNOS expression was also induced (Figure 2d), in line with NO output. To confirm that C protein regulates iNOS expression, RAW264.7 cells were transfected with a plasmid encoding C protein and a plasmid encoding luciferase under the control of the iNOS-promoter. These cells were then infected with SeV 4C(–) (Figure 2e). Lack of luciferase activity indicated that the iNOS promoter was not activated, suggesting that exogenously expressed C protein is able to suppress mutant virus-induced iNOS expression and NO output.
Figure 2.
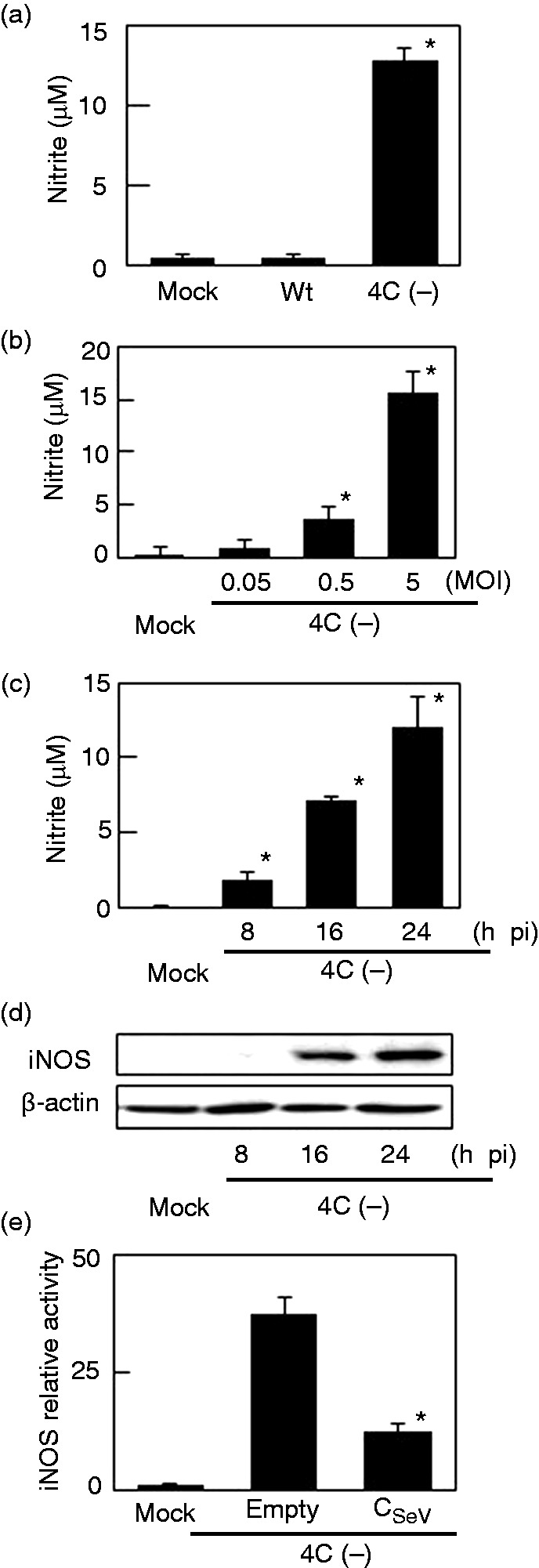
NO and iNOS production in RAW264.7 cells infected with SeV 4C(–). (a–d) Cells were mock-infected or infected with SeV strains at MOI 5 (a, c, d), or as indicated (b). Culture media were collected 24 h postinfection (a, b) or at the indicated time points (c) and assayed for nitrite. Cells were also harvested at the indicated time points and immunoblotted with anti-iNOS (d). (e) Cells were transfected with pCA7HA vector with (CSeV) or without (Empty) C protein, along with pRL-TK or piNOS-Luc, a firefly luciferase reporter plasmid driven by the iNOS promoter. At 24 h posttransfection, cells were mock-infected or infected with SeV 4C(–) at MOI 5, harvested eight h thereafter, and assayed by a dual-luciferase assay system to evaluate activation of the iNOS promoter. *P < 0.01 vs infection with wtSeV (a), vs mock infection (b,c), or vs pCA7HA empty vector.iNOS: inducible NO synthase; SeV: Sendai virus; TK: thymidine kinase; wt: wild type.
