

Abstract
Supplemental Digital Content is Available in the Text.
To the Editors:
Transmitted drug resistance (TDR) in HIV-1 isolates to nonnucleoside reverse transcriptase inhibitors (NNRTIs) has an estimated prevalence of 7% in the United States.1 Internationally, NNRTI TDR mutations are estimated to have a prevalence of 11% in southern Africa, 10% in western Africa, 7.2% in central and eastern Africa, 9.4% in Latin America, 4.1% in Europe, and 3.2% in Asia.2,3 The most frequent NNRTI TDR mutations include the reverse transcriptase (RT) amino acid substitutions K103N, Y181C, and G190A.4 Current guidelines recommend that HIV-1 genotyping is performed before initiating therapy to determine whether TDR mutations are present to assist clinicians in selecting an effective antiviral treatment regimen.5,6
Doravirine (DOR) is a novel NNRTI for the treatment of HIV-1.7,8 DOR is active in vitro against both wild-type HIV-1 and most common NNRTI-resistant variants (including K103N, Y181C, and G190A) at concentrations achieved with 100 mg once-daily dosing.9 DOR has a unique in vitro resistance profile among NNRTIs, with lower IC50 values against NNRTI resistant variants as compared to other NNRTIs.9–11 This trial was designed to evaluate the clinical efficacy of DOR against the HIV-1 NNRTI TDR mutants K103N, Y181C, and G190A.
METHODS
This trial (NCT02629822) was a phase 2, multicenter, open-label, single-arm trial of DOR/3TC/TDF once daily in HIV-1–infected adults with a single NNRTI TDR mutation. The trial was conducted at 7 centers in 5 countries (Canada, France, Spain, United States, and United Kingdom) in accordance with principles of Good Clinical Practice and was approved by institutional review boards and regulatory agencies.
Participants were antiretroviral-naive HIV-1–infected adults (≥18 years) with baseline plasma HIV-1 RNA ≥1000 copies/mL and CD4+ T-cell count ≥100 cells/mm3 within 45 days before initiation of study treatment. Eligibility required a genotype-confirmed, single NNRTI mutation consisting of RT K103N, Y181C, or G190A. Participants were required to be clinically stable as determined by the investigator, have a calculated creatinine clearance ≥ 50 mL/min, alkaline phosphatase ≤3.0x upper normal limit, AST (SGOT) and ALT (SGPT) ≤5.0x upper normal limit, and hemoglobin ≥9.0 g/dL for females or ≥10.0 g/dL for males. Participants were excluded for any of the following: previous treatment for a viral infection such as hepatitis B with an agent active against HIV-1; documented genotypic resistance to study drugs; any medical condition requiring the use of systemic immunosuppressive therapy or immune modulators; acute hepatitis, decompensated liver disease, or cirrhosis.
This was a single-arm, open-label trial. All participants were treated with a single fixed-dose combination tablet containing 100 mg doravirine, 300 mg lamivudine, and 300 mg tenofovir disoproxil fumarate (DOR/3TC/TDF) taken once daily. Participants were screened within 45 days before study entry. Blood samples for HIV-1 RNA quantification were collected at all study visits (week 4, 8, 12, 16, 24, 36, 48, 60, 72, 84, and 96), as well as the virologic failure visit or early discontinuation visit, if applicable. HIV-1 RNA quantification was performed using the Abbott Real-Time HIV-1 assay. Before screening, participants were required to have documentation of a single genotypic HIV-1 resistance mutation consisting of either RT K103N, Y181C, or G190A. An additional screening sample was obtained to confirm the previously documented mutation by the central laboratory; if the required mutation was not confirmed, that participant was excluded from the efficacy analysis, but allowed to remain in the trial. CD4+ T-cell counts were determined at screening, at day 1, and at weeks 24, 48, 72, and 96. Protocol-defined virologic failure (PDVF) was defined as confirmed HIV-1 RNA ≥50 copies/mL after initial response of HIV-1 RNA <50 copies/mL at any time during the study; HIV-1 RNA ≥200 copies/mL at week 24 or week 36; or HIV-1 RNA ≥50 copies/mL at or after week 48. Confirmation of PDVF required an additional measurement of HIV-1 RNA taken at least 1 week apart. Participants who met these criteria were discontinued from the trial. Safety was monitored by adverse event (AE) reporting, evaluation of treatment-emergent laboratory abnormalities, and physical examinations. Clinical AEs were assessed by the investigator for intensity and relationship to study therapy. Laboratory values were graded in severity based on the Division of AIDS criteria for grading adverse events.12
