

Short abstract
Dendritic cells (DCs) are professional Ag-presenting cells that play a critical role in both innate and adaptive immune responses. DCs recognize and respond to bacteria through multiple PRRs, including TLRs. Heat shock protein gp96/grp94 is a master essential chaperone for TLRs in the endoplasmic reticulum. We generated DC-specific gp96-knockout (KO) mice and showed that gp96 KO DCs were unable to respond to multiple TLR ligands. TLR-mediated hyperinflammatory response can lead to sepsis. However, the roles of neither DCs nor the DC-intrinsic gp96 in the process are completely understood. In a LPS-induced sepsis model, we hereby found that deletion of gp96 in DCs significantly reduced serum TNF-α levels and improved survival. Furthermore, using the well-defined polymicrobial sepsis model of cecal ligation and puncture, we found that DC-specific ablation of gp96 improved survival with significantly attenuated liver and renal injuries, decreased circulating inflammatory cytokines, altered DC maturation and activation, and increased serum Ig. Collectively, we demonstrate that deletion of gp96 in DCs is beneficial in protecting mice against sepsis induced by both endotoxemia and polymicrobial infections. We conclude that targeting gp96 in DCs may provide a potential novel approach for reducing the morbidity and mortality of sepsis.
Keywords: Dendritic cell, chaperone, gp96/grp94, sepsis, TLR
Introduction
Sepsis is a life-threatening condition caused by a dysregulated host response to microbial infections or bacterial products, such as LPS. The cellular components of the innate immune system serve as the first line of defense against invading pathogens; however, excessive activation of various immune cells, including B cells, T cells, macrophages, and dendritic cells (DCs), which leads to release of inflammatory cytokines including TNF-α and IL-12, is a hallmark of sepsis.1
Among professional Ag-presenting cells, DCs serve as an essential interface between innate and adaptive immune responses.2–4 DCs recognize and respond to microbes such as bacteria and virus through multiple PRRs, including TLRs, NLRs, RIG I-like receptors, C-type lectins, and mannose receptors.5–7 During sepsis, DCs can be activated by TLRs and this activation contributes to sepsis-associated immunosuppression, organ injury and mortality.8–10 Activation of adenosine monophosphate kinase (AMPK), which suppresses the activation of DCs, has been reported to prevent the inflammation and organ damage during sepsis.9,11,12 Targeting DCs was therefore thought to be promising for the management of sepsis via both an immunotherapeutic and an immunomodulatory approach.9 Thus, it becomes very important to understand the function and related mechanisms of DCs in the immunopathogenesis of sepsis.
Heat shock protein (HSP) gp96,13 also known as grp94,14 endoplasmin,15 ERp99,16 and HSP90b1,17 is an endoplasmic reticulum (ER) paralogue of HSP90. As a molecular chaperone, gp96 is the most abundant and ubiquitous protein in the ER lumen and is induced by the accumulation of misfolded proteins.18 Genetic studies have demonstrated that gp96 is an essential master molecular chaperone for most of the TLRs,19–24 multiple integrins,20,23 GARP,25–27 and Wnt co-receptor LRP6.28,29 gp96 can bind to and act as a master chaperone for TLRs on macrophages and DCs, while it itself might also stimulate pro-inflammatory cytokines (TNF-α and IL-12) secretion.20,30 Our recent study demonstrated that macrophage-specific gp96-knockout mice have significantly less inflammation in the colon and lower percentages of Th17 and Th1 cells in colonic lamina propria (cLP) compared with their wild type (WT) littermates.31 Furthermore, we generated a unique CD11c+ cell-specific gp96-deficient mouse model (abbreviated as KO mice hereafter) with selective deletion of gp96 in DCs and demonstrated that gp96-deficient DCs were unable to respond to TLR ligands,32 suggesting a critical role of gp96 in DC activation and in exacerbating inflammation. This constellation of findings led us to explore the role of DC-intrinsic gp96 in sepsis.
In this study, we investigated whether DC-intrinsic gp96 contributes to the pathogenesis of sepsis. First, we challenged WT and KO mice with a lethal dose of LPS, and we found that WT mice produced significantly more serum TNF-α than did KO mice. Consistent with the detrimental effect of this cytokine in sepsis, we found that the WT mice were more susceptible to LPS-induced endotoxemia than the KO mice. Furthermore, using a well-defined sepsis model of cecal ligation and puncture (CLP),33–35 we found that KO mice had significantly attenuated liver and renal injury, decreased circulation inflammatory cytokines, and improved survival. Interestingly, we found that KO mice had reduced DC maturation but increased serum level of Ig after CLP, which may contribute to the protection against sepsis. Our results demonstrate that deletion of gp96 in DCs is protective against sepsis induced by both endotoxemia and polymicrobial infection. This work supports the notion of targeting gp96 in DCs as a potential novel approach for the treatment of sepsis.
