

Abstract
Previous studies have reported that lipoxin A4 (LXA4) and the angiotensin I-converting enzyme 2 (ACE2), angiotensin-(1-7) [Ang-(1-7)], and its receptor Mas [ACE2-Ang-(1-7)-Mas] axis play important protective roles in acute lung injury (ALI). However, there is still no direct evidence of LXA4-mediated protection via the ACE2-Ang-(1-7)-Mas axis during ALI. This work was performed using an LPS-induced ALI mouse model and the data indicated the following. First, the animal model was established successfully and LXA4 ameliorated LPS-induced ALI. Second, LXA4 could increase the concentration and activity of ACE2 and the levels of Ang-(1-7) and Mas markedly. Third, LXA4 decreased the levels of TNF-α, IL-1β, and reactive oxygen species while increasing IL-10 levels. Fourth, LXA4 inhibited the activation of the NF-κB signal pathway and repressed the degradation of inhibitor of NF-κB, the phosphorylation of NF-κB, and the translocation of NF-κB. Finally, and more importantly, BOC-2 (LXA4 receptor inhibitor), MLN-4760 (ACE2 inhibitor), and A779 (Mas receptor antagonist) were found to reverse all of the effects of LXA4. Our data provide evidence that LXA4 protects the lung from ALI through regulation of the ACE2-Ang-(1-7)-Mas axis.
Keywords: Acute lung injury, lipoxins, RAAS, inflammation
Introduction
Acute lung injury (ALI) and acute respiratory distress syndrome (ARDS) are inflammatory responses that take place in the lung and can cause severe hypoxemia and noncardiogenic pulmonary edema.1 ALI and ARDS are major causes of acute respiratory failure in critically ill patients. Despite improvements in supportive care, mortality rates due to ALI and ARDS are high, with approximately 2.82 deaths per 100,000 persons in the United States in 2013.2 For these reasons, it is necessary to explore the mechanisms underlying ALI and ARDS, which may spur the discovery of efficacious therapeutic interventions. It is generally accepted that the vital pathogenesis of ALI is inflammation, which is accompanied by infiltration and accumulation of inflammatory cells, pro-inflammatory cytokines, reactive oxygen species (ROS), and activation of the NF-κB signaling pathway.3–5
Lipoxins (LXs) are products of arachidonic acid metabolism secreted mainly through cell-to-cell interactions during lipoxygenase-dependent processes.6,7 The interaction of lipoxin A4 (LXA4) and its receptor has a direct anti-inflammatory effect and other activities that promote inflammation resolution in several inflammatory models.8–10 Several studies have suggested that LXs and their analogs display protective effects in ALI.11–13 However, the mechanisms that mediate the beneficial effect of LXs in ALI have not been elucidated fully.
The renin–angiotensin–aldosterone system (RAAS), which comprises the angiotensin I-converting enzyme (ACE)-AngII-AT1R axis and the angiotensin I-converting enzyme 2 (ACE2)-Ang-(1-7)-Mas axis, is well known for its vital and bidirectional roles in the urinary and cardiovascular systems. Recent reports indicate that ACE2, angiotensin-(1-7) [Ang-(1-7)], and its receptor Mas are detectable in normal lung tissue and, upon lung injury, their expressions change and are positively correlated with the degree of lung injury.14,15 Furthermore, some studies have suggested that the ACE2-Ang-(1-7)-Mas axis has a protective effect in ALI, so ACE2 and Ang-(1-7) are considered some of the most promising new targets for intervention during ALI.16–18
In our previous study, we observed that BML-111, the agonist of the LXA4 receptor, could up-regulate and activate the ACE2-Ang-(1-7)-Mas axis in the liver.19 However, whether the ACE2-Ang-(1-7)-Mas axis is involved in the LXA4-mediated protective process against ALI is still unclear. Here, we used a LPS-induced ALI mouse model to explore the role and interaction of LXA4 and the ACE2-Ang-(1-7)-Mas axis in ALI. Finally, to gain a better understanding of the mechanisms that underlie the action of LXA4, we also investigated whether BOC-2 (LXA4 receptor inhibitor), MLN-4760 (ACE2 inhibitor), and A779 (Mas receptor antagonist) attenuated the protective effects of LXA4.