IFN-β induced by SeV 4C(–) is only marginally involved in iNOS and NO induction
The iNOS promoter contains response elements to NF-κB and to ISGF3. This complex is formed when type I IFN engages specific cell surface receptors, activates JAK-STAT signaling, and elicits phosphorylation of tyrosines in STAT1 and STAT2. As previously observed,9 SeV 4C(–) induced significantly higher production of IFN-β (∼103 IU/ml) in RAW264.7 cells in comparison to wtSeV (Figure 3a). Accordingly, the downstream genes STAT1 and STAT2 were also more strongly expressed (Figure 3c) and phosphorylated (Figure 3b).
Figure 3.

Role of IFN-β in NO and iNOS production in RAW264.7 cells infected with SeV 4C(–). (a–c) Cells were mock-infected or infected with wt or SeV 4C(–) at MOI 5. Culture media were collected 24 h postinfection and assayed for IFN-β (a), whereas cells were harvested at five h (b) or 24 h postinfection (c) and immunoblotted with Abs to unphosphorylated and phosphorylated STAT1 and STAT2. (d) Cells were infected with SeV 4C(–) at MOI 5 and incubated for 24 h in the presence or absence of neutralizing Abs to IFN-β. Culture media were then assayed for nitrite. Cells were transfected with pRL-TK or pISRE-Luc, a firefly luciferase reporter plasmid driven by the ISRE promoter. At 24 h posttransfection, cells were mock-infected or infected with SeV 4C(–) at MOI 5, harvested after 8 h, and assayed by a dual-luciferase assay system to evaluate activation of the iNOS promoter. (e, f) Cells were mock-infected or infected with wtSeV at MOI 5. Cells were then mock-treated or treated with 103 or 104 IU/ml IFN-β for 20 h, beginning at 4 h postinfection. Culture media were then assayed for nitrite (e), whereas cells were immunoblotted with anti-iNOS (f). *P < 0.01 vs mock treatment with a-IFN-B antibody (d) or vs treatment with 104 IU/ml IFN-B after mock infection (e).iNOS: inducible NO synthase; ISRE: IFN-stimulated response element; SeV: Sendai virus; TK: thymidine kinase; wt: wild type.
To determine whether IFN-β expression induced by SeV 4C(–) amplifies NO output, we assessed the effects of neutralizing Abs against IFN-β. However, the Abs inhibited NO production only marginally in RAW264.7 cells infected with the virus (Figure 3d), suggesting that IFN-β is not extensively involved. The blocking of SeV 4C(–)-induced IFN-β by the Abs was confirmed with an ISRE luciferase reporter vector, which is a JAK-STAT pathway-responsive reporter. Conversely, addition of an equivalent amount of exogenous IFN-β (103 IU/ml) as induced by the virus did not elicit iNOS expression and NO production as shown in Figure 3d and e. Indeed, a higher concentration of exogenous IFN-β (104 IU/ml) was required to elicit iNOS expression and NO output (Figure 3e, f). Nevertheless, prior infection by wtSeV suppresses iNOS expression and NO production even in this extreme case. Taken together, these results suggest that the IFN-β induced SeV 4C(–) is only marginally involved in iNOS induction and subsequent NO output.