The primary efficacy endpoint was the proportion of participants achieving HIV-1 RNA <50 copies/mL at week 48. An additional objective was to evaluate the safety and tolerability of DOR/3TC/TDF, as assessed by review of safety data. Secondary and exploratory efficacy endpoints included HIV-1 RNA <50 copies/mL by week 96 and the change from baseline in CD4+ T-cell count. Only descriptive summary statistics and confidence intervals were performed for both efficacy and safety endpoints with no formal hypothesis testing and no missing data approach applied. The 95% confidence interval was calculated with the Clopper and Pearson method.13
RESULTS
Planned enrollment was 60 participants. However, recruitment was much slower than anticipated (only 10 enrolled after 13.5 months). Therefore, further enrollment was halted due to projected inability to recruit the planned number of participants. Eighteen total participants were screened, of whom 10 were enrolled, 9 met criteria for inclusion in the efficacy analysis (1 did not have previous K103N resistance mutation confirmed at screening by the central laboratory), 8 completed the study through week 48, and 7 completed the study through week 96. All 10 enrolled participants took at least one dose of DOR/3TC/TDF and were included in the safety analysis. Two participants were discontinued before week 48, one of whom was lost to follow-up after week 16. This participant was excluded from efficacy analysis due to nonconfirmed NNRTI mutation. The other participant discontinued at week 36 for poor adherence; this participant met PDVF criteria and was included in the efficacy analyses. One additional participant was lost to follow-up between week 48 and week 96 (see Table 1, Supplemental Digital Content, http://links.lww.com/QAI/B370). Participants were 80.0% male with median age 32.5 years (range 25–56). The baseline median CD4+ T-cell count was 407.5 cells/mm3 (range 213–607), and the median plasma HIV-1 RNA was 17,281 copies/mL (range 1366–295,604). One participant had baseline HIV-1 RNA >100,000 copies/mL. Eight participants had RT K103N and 2 participants had RT G190A. Adherence with study medication, as measured by participant medication diaries, was high with 8 of 10 participants reporting ≥90% adherence.
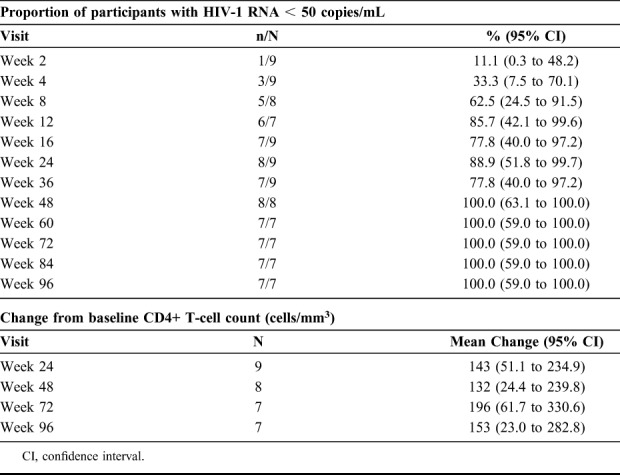
All 8 participants who completed the week-48 visit and all 7 participants who completed the week-96 visit achieved HIV-1 RNA <50 copies/mL (Table 1). Participants had a mean increase from baseline in CD4+ T-cells of 153 cells/mm3 at week 96 (Table 1). One participant with baseline G190A discontinued at week 36 with PDVF (poor adherence determined by self-reporting and pill count). This participant did not take DOR/3TC/TDF for 16 days before the week 24 visit and had viral rebound with HIV-1 RNA of 5393 copies/mL at week 24 after previously achieving viral suppression. After restarting treatment, HIV-1 RNA at the viral failure confirmation visit was 55 copies/mL. No additional drug resistance mutations were identified at the time of viral rebound. The participants who were lost to follow-up at week 16 (no confirmatory genotype) and at week 48 also had HIV-1 RNA <50 copies/mL at their previous visit.
TABLE 1.
Efficacy Outcomes for DOR/3TC/TDF Treatment
AEs were reported for 9 of the 10 participants who received DOR/3TC/TDF over 96 weeks. There were no deaths. One serious AE was reported, allergy to an arthropod sting, and was judged as not drug related by the investigator. AEs experienced by >1 participant were abdominal discomfort, diarrhea, nausea, fatigue, and back pain, each reported in 2 participants. Six drug-related AEs were reported, all were nonserious, and none resulted in discontinuation. Drug-related AEs experienced by >1 participant were abdominal discomfort and fatigue.