Materials and methods
Mice
DC-specific gp96-deficient mice (CD11cCre+ Hsp90b1flox/flox) and control littermates (CD11cCre−Hsp90b1flox/flox) were generated by crossing Hsp90b1flox/flox mice with CD11c-Cre transgenic mice.32 All animal experimental protocols were approved by the Medical University of South Carolina Institutional Animal Care and Use Committee (IACUC). All procedures complied with the standards for care and use of animal subjects as stated in the Guide for the Care and Use of Laboratory Animals (Institute of Laboratory Resources, National Academy of Sciences, Bethesda, MD, USA) as well as established institutional guidelines and regulations.
Reagents
Abs used for flow cytometry were obtained from BD Biosciences (Mountain View, CA) and eBioscience (San Diego, CA). LPS (055: B5) was purchased from Sigma-Aldrich (St Louis, MO). All other chemicals were obtained from Sigma-Aldrich (St Louis, MO) and Fisher Scientific (Pittsburgh, PA).
LPS-induced endotoxemia and survival study
Endotoxemia was induced in DC-specific gp96 KO mice and their WT littermates (8–12 wk old) by i.p. injection of LPS (25 mg/kg body mass; Escherichia coli LPS 055: B5; Sigma-Aldrich, St. Louis, MO) dissolved in sterile saline, as described previously.20,33 The sera were collected at 1.5 h after LPS administration for cytokine analyses. For the survival study, mouse survival was monitored every 12 h for a total of 3 d.
CLP-Induced sepsis and survival study
DC-specific gp96 KO mice and their WT littermates (12–16 wk old) were housed in a specific pathogen-free environment. All surgery was performed under anesthesia. CLP was performed as described previously.33 Briefly, the cecum was ligated at the colon juncture and punctured once with a 22-gauge needle. All animals were fluid-resuscitated subcutaneously with sterile normal saline. Sham operations were performed in the same way as CLP, but without ligation and puncture of the cecum. Mice were sacrificed at 24 h after CLP, and serum was collected for biochemical analyses. The spleen was isolated for DC maturation analysis by flow cytometry. For the survival study, mice were monitored every 24 h for a total of 7 d.
ELISA
IL-12p40 and TNF-α levels in the serum were measured with ELISA kits from BD Biosciences (San Diego, CA) according to the manufacturer’s protocol. IL-10 levels in the serum were measured using a mouse IL-10 ELISA kit (eBioscience, San Diego, CA) according to the manufacturer’s protocol. The serum alanine transaminase (ALT), aspartate aminotransferase (AST), blood urea nitrogen (BUN), and creatinine levels were measured using appropriate mouse ELISA kits (BioAssay Systems, Atlanta, GA). Ig levels in the sera were determined by a sandwich ELISA kit from Southern Biotechnology Associates (Birmingham, AL).
Flow cytometry
Surface staining of cells and flow cytometry were done as described previously.20,36 Briefly, splenocytes were isolated and RBCs were lysed. After washing with FACS buffer (PBS with 2% FBS and 0.09% NaN3), cells were pelleted and blocked with FcR Ab for 10 min, followed by incubation with fluorochrome-labeled Abs at the appropriate dilution for 30 min at 4°C. After staining, cells were then washed with FACS buffer and acquired on FACSVerse (Becton Dickinson, Franklin Lakes, NJ). Dead cells were always gated out by 7AAD exclusion. The results were analyzed with the FlowJo software (Tree Star, Ashland, OR).
Culture of bone marrow-derived DCs
WT and KO bone marrow (BM) cells were isolated from femurs and tibias. BM cells were cultured in RPMI 1640 medium supplemented with 10% FCS, 100 U/mL of penicillin, 100 mg/mL of streptomycin, 20 ng/mL GM-CSF, and 10 ng/mL IL-4 for 6 d. The floating cells were harvested, which were mostly BM-derived DCs (BMDCs) by phenotypic analysis.
Microarray and pathway analysis
WT and KO BMDCs were stimulated with 200 ng/mL LPS (E. coli 055:B5) from Sigma for 6 h. Total RNA was extracted from WT and gp96 KO BMDCs using an RNeasy Mini Kit (Qiagen, Valencia, CA). Fluorescent antisense amplified RNA (aRNA) target preparation was performed using an Eberwine-based amplification method with an Amino Allyl MessageAmp II aRNA Amplification Kit. Cy5-Labeled RNA targets were hybridized to Mouse Whole Genome OneArray® v2 (Phalanx Biotech Group, Belmont, CA), and slides were scanned by the Axon 4000 scanner (Molecular Devices, Sunnyvale, CA). The Cy5 fluorescent intensity of each spot was analyzed by Genepix 4.1 software (Molecular Devices, Sunnyvale, CA). Genes with log2 ratio ≥ 2.0 or log2 ratio ≤ 2.0 and P value < 0.05 are further analyzed. Genes were classified using KEGG Pathway Database (http://www.genome.jp/kegg/pathway.html) and those involved in inflammation were highlighted. A heat map was generated to demonstrate differential expression of genes in the WT and KO DCs. In the heat map, the given gene is presented as compared to the median value for that gene in the WT and KO DC data sets.