Materials and methods
Reagents
LPS (Escherichia coli 055:B5) was from Sigma-Aldrich (St. Louis, MO, USA). Lipoxin A4 (LXA4) (5S, 6R, 15S-trihydroxy-7E, 9E, 11Z, 13E-eicosa-tetraenoic acid) was from Cayman Chemical (Ann Arbor, MI, USA), BOC-2 (LXA4 receptor antagonist) was from GenScript (Piscataway, NJ, USA), A779 (selective inhibitor of the Mas receptor) was from Abcam (Cambridge, MA, USA), and MLN-4760 (ACE2 antagonist) was from Merck Millipore (Cambridge, UK). The myeloperoxidase (MPO) kit was from Nanjing Jiancheng Bioengineering Institute (Nanjing, China). The ACE2 activity assay kit was from Genmed Scientifics (Arlington, VA, USA). ELISA kits for ACE2, Ang-(1-7), inflammatory factors (TNF-α, IL-1β, and IL-10), and ROS were from Cloud-Clone (Houston, TX, USA). Nuclear Transfer Assay Kit of NF-κB Activation was from Beyotime Biotechnology (Shanghai, China). The primary Abs against Ang-(1-7), Mas, and β-actin were from Cloud-Clone. Primary Abs against NF-κB p65, p-p65, and inhibitor of NF-κB (IκB-α) were from Cell Signaling Technology (Danvers, MA, USA). The Eastep Super Total RNA Extraction Kit, Easy Script First-Strand cDNA Synthesis Super Mix Kit, and TransStarat Tip Green qPCR Super Mix Kit were all from Promega (Madison, WI, USA).
Animals
All experimental procedures were performed according to institutional animal guidelines. The experimental protocols were approved by the institutional animal care and use committee and appropriate measures were taken to minimize pain and stress to the animals. Sixty adult male Kun Ming (KM) mice (4–6 wk old, 20–30 g) were obtained from the Animal Center of Nanchang University (Nanchang, China), raised in an environmentally controlled room with a 12/12 h light/dark cycle, and allowed free access to food and water in temperature-controlled rooms (25 ± 1℃).
Experimental groups
Sixty mice were randomly divided into six groups (n = 10) as follows: (1) in the control group, animals received 10 ml/kg saline by intraperitoneal (i.p.) injection; (2) in the LPS group, animals received 10 mg/kg LPS by i.p. injection; in the (3) LPS + LXA4 group, animals received 100 µg/kg LXA4 by tail vein injection 30 min prior to receiving 10 mg/kg LPS i.p.; (4) in the LPS + LXA4 + BOC-2 group, animals received 50 µg/kg BOC-2 by tail vein injection prior to receiving 100 µg/kg LXA4 by tail vein injection, followed by 10 mg/kg LPS after 30 min after LXA4 administration; (5) in the LPS + LXA4 + A779 group, animals received 10 µg/kg A779 by tail vein injection prior to receiving 100 µg/kg LXA4 by tail vein injection, followed by 10 mg/kg LPS after 30 min after LXA4 administration; and (6) in the LPS + LXA4 + MLN-4760 group, animals received 1 mg/kg MLN-4760 by tail vein injection prior to receiving 100 µg/kg LXA4 by tail vein injection, followed by 10 mg/kg LPS 30 min after LXA4 administration.
Morphological evaluation
Lung tissues that were not subjected to bronchoalveolar lavage fluid (BALF) collection were selected and washed with cold PBS, fixed with 10% paraformaldehyde, dehydrated in a series of graded ethanols, embedded in paraffin wax, cut into 5 µm slices, and stained with hematoxylin and eosin. Histopathological features of the samples were assessed using light microscopy.
Estimation of wet/dry ratio of the lungs
The inferior lobe of the right lung was excised immediately upon sacrifice, rinsed in PBS, and weighed to determine the wet mass. The lung was then desiccated for 72 h at 60℃ in an oven. Subsequently, the lung was weighed and the procedure was repeated several times until the stable mass value was considered the dry mass. The ratio of the wet-to-dry mass of lung was calculated to quantify the extent of pulmonary edema.
Measurement of pulmonary MPO activity
Tissue-associated MPO activity was determined as an indicator of neutrophil infiltration. Briefly, liver tissues were homogenized and then centrifuged at 30,000 g for 30 min. The pellet was re-suspended in another potassium phosphate buffer (50 mmol/l, pH 6.0) with 0.5% hexadecyltrimethyl ammonium bromide. Samples were centrifuged at 20,000 g for 15 min at 4℃ and supernatants were collected. The activities of MPO in the livers were assayed by measuring absorbance at 460 nm. Results were expressed as units of MPO per gram of wet tissue.
ACE2 activity detection
Lung tissues were prepared to determine extent of ACE2 activation according to manufacturer’s instructions (Genmed Scientifics, Wilmington, DE, USA). First, 50 mg of frozen lung tissue was washed with 1 ml of reagent A and the lung tissue samples were homogenized in 500 µl of reagent B. Then, the supernatant of the samples was obtained by centrifugation at 1500 g for 10 min at 4℃ and the concentration of protein was measured using a bicinchoninic acid (BCA) kit. Next, the supernatant of the samples was mixed thoroughly with 80 µl of reagent B. Subsequently, 180 µl of reagent C and 50 µl of reagent E were added to the tubes and samples were incubated at 37℃ for 15 min in the dark. Finally, 20 µl of reagent D or 100 µg of protein was added to the tubes and mixed for 10 s and the optical density was measured at 340 nm using a microplate reader.
