

Short abstract
Over the past few years the NAD-related compounds nicotinamide (NAM), nicotinamide riboside (NR) and 1-methylnicotinamide (MNA) have been established as important molecules in signalling pathways that contribute to metabolic functions of many cells, including those of the immune system. Among immune cells, monocytes/macrophages, which are the major players of inflammatory processes, are especially susceptible to the anti-inflammatory action of NAM. Here we asked whether NAM and the two other compounds have the potential to regulate differentiation and LPS-induced biological answers of the monocytic cell line THP-1. We show that treatment of THP-1 cells with NAM, NR and MNA resulted in growth retardation accompanied by enrichment of cells in the G0/G1-phase independent of p21 and p53. NAM and NR caused an increase in intracellular NAD concentrations and SIRT1 and PARP1 mRNA expression was found to be enhanced. The compounds failed to up-regulate the expression of the cell surface differentiation markers CD38, CD11b and CD14. They modulated the reactive oxygen species production and primed the cells to respond less effectively to the LPS induced TNF-α production. Our data show that the NAD metabolites interfere with early events associated with differentiation of THP-1 cells along the monocytic path and that they affect LPS-induced biological responses of the cell line.
Keywords: 1-Methylnicotinamide, nicotinamide, nicotinamide riboside, pro-inflammatory cytokines, reactive oxygen species, THP-1 differentiation
Introduction
Nicotinamide (NAM) and nicotinic acid belong to the vitamin B3 group, also known as niacin.1 There is increasing evidence that NAM, a natural occurring compound of the human diet, can be used as a potential therapeutic drug to treat numerous diseases such as metabolic and immunological disorders.2,3
In most mammalian cells, NAM is the predominant precursor of NAD, a molecule playing a key role in energy metabolism and electron transfer. NAD and its reduced form NADH function as cofactors in multiple redox reactions. Electrons derived from NADH are transferred to O2 to generate ATP. Unlike NAD-dependent redox reactions, NAD-dependent signalling processes are catalysed by enzymes that continuously consume NAD. NAD-Degrading enzymes regulate various biological processes, such as transcription, cell cycle progression, DNA repair and intracellular communication.4,5
Enzymatic cleavage of NAD, which is catalysed by CD38, ADP-ribosyltransferases (ARTs), poly(ADP-ribose) polymerases (PARPs) and sirtuins, results in the release of NAM and the ADP-ribose moiety (ADPR). While NAM serves as a feedback inhibitor of these enzymes,6,7 ADPR is transferred onto an acceptor or used to facilitate Ca2+ influx.8 To maintain normal tissue levels of NAD and to relieve NAM inhibition of NAD-consuming enzymes, a salvage pathway recycles NAM back to NAD (Figure 1). Nicotinamide phosphoribosyltransferase (NAMPT), the first enzyme in this pathway, catalyses the production of nicotinamide mononucleotide (NMN) from NAM and phosphoribosyl pyrophosphate (PRPP). In a second step, NMN together with ATP is then used by nicotinamide mononucleotide adenylyltransferases (NMNAT 1-3) to yield NAD.9 Only recently nicotinamide riboside (NR), a natural product found in milk, has also been identified as a NAD precursor.10 NR is metabolised to NMN, which in turn is converted into NAD, a reaction catalysed by NMNAT 1-3 (Figure 1). Apart from being recycled to NAD, NAM is irreversibly methylated by nicotinamide-N-methyltransferase (NNMT) to yield 1-methylnicotinamide (MNA).11 MNA serves as a substrate of aldehyde oxidase 1 (AOX1) to generate N-methyl-2-pyridone-5-carboxamide (2PY) as well as N-methyl-4-pyridone-3-carboxamide (4PY) and hydrogen peroxide which, at low physiological concentrations, has been suggested to exert lifespan extending effects.11 Considering the crucial role of NAD in regulating metabolic processes, much attention has been paid to the use of the NAD precursors NAM and NR,3,10,12–15 and the NAD degradation product MNA,16 as agents for therapeutic intervention. These are partly based on the well described anti-inflammatory effects of NAM and NR.7,17–19 In contrast, little is known about a role of NAD metabolites in regulating differentiation of myeloid leukaemia cells such as HL60, U937 and THP-1. When exposed to vitamin D3 (VitD), these cells undergo a cell cycle arrest and express surface markers of monocytic cells including CD38,20 CD11b and CD14.21–23
Figure 1.

Intracellular NAD metabolism. NAM is a product of deacetylation and ADP-ribosylation reactions, which are catalysed by NAD-consuming enzymes such as sirtuins, PARPs, ARTs and CD38. NAMPT, the rate-limiting step in the NAD salvage pathway, catalyses the formation of NMN from NAM and 5′-phosphoribosyl-1-pyrophosphate. NMN is then converted into NAD by NMNAT 1-3. NAM can also be methylated by NNMT leading to the formation of MNA. NR enters the NAD biosynthesis pathway by phosphorylation to NMN, a step catalysed by NRKs. Other NAD biosynthesis pathways include the de novo synthesis starting from tryptophan and the Preiss–Handler route by which nicotinic acid enters the de novo pathway.
CD38 is a type II glycoprotein that transforms NAD into cyclic ADP-ribose (cADPR), a powerful mobiliser of intracellular Ca2+.24,25 During differentiation, myeloid cells strongly up-regulate CD38 expression and there is some evidence that CD38 and the consequential accumulation of cADPR play a causal role in mediating differentiation.26 In addition to CD38, CD11b is another classical marker of monocyte differentiation.27 CD11b associated with CD18, is involved in cell adhesion and functions as a complement receptor and part of the multimeric LPS-receptor complex.28,29 CD14 binds LPS and transfers it to myeloid differentiation factor 2 (MD2) to initiate TLR4 dimerisation and signalling. Its expression has been shown to increase dramatically during the differentiation of monocytic precursors into monocytes.21–23
In the present study, we asked whether the NAD related compounds NAM, NR and MNA induce cellular processes that affect maturation and functional properties of THP-1 cells. We identified all three compounds as agents with anti-proliferative effects that, to a limited extend, are able to induce THP-1 cells to differentiate along the monocytic path. They prime the cells to modulate the release of reactive oxygen species (ROS) and to suppress LPS-induced TNF-α production, most likely in a CD14- and TLR4-independent manner.
Material and methods
Reagents
NAM, MNA and resveratrol were purchased form Sigma-Aldrich (Taufkirchen, Germany), NR from ChromaDex (Irvine, CA, USA) and VitD (1,25-(OH)2-D3) from Cayman Chemical (Ann Arbor, MI, USA). VitD was dissolved in 40% ethanol (stock solution of 2.5 mM), resveratrol in DMSO (stock solution 5 mg/ml), NAM in distilled water (stock solution 1M) or NR and MNA in media (stock solution 1M each).