NF-κB mediates NO induction in RAW264.7 cells infected with SeV 4C(–)
NF-κB is an important transcription factor that activates iNOS as well as cytokines such as TNF-α and IL-6, which, in turn, up-regulate COX-2. As shown in Figure 4a–c, notable amounts of TNF-α, IL-6, and COX-2 were produced after infection with SeV 4C(–), but not after infection with the wt virus. Phosphorylation of the p65 subunit in NF-κB was also observed in cells infected with the mutant virus (Figure 4d). Strikingly, a chemical inhibitor of NF-κB, Bay, that selectively and irreversibly inhibits NF-κB activation by blocking the cytokine-induced phosphorylation of IκB-α, inhibited NO production induced by the mutant virus without affecting the replication of SeV 4C(–) (Figure 4e). Collectively, the results suggest that NF-κB is largely responsible for NO induction in RAW264.7 cells infected with SeV 4C(–).
Figure 4.
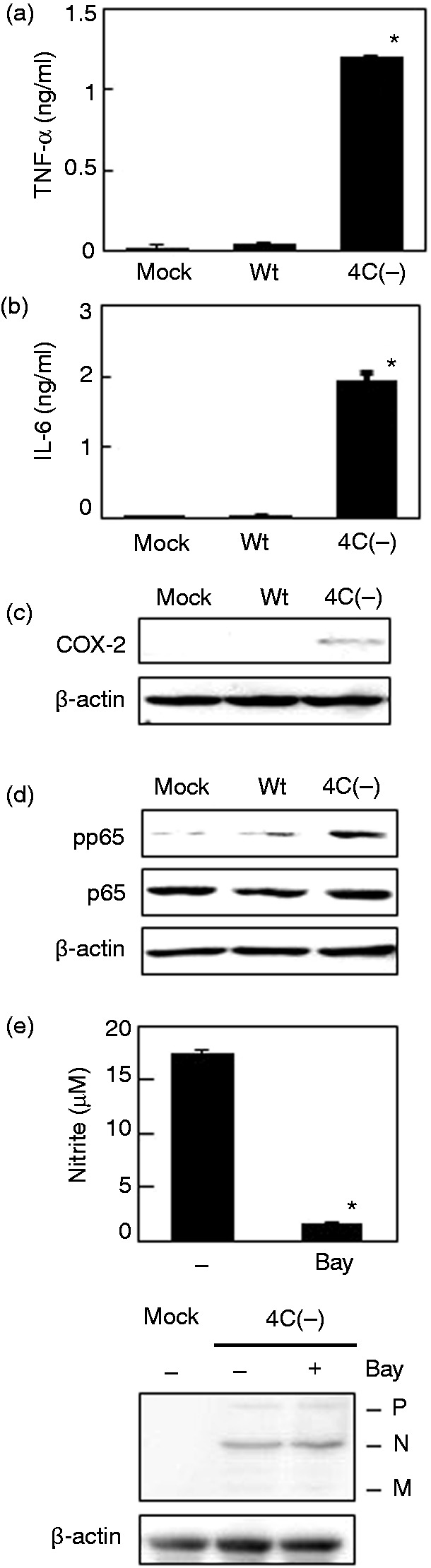
Activation of NF-κB in RAW264.7 cells infected with SeV 4C(–). (a, b) Cells were mock-infected or infected with wt or SeV 4C(–) at MOI 5. Culture media were collected 5 h (a) and 24 h (b) thereafter and assayed for TNF-α and IL-6, whereas cells were harvested at 5 h (d) and 24 h (c) and immunoblotted with Abs to phosphorylated p65, p65, and COX-2. (e) Cells were mock-treated or treated with Bay 11-7082. (Bay) for 30 min and then infected with SeV 4C(–) at MOI 5 for 24 h. Culture media were then assayed for nitrite (upper panel), whereas cells were immunoblotted with rabbit serum against SeV (lower panel). Viral proteins P, N, and M are marked in the lower panel. *P < 0.01 vs infection with wtSeV (a, b) or vs mock treatment with Bay (e). COX-2: cyclooxygenase-2; SeV: Sendai virus; wt: wild type.