Statistical analysis
Statistical significance was determined by analysis of variance (ANOVA) with Fisher’s probable least-squares difference test, and Student's t-test or log-rank (Mantel–Cox) test using GraphPad Prism software. Error bars represent SEM. A value of P < 0.05 was considered statistically significant.
Results
Attenuation of LPS-induced endotoxemia in dendritic cell-specific gp96-deficient mice
Recently, we have generated DC-specific gp96-deficient mice by crossing Hsp90b1flox/floxmice with CD11cCre mice.20,21,32,37 We found that deletion of gp96 in DCs rendered them unresponsive to stimulation by TLR2, TLR4, and TLR9 ligands.32 Thus, our DC-specific gp96-deficient mice present a DC-specific pan TLR KO mouse model. To determine the effect of DC-intrinsic gp96 in endotoxin shock, WT and KO mice were administrated intraperitoneally with a lethal dose of LPS. Mouse survival was monitored every 12 h. By d 2, 100% of the WT mice succumbed to endotoxemia; in contrast, 40% of DC gp96 KO mice survived after LPS administration (P < 0.0001; Figure 1a). Compared to the LPS treated WT mice, deletion of gp96 markedly reduced serum levels of TNF-α (WT: 22.3 ± 1.9 ng/mL, KO: 4.3 ± 0.9 ng/mL; P < 0.0001; Figure 1b). This result demonstrates that DCs play a significant role during the hyperinflammatory phase of LPS-induced endotoxemia and deletion of gp96 in DCs attenuates the TLR-induced inflammatory response.
Figure 1.

Attenuation of LPS-induced endotoxemia in DC-specific gp96-deficient mice. (a) 8–12-wk-old KO mice (n = 7) and control littermates (n = 6) were administered i.p. with 25 mg/kg body mass LPS, and mouse survival was monitored for 72 h. (b) Mice were bled at 1.5 h after LPS injection. Serum TNF-α was measured by ELISA. ***P < 0.001, ****P < 0.0001.
Deletion of gp96 in DCs protects against CLP-Induced polymicrobial sepsis
To further investigate whether DC-intrinsic gp96 plays a pivotal role in sepsis, we used a well-defined sepsis model of CLP. We found that KO mice exhibited significantly increased the survival rates in comparison with their WT littermates (n = 21, P < 0.01; Figure 2a). In addition, deletion of gp96 dramatically lowered CLP-Induced serum levels of IL-12p40 (WT: 903.9 ± 42.7 pg/mL, KO: 341.6 ± 55.8 pg/mL; n = 5, P < 0.001; Figure 2b) and IL-10 (WT: 5.1 ± 1.4 ng/mL, KO: 0.5 ± 0.1 ng/mL; n = 4, P < 0.05; Figure 2c). Liver function was significantly improved in KO mice after CLP surgery evidenced by reduced circulating ALT (WT: 38.8 ± 3.5 U/l, KO: 24.4 ± 2.5 U/l; n = 3–6, P < 0.05; Figure 2d) and AST (WT: 65.3 ± 8.1 U/l, KO: 36.0 ± 3.9 U/l; n = 3–6, P < 0.05; Figure 2e). However, the ALT (WT: 18.3 ± 0.6 U/l, KO: 24.6 ± 4.8 U/l) and AST levels (WT: 21.7 ± 2.1 U/l, KO: 21.1 ± 5.8 U/l) were comparable between WT and KO mice after Sham operation (Figure 2d and e). Moreover, two important renal functional indicators, serum creatinine (WT: 0.7 ± 0.1 mg/dl, KO: 0.4 ± 0.02 mg/dl; n ≥ 4, P < 0.001; Figure 2f) and BUN levels (WT: 134.7 ± 11.0 mg/dl, CLP KO: 59.6 ± 11.1 mg/dl; n ≥ 4, P < 0.01; Figure 2g) were also significantly attenuated in KO mice after CLP surgery, in comparison with WT mice. These data indicated that deletion of gp96 in DCs protected against CLP-Induced polymicrobial sepsis.
Figure 2.