Cell culture
THP-1 cells were cultured in RPMI 1640 (contains 0.008 mM NAM; GE Healthcare, Little Chalfont, UK) supplemented with 10% FCS (Sigma-Aldrich), 1% penicillin (10,000 U/ml) and streptomycin (10,000 µg/ml) (Seromed Biochrom KG, Berlin, Germany) and 0.05 mM 2-mercaptoethanol (Life Technologies, Darmstadt, Germany). Cells (0.2 × 106/ml) were treated in a humidified atmosphere of 5% CO2 at 37°C with 8 mM NAM, 8 mM NR, 3.2 mM MNA, 0.1 µM VitD and the corresponding solvent controls for several times (24–96 h). The cell numbers were counted with a Neubauer chamber and the viability was determined by trypan blue (Sigma-Aldrich) exclusion.
Detection of apoptosis/necrosis
Apoptosis and necrosis of THP-1 cells were determined using a FITC Annexin V/PI kit (BioLegend, San Diego, CA, USA) according to the manufacturer’s instruction. Analyses were performed using a FACS Canto II flow cytometer (Becton Dickinson, Franklin Lakes, NY, USA).
Determination of intracellular total NAD
Cells (2 × 106) were harvested by centrifugation (400 g, 6 min, room temperature (RT)). The pellet was re-suspended in 200 μL 1 M perchloric acid and homogenised by sonication (3 strokes, output 30%, Bandelin Sonopuls GM70, Bandelin, Berlin, Germany). After centrifugation (18,000 g, 5 min, 4°C) the pellet of each sample was used for protein determination (Bio-Rad DC Protein Assay, Hercules, CA, USA). Supernatants were neutralised with 3M potassium carbonate, centrifuged again (18,000 g, 10 min, 4°C) and frozen at −80°C. NAD concentrations (NAD plus oxidised NADH) were determined by HPLC analysis, as described previously.30,31 The NAD levels were normalised to the total protein content.
Detection of cell surface markers
After 96 h THP-1 cells were harvested by centrifugation (400 g, 6 min, RT). Cells (1.5 × 105) were incubated at 4°C with 10% human AB-serum (Institute of Transfusions Medicine, University Hospital Leipzig) to saturate Fc receptors and block unspecific bindings. After 15 min the cells were washed (PBS + 10% Emagel (Pirmal Healthcare, Morpeth, Northumberland, UK + 0.1% NaN3)) and the direct dye labelled Abs anti-CD14-APC (BioLegend), anti-TLR4-PE (R&D systems, Minneapolis, MO, USA) or the non-labelled anti-CD11b (BL-M/G1), anti-CD14 (BL-M/G14) and anti-CD38 (BL-AC38) Abs (all from DiaMak, Leipzig, Germany) as well as the corresponding isotypes were added for 20 min at 4°C. Cells were then washed and the non-labelled probes further incubated for 15 min with a FITC-labelled goat-anti-mouse Ab. After washing and fixation the cells were analysed on a FACS Canto II flow cytometer (Becton Dickinson). For data analysis median fluorescence intensity (MFI) values of the isotype were subtracted from MFI values of the sample.
Detection of TNF-α in supernatants
THP-1 cells (1 × 106/ml) were cultured for 96 h before they were incubated in the presence and absence of 100 ng/ml LPS (Escherichia coli, serotype 055:B5, Sigma-Aldrich) for 4 h. TNF-α levels in the supernatants were determined using a commercially available ELISA-based assay system (PeproTech, Rocky Hill, NJ, USA) according to the manufacturer’s protocol.
Detection of intracellular ROS
THP-1 cells cultured for 96 h were harvested by centrifugation (400 g, 6 min, RT). Cells (1.5 × 105) were re-suspended in 250 µl PBS and incubated for 30 min at 37°C, 5% CO2 before 100 ng/ml LPS and 20 µM 2′,7′-dichlorodihydrofluorescein diacetate (DCF-DA) (Sigma-Aldrich) were added. After 30 min intracellular ROS production was measured by oxidation of intracellular fluorescent DCF-DA by flow cytometry on a FACS Canto II flow cytometer (Becton Dickinson). The shift in fluorescence intensity was used to assess the percentage of cells that were able to generate ROS. Gates to determine positive cells were set to exclude 95% of control cells.
Cell cycle analyses
THP-1 cells were harvested after 96 h by centrifugation (400 g, 6 min, RT). Cells (1 × 106) were re-suspended in 0.5 ml PBS/EDTA and fixed by addition of 1.5 ml ice-cold ethanol. After centrifugation, the cells were re-suspended in PBS/EDTA treated with 10 µg/ml RNase A (Life Technologies) and passed through a cell filter (30 µm). Cell cycle analysis was performed by flow cytometry on a FACS Canto II flow cytometer (Becton Dickinson) 15 min after the addition of 100 µg/ml propidium iodide (PI).
Western blot analysis
Western blot analysis was carried out as described previously.32 THP-1 cells (1 × 106) were suspended in RIPA buffer (50 mM Tris, 150 mM NaCl, 1% Nonidet P 40, 0.5% deoxycholate, 0.1% SDS; pH 7.5) supplemented with cOmplete™ EDTA-free protease inhibitor cocktail (Roche Diagnostics, Mannheim, Germany). After sonication, the samples were centrifuged for 5 min at 15,000 g and 4°C. The protein concentrations in the supernatants were determined using a detergent compatible protein assay (DC™ protein assay, Bio-Rad, Hercules, CA, USA) according to the manufacturer’s protocol. Cell lysates (20 µg) boiled in 1× Laemmli sample buffer were run on a 12% SDS-polyacrylamide gel (Protean II, Bio-Rad GmbH) and transferred to polyvinylidene difluoride (PVDF) membranes (Amersham Biosciences, Munich, Germany). Membranes were probed with anti-SIRT1 Ab (C14H4, 1:1000, Cell Signaling, Frankfurt am Main, Germany), anti-PARP1 Ab (46D11, 1:1000, Cell Signaling) and anti-β-actin Ab (AC74, 1:2000, Sigma-Aldrich). The following POD-conjugated secondary Abs were used: goat-anti-rabbit IgG Ab (1:2000, Cell Signaling) or goat-anti-mouse IgG Ab (1:8000, Sigma-Aldrich).
Chemiluminescent detection was achieved by using the ECL-A/ECL-B reagents (both from Sigma Aldrich) and the Luminescent Image Analyzer Stella (raytest, Straubenhardt, Germany).
RNA isolation, reverse transcription and real-time PCR (qPCR)
Total RNA was isolated from THP-1 cells (1 × 106) using the RNeasy Mini Kit (Qiagen, Hilden, Germany) according to manufacturer’s instruction. DNase I treatment and reverse transcription were performed as previously described.32 Real-time PCR was performed using the following reaction mixture: 7.5 µL SYBR Green PCR mastermix (Bio-Rad), 250 nM forward and reverse primers (see below) and 1.5 µL of cDNA template in a final volume of 15 µL. The following primers were used for the qPCR:
| GNB2L1 | fwd 5′-GAGTGTGGCCTTCTCCTCTG-3′ |
| rev 5′-GCTTGCAGTTAGCCAGGTTC-3′ | |
| U6 | fwd 5′ -CTCGCTTCGGCAGCACA-3′ |
| rev 5′-AACGCTTCACGAATTTGCGT-3′ | |
| CDKN1A/p21 | fwd 5′-GGAAGACCATGTGGACCTGT-3′ |
| rev 5′-GGATTAGGGCTTCCTCTTGG-3′ | |
| SIRT1 | fwd 5′-TGAGTGGCTGGAACAGTGAG-3′ |
| rev 5′ -AGCGCCATGGAAAATGTAAC-3′ | |
| PARP1 | fwd 5′-CCCGTGACAGGCTACATGTT-3′ |
| rev 5′ -CTTTGACACTGTGCTTGCCC-3′ |
The reactions were performed as previously described,32 using GNB2L1 and U6 as the reference genes.