ELISA
BALF and serum samples were collected for measurement of TNF-α, IL-1β, and IL-10 levels using ELISA kits. In addition, the concentrations of ACE2 and Ang-(1-7) and the level of ROS in the lung tissues were also assessed using ELISA kits. Serum was acquired by centrifugation of blood samples at 550 g for 10 min. After preparation of the standard reagents by serial dilution, 50 µl of standard solution or sample and 50 µl of detection reagent A were added to the tubes, mixed thoroughly, and incubated for 1 h at 37℃. Thereafter, the microplates were washed three times. Next, 100 µl of detection reagent B was added, samples were mixed thoroughly, incubated for 0.5 h at 37℃, and washed five times. Soon afterward, 90 µl of tetramethylbenzidine substrate was added to the samples, mixed, and incubated for 10 min in the dark. Finally, 50 µl of stopping buffer was added and the optical density was measured at 450 nm. The concentration was assessed in reference to the standard curve.
Immunofluorescence analysis
Immunofluorescence was used to assess the activation of NF-κB in optimal cutting temperature compound-embedded tissue sections (6 µm thickness). The samples were prepared according to the instructions included in the nuclear transfer assay kit (Beyotime Biotechnology). The lung sections were incubated with primary Abs against NF-κB and then with biotin secondary Abs. Finally, the handled slices were observed under a confocal microscope.
Western blot analysis
Lung tissue samples (100 mg) were lysed and homogenized in 1 ml of radioimmunoprecipitation assay buffer with phenylmethanesulfonyl fluoride to obtain the total protein [to assay the expression of Ang-(1-7), p65, p-p65, and IκBα] and the membrane protein (to assay the expression of Mas). The protein concentrations were measured using a BCA protein assay kit and proteins (30 µg) were transferred electrophoretically onto polyvinylidene fluoride membranes (200 mA for 2 h at 4℃) following separation on a 10% sodium dodecyl sulfate-polyacrylamide gel. The membranes were blocked in Tris-buffered saline plus Tween 20 (TBST) containing 5% nonfat dry milk at 25 ± 1℃ (room temperature) for 2 h, followed by an overnight (≥12 h) incubation at 4℃ with specific primary Abs [Ang-(1-7), Mas, p65, p-p65, or IκBα]. The following day, the membranes were washed thoroughly with TBST three times and then incubated for an additional 1.5 h with horseradish peroxidase-conjugated secondary Abs (1:5000 dilution) at room temperature. Subsequently, membranes were washed three times with TBST and bands were visualized with enhanced chemiluminescence according to the manufacturer's protocol (Pierce, Rockford, IL, USA).
RNA isolation and real-time PCR analysis
For PCR analysis, lung tissues were prepared for RNA isolation using an Eastep Super Total RNA Extraction Kit and then cDNAs were synthesized by reverse transcription of 1 µg total RNA using an Easy Script First-Strand cDNA Synthesis SuperMix Kit Transgen. Subsequently, the mRNA level of Mas was quantified using the Bio-Rad iCyCler real-time PCR7500 machine with SYBR Green technology. Transgen and the primers for Mas and internal control GAPDH were synthesized by Kingsbury Biological Technology Company. Gene expression was measured using the ΔΔCT method and was normalized to internal control GAPDH mRNA levels. The primer sequences were as follows: GAPDH forward primer: 5′-AGAGGGATGCTGCCCTTACC-3′; GAPDH reverse primer: 5′-ATCCGTTCACACCGACCTTC-3′; Mas forward primer: 5′-CCATGAGGAACTGGGAGGAG-3′; and Mas reverse primer: 5′-GACAGAGGGCTAGAGGCTTG-3′.
Statistical analysis
The data are presented as the means ± SEM from three or more independent experiments unless specified otherwise. The differences between groups were evaluated using one-way ANOVA with SPSS version 17.0 (IBM, Armonk, NY, USA). In all analyses, P values < 0.05 were considered statistically significant.
Results
LXA4 protects lungs from LPS-induced ALI
To assess the effect of LXA4 on lung injury and the relationship between LXA4 and the ACE2-Ang-(1-7)-Mas axis on ALI, histopathological changes, wet/dry ratio, total protein in BALF, white cell count in the blood, and MPO activities were examined. LPS stimulation induced the infiltration of inflammatory cells in the lungs with markedly swollen sections and broadening tissue gap (Figure 1a). In addition, higher wet/dry ratios (Figure 1b), total protein in BALF (Figure 1c), white cell count in the blood, and MPO activities (Figure 1d) were observed with LPS stimulation compared with controls. However, treatment with LXA4 inhibited the inflammatory response (Figure 1a, d, and e), reduced the degree of lung edema (Figure 1a and b), and attenuated vascular damage (Figure 1d). Conversely, treatment with BOC-2 (a specific inhibitor of the receptor of LXA4), A779 (a selective Mas receptor inhibitor), and MLN-4760 (ACE2 antagonist) could abolish all of the protective effects of LXA4 (Figure 1). These data indicated that LXA4 protected lungs from LPS-induced ALI effectively and the ACE2-Ang-(1-7)-Mas axis may play an important role in this process.