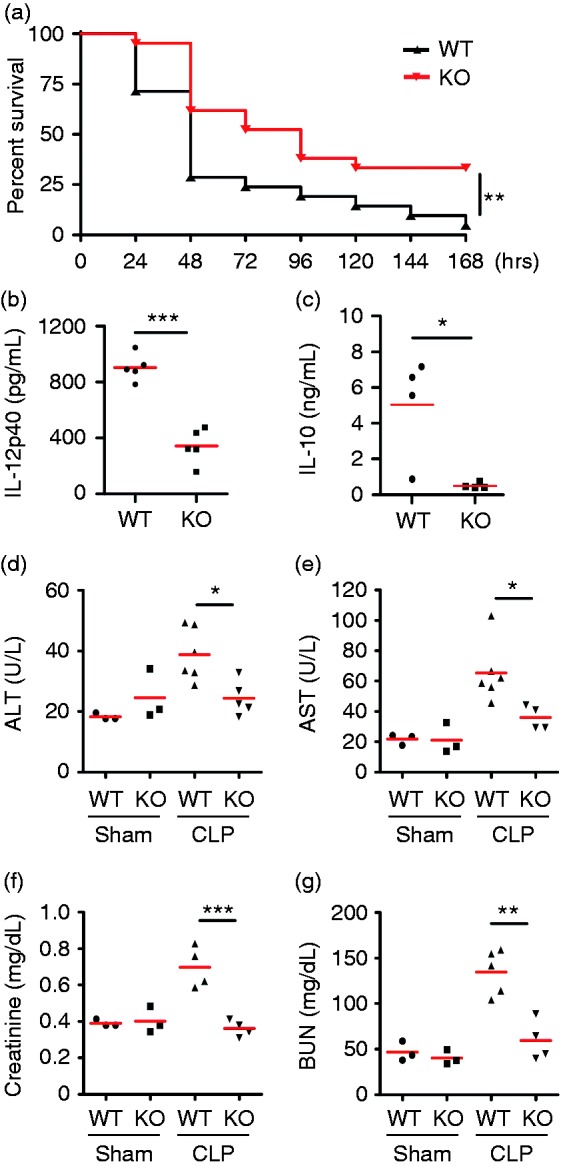
Deletion of gp96 in DCs protects mice against CLP-Induced polymicrobial sepsis. (a) KO mice (n = 21) and WT littermates (n = 21) were subjected to CLP and monitored for survival. (b, c) Sera were collected at 24 h after CLP from KO and WT littermates (n = 5), and cytokine IL-12p40 (b) and IL-10 (c) were measured by ELISA. (d–g) Sera were collected at 24 h from KO and WT littermates after a sham operation (n = 3) or CLP (n ≥ 4), and ALT (d), AST (e), creatinine (f), and BUN (g) levels were measured by ELISA. *P < 0.05, **P < 0.01, ***P < 0.001.
Deletion of gp96 in DCs impairs DC maturation in CLP-Induced septic mice
We reported previously that gp96-deficient DCs were unable to respond to TLR ligands.32 We next examined whether gp96 deletion in DCs has an impact on the DC maturation and activation in CLP-Induced septic mice as well as sham mice in vivo. We isolated splenic cells from WT and KO mice after CLP or Sham procedure and performed comparative phenotypic analysis of DCs. Previous studies demonstrated that gp96 is an essential molecular chaperone for multiple integrins.20,21,23 Deletion of gp96 from DCs resulted in decreased surface expression of CD11c. Thus, we used a negative gating strategy (B220-MHC class II+) to define DC lineages (Figure 3a, left),32,38 and further checked surface expression of CD83 and CD86 in DCs (Figure 3a, right), which are characteristic cell surface markers for fully matured DCs and are important for providing costimulatory signals for T cell activation and survival. We found that deletion of gp96 in DCs completely abrogated up-regulation of CD83 and CD86 in the CLP-Induced septic mice (n = 6–7, P < 0.001; Figure 3b and c). This result suggested that DC-intrinsic gp96 is essential for the maturation and activation of DCs during the CLP-Induced sepsis.
Figure 3.

Deletion of gp96 in DCs impairs DC maturation in CLP-induced septic mice. (a) Flow cytometry analysis of splenic DCs by negative gating strategy to define DCs (B220−MHCII+, left panel), followed by examining cell surface maturation markers CD83 and CD86 (right panel, open histogram with solid line). Gray-shaded histograms represent isotype controls. (b, c) Quantification of CD83+ (b) and CD86+ (c) splenic DCs as defined in (a) in WT and KO mice after Sham treatment (n = 3) or CLP (n = 7). ***P < 0.001.
Deletion of gp96 in DCs significantly increases the systemic Ig production
A recent study reported that commensal bacteria-induced systemic IgA contributed to protecting against polymicrobial sepsis.39 Previously we have demonstrated that DC-specific gp96-deficient mice developed spontaneous colitis with age. Consistent with the report that intestinal IgA coating represents ongoing inflammatory conditions, we also found that DC-specific gp96 KO mice had significantly higher levels of IgA and IgG1 in the sera compared with WT controls at baseline, and the level of fecal IgA was also significantly higher in KO mice.32 Next, we examined whether deletion of gp96 in DCs affects systemic Ig production in CLP-Induced septic mice. Indeed, we found that sham KO mice had significantly higher levels of IgA (Figure 4a), IgG1 (Figure 4b), and IgG2c (Figure 4d) but not IgG2b (Figure 4c), IgG3 (Figure 4e), and IgM (Figure 4f) compared with sham WT controls. Moreover, the levels of IgA (Figure 4a) and IgG1 (Figure 4b) were also significantly increased in the CLP KO mice compared with the CLP WT controls, while the levels of IgG2b and IgM remained the same. However, IgG2c and IgG3 levels were significantly reduced in the CLP KO mice compared with the CLP WT controls (Figure 4d and e). Our data suggest that systemic IgA and IgG1 contribute to protecting against polymicrobial sepsis. Thus, targeting gp96 in DCs will provide a potential therapeutic for the treatment of sepsis.
Figure 4.