Statistical analyses
All statistical analyses were performed using SigmaPlot software (Systat Software GmbH, Erkrath, Germany). Statistical significance was calculated with the paired Student’s t-test and classified as follows: *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001.
Results
NAD metabolites inhibit proliferation of THP-1 cells
To assess the influence of the NAD metabolites NAM (8 mM), NR (8 mM) and MNA (3.2 mM) on the growth of THP-1 cells, the cells were counted 24 h, 48 h, 72 h and 96 h after exposure to the compounds. Concentrations exceeding 8 mM (NAM, NR) or 3.2 mM (MNA) strongly reduced the viability of cells (data not shown). Of the three compounds tested, NAM was most effective in reducing cell growth followed by NR and MNA (Figure 2a). The cell numbers determined after 96 h were diminished by 40%, 31% and 12%, respectively (Figure 2a). A loss of cell viability as judged by annexin V/ PI staining could be excluded (Figure 2b). The reduction in cell growth induced by NAM was similar to that seen after treatment with VitD (Figure 2a insert), a vitamin well known to regulate differentiation and proliferation of human myeloblastic leukaemia cells.20,33,34
Figure 2.
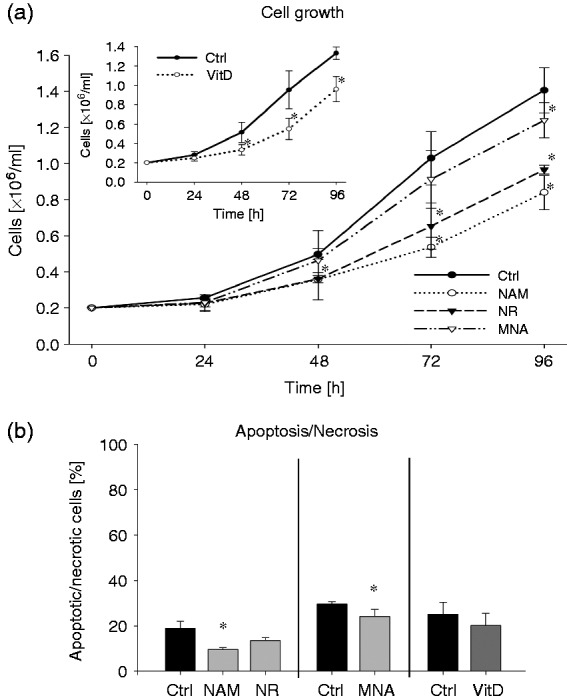
Cell growth and apoptosis/necrosis of THP-1 cells after treatment with NAM, NR, MNA and VitD. THP-1 cells (0.2 × 106/ml) were treated with 8 mM NAM, 8 mM NR, 3.2 mM MNA, 0.1 mM VitD or the respective solvent controls (ctrl). (a) Determination of the cell number was performed 24 h, 48 h, 72 h and 96 h after treatment. Shown are the cell numbers ± SD (n = 3). (b) After 96 h of treatment apoptosis/necrosis was assessed by FITC Annexin V/PI staining. Shown are means of apoptotic/necrotic cells [%] ± SD (n = 3). Statistics were performed with the paired Student’s t-test calculated to the respective control.
NAM, NR and MNA influence the cell cycle progression
Based on the observation that the NAD derivatives lead to reduced proliferation rates of THP-1 cells, we analysed the influence of these compounds on cell cycle progression. The cells were synchronised in G0/G1 by serum deprivation for 48 h. Simultaneous to the treatment with NAM, NR and MNA, cells were stimulated to re-enter the cell cycle by serum addition and DNA amounts were analysed after 24 h and 48 h. Serum deprivation led to an enrichment of cells in G0/G1-phases (Figure 3a). Addition of serum for 24 h and 48 h caused a significant decrease in G0/G1 cells. Exposure to NAM in parallel to serum re-stimulation resulted in an enrichment of cells in G0/G1 and in an equivalent decrease in S-phase, indicating that NAM caused the cells to undergo a G0/G1 cell cycle arrest or a prolonged G1-phase. Similar but less distinct effects were observed in response to NR and MNA (Figure 3b).
Figure 3.

Cell cycle analyses after treatment with NAM, NR and MNA. THP-1 cells (0.2 × 106/ml) were serum-starved (SS) for 48 h. Upon addition of serum, the cells were cultured in the presence of NAM, 8 mM NR, 3.2 mM MNA or the respective solvent control (ctrl) and cell cycle analyses were performed after 24 h and 48 h by flow cytometry. (a) One representative experiment of NAM treated cells is shown. Cell cycle phases were defined by DNA staining with propidium iodide (PI). (b) Percentages of cells in G0/G1- and S-phases. Shown are mean values ± SD (n = 3). Statistics were performed with the paired Student’s t-test calculated to the respective control.
Stress-induced arrest of eukaryotic cells is often mediated by accumulation of the transcription factor and tumour suppressor p53, which results in an increased transcription of the CDKN1A gene. This gene encodes for the cyclin dependent kinase (CDK) inhibitor p21WAF1/CIP1, a potent mediator of cell cycle arrest.35 Thus, we next tested whether expressions of p53 and p21 were increased by NAM. We did not detect any p53 or p21 protein expression neither in unstimulated nor in NAM-treated cells (data not shown). Furthermore, CDKN1A/p21 mRNA expression was not activated in response to NAM (SupplementalFigure 1). These findings indicate that accumulation of NAM-treated cells in the G0/G1-phase is p53 and p21-independent.
We next tested the effect of NAM on the cell cycle distribution of THP-1 cells stimulated with the sirtuin activator resveratrol.36 Sirtuins are NAD-dependent histone-deacetylases that regulate the passage of eukaryotic cells through the cell cycle. Furthermore, NAM has been described as an inhibitor of sirtuins.37 As shown in Figure 4, exposure to resveratrol for 48 h resulted in a decrease in G0/G1 positive cells, concomitant with an equivalent increase of cells in S-phase. In the presence of resveratrol alone and together with NAM, a similar percentage of cells was found in G0/G1- and S-phase, while in the absence of NAM, more cells had entered G2/M-phase. Thus, these data suggest that resveratrol overcomes the NAM-induced growth retardation.
Figure 4.

Cell cycle analyses after treatment with NAM and resveratrol. THP-1 cells (0.2 × 106/ml) were serum-starved (SS) for 48 h. Upon addition of serum, the cells were cultured in the presence of 8 mM NAM and/or 10 µM resveratrol (Res) or the respective solvent control (ctrl) and cell cycle analyses were performed after 48 h by flow cytometry. (a) One representative experiment is shown. Cell cycle phases were defined by DNA staining with propidium iodide (PI). (b) Percentages of cells in G0/G1-, S- and G2/M-phase. Shown are mean values ± SD (n = 3). Statistics were performed with the paired Student’s t-test calculated to the respective control.