Figure 1.

LXA4 protects against LPS-induced ALI effectively. Shown are: (a) Histological changes of lung tissue (hematoxylin and eosin, 200×), (b) wet/dry ratio, (c), total protein concentration in BALF, (d) white cell count in the blood, and (e) MPO activity. Data are presented as means ± SEM. *P < 0.05 and **P < 0.01 compared with control group, #P < 0.05 and ##P < 0.01 compared with LPS group, ΔP < 0.05 and ΔΔP < 0.01 compared with LPS + LXA4 group.
LXA4 up-regulates and activates the ACE2-Ang-(1-7)-Mas axis in LPS-induced ALI
To determine whether the ACE2-Ang-(1-7)-Mas axis was involved in the LXA4-mediated protection during LPS-induced ALI, we evaluated the concentrations and activities of ACE2 and gene and protein expression levels of Ang-(1-7) and its receptor Mas in lung tissues through Western blot, ELISA, and quantitative PCR. Compared with the control group, LPS stimulation decreased ACE2 activities and contents in lung tissues significantly (Figure 2a and b), whereas the changes of Ang-(1-7) and Mas were negligible except for the results of Western blot (Figure 2c–f). Additionally, administration of LXA4 up-regulated and activated the ACE2-Ang-(1-7)-Mas axis significantly, including increasing the content and activity of ACE2 and up-regulating the mRNA and protein expression levels of Ang-(1-7) and Mas in lung tissues (Figure 2). However, BOC-2, A779, and MLN-4760 counteracted all of the effects of LXA4 (Figure 2).
Figure 2.

LXA4 up-regulate and activates the ACE2-Ang-(1-7)-Mas axis in LPS-induced ALI. (a) The activities of ACE2 in lung tissue were observed via kits. (b and c) The contents of ACE2 (b) and Ang-(1-7) (b) in the lung tissue were examined via ELISA. (d and e) The expression levels of Ang-(1-7) and Mas in lung tissue were examined via Western blot. (f) Mas mRNA expression levels were determined via quantitative PCR. Data are presented as means ± SEM. *P < 0.05 and **P < 0.01 compared with control group, ##P < 0.01 compared with LPS group, ΔP < 0.05 and ΔΔP < 0.01 compared with LPS + LXA4 group.
LXA4 regulates LPS-induced inflammation via the ACE2-Ang-(1-7)-Mas axis in ALI
Inflammation is known to play critical roles in LPS-induced ALI. To determine whether the mechanism by which LXA4 exerts its protective effect on LPS-induced lung injury was related to the ACE2-Ang-(1-7)-Mas axis, we examined the concentrations of pro-inflammatory factors, including TNF-α and IL-1β, and the levels of anti-inflammatory cytokine IL-10 in the serum and BALF. The data indicated that LXA4 decreased LPS-induced TNF-α and IL-1β (Figure 3a–d) while increasing the level of expression of IL-10 (Figure 3e and f) in the serum and BALF. We also evaluated the levels of ROS in the lungs. LPS stimulation rendered ROS expression significantly more abundant than in the control group, but the ROS levels were reduced upon treatment with LXA4 (Figure 4). Nevertheless, A779 and MLN-4760, inhibitors of the ACE2-Ang-(1-7)-Mas axis, offset the protective effects of LXA4 (Figures 3 and 4). Expectedly, BOC-2, an inhibitor of the LXA4 receptor, also blocked the effect of LXA4 (Figures 3 and 4). Collectively, these data indicated that LXA4 could protect lungs from LPS-induced ALI through the ACE2-Ang-(1-7)-Mas axis.
Figure 3.

LXA4 regulates LPS-induced inflammation via the ACE2-Ang-(1-7)-Mas axis in ALI. (a and b) The concentrations of TNF-α in the BALF (a) and serum (b). (c and d) The concentrations of IL-1β in the BALF (c) and serum (d). (e and f) The concentrations of IL-10 in the BALF (e) and serum (f). Data are presented as means ± SEM. *P < 0.05 and **P < 0.01 compared with control group, #P < 0.05 and ##P < 0.01 compared with LPS group, ΔP < 0.05 and ΔΔP < 0.01 compared with LPS + LXA4 group.
Figure 4.
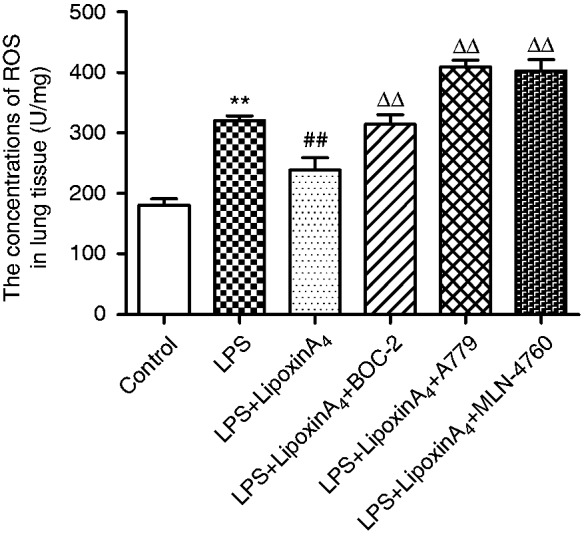
LXA4 regulates LPS-induced ROS via the ACE2-Ang-(1-7)-Mas axis in ALI. The expression of ROS in the lung tissue was determined via assay kits according to the manufacturer’s instructions. Data are presented as means ± SEM. **P < 0.01 compared with control group, ##P < 0.01 compared with LPS group, ΔΔP < 0.01 compared with LPS + LXA4 group.