Influenza virus NS1 inhibits NO production induced by SeV 4C(–) by sequestering dsRNA
As SeV 4C(–), but not the wt, strongly activates NF-κB (Figure 4), C protein may block signaling pathways that converge at NF-κB. Thus, we investigated the role of TLR4, which is required for one such pathway (Figure 5a). Exposure of RAW264.7 cells to LPS, a TLR4 ligand, induced expression of COX-2. In agreement with our previous report, however, these effects were unaffected by prior infection with wt or SeV 4C(–), indicating that C protein does not block TLR4-dependent activation of NF-κB.
Figure 5.
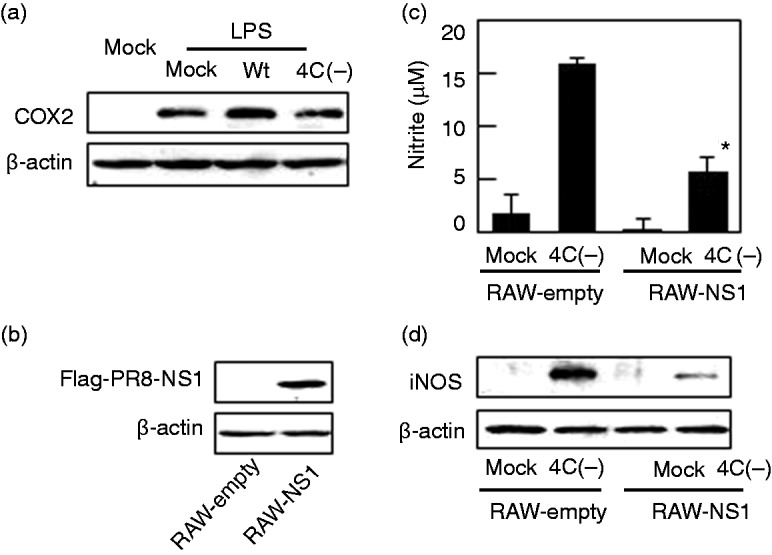
Effect of influenza virus NS1 on NO production induced by SeV 4C(–). (a) RAW264.7 cells were mock-infected or infected with wt or SeV 4C(–) at MOI 5. At 4 h postinfection, cells were mock-treated or treated with 100 ng/ml LPS, harvested 24 h (a) thereafter, and immunoblotted using Abs to COX-2. (b) RAW264.7 cells stably expressing Flag-tagged NS1 (RAW-NS1) were immunoblotted with anti-Flag Ab. (c, d) RAW264.7 cells stably transfected with the empty vector (RAW-empty) or the vector expressing NS1 protein (RAW-NS1) were mock-infected or infected with SeV 4C(–) at MOI 5. Culture media (c) and cells (d) were collected 24 h postinfection and assayed for nitrite and iNOS, respectively. *P < 0.01 vs cells stably transfected with the empty vector (RAW-empty). COX-2: cyclooxygenase-2; NS1: nonstructural protein 1; SeV: Sendai virus; wt: wild type.
Previously, we demonstrated in human glioblastoma U118 cells that SeV 4C(–), but not the wt, generates ds viral RNA during replication and transcription.11 In another report using influenza virus, dsRNA were reported to induce NO production through protein kinase (PKR)-dependent activation of NF-κB.28 In addition, dsRNA may engage TLR3 and retinoic acid-inducible gene-I-like receptors, both of which also activate NF-κB.29,30 Thus, we tested whether activation of NF-κB by SeV 4C(–), but not by the wt, results from production of dsRNA. Accordingly, we established a RAW264.7 cell line stably expressing influenza virus NS1 (RAW-NS1), which is well known to sequester dsRNA and thereby inhibit IFN production.19 As shown in Figure 5d and e, expression of NS1 significantly inhibited NO production and iNOS expression after infection with SeV 4C(–). Hence, C protein appears to suppress NO induction by restricting the generation of dsRNA during viral replication and transcription.