NAM and NR enhance intracellular NAD concentrations
Next, we asked whether the general perception that intracellular NAD is synthesised from NAM and NR applies to the here described cell system.10,38 As seen in Figure 5, treatment of THP-1 cells with 8 mM NAM resulted in an increase in intracellular NAD concentrations, which peaks at 24 h, declines thereafter and remains elevated up to 96 h. Strikingly, the ability of NR (8 mM) to enhance intracellular NAD levels was very similar to that seen of NAM. However, values reached at 24 h were lower and did not decline thereafter. As expected, MNA did not induce a rise in intracellular NAD concentrations. On the contrary, it even led to a slight, but significant decrease.
Figure 5.
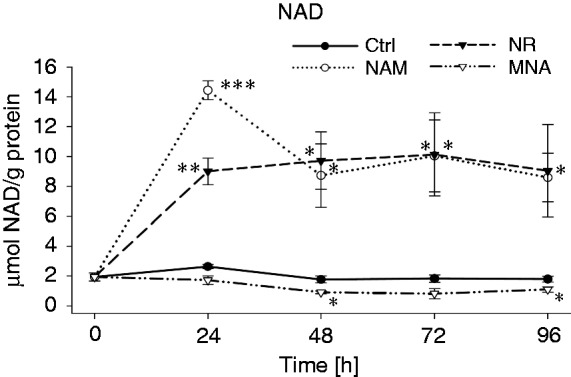
NAD concentrations after treatment with NAM, NR and MNA. THP-1 cells (0.2 × 106/ml) were treated with 8 mM NAM, 8 mM NR, 3.2 mM MNA or the respective solvent control (ctrl). NAD values were determined at 0 h, 24 h, 48 h, 72 h and 96 h and normalised to the total protein content. Shown are means ± SD (n = 3). Statistics were performed with the paired Student’s t-test calculated to the respective control.
Together these data demonstrate that NAM and NR are metabolised to NAD and that a single dose is sufficient to maintain a constant, elevated NAD concentration for up to 3 d. During the first 24 h the activity of NAD synthesising enzymes very likely prevails that of NAD consuming enzymes (i.e. PARPs, CD38, sirtuins) and at 48 h regeneration and maintenance of a cellular NAD pool is reached.
Effect of NAM on SIRT1 and PARP1 expression
In light of the NAM-induced rise in intracellular NAD levels, we determined expression levels of SIRT1 and PARP1, two mayor NAD consuming enzymes.7 Time course experiments (Figure 6a) showed that 24 h after treatment with NAM, SIRT1 mRNA expression was increased and that the values remained elevated up to 96 h. In line with the mRNA values, SIRT1 protein expression (Figure 6b) was found to be slightly increased at 48 h and 72 h in response to NAM. Western blot analysis showed three bands, one of them corresponding to the full-length protein (∼120 kDa) whereas the faster migrating band very likely belongs to an alternatively spliced isoform recently described in mice.39
Figure 6.

SIRT1 and PARP1 expression after treatment with NAM. THP-1 cells (0.2 × 106/ml) were treated with or without 8 mM NAM. After 24 h, 48 h, 72 h and 96 h mRNA values of (a) SIRT1 and (c) PARP1 were determined by real-time PCR. Data represent mRNA levels of NAM-treated cells relative to the untreated control at each time point analysed (-NAM = 1, black bar) ± SD (n = 3). (b) SIRT1 and (d) PARP1 protein were analysed by Western blot after 24 h, 48 h, 72 h, 96 h and 96 h, respectively. Densitometry data of proteins were normalised to the corresponding β-actin control. Data represent protein levels of NAM-treated cells relative to the untreated control at each time point analysed (-NAM = 100%, black bar) ± SD ((b) n = 3; (d) n = 4). Statistics were performed with the paired Student’s t-test calculated to the respective control.
Unlike SIRT1, PARP1 mRNA (Figure 6c) did not rise significantly until 96 h after incubation. An increase of PARP1 protein could not be detected at that time point (Figure 6d).
Effect of NAD metabolites on CD38, CD11b and CD14 expression
To test whether NAM, NR and MNA were able to induce monocytic differentiation, the expression of classical monocytic differentiation markers such as CD38, CD11b and CD14 was measured. As seen in Figure 7, THP-1 cells express little CD11b and CD38 on their surface and CD14 is hardly detectable. Expression levels of CD11b and CD14 were slightly elevated in response to NAM while CD11b expression was increased in MNA treated cells and CD38 levels remained unaltered. Compared to the expression levels of the three surface molecules induced by VitD, which is well known to promote maturation of myeloid cells,40–42 the effects on CD11b and CD14 are minor, indicating an absence of maturation pressure.
Figure 7.
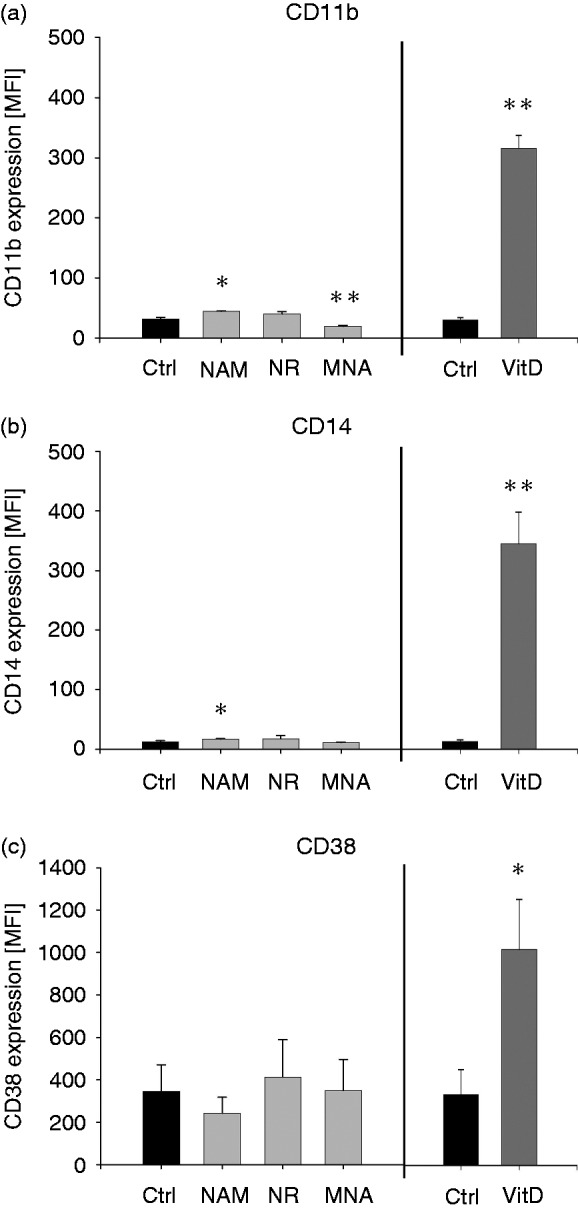
Expression of cell surface markers after treatment with NAM, NR, MNA and VitD. THP-1 cells (0.2 × 106/ml) were treated with 8 mM NAM, 8 mM NR, 3.2 mM MNA, 0.1 mM VitD or the respective solvent control (ctrl). After 96 h the expressions of the cell surface markers (a) CD11b, (b) CD14 and (c) CD38 were measured by flow cytometry. Shown are mean values of the median fluorescence intensity ± SD (n = 3). Statistics were performed with the paired Student’s t-test calculated to the respective control.