LXA4 inhibits NF-κB pathway via the ACE2-Ang-(1-7)-Mas axis in LPS-induced ALI
The NF-κB pathway controls the initiation and development of inflammation, so we examined pathway activation, including the levels of IκB, NF-κB p65, and p-p65, via Western blot and immunofluorescence analysis. The results indicated that LPS administration could activate the NF-κB pathway, including inducing the degradation of IκB (Figure 5a and b), the phosphorylation of NF-κB p65 (Figure 5c and d), and the transposition of p65 (Figure 6). However, treatment with LXA4 repressed the effects of LPS (Figures 5 and 6). Nevertheless, treatment with A779 and MLN4760, inhibitors of the ACE2-Ang-(1-7)-Mas axis, and BOC-2, an inhibitor of the LXA4 receptor, could all abolish the effects of LXA4 (Figures 5 and 6).
Figure 5.

LXA4 inhibits NF-κB pathways via the ACE2-Ang-(1-7)-Mas axis in LPS-induced ALI. (a and b) The expression levels of IκBα in lung tissues were examined by Western blot (a) and optical density (b) analysis. (c and d) The expression levels of p65 and p-p65 in lung tissues were examined by Western blot (c) and optical density (d) analysis. Data are presented as means ± SEM. *P < 0.05 and **P < 0.01 compared with control group, ##P < 0.01 compared with LPS group, ΔP < 0.05 and ΔΔP < 0.01 compared with LPS + LXA4 group.
Figure 6.

LXA4 inhibits NF-κB translocation via the ACE2-Ang-(1-7)-Mas axis in LPS-induced ALI. Shown is immunofluorescence analysis of NF-κB translocation. Immunostaining was performed with P65 (in red) and nuclei were stained with DAPI (in blue). The images were observed under a magnification of 630×.
Discussion
LXA4 was the first lipid mediator identified and has dual effects of anti-inflammation and pro-resolution of inflammation.20,21 LXA4 is produced at sites of lung inflammation in the upper and lower airways and the vasculature.22,23 In response to inflammation, in airway epithelial cells, 15-LOX-derived 15-S-hydroxyeicosatetraenoic acid can be transformed by neutrophil 5-LOX to an unstable epoxytetraene intermediate, which is converted to LXA4 and LXB4 by enzymatic hydrolysis.24 LXA4 works by binding specifically and reversibly to ALX/FPR2, a G-protein-coupled receptor.24–26
Our data demonstrated clearly that LXA4 exerts potent protective effects in lung after LPS challenge, which was consistent with the changes observed in histopathologic evaluation (Figure 1a), pulmonary edema (Figure 1b), and microvascular permeability (Figure 1c), in addition to neutrophil infiltration, including white blood cells in BALF and lung MPO activity (Figure 1d and e). We also found that treatment of mice with LXA4 could increase the activity and concentration of ACE2 (Figure 2) and the expression levels of Ang-(1-7) and its receptor Mas (Figure 2) markedly. Third, LXA4 was found capable of increasing the expression of anti-inflammatory IL-10 (Figure 3), but decreased the levels of pro-inflammatory TNF-α, IL-1β, and ROS (Figures 3 and 4) and inhibited the NF-κB signal pathway, including repressing the degradation of I-κB, the phosphorylation of NF-κB, and the translocation of NF-κB (Figures 5 and 6). Finally, our data provided evidence that A779 and MLN4760, inhibitors of the ACE2-Ang-(1-7)-Mas axis, and BOC-2, an inhibitor of the LXA4 receptor, could abolish the effects of LXA4 (Figures 1–6), suggesting that the ACE2-Ang-(1-7)-Mas axis played an essential role in the protective effects of LXA4 on ALI in mice and this was ALX/FPR2 dependent.