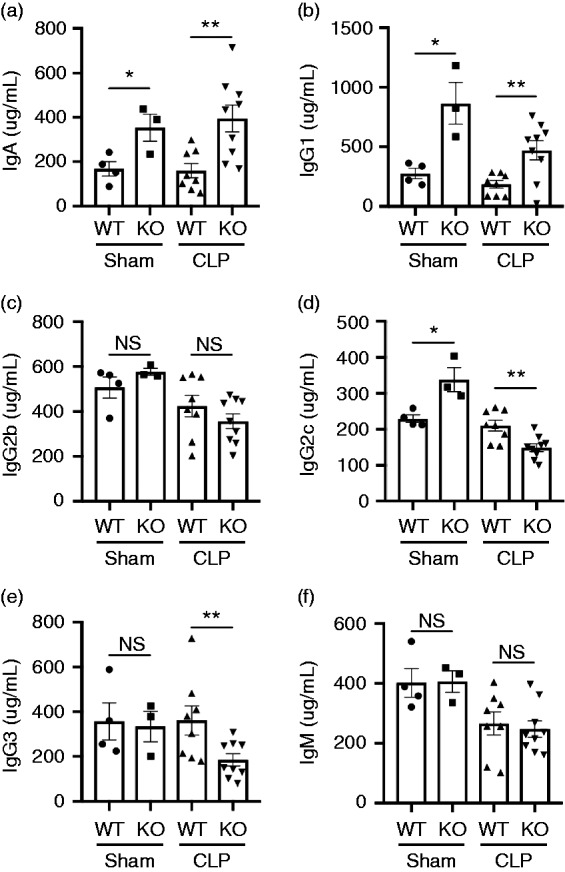
Deletion of gp96 in DCs results in altered the systemic Ig production. (a–f) KO mice and WT littermates were subjected to Sham (n = 3–4) or CLP (n = 8). Serum was collected at 24 h after the surgery, and then the Ig levels of IgA (a), IgG1 (b), IgG2b (c), IgG2c (d), IgG3 (e), and IgM (f) in the sera were measured by ELISA. Error bars indicate SEM. *P < 0.05; **P < 0.01.
Microarray analysis of LPS-induced inflammatory pathways in BMDCs
We so far have shown that deletion of gp96 in DCs in mice significantly reduced the level of systemic inflammatory cytokines, improved their survival in response to LPS-induced endotoxemia and CLP-Induced polymicrobial sepsis (Figures 1 and 2). Also, deletion of gp96 attenuated DC maturation and activation after CLP. By using a gene expression microarray analysis, we next investigated what were the LPS-inducible genes in DCs that were altered by gp96 deletion. We treated WT and KO BMDCs with or without LPS for 6 h, and then quantified the levels of mRNA encoded by genes involved in inflammatory pathways. We found that multiple inflammatory cytokine levels were significantly decreased in the KO BMDCs including Il1a, Il1f6, Il1rn, Il6, Il12b, and Tnfsf4 along with inflammation-related genes (Cxcl1, Cxcl5, Cxcl9, Cxcl12, Nr1h3) (Figure 5) after LPS treatment. However, the levels of inflammatory cytokines in unstimulated DCs are very low or undetectable compared with LPS stimulated DCs. Also, the expression levels of these genes are comparable between unstimulated WT and KO DCs (data not shown). This result is consistent with that gp96 chaperone TLRs.20,21,23,32 This data suggests that gp96 deletion offers a unique advantage in treating sepsis over existing approaches by down-regulating multiple inflammatory pathways.40–43
Figure 5.

Microarray analysis of mRNAs levels in WT and gp96 KO BMDCs in response to LPS. WT or KO BMDCs were stimulated with LPS (200 ng/mL) for 6 h followed by mRNA extraction, reverse transcription, and cDNA microarray analysis using the whole mouse cDNA array. The heat map shows the genes involved in the inflammatory pathway using the KEGG Pathway Database analysis. Comparing with WT BMDCs, KO cells expressed significantly less inflammatory cytokines in response to LPS.
Discussion
DCs are professional Ag-presenting cells that play an important role in both innate and adaptive immunity. DCs have been shown to be significantly involved in all phases of the pathological development of sepsis from the initial inflammatory response to the immune suppressive stage; however, the extent of their contributions has not been fully elucidated. DCs recognize microbes and pathogens through PRRs, including TLRs and NLRs.5–7 We previously reported that heat shock protein gp96 is an essential immune chaperone for TLRs including TLR1, TLR2, TLR4, TLR5, TLR6, TLR7, TLR8, and TLR9, integrins, and other vital innate receptors.20,21,23,44 Consistent with gp96 chaperone function, gp96 KO DCs failed to respond to stimulation by TLR2, TLR4, and TLR9 ligands in vitro.32 In this study, we found that specific deletion of gp96 in DCs protected against both LPS-induced endotoxemia and CLP-Induced polymicrobial sepsis, evidenced by decreased circulating inflammatory cytokines (TNF-α and IL-12), attenuated liver and renal injury, and reduced mortality. Moreover, deletion of gp96 in DCs completely abrogated up-regulation of CD83 and CD86 in the CLP mice (Figure 3b and c), which suggested that DC-intrinsic gp96 is essential for the maturation and activation of DCs during multi-microbial sepsis. Our study suggested that DCs are a major source of inflammatory cytokines during sepsis and that DC-intrinsic TLR signaling including TLR2 and TLR4 may contribute to mortality associated with sepsis.