NAD metabolites modulate the generation of ROS and TNF-α production
A key feature of terminally differentiated myeloid cells is their ability to eliminate pathogens by producing a variety of toxic products including ROS. To test whether THP-1 cells exposed to NAM, NR and MNA display cellular functional responses that reflect monocytic properties, we measured the production of ROS in response to LPS. As demonstrated in Figure 8, THP-1 cells treated with VitD for 96 h produce considerable amounts of ROS when exposed to LPS. A similar increase in ROS production was also seen in response to treatment with NAM and NR. MNA however caused a small decrease of ROS production under the same experimental conditions.
Figure 8.
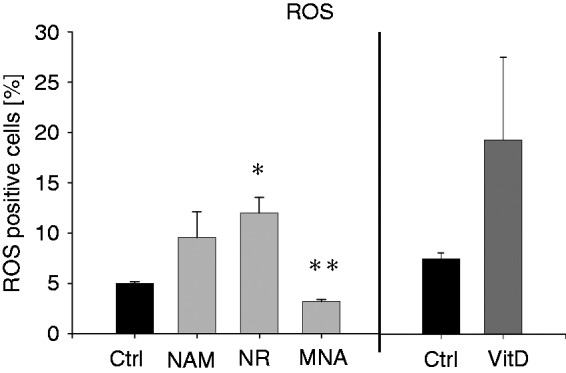
ROS production after treatment with NAM, NR, MNA and VitD. THP-1 cells (0.2 × 106/ml) were treated with 8 mM NAM, 8 mM NR, 3.2 mM MNA, 0.1 mM VitD or the respective control (ctrl). After 96 h the cells (1 × 106/ml) were stimulated with 100 ng/ml LPS for 30 min and intracellular ROS production was measured by flow cytometry. Shown are the percentages of the ROS (DCF)-positive cells ± SD (n = 3). Statistics were performed with the paired Student’s t-test calculated to the respective control.
In light of the importance of monocytic cells in mounting a protective immune response against pathogens, which includes the release of pro-inflammatory mediators, we tested whether NAM and its derivatives influence the LPS-induced production of TNF-α, one of the most prominent pro-inflammatory cytokines. As illustrated in Figure 9a, treatment with LPS resulted in an increase in TNF-α production which declined in response to NAM, NR and MNA. Exposure to VitD exerted similar effects (Figure 9b). Recent reports have revealed a link between intracellular NAD levels and LPS-induced TNF-α production in THP-1-derived macrophages.43 To investigate whether such a link exists in THP-1 cells, we determined both intracellular NAD levels and TNF-α concentrations 96 h after treatment with the NAD metabolites and after 4 h exposure to LPS. Before adding LPS, the cells were taken up in fresh media (without NAD metabolites) and the cell numbers were adjusted. As seen in Figure 9c, adding fresh media to either control or MNA pre-treated cells had no effect on NAD concentrations whereas in NAM and NR pre-treated cells NAD levels declined. Independent of the presence of LPS identical NAD concentrations were reached (Figure 9d), indicating that NAD has no role in the NAM-, NR- and MNA-mediated decrease in TNF-α production.
Figure 9.

LPS-induced TNF-α production and NAD-content. THP-1 cells (0.2 × 106/ml) were treated with (a) 8 mM NAM, (c) 8 mM NR, (d) 3.2 mM MNA and (b) 0.1 mM VitD or the respective control (ctrl). After 96 h the cells (1 × 106/ml) were incubated for further 4 h in the absence and presence of 100 ng/ml LPS. (a, b) TNF-α concentrations in the supernatants were measured by ELISA. Shown are mean values ± SD ((a) n = 6; (b) n = 3). NAD values of (c) unstimulated and (d) LPS-stimulated cells were normalised to the total protein content. Shown are means ± SD (n = 3). Statistics were performed with the paired Student’s t-test calculated to the respective control.
Given that CD14 and TLR4 are major players in facilitating LPS-induced TNF-α production, we next examined the influence of NAM and VitD on expression levels of the two cell surface proteins. We found that CD14 and TLR4 are hardly detectable on the surface of both stimulated and unstimulated THP-1 cells and that treatment with VitD resulted in a strong up-regulation of CD14 but had no effect on TLR4 expression (Figure 10). When exposed to NAM the expression levels of CD14 and TLR4 remained almost unchanged (Figure 10). Thus, the two surface molecules are unlikely to control the NAM-dependent down-regulation of the LPS-induced TNF-α production.
Figure 10.
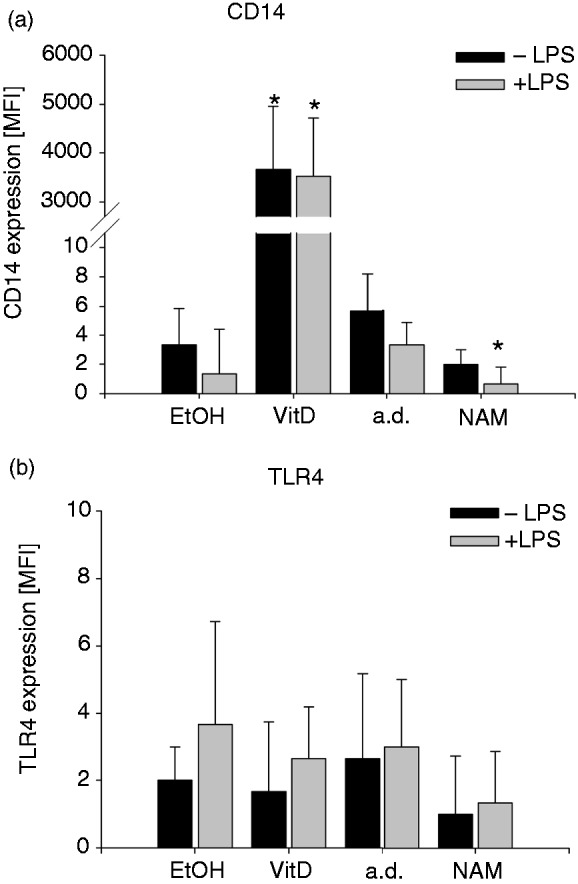
Expression of CD14 and TLR4 after treatment with NAM and VitD. THP-1 cells (0.2 × 106/ml) were treated with 8 mM NAM, 0.1 mM VitD or the respective control (a.d./EtOH). After 96 h the cells were incubated for 4 h in the absence and presence of 100 ng/ml LPS. The expressions of the cell surface markers (a) CD14 and (b) TLR4 were measured by flow cytometry. Shown are mean values of the median fluorescence intensity ± SD (n = 3). Statistics were performed with the paired Student’s t-test calculated to the respective control (a.d./EtOH).