ACE2 is a major member of the renin–angiotensin system.27 Renin induces the production of angiotensin I (Ang I), and (Ang I) can transfer to angiotensin II (Ang II) through ACE [27]. ACE2 is the homolog of ACE, but it negates the activity of ACE via induction of Ang II degradation to Ang-(1-7), which binds to its receptor, Mas, and counteracts the effects of Ang II.27 The protective roles of ACE2 and Ang-(1-7) in lung disease have been confirmed in previous works and have been reported to occur mainly via anti-inflammation and anti-oxidation effects.16–18,28–34 It is well known that inflammatory cytokines, oxidative stress, and the upstream NF-κB signal pathway activation are vital aspects of ALI. It has been reported recently that the ACE2-Ang-(1-7)-Mas axis could repress inflammation, reduce the production of ROS, and inhibit NF-κB signal pathway.35–39 Further, various studies have demonstrated that LXA4 can repress the inflammatory response,40–42 decrease the production of ROS,43,44 and control the NF-κB signal pathway.45,46 Consistent with other reports, our results demonstrate that LXA4 was capable of increasing the amount of IL-10 (Figure 3), but decreased TNF-α, IL-1β, and ROS levels (Figures 3 and 4) and inhibited the degradation of IκB, the phosphorylation of NF-κB, and the translocation of NF-κB (Figures 5 and 6). Furthermore, the data demonstrated that LXA4 could up-regulate and activate the ACE2-Ang-(1-7)-Mas axis (Figure 2). More importantly, A779 and MLN4760, inhibitors of the ACE2-Ang-(1-7)-Mas axis, abolished all effects of LXA4, including protection against ALI, regulation of inflammation, and inhibition of the NF-κB signal pathway. These results suggested that LXA4 protected lungs from ALI through regulating the ACE2-Ang-(1-7)-Mas axis and the ACE2-Ang-(1-7)-Mas axis might be upstream of the NF-κB signal pathway. The possible underlying mechanisms of LXA4 in controlling ALI are depicted in Figure 7. We posit that the ACE2-Ang-(1-7)-Mas axis may be a novel and key target of LXs.
Figure 7.
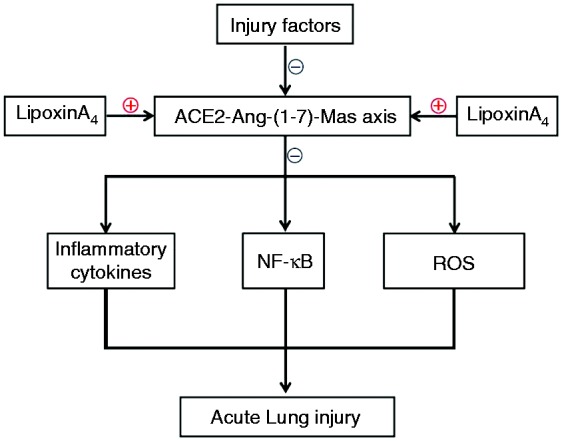
The summary of this research.
In summary, the present study provides evidence that LXA4 can promote the resolution of ALI in mice through the ACE2-Ang-(1-7)-Mas axis in part via FPR2/ALX. This research increases our understanding of the functions and mechanisms of LXs, which is beneficial because it supports the early application of LXs in clinical treatment in ALI and other inflammatory diseases. In addition to LXs themselves, the pro-resolving mediators ACE/Ang-(1-7)/Mas have potential as new therapeutic approaches for patients with ALI and ARDS.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (No. 81760117 and No. 81460126) and the Science and Technology Support Plan of Jiangxi Province (No.20151BBG70246). We thank LetPub (www.letpub.com) for language assistance during the preparation of this manuscript.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
References
- 1.Rubenfeld GD, Caldwell E, Peabody E, et al. Incidence and outcomes of acute lung injury. N Engl J Med 2005; 353: 1685–1693. [DOI] [PubMed] [Google Scholar]
- 2.Cochi SE, Kempker JA, Annangi S, et al. Mortality trends of acute respiratory distress syndrome in the United States from 1999-2013. Ann Am Thorac Soc 2016; 13: 1742–1751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chopra M, Reuben JS, Sharma AC. Acute lung injury: apoptosis and signaling mechanisms. Exp Biol Med (Maywood) 2009; 234: 361–371. [DOI] [PubMed] [Google Scholar]
- 4.Mendelson CR. Role of transcription factors in fetal lung development and surfactant protein gene expression. Annu Rev Physiol 2000; 62: 875–915. [DOI] [PubMed] [Google Scholar]
- 5.Rooney SA. Regulation of surfactant secretion. Comp Biochem Physiol A Mol Integr Physiol 2000; 129: 233–243. [DOI] [PubMed] [Google Scholar]
- 6.Serhan CN. Lipoxins and aspirin-triggered 15-epi-lipoxins are the first lipid mediators of endogenous anti-inflammation and resolution. Prostaglandins Leukot Essent Fatty Acids 2005; 73: 141–162. [DOI] [PubMed] [Google Scholar]