Discussion
In this study, we expanded on our previous findings that SeV C protein overcomes the type I IFN system in HeLa cells and demonstrated that SeV C protein limits NO production in infected RAW264.7 macrophages.
We found that SeV 4C(–) activates IFN-β in RAW264.7 cells, although this cytokine was only minimally involved in the associated increase in iNOS and NO (Figure 3). Rather, activation of NF-κB is largely responsible (Figure 4). Indeed, this effect is blocked by Bay, a chemical inhibitor of NF-κB, but not by neutralizing Abs against IFN-β. Strikingly, influenza virus NS1, which sequesters ds viral RNA,19 also suppresses NF-κB activation and subsequent NO production in cells infected with SeV 4C(–) (Figure 5). SeV 4C(–), but not the wt, generates dsRNA during replication and transcription in human glioblastoma U118 cells.11
Based on these multiple lines of evidence, the SeV C protein seems to limit the production of dsRNA and thereby prevent NF-κB activation, iNOS expression, and NO output. Nevertheless, we cannot at present exclude the possibility that NS1 inhibits NO induction by interacting with the ubiquitin ligase TRIM25 to ubiquitinate and inactivate retinoic acid-inducible genes.31
In a previous study,18 wtSeV was found to inhibit LPS-induced production of NO, but not of TNF-α and IL-6. This inhibition was associated with suppressed activation of STAT1 and STAT2, but not of NF-κB, as well as with SeV C protein. Indeed, the ability of the C protein to block LPS-induced NO production was abolished by an F170S mutation, which also blocked the ability to activate STAT proteins. Thus, C protein appears to suppress LPS-induced NO production by inhibiting JAK-STAT signaling. In contrast, we have now observed that SeV 4C(–), but not the wt, induces production of not only NO but also TNF-α and IL-6. The differences in the mechanisms underlying the inhibitory effects of the C protein on LPS- and virus-induced NO production may be attributable to the differences in ligands that activate NF-κB. Moreover, IL-6 induction by SeV 4C(–)-infected cells could also induce NO production in the context of SeV 4C(–) infection because IL-6 is a powerful inducer of NO production.5
Regarding whether C protein in paramyxoviruses prevents NO production by limiting dsRNA production, C-deficient measles virus also induces type I IFN, but not when ds viral RNAs are sequestered by influenza virus NS1.19 Similarly, the C protein in human parainfluenza virus type 1 suppresses dsRNA-dependent PKR, melanoma-associated differentiation gene 5 (MDA5), and type I IFN by limiting dsRNA.32 Collectively, the data strongly suggest that the C protein in paramyxoviruses suppresses NO production in the host by restricting dsRNA expression, although the C protein from measles virus is at most 20% homologous to the SeV C protein.
A critical question raised by these findings is whether the NO antagonism induced by the SeV C protein, which decreases the production of NO, contributes to the pathogenicity of this virus. Although the role of NO in influenza virus infection is not well understood in vitro, NO clearly plays a role in fighting influenza virus infection. The addition of SNAP to Madin-Darby canine kidney (MDCK) cells immediately after infection with the influenza A and B viruses inhibited the replication of both viruses in a dose-dependent manner during the initial stages of infection.7 Similarly, when MDCK cells were exposed to gaseous NO before or after infection with influenza A and B viruses, the inactivation of viral neuraminidase activity and inhibition of viral infectivity was observed both with pre- and postinfection NO exposure.4 Although NO has been shown to have no apparent antiviral effect on SeV replication, we demonstrated that NO reduces viral infectivity. Therefore, antagonism of the NO system, which decreases NO production, may be involved in viral pathogenicity.