DISCUSSION
DOR/3TC/TDF demonstrated antiretroviral efficacy through 96 weeks of treatment in a small number of treatment-naive HIV-1–infected participants with baseline NNRTI resistance mutations of K103N and G190A. The 8 participants who completed the study through week 48 and the 7 participants who completed the study through week 96 all achieved virologic suppression (HIV-1 RNA <50 copies/mL) demonstrating sustained antiviral efficacy. In addition, the 2 participants who discontinued before week 48, and the one participant who discontinued before week 96, achieved virologic suppression before discontinuation. This trial also demonstrated that DOR/3TC/TDF was well tolerated in the limited number of study participants.
Preclinical studies showed that DOR had an inhibitory quotient [IQ, defined as ratio of clinical trough concentration to the 50% inhibitory concentration (Ctrough/IC50)] of 50 for wild-type HIV-1, and 39, 27, and 15 for RT K103N, Y181C, G190A mutants, respectively, and predict that DOR would be clinically effective against these mutants.9 Although participants infected with HIV-1 harboring the Y181C mutation were not enrolled in this study, the IQ of Y181C being between the IQs of K103N and G190A suggests that DOR would be clinically effective for this mutation. In a separate trial (DRIVE-SHIFT), evaluating antiviral efficacy in virologically suppressed participants switching from a baseline therapy to DOR/3TC/TDF, genotyping before initial treatment indicated that 24 participants had HIV-1 with NNRTI resistance mutations K103N, Y181C, and/or G190A. Of those 24 participants, 23 switched to DOR/3TC/TDF, and 21 of those who switched regimens remained suppressed through week 48; the other 2 discontinued early but were still suppressed at their last study visit.14 Overall, this trial supports previous in vitro findings that DOR is active against HIV-1 with K103N and G190A mutations based on the observed antiretroviral efficacy in a small number of participants.
Supplementary Material
ACKNOWLEDGMENTS
The authors thank the patients and their families and caregivers for participating in this trial, along with all investigators and site personnel. Medical writing assistance was provided by Dean Campbell, PhD, and editorial assistance was provided by Carol Zecca, BS, both of Merck & Co., Inc., Kenilworth, NJ, USA.
Footnotes
Supported by Merck, Sharp, & Dohme Corp, a subsidiary of Merck & Co., Inc., Kenilworth, NJ.
The authors have no funding or conflicts of interest to disclose.
A.R., V.T., D.H., S.K., P.S., G.J.H., C.H., C.B., and H.T. are employees of Merck & Co., Inc., Kenilworth, NJ, and may own stock and/or hold stock options in the Company. J.-M.M. has received honoraria from Gilead Sciences, Inc, Foster City, CA, and Merck, Sharp, & Dohme Corp, a subsidiary of Merck & Co., Inc., Kenilworth, NJ for participating in advisory boards and has received a research grant from Gilead Sciences Inc. The remaining authors have no conflicts of interest to disclose.
Supplemental digital content is available for this article. Direct URL citations appear in the printed text and are provided in the HTML and PDF versions of this article on the journal's Web site (www.jaids.com).
REFERENCES
- 1.Buchacz K, Young B, Palella FJ, Jr, et al. Trends in use of genotypic resistance testing and frequency of major drug resistance among antiretroviral-naive persons in the HIV Outpatient Study, 1999–2011. J Antimicrob Chemother. 2015;70:2337–2346. [DOI] [PubMed] [Google Scholar]
- 2.Gupta RK, Gregson J, Parkin N, et al. HIV-1 drug resistance before initiation or re-initiation of first-line antiretroviral therapy in low-income and middle-income countries: a systematic review and meta-regression analysis. Lancet Infect Dis. 2018;18:346–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Frentz D, Van de Vijver DA, Abecasis AB, et al. Increase in transmitted resistance to non-nucleoside reverse transcriptase inhibitors among newly diagnosed HIV-1 infections in Europe. BMC Infect Dis. 2014;14:407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zazzi M, Hu H, Prosperi M. The global burden of HIV-1 drug resistance in the past 20 years. PeerJ. 2018;6:e4848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.British HIV Association. BHIVA Guidelines for the Treatment of HIV-1-Positive Adults With Antiretroviral Therapy 2015 (2016 Interim Update) Available at: http://www.bhiva.org/HIV-1-treatment-guidelines.aspx. Accessed October 29, 2018. [Google Scholar]
- 6.Department of Health and Human Services, Panel on Antiretroviral Guidelines for Adults and Adolescents. Guidelines for the Use of Antiretroviral Agents in Adults and Adolescents with HIV; 2018. Available at: https://aidsinfo.nih