- 7.Chiang N, Takano T, Arita M, et al. A novel rat lipoxin A4 receptor that is conserved in structure and function. Br J Pharmacol 2003; 139: 89–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Haworth O, Cernadas M, Yang R, et al. Resolvin E1 regulates interleukin 23, interferon-gamma and lipoxin A4 to promote the resolution of allergic airway inflammation. Nat Immunol 2008; 9: 873–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chan MM, Moore AR. Resolution of inflammation in murine autoimmune arthritis is disrupted by cyclooxygenase-2 inhibition and restored by prostaglandin E2-mediated lipoxin A4 production. J Immunol 2010; 184: 6418–6426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen M, Divangahi M, Gan H, et al. Lipid mediators in innate immunity against tuberculosis: opposing roles of PGE2 and LXA4 in the induction of macrophage death. J Exp Med 2008; 205: 2791–2801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cheng X, He S, Yuan J, et al. Lipoxin A4 attenuates LPS-induced mouse acute lung injury via Nrf2-mediated E-cadherin expression in airway epithelial cells. Free Radic Biol Med 2016; 93: 52–66. [DOI] [PubMed] [Google Scholar]
- 12.Ortiz-Muñoz G, Mallavia B, Bins A, et al. Aspirin-triggered 15-epi-lipoxin A4 regulates neutrophil-platelet aggregation and attenuates acute lung injury in mice. Blood 2014; 124: 2625–2634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fang X, Abbott J, Cheng L, et al. Human mesenchymal stem (stromal) cells promote the resolution of acute lung injury in part through lipoxin A4. J Immunol 2015; 195: 875–881. [DOI] [PubMed] [Google Scholar]
- 14.Xiao HL, Li CS, Zhao LX, et al. Captopril improves postresuscitation hemodynamics protective against pulmonary embolism by activating the ACE2/Ang-(1-7)/Mas axis. Naunyn Schmiedebergs Arch Pharmacol 2016; 389: 1–11. [DOI] [PubMed] [Google Scholar]
- 15.Hao Y, Liu Y. Osthole alleviates bleomycin-induced pulmonary fibrosis via modulating angiotensin-converting enzyme 2/angiotensin-(1-7) axis and decreasing inflammation responses in rats. Biol Pharm Bull 2016; 39: 457–465. [DOI] [PubMed] [Google Scholar]
- 16.Zambelli V, Bellani G, Borsa R, et al. Angiotensin-(1-7) improves oxygenation, while reducing cellular infiltrate and fibrosis in experimental acute respiratory distress syndrome. Intensive Care Med Exp 2015; 3: 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Supé S, Kohse F, Gembardt F, et al. Therapeutic time window for angiotensin-(1-7) in acute lung injury. Br J Pharmacol 2016; 173: 1618–1628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li Y, Zeng Z, Cao Y, et al. Angiotensin-converting enzyme 2 prevents lipopolysaccharide-induced rat acute lung injury via suppressing the ERK1/2 and NF-κB signaling pathways. Sci Rep 2016; 6: 27911–27911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hu QD, Hu ZZ, Chen QF, et al. BML-111 equilibrated ACE-AngII-AT1R and ACE2-Ang-(1-7)-Mas axis to protect hepatic fibrosis in rats. Prostaglandins Other Lipid Mediat 2017; 131: 75–82. [DOI] [PubMed] [Google Scholar]
- 20.Levy BD, Clish CB, Schmidt B, et al. Lipid mediator class switching during acute inflammation: signals in resolution. Nat Immunol 2001; 2: 612–619. [DOI] [PubMed] [Google Scholar]
- 21.Serhan CN, Takano T, Maddox JF. Aspirin-triggered 15-epilipoxin A4 and stable analogs on lipoxin A4 are potent inhibitors of acute inflammation. Receptors and pathways. Adv Exp Med Biol 1999; 447: 133–149. [DOI] [PubMed] [Google Scholar]
- 22.Levy BD, Romano M, Chapman HA, et al. Human alveolar macrophages have 15-lipoxygenase and generate 15(S)-hydroxy-5,8,11-cis-13-trans-eicosatetraenoic acid and lipoxins. J Clin Invest 1993; 92: 1572–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Serhan CN. Lipoxins and aspirin-triggered 15-epi-lipoxins are the first lipid mediators of endogenous anti-inflammation and resolution. Prostaglandins Leukot Essent Fatty Acids 2005; 73: 141–162. [DOI] [PubMed] [Google Scholar]
- 24.Levy BD, Serhan CN. Resolution of acute inflammation in the lung. Annu Rev Physiol 2014; 76: 467–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fiore S, Ryeom SW, Weller PF, et al. Lipoxin recognition sites. Specific binding of labeled lipoxin A4 with human neutrophils. J Biol Chem 1992; 267: 16168–16176. [PubMed] [Google Scholar]
- 26.Filep JG. Biasing the lipoxin A4/formyl peptide receptor 2 pushes inflammatory resolution. Proc Natl Acad Sci USA 2013; 110: 18033–18034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Imai Y, Kuba K, Penninger JM. The discovery of angiotensin-converting enzyme 2 and its role in acute lung injury in mice. Exp Physiol 2008; 93: 543–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen D, Jiao G, Ma T, et al. The mechanism of rapamycin in the intervention of paraquat-induced acute lung injury in rats. Xenobiotica 2015; 45: 538–546. [DOI] [PubMed] [Google Scholar]