In summary, we have studied the role of SeV C protein in limiting NO production in infected RAW264.7 macrophages. Our findings reveal that the ability of the C protein to limit dsRNA generation is more important than its anti-NO antagonistic ability in terms of limiting NO production in the context of viral infection. This finding indicates that the NF-κB pathway contributes to SeV-induced NO production more than the JAK-STAT pathway.
Acknowledgements
We thank Y. Nakatsu and M. Takeda for providing pCXN2-Flag-NS1 and pCXN2, and K Takahashi for excellent technical assistance.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by a grant from the MEXT-Supported Program for Strategic Research Foundation at Private Universities, 2011–2015 (S1101027) and in part by a KAKENHI Grant-in-Aid for Scientific Research from the Japan Society for the Promotion of Science (C17K08855).
References
- 1.Vareille M, Kieninger E, Edwards MR, et al. The airway epithelium: Soldier in the fight against respiratory viruses. Clin Microbiol Rev 2011; 24: 210–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Villalón-Letelier F, Brooks AG, Saunders PM, et al. Host cell restriction factors that limit influenza A infection. Viruses 2017; 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cline TD, Beck D, Bianchini E. Influenza virus replication in macrophages: Balancing protection and pathogenesis. J Gen Virol 2017; 98: 2401–2412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Regev-Shoshani G, Vimalanathan S, McMullin B, et al. Gaseous nitric oxide reduces influenza infectivity in vitro. Nitric Oxide 2013; 31: 48–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Abdul-Cader MS, Amarasinghe A, Abdul-Careem MF. Activation of toll-like receptor signaling pathways leading to nitric oxide-mediated antiviral responses. Arch Virol 2016; 161: 2075–2086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bi Z, Reiss CS. Inhibition of vesicular stomatitis virus infection by nitric oxide. J Virol 1995; 69: 2208–2213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rimmelzwaan GF, Baars MM, de Lister P, et al. Inhibition of influenza virus replication by nitric oxide. J Virol 1999; 73: 8880–8883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stark JM, Khan AM, Chiappetta CL, et al. Immune and functional role of nitric oxide in a mouse model of respiratory syncytial virus infection. J Infect Dis 2005; 191: 387–395. [DOI] [PubMed] [Google Scholar]
- 9.Komatsu T, Takeuchi K, Yokoo J, et al. C and V proteins of Sendai virus target signaling pathways leading to IRF-3 activation for the negative regulation of interferon-beta production. Virology 2004; 325: 137–148. [DOI] [PubMed] [Google Scholar]
- 10.Komatsu T, Takeuchi K, Gotoh B. Bovine parainfluenza virus type 3 accessory proteins that suppress beta interferon production. Microbes Infect 2007; 9: 954–962. [DOI] [PubMed] [Google Scholar]
- 11.Takeuchi K, Komatsu T, Kitagawa Y, et al. Sendai virus C protein plays a role in restricting PKR activation by limiting the generation of intracellular double-stranded RNA. J Virol 2008; 82: 10102–10110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kitagawa Y, Yamaguchi M, Zhou M, et al. A tryptophan-rich motif in the human parainfluenza virus type 2 V protein is critical for the blockade of toll-like receptor 7 (TLR7)- and TLR9-dependent signaling. J Virol 2011; 85: 4606–4611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yamaguchi M, Kitagawa Y, Zhou M, et al. An anti-interferon activity shared by paramyxovirus C proteins: Inhibition of Toll-like receptor 7/9-dependent alpha interferon induction. FEBS Lett 2014; 588: 28–34. [DOI] [PubMed] [Google Scholar]