Recent studies demonstrate that DCs can adopt tolerogenic functions in the presence of Wnt3a and Wnt5a, and that these DCs produce high levels of IL-10, TGF-β, retinoic acid (RA), IL-27, and vascular endothelial growth factor in response to TLR ligands.45–47 In addition, DC-intrinsic TLR2 signaling activated β-catenin and induced IL-10 and RA production.48 We have demonstrated that gp96 is an essential molecular chaperone for Wnt co-receptor LRP6. Consistent with the loss of LRP6 and Wnt signaling, gp96 KO cells failed to up-regulate Axin2 mRNA in response to Wnt-3a.28 In the present study, we found that gp96 KO mice produced a low level of IL-10 during CLP-Induced polymicrobial sepsis (Figure 2c), which may be due to loss of both TLR and Wnt signaling. Further studies are needed to clarify the relative contribution by each of the two pathways in sepsis.
Low levels of circulating IgA, IgG1, and IgM have been associated with low survival rates in patients with severe sepsis or septic shock.49,50 Treatment with combined i.v. IgG/IgA/IgM decreased the risk of death after 28 d in patients with severe sepsis and/or septic shock.51 In addition, a recent study reported that commensal bacteria-induced systemic IgA contributed to protecting against polymicrobial sepsis.39 We found that CLP KO mice had significantly higher levels of IgA and IgG1 (Figure 4) compared with the CLP WT controls; while the levels of IgG2b and IgM remained the same. However, IgG2c and IgG3 levels were significantly reduced in the CLP KO mice compared with the CLP WT controls (Figure 4d and e). Our data indicate that systemic IgA and IgG1 contribute to protecting against polymicrobial sepsis.
In summary, our results demonstrate that DCs are one of the major cellular sources of inflammatory cytokines during sepsis. Deletion of gp96 in DCs is beneficial in both LPS-induced endotoxemia and CLP-Induced polymicrobial sepsis through a variety of mechanisms including blocking TLR signaling, inhibiting Wnt signaling, and increasing systemic IgA and IgG1. Therefore, targeting gp96 in DCs either genetically or pharmacologically29,52–54 may provide a potential novel approach for the sepsis treatment.
Acknowledgements
The authors thank Dr Gary Hardiman for assistance with microarray data analysis. The authors also thank the past and present laboratory members of HF, ZL and BL for their input throughout the course of this work. This study was partially supported by multiple National Institutes of Health grants.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: BL is supported by the National Institutes of Health (NIH) (Grant Numbers R01CA193939 and U01AI125859), ZL is supported by NIH (Grant Numbers P01CA186866, R01CA188419, R01AI077283, R01CA213290, and R01DK105033), and HF is supported by NIH (Grant Numbers R01GM113995, R01GM130653, and NIH.NCATS SCTR UL1TR001450).
References
- 1.Kumar V, Sharma A. Innate immunity in sepsis pathogenesis and its modulation: new immunomodulatory targets revealed. J Chemother 2008; 20: 672–683. [DOI] [PubMed] [Google Scholar]
- 2.Vega-Ramos J, Roquilly A, Asehnoune K, et al. Modulation of dendritic cell antigen presentation by pathogens, tissue damage and secondary inflammatory signals. Curr Opin Pharmacol 2014; 17: 64–70. [DOI] [PubMed] [Google Scholar]
- 3.Bouras M, Asehnoune K, Roquilly A. Contribution of dendritic cell responses to sepsis-induced immunosuppression and to susceptibility to secondary pneumonia. Front Immunol 2018; 9: 2590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Steinman RM, Hawiger D, Nussenzweig MC. Tolerogenic dendritic cells. Annu Rev Immunol 2003; 21: 685–711. [DOI] [PubMed] [Google Scholar]
- 5.Lavelle EC, Murphy C, O’Neill LA, et al. The role of TLRs, NLRs, and RLRs in mucosal innate immunity and homeostasis. Mucosal Immunol 2010; 3: 17–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Figdor CG, van Kooyk Y, Adema GJ. C-type lectin receptors on dendritic cells and Langerhans cells. Nat Rev Immunol 2002; 2: 77–84. [DOI] [PubMed] [Google Scholar]
- 7.Geremia A, Biancheri P, Allan P, et al. Innate and adaptive immunity in inflammatory bowel disease. Autoimmun Rev 2014; 13: 3–10. [DOI] [PubMed] [Google Scholar]
- 8.Bruns S, Pastille E, Wirsdorfer F, et al. Lipopeptides rather than lipopolysaccharide favor the development of dendritic cell dysfunction similar to polymicrobial sepsis in mice. Inflamm Res 2013; 62: 627–636. [DOI] [PubMed] [Google Scholar]
- 9.Kumar V. Dendritic cells in sepsis: potential immunoregulatory cells with therapeutic potential. Mol Immunol 2018; 101: 615–626. [DOI] [PubMed] [Google Scholar]
- 10.Wilson NS, Behrens GM, Lundie RJ, et al. Systemic activation of dendritic cells by Toll-like receptor ligands or malaria infection impairs cross-presentation and antiviral immunity. Nat Immunol 2006; 7: 165–172. [DOI] [PubMed] [Google Scholar]