- 29.Liu W, Shan LP, Dong XS, et al. Toll-like receptor 4 implicated in acute lung injury induced by paraquat poisoning in mice. Int J Clin Exp Med 2014; 7: 3392–3397. [PMC free article] [PubMed] [Google Scholar]
- 30.Liu F, Gao F, Li Q, et al. The functional study of human umbilical cord mesenchymal stem cells harbouring angiotensin-converting enzyme 2 in rat acute lung ischemia-reperfusion injury model. Cell Biochem Funct 2014; 32: 580–589. [DOI] [PubMed] [Google Scholar]
- 31.Gao F, Li Q, Hou L, et al. Mesenchymal stem cell-based angiotensin-converting enzyme 2 in treatment of acute lung injury rat induced by bleomycin. Exp Lung Res 2014; 40: 392–403. [DOI] [PubMed] [Google Scholar]
- 32.Liu D, Dong Y, Liu Z, et al. Impact of trem-2 gene silencing on inflammatory response of endotoxin-induced acute lung injury in mice. Mol Cell Biochem 2014; 394: 155–161. [DOI] [PubMed] [Google Scholar]
- 33.Min F, Gao F, Li Q, et al. Therapeutic effect of human umbilical cord mesenchymal stem cells modified by angiotensin-converting enzyme 2 gene on bleomycin-induced lung fibrosis injury. Mol Med Rep 2015; 11: 2387–2396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang X, Gao F, Li Q, et al. Mscs with ace ii gene affect apoptosis pathway of acute lung injury induced by bleomycin. Exp Lung Res 2015; 41: 32–43. [DOI] [PubMed] [Google Scholar]
- 35.Lu W, Kang J, Hu K, et al. Angiotensin-(1-7) inhibits inflammation and oxidative stress to relieve lung injury induced by chronic intermittent hypoxia in rats. Braz J Med Biol Res 2016; 49: e5431–e5431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Simoes e Silva AC, Silveira KD, et al. ACE2, angiotensin-(1-7) and Mas receptor axis in inflammation and fibrosis. Br J Pharmacol 2013; 169: 477–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Meng Y, Li T, Zhou GS, et al. The angiotensin-converting enzyme 2/angiotensin (1-7)/Mas axis protects against lung fibroblast migration and lung fibrosis by inhibiting the NOX4-derived ROS-mediated RhoA/Rho kinase pathway. Antioxid Redox Signal 2015; 22: 241–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Morales MG, Abrigo J, Meneses C, et al. The Ang-(1-7)/Mas-1 axis attenuates the expression and signalling of TGF-β1 induced by AngII in mouse skeletal muscle. Clin Sci (Lond) 2014; 127: 251–264. [DOI] [PubMed] [Google Scholar]
- 39.Meng Y, Yu CH, Li W, et al. Angiotensin-converting enzyme 2/angiotensin-(1-7)/Mas axis protects against lung fibrosis by inhibiting the MAPK/NF-κB pathway. Am J Respir Cell Mol Biol 2014; 50: 723–736. [DOI] [PubMed] [Google Scholar]
- 40.Planaguma A, Kazani S, Marigowda G, et al. Airway lipoxin A4 generation and lipoxinA4 receptor expression are decreased in severe asthma. Am J Respir Crit Care Med 2008; 178: 574–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Medeiros R, Rodrigues GB, Figueiredo CP, et al. Molecular mechanisms of topical anti-inflammatory effects of lipoxinA(4) in endotoxin-induced uveitis. Mol Pharmacol 2008; 74: 154–161. [DOI] [PubMed] [Google Scholar]
- 42.Kieran NE, Maderna P, Godson C. Lipoxins: potential anti-inflammatory, proresolution, and antifibrotic mediators in renal disease. Kidney Int 2004; 65: 1145–1154. [DOI] [PubMed] [Google Scholar]
- 43.Zhou XY, Li YS, Wu P, et al. Lipoxin A(4) inhibited hepatocyte growth factor-induced invasion of human hepatoma cells. Hepatol Res 2009; 39: 921–930. [DOI] [PubMed] [Google Scholar]
- 44.Xia J, Zhou XL, Zhao Y, et al. Roles of lipoxin A4 in preventing paracetamol-induced acute hepatic injury in a rabbit model. Inflammation 2013; 36: 1431–1439. [DOI] [PubMed] [Google Scholar]
- 45.Zhou XY, Wu P, Zhang L, et al. Effects of lipoxin A(4) on lipopolysaccharide induced proliferation and reactive oxygen species production in RAW264.7 macrophages through modulation of G-CSF secretion. Inflamm Res 2007; 56: 324–333. [DOI] [PubMed] [Google Scholar]
- 46.Nascimento-Silva V, Arruda MA, Barja-Fidalgo C, et al. Aspirin-triggered lipoxin A4 blocks reactive oxygen species generation in endothelial cells: a novel antioxidative mechanism. Thromb Haemost 2007; 97: 88–98. [PubMed] [Google Scholar]