- 14.Farlik M, Reutterer B, Schindler C, et al. Nonconventional initiation complex assembly by STAT and NF-kappaB transcription factors regulates nitric oxide synthase expression. Immunity 2010; 33: 25–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Garcin D, Latorre P, Kolakofsky D. Sendai virus C proteins counteract the interferon-mediated induction of an antiviral state. J Virol 1999; 73: 6559–6565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Komatsu T, Takeuchi K, Yokoo J, et al. Sendai virus blocks alpha interferon signaling to signal transducers and activators of transcription. J Virol 2000; 74: 2477–2480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gotoh B, Takeuchi K, Komatsu T, et al. The STAT2 activation process is a crucial target of Sendai virus C protein for the blockade of alpha interferon signaling. J Virol 2003; 77: 3360–3370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Odkhuu E, Komatsu T, Naiki Y, et al. Sendai virus C protein inhibits lipopolysaccharide-induced nitric oxide production through impairing interferon-β signaling. Int Immunopharmacol 2014; 23: 267–272. [DOI] [PubMed] [Google Scholar]
- 19.Nakatsu Y, Takeda M, Ohno S, et al. Translational inhibition and increased interferon induction in cells infected with C protein-deficient measles virus. J Virol 2006; 80: 11861–11867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kurotani A, Kiyotani K, Kato A, et al. Sendai virus C proteins are categorically nonessential gene products but silencing their expression severely impairs viral replication and pathogenesis. Genes Cells 1998; 3: 111–124. [DOI] [PubMed] [Google Scholar]
- 21.Kato A, Kiyotani K, Sakai Y, et al. The paramyxovirus, Sendai virus, V protein encodes a luxury function required for viral pathogenesis. EMBO J 1997; 16: 578–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gotoh B, Takeuchi K, Komatsu T, et al. Knockout of the Sendai virus C gene eliminates the viral ability to prevent the interferon-alpha/beta-mediated responses. FEBS Lett 1999; 459: 205–210. [DOI] [PubMed] [Google Scholar]
- 23.Takeuchi K, Komatsu T, Yokoo J, et al. Sendai virus C protein physically associates with Stat1. Genes Cells 2001; 6: 545–557. [DOI] [PubMed] [Google Scholar]
- 24.Green LC, Wagner DA, Glogowski J, et al. Analysis of nitrate, nitrite, and [15N]nitrate in biological fluids. Anal Biochem 1982; 126: 131–138. [DOI] [PubMed] [Google Scholar]
- 25.Yoshitake J, Akaike T, Akuta T, et al. Nitric oxide as an endogenous mutagen for Sendai virus without antiviral activity . J Virol 2004; 78: 8709–8719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Croen KD. Evidence for an antiviral effect of nitric oxide. Inhibition of herpes simplex virus type 1 replication. J Clin Invest 1993; 91: 2446–2452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Aktan F. iNOS-mediated nitric oxide production and its regulation. Life Sci 2004; 75: 639–653. [DOI] [PubMed] [Google Scholar]
- 28.Uetani K, Der SD, Zamanian-Daryoush M, et al. Central role of double-stranded RNA-activated protein kinase in microbial induction of nitric oxide synthase. J Immunol 2000; 165: 988–996. [DOI] [PubMed] [Google Scholar]
- 29.Alexopoulou L, Holt AC, Medzhitov R, et al. Recognition of double-stranded RNA and activation of NF-kappa B by Toll-like receptor 3. Nature 2001; 413: 732–738. [DOI] [PubMed] [Google Scholar]
- 30.Yoneyama M, Kikuchi M, Natsukawa T, et al. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat Immunol 2004; 5: 730–737. [DOI] [PubMed] [Google Scholar]
- 31.Gack MU, Albrecht RA, Urano T, et al. Influenza A virus NS1 targets the ubiquitin ligase TRIM25 to evade recognition by the host viral RNA sensor RIG-I. Cell Host Microbe 2009; 5: 439–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Boonyaratanakornkit J, Bartlett E, Schomacker H, et al. The C proteins of human parainfluenza virus type 1 limit double-stranded RNA accumulation that would otherwise trigger activation of MDA5 and protein kinase R. J Virol 2011; 85: 1495–1506. [DOI] [PMC free article] [PubMed] [Google Scholar]