- 11.Tan PH, Tyrrell HE, Gao L, et al. Adiponectin receptor signaling on dendritic cells blunts antitumor immunity. Cancer Res 2014; 74: 5711–5722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Escobar DA, Botero-Quintero AM, Kautza BC, et al. Adenosine monophosphate-activated protein kinase activation protects against sepsis-induced organ injury and inflammation. J Surg Res 2015; 194: 262–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Srivastava PK, DeLeo AB, Old LJ. Tumor rejection antigens of chemically induced sarcomas of inbred mice. Proc Natl Acad Sci USA 1986; 83: 3407–3411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lee AS, Delegeane A, Scharff D. Highly conserved glucose-regulated protein in hamster and chicken cells: preliminary characterization of its cDNA clone. Proc Natl Acad Sci USA 1981; 78: 4922–4925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Koch G, Smith M, Macer D, et al. Endoplasmic reticulum contains a common, abundant calcium-binding glycoprotein, endoplasmin. J Cell Sci 1986; 86: 217–232. [DOI] [PubMed] [Google Scholar]
- 16.Lewis MJ, Mazzarella RA, Green M. Structure and assembly of the endoplasmic reticulum. The synthesis of three major endoplasmic reticulum proteins during lipopolysaccharide-induced differentiation of murine lymphocytes . J Biol Chem 1985; 260: 3050–3057. [PubMed] [Google Scholar]
- 17.Chen B, Piel WH, Gui L, et al. The HSP90 family of genes in the human genome: insights into their divergence and evolution. Genomics 2005; 86: 627–637. [DOI] [PubMed] [Google Scholar]
- 18.Kozutsumi Y, Segal M, Normington K, et al. The presence of malfolded proteins in the endoplasmic reticulum signals the induction of glucose-regulated proteins. Nature 1988; 332: 462–464. [DOI] [PubMed] [Google Scholar]
- 19.Randow F, Seed B. Endoplasmic reticulum chaperone gp96 is required for innate immunity but not cell viability. Nat Cell Biol 2001; 3: 891–896. [DOI] [PubMed] [Google Scholar]
- 20.Yang Y, Liu B, Dai J, et al. Heat shock protein gp96 is a master chaperone for toll-like receptors and is important in the innate function of macrophages. Immunity 2007; 26: 215–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu B, Li Z. Endoplasmic reticulum HSP90b1 (gp96, grp94) optimizes B-cell function via chaperoning integrin and TLR but not immunoglobulin. Blood 2008; 112: 1223–1230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu K, Nussenzweig MC. Origin and development of dendritic cells. Immunol Rev 2010; 234: 45–54. [DOI] [PubMed] [Google Scholar]
- 23.Staron M, Yang Y, Liu B, et al. gp96, an endoplasmic reticulum master chaperone for integrins and Toll-like receptors, selectively regulates early T and B lymphopoiesis. Blood 2010; 115: 2380–2390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu B, Yang Y, Qiu Z, et al. Folding of Toll-like receptors by the HSP90 paralogue gp96 requires a substrate-specific cochaperone. Nat Commun 2010; 1: 79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang Y, Wu BX, Metelli A, et al. GP96 is a GARP chaperone and controls regulatory T cell functions. J Clin Invest 2015; 125: 859–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Metelli A, Wu BX, Fugle CW, et al. Surface expression of TGFbeta docking receptor GARP promotes oncogenesis and immune tolerance in breast cancer. Cancer Res 2016; 76: 7106–7117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rachidi S, Metelli A, Riesenberg B, et al. Platelets subvert T cell immunity against cancer via GARP-TGFbeta axis. Sci Immunol 2017; 2: eaai7911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu B, Staron M, Hong F, et al. Essential roles of grp94 in gut homeostasis via chaperoning canonical Wnt pathway. Proc Natl Acad Sci USA 2013; 110: 6877–6882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hua Y, White-Gilbertson S, Kellner J, et al. Molecular chaperone gp96 is a novel therapeutic target of multiple myeloma. Clin Cancer Res 2013; 19: 6242–6251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vabulas RM, Braedel S, Hilf N, et al. The endoplasmic reticulum-resident heat shock protein Gp96 activates dendritic cells via the Toll-like receptor 2/4 pathway. J Biol Chem 2002; 277: 20847–20853. [DOI] [PubMed] [Google Scholar]