Discussion
It is well known that VitD induces immature leukaemia cells to differentiate into mature monocytic cells40–42 and to cause macrophages to shift to a M2-phenotype.44 According to recent reports, the capability to influence proliferation and differentiation processes has also been attributed to another vitamin namely vitamin B3 (niacin). Niacin-related compounds including NAM have been shown to induce differentiation of promyelocytic leukaemia HL-60 cells,45 and to regulate retinoic acid (RA) and VitD induced differentiation and cell cycle arrest of these cells.20 An inhibitory effect of NAM on RA induced CD38 expression and differentiation was described by Munshi et al.26 Studies by Skokowa et al. revealed a decisive role of metabolites of the NAD salvage pathway in G-CSF triggered granulopoiesis.46
In contrast to NAM, information about NR and MNA as potential modulators of proliferation and differentiation are rare. NR has been mainly looked upon as a compound enhancing oxidative metabolism and protecting against metabolic and mitochondrial diseases.15 MNA has been tested in clinical trials for various applications, including treatment of hypertriglyceridemia,16,47 which contributes to cardiovascular disease and reduced lifespan.47
We here show that all three NAD metabolites inhibit the proliferation of the myeloid cell line THP-1. Growth attenuation was most pronounced in response to NAM and equalled that induced by VitD. These data are in line with publications describing NAM as an agent that through down-regulation of SIRT1 activity inhibits proliferation of chronic lymphocytic leukaemia and pancreatic cancer cells.48,49 The NAM-induced reduction in proliferation was found to be accompanied by activation of cell cycle arrest via p53 and p21 expression,48 as well as by induction of apoptosis.48,49 We did not observe any enhanced apoptosis rate nor did we detect any p53 and p21 protein expression in THP-1 cells treated with or without NAM (data not shown), indicating that retardation of cell growth is most likely due to prolonged cell cycle rather than to cell cycle arrest.
As NAM and NR lead to increased intracellular NAD levels and cell cycle progression is regulated by intertwined redox oscillators like NAD/NADH, it is possible that, as suggested by da Veiga Moreira et al.,50 intracellular NAD concentrations may influence the cell cycle phases.
NAD has emerged as an important mediator of signalling mainly by serving as a substrate of NAD-consuming enzymes such as SIRTs and PARPs. Most sirtuins catalyse the NAD-dependent deacetylation of an acetyl lysine residue, giving rise to the deacetylated protein, NAM and O-acetyl-ADP-ribose while the enzymatic cleavage of NAD by PARPs results in formation of NAM and the transfer of multiple ADPR-moieties to their specific target protein. Both enzyme families regulate fundamental events such as apoptosis, cell-cycle progression, DNA repair and transcription.51–54 They compete for the same intracellular NAD-pool and NAM is a well-known inhibitor of these NAD-depleting enzymes.7,55,56 The relation between sirtuin and PARP activity is rather complex. For example, activation of SIRT1 is associated with reduction of PARP1 activity whereas deletion of SIRT1 increases the production of PAR (ADP-ribose-polymer).7 In line with previous reports, we here show that NAM and NR supplementation enhance intracellular NAD-levels which remain elevated up to 96 h. NAM also induces the expression of SIRT1 and PARP1 at the mRNA and SIRT1 only weakly at the protein level. Although both enzymes cleave NAD, the degradation seems to be compensated by NAD synthesis so that steady state levels of NAD are reached. Since growth retardation is not accompanied by NAD depletion and independent of p21 and p53, it might very well be related to the down-regulation of other metabolic events such as protein synthesis.
In contrast to the here described tumour cell line, NAM has been shown to accelerate cell proliferation of human pluripotent stem cells.57 The authors speculate that the effects of NAM on pluripotency may depend on its role as a NAD precursor and on its ability to enhance resistance to cellular stress.57 Enhanced DNA synthesis and proliferation has also been shown to occur in rat liver cells in response to MNA.58
MNA, by serving as a substrate for the enzyme AOX1/GAD-3, has recently been described as a compound able of generating a H2O2-mediated ROS signal that, at least in Drosophila, promotes lifespan and metabolic health.11 The authors speculate that sirtuins play a key role in lifespan regulation by providing increased amounts of the MNA precursor NAM.
Besides reducing proliferation of THP-1 cells, the compounds also interfere with LPS-induced monocytic properties like ROS and TNF-α production. While NAM and NR stimulate ROS production in THP-1 cells, MNA, unlike in Drosophila,11 displayed inhibitory effects. These dissimilar findings clearly reflect the complexity of the biological functions of MNA and significance of the experimental settings when defining functional properties. Common to all three compounds is their ability to inhibit LPS-induced TNF-α release. Among immune cells, monocytes/macrophages which are the major players of inflammatory processes are especially susceptible to the anti-inflammatory action of NAM.18,59 Based on recent data showing that LPS caused a substantial increase in NAD levels and TNF-α in pro-inflammatory (M1) macrophages but not in anti-inflammatory (M2) macrophages, a link between NAD levels and LPS-induced TNF-α release has been suggested.43 We found that stimulation of THP-1 cells with LPS (ctrl) resulted in an increase in TNF-α production but had no effect on NAD concentrations. The diminished TNF-α release in response to pre-treatment with NAD metabolites was not associated with changes in NAD levels induced by LPS.
Although TLR4 and CD14, two important molecules facilitating LPS signalling, are expressed at very low levels, they seem to be sufficient to initiate signal transduction. Having shown that the NAM-induced decrease in TNF-α production is not accompanied by a diminished expression of CD14 and TLR4, we can exclude a role of the two surface proteins in down-regulating the inflammatory response. Suppression of the TNF-α production is most likely regulated via modulation of the transcription factors NF-κB and FoxO3.17,60,61
Taken together our data clearly indicate that NAM and NR, two naturally occurring nutrients and MNA, the methylation product of NAM, have the potential to retard the proliferation of a myeloid leukaemia cell line by arresting the cells in G0/G1-phase. Apart from their anti-proliferative properties, the compounds exert anti-inflammatory effects and especially NAM and NR prime the cells to generate ROS. Although the retardation of proliferation is not accompanied by differentiation into functional mature cells, vitamin B3 and its derivatives strongly influence immunological properties of monocytic cells.
Supplemental Material
Supplemental Material for NAD metabolites interfere with proliferation and functional properties of THP-1 cells by Katharina Petin, Ronald Weiss, Gerd Müller, Antje Garten, Anja Grahnert, Ulrich Sack and Sunna Hauschildt in Innate Immunity
Declaration conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: K Petin was supported by a scholarship of Leipzig University, Germany. A Garten has received funding from the European Union’s Horizon 2020 research and innovation programme under the Marie Sklodowska-Curie grant agreement (Grant Number 705869).
Supplemental material
Supplemental material is available for this article online.
References
- 1.Bogan KL, Brenner C. Nicotinic acid, nicotinamide, and nicotinamide riboside: a molecular evaluation of NAD+ precursor vitamins in human nutrition. Annu Rev Nutr 2008; 28: 115–130. [DOI] [PubMed] [Google Scholar]
- 2.Benavente CA, Jacobson MK, Jacobson EL. NAD in skin: therapeutic approaches for niacin. Curr Pharm Des 2009; 15: 29–38. [DOI] [PubMed] [Google Scholar]
- 3.Cabrera-Rode E, Molina G, Arranz C, et al. Effect of standard nicotinamide in the prevention of type 1 diabetes in first degree relatives of persons with type 1 diabetes. Autoimmunity 2006; 39: 333–340. [DOI] [PubMed] [Google Scholar]
- 4.Chiarugi A, Dölle C, Felici R, et al. The NAD metabolome–a key determinant of cancer cell biology. Nat Rev Cancer 2012; 12: 741–752. [DOI] [PubMed] [Google Scholar]
- 5.Berger F, Ramírez-Hernández MH, Ziegler M. The new life of a centenarian: signalling functions of NAD(P). Trends Biochem Sci 2004; 29: 111–118. [DOI] [PubMed] [Google Scholar]
- 6.Magni G, Amici A, Emanuelli M, et al. Enzymology of NAD+ synthesis. Adv Enzymol Relat Areas Mol Biol 1999; 73: 135. [DOI] [PubMed] [Google Scholar]
- 7.Belenky P, Bogan KL, Brenner C. NAD+ metabolism in health and disease. Trends Biochem Sci 2007; 32: 12–19. [DOI] [PubMed] [Google Scholar]
- 8.Gerth A, Nieber K, Oppenheimer NJ, et al. Extracellular NAD+ regulates intracellular free calcium concentration in human monocytes. Biochem J 2004; 382: 849–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Grahnert A, Grahnert A, Klein C, et al. NAD+: a modulator of immune functions. Innate Immun 2011; 17: 212–233. [DOI] [PubMed] [Google Scholar]
- 10.Bieganowski P, Brenner C. Discoveries of nicotinamide riboside as a nutrient and conserved NRK genes establish a Preiss-Handler independent route to NAD+ in fungi and humans. Cell 2004; 117: 495–502. [DOI] [PubMed] [Google Scholar]
- 11.Schmeisser K, Mansfeld J, Kuhlow D, et al. Role of sirtuins in lifespan regulation is linked to methylation of nicotinamide. Nat Chem Biol 2013; 9: 693–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vague P, Picq R, Bernal M, et al. Effect of nicotinamide treatment on the residual insulin secretion in type 1 (insulin-dependent) diabetic patients. Diabetologia 1989; 32: 316–321. [DOI] [PubMed] [Google Scholar]
- 13.Ungerstedt JS, Blomback M, Soderstrom T. Nicotinamide is a potent inhibitor of proinflammatory cytokines. Clin Exp Immunol 2003; 131: 48–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Niren NM. Pharmacologic doses of nicotinamide in the treatment of inflammatory skin conditions: a review. Cutis 2006; 77: 11–16. [PubMed] [Google Scholar]
- 15.Cantó C, Houtkooper RH, Pirinen E, et al. The NAD(+) precursor nicotinamide riboside enhances oxidative metabolism and protects against high-fat diet-induced obesity. Cell Metab 2012; 15: 838–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gebicki J, Chlopicki S. Method for treating hypertriglyceridemia, dyslipidemia und hypercholesterolemia with a 1-methylnicotinamide salt. Patent application 7935717B2, USA, 2011.
- 17.Lappas M, Permezel M. The anti-inflammatory and antioxidative effects of nicotinamide, a vitamin B(3) derivative, are elicited by FoxO3 in human gestational tissues: implications for preterm birth. J Nutr Biochem 2011; 22: 1195–1201. [DOI] [PubMed] [Google Scholar]
- 18.Weiss R, Schilling E, Grahnert A, et al. Nicotinamide: a vitamin able to shift macrophage differentiation toward macrophages with restricted inflammatory features. Innate Immun 2015; 21: 813–826. [DOI] [PubMed] [Google Scholar]
- 19.Lee HJ, Hong Y-S, Jun W, et al. Nicotinamide riboside ameliorates hepatic metaflammation by modulating NLRP3 inflammasome in a rodent model of type 2 diabetes. J Med Food 2015; 18: 1207–1213. [DOI] [PubMed] [Google Scholar]
- 20.Shen M, Yen A. Nicotinamide cooperates with retinoic acid and 1,25-dihydroxyvitamin D(3) to regulate cell differentiation and cell cycle arrest of human myeloblastic leukemia cells. Oncology 2009; 76: 91–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ziegler-Heitbrock HW, Ulevitch RJ. CD14: cell surface receptor and differentiation marker. Immunol Today 1993; 14: 121–125. [DOI] [PubMed] [Google Scholar]
- 22.Antal-Szalmas P, Strijp JA, Weersink AJ, et al. Quantitation of surface CD14 on human monocytes and neutrophils. J Leukoc Biol 1997; 61: 721–728. [DOI] [PubMed] [Google Scholar]
- 23.Zhang DE, Hetherington CJ, Gonzalez DA, et al. Regulation of CD14 expression during monocytic differentiation induced with 1alpha,25-dihydroxyvitamin D3. J Immunol 1994; 153: 3276–3284. [PubMed] [Google Scholar]
- 24.Perraud AL, Fleig A, Dunn CA, et al. ADP-ribose gating of the calcium-permeable LTRPC2 channel revealed by Nudix motif homology. Nature 2001; 411: 595–599. [DOI] [PubMed] [Google Scholar]
- 25.Young GS, Choleris E, Lund FE, et al. Decreased cADPR and increased NAD+ in the Cd38-/- mouse. Biochem Biophys Res Commun 2006; 346: 188–192. [DOI] [PubMed] [Google Scholar]
- 26.Munshi CB, Graeff R, Lee HC. Evidence for a causal role of CD38 expression in granulocytic differentiation of human HL-60 cells. J Biol Chem 2002; 277: 49453–49458. [DOI] [PubMed] [Google Scholar]
- 27.Liu Q, Ning W, Dantzer R, et al. Activation of protein kinase C-zeta and phosphatidylinositol 3'-kinase and promotion of macrophage differentiation by insulin-like growth factor-I. J Immunol 1998; 160: 1393–1401. [PubMed] [Google Scholar]
- 28.Harris ES, McIntyre TM, Prescott SM, et al. The leukocyte integrins. J Biol Chem 2000; 275: 23409–23412. [DOI] [PubMed] [Google Scholar]
- 29.Pfeiffer A, Böttcher A, Orsó E, et al. Lipopolysaccharide and ceramide docking to CD14 provokes ligand-specific receptor clustering in rafts. Eur J Immunol 2001; 31: 3153–3164. [DOI] [PubMed] [Google Scholar]
- 30.Grohmann T, Penke M, Petzold-Quinque S, et al. Inhibition of NAMPT sensitizes MOLT4 leukemia cells for etoposide treatment through the SIRT2-p53 pathway. Leuk Res 2018; 69: 39–46. [DOI] [PubMed] [Google Scholar]
- 31.Schuster S, Penke M, Gorski T, et al. FK866-induced NAMPT inhibition activates AMPK and downregulates mTOR signaling in hepatocarcinoma cells. Biochem Biophys Res Commun 2015; 458: 334–340. [DOI] [PubMed] [Google Scholar]
- 32.Schilling E, Weiss R, Grahnert A, et al. Molecular mechanism of LPS-induced TNF-α biosynthesis in polarized human macrophages. Mol Immunol 2018; 93: 206–215. [DOI] [PubMed] [Google Scholar]