- 31.Morales C, Rachidi S, Hong F, et al. Immune chaperone gp96 drives the contributions of macrophages to inflammatory colon tumorigenesis. Cancer Res 2014; 74: 446–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hua Y, Yang Y, Sun S, et al. Gut homeostasis and regulatory T cell induction depend on molecular chaperone gp96 in CD11c(+) cells. Sci Rep 2017; 7: 2171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li P, Guo Y, Bledsoe G, et al. Kallistatin treatment attenuates lethality and organ injury in mouse models of established sepsis. Crit Care 2015; 19: 200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Guo L, Zheng Z, Ai J, et al. Hepatic scavenger receptor BI protects against polymicrobial-induced sepsis through promoting LPS clearance in mice. J Biol Chem 2014; 289: 14666–14673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gao M, Wang X, Zhang X, et al. Attenuation of cardiac dysfunction in polymicrobial sepsis by microRNA-146a is mediated via targeting of IRAK1 and TRAF6 expression. J Immunol 2015; 195: 672–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu B, Staron M, Li Z. Murine but not human basophil undergoes cell-specific proteolysis of a major endoplasmic reticulum chaperone. PLoS One 2012; 7: e39442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Caton ML, Smith-Raska MR, Reizis B. Notch-RBP-J signaling controls the homeostasis of CD8- dendritic cells in the spleen. J Exp Med 2007; 204: 1653–1664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tan SY, Cavanagh LL, d'Advigor W, et al. Phenotype and functions of conventional dendritic cells are not compromised in aged mice. Immunol Cell Biol 2012; 90: 722–732. [DOI] [PubMed] [Google Scholar]
- 39.Wilmore JR, Gaudette BT, Gomez Atria D, et al. Commensal microbes induce serum IgA responses that protect against polymicrobial sepsis. Cell Host Microbe 2018; 23: 302–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Brown RM, Semler MW. Fluid management in sepsis. J Intensive Care Med 2019; 34: 364–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rello J, van Engelen TSR, Alp E, et al. Towards precision medicine in sepsis: a position paper from the European Society of Clinical Microbiology and Infectious Diseases. Clin Microbiol Infect 2018; 24: 1264–1272. [DOI] [PubMed] [Google Scholar]
- 42.Thampy LK, Remy KE, Walton AH, et al. Restoration of T cell function in multi-drug resistant bacterial sepsis after interleukin-7, anti-PD-L1, and OX-40 administration. PLoS One 2018; 13: e0199497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Peters van Ton AM, Kox M, Abdo WF, et al. Precision immunotherapy for sepsis. Front Immunol 2018; 9: 1926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ansa-Addo EA, Thaxton J, Hong F, et al. Clients and oncogenic roles of molecular chaperone gp96/grp94. Curr Top Med Chem 2016; 16: 2765–2778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Oderup C, LaJevic M, Butcher EC. Canonical and noncanonical Wnt proteins program dendritic cell responses for tolerance. J Immunol 2013; 190: 6126–6134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shen CC, Kang YH, Zhao M, et al. WNT16B from ovarian fibroblasts induces differentiation of regulatory T cells through beta-catenin signal in dendritic cells. Int J Mol Sci 2014; 15: 12928–12939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Suryawanshi A, Manoharan I, Hong Y, et al. Canonical wnt signaling in dendritic cells regulates Th1/Th17 responses and suppresses autoimmune neuroinflammation. J Immunol 2015; 194: 3295–3304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Silva-Garcia O, Valdez-Alarcon JJ, Baizabal-Aguirre VM. The Wnt/beta-catenin signaling pathway controls the inflammatory response in infections caused by pathogenic bacteria. Mediators Inflamm 2014; 2014: 310183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bermejo-Martin JF, Rodriguez-Fernandez A, Herran-Monge R, et al. Immunoglobulins IgG1, IgM and IgA: a synergistic team influencing survival in sepsis. J Intern Med 2014; 276: 404–412. [DOI] [PubMed] [Google Scholar]
- 50.Giamarellos-Bourboulis EJ, Apostolidou E, Lada M, et al. Kinetics of circulating immunoglobulin M in sepsis: relationship with final outcome. Crit Care 2013; 17: R247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kreymann KG, de Heer G, Nierhaus A, et al. Use of polyclonal immunoglobulins as adjunctive therapy for sepsis or septic shock. Crit Care Med 2007; 35: 2677–2685. [PubMed] [Google Scholar]
- 52.Patel HJ, Patel PD, Ochiana SO, et al. Structure–activity relationship in a purine-scaffold compound series with selectivity for the endoplasmic reticulum Hsp90 paralog Grp94. J Med Chem 2015; 58: 3922–3943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Que NLS, Crowley VM, Duerfeldt AS, et al. Structure based design of a Grp94-selective inhibitor: exploiting a key residue in Grp94 to optimize paralog-selective binding. J Med Chem 2018; 61: 2793–2805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wang J, Grishin AV, Ford HR. Experimental anti-inflammatory drug semapimod inhibits TLR signaling by targeting the TLR chaperone gp96. J Immunol 2016; 196: 5130–5137. [DOI] [PMC free article] [PubMed] [Google Scholar]