- 33.Daigneault M, Preston JA, Marriott HM, et al. The identification of markers of macrophage differentiation in PMA-stimulated THP-1 cells and monocyte-derived macrophages. PLoS One 2010; 5: e8668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yen A, Brown D, Fishbaugh J. Control of HL-60 monocytic differentiation. Different pathways and uncoupled expression of differentiation markers. Exp Cell Res 1987; 168: 247–254. [DOI] [PubMed] [Google Scholar]
- 35.Dulić V, Kaufmann WK, Wilson SJ, et al. p53-dependent inhibition of cyclin-dependent kinase activities in human fibroblasts during radiation-induced G1 arrest. Cell 1994; 76: 1013–1023. [DOI] [PubMed] [Google Scholar]
- 36.Villalba JM, Alcaín FJ. Sirtuin activators and inhibitors. Biofactors 2012; 38: 349–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Inoue T, Hiratsuka M, Osaki M, et al. The molecular biology of mammalian SIRT proteins: SIRT2 in cell cycle regulation. Cell Cycle 2007; 6: 1011–1018. [DOI] [PubMed] [Google Scholar]
- 38.Magni G, Amici A, Emanuelli M, et al. Enzymology of NAD+ homeostasis in man. Cell Mol Life Sci 2004; 61: 19–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Deota S, Chattopadhyay T, Ramachandran D, et al. Identification of a tissue-restricted isoform of SIRT1 defines a regulatory domain that encodes specificity. Cell Rep 2017; 18: 3069–3077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Abe E, Miyaura C, Sakagami H, et al. Differentiation of mouse myeloid leukemia cells induced by 1alpha,25-dihydroxyvitamin D3. Proc Natl Acad Sci USA 1981; 78: 4990–4994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Oberg F, Botling J, Nilsson K. Functional antagonism between vitamin D3 and retinoic acid in the regulation of CD14 and CD23 expression during monocytic differentiation of U-937 cells. J Immunol 1993; 150: 3487–3495. [PubMed] [Google Scholar]
- 42.Schwende H, Fitzke E, Ambs P, et al. Differences in the state of differentiation of THP-1 cells induced by phorbol ester and 1,25-dihydroxyvitamin D3. J Leukoc Biol 1996; 59: 555–561. [PubMed] [Google Scholar]
- 43.Al-Shabany AJ, Moody AJ, Foey AD, et al. Intracellular NAD+ levels are associated with LPS-induced TNF-α release in pro-inflammatory macrophages. Biosci Rep 2016; 36: e00301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang X, Zhou M, Guo Y, et al. 1,25-Dihydroxyvitamin D3 promotes high glucose-induced M1 macrophage switching to M2 via the VDR-PPARγ signaling pathway. Biomed Res Int 2015; 2015: 157834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Iwata K, Ogata S, Okumura K, et al. Induction of differentiation in human promyelocytic leukemia HL-60 cell line by niacin-related compounds. Biosci Biotechnol Biochem 2003; 67: 1132–1135. [DOI] [PubMed] [Google Scholar]
- 46.Skokowa J, Lan D, Thakur BK, et al. NAMPT is essential for the G-CSF-induced myeloid differentiation via a NAD(+)-sirtuin-1-dependent pathway. Nat Med 2009; 15: 151–158. [DOI] [PubMed] [Google Scholar]
- 47.Nordestgaard BG, Benn M, Schnohr P, et al. Nonfasting triglycerides and risk of myocardial infarction, ischemic heart disease, and death in men and women. JAMA 2007; 298: 299–308. [DOI] [PubMed] [Google Scholar]
- 48.Audrito V, Vaisitti T, Rossi D, et al. Nicotinamide blocks proliferation and induces apoptosis of chronic lymphocytic leukemia cells through activation of the p53/miR-34a/SIRT1 tumor suppressor network. Cancer Res 2011; 71: 4473–4483. [DOI] [PubMed] [Google Scholar]
- 49.Zhang J-G, Zhao G, Qin Q, et al. Nicotinamide prohibits proliferation and enhances chemosensitivity of pancreatic cancer cells through deregulating SIRT1 and Ras/Akt pathways. Pancreatology 2013; 13: 140–146. [DOI] [PubMed] [Google Scholar]
- 50.da Veiga Moreira J, Peres S, Steyaert J-M, et al. Cell cycle progression is regulated by intertwined redox oscillators. Theor Biol Med Model 2015; 12: 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bouchard VJ, Rouleau M, Poirier GG. PARP-1, a determinant of cell survival in response to DNA damage. Exp Hematol 2003; 31: 446–454. [DOI] [PubMed] [Google Scholar]
- 52.Bürkle A. Physiology and pathophysiology of poly(ADP-ribosyl)ation. Bioessays 2001; 23: 795–806. [DOI] [PubMed] [Google Scholar]
- 53.Michan S, Sinclair D. Sirtuins in mammals: insights into their biological function. Biochem J 2007; 404: 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.d'Amours D, Desnoyers S, d'Silva I, et al. Poly(ADP-ribosyl)ation reactions in the regulation of nuclear functions. Biochem. J. 1999; 342: 249–268. [PMC free article] [PubMed] [Google Scholar]
- 55.Avalos JL, Bever KM, Wolberger C. Mechanism of sirtuin inhibition by nicotinamide: altering the NAD(+) cosubstrate specificity of a Sir2 enzyme. Mol Cell 2005; 17: 855–868. [DOI] [PubMed] [Google Scholar]
- 56.Gallo CM, Smith DL, Smith JS. Nicotinamide clearance by Pnc1 directly regulates Sir2-mediated silencing and longevity. Mol Cell Biol 2004; 24: 1301–1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Son MJ, Son M-Y, Seol B, et al. Nicotinamide overcomes pluripotency deficits and reprogramming barriers. Stem Cells 2013; 31: 1121–1135. [DOI] [PubMed] [Google Scholar]
- 58.Hoshino J, Kühne U, Kröger H. Enhancement of DNA synthesis and cell proliferation by 1-methylnicotinamide in rat liver cells in culture: implication for its in vivo role. Biochem Biophys Res Commun 1982; 105: 1446–1452. [DOI] [PubMed] [Google Scholar]
- 59.La Montserrat-de Paz S, Naranjo MC, Lopez S, et al. Niacin and its metabolites as master regulators of macrophage activation. J Nutr Biochem 2017; 39: 40–47. [DOI] [PubMed] [Google Scholar]
- 60.Grange PA, Raingeaud J, Calvez V, et al. Nicotinamide inhibits Propionibacterium acnes-induced IL-8 production in keratinocytes through the NF-kappaB and MAPK pathways. J Dermatol Sci 2009; 56: 106–112. [DOI] [PubMed] [Google Scholar]
- 61.Crowley CL, Payne CM, Bernstein H, et al. The NAD+ precursors, nicotinic acid and nicotinamide protect cells against apoptosis induced by a multiple stress inducer, deoxycholate. Cell Death Differ 2000; 7: 314–326. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Material for NAD metabolites interfere with proliferation and functional properties of THP-1 cells by Katharina Petin, Ronald Weiss, Gerd Müller, Antje Garten, Anja Grahnert, Ulrich Sack and Sunna Hauschildt in Innate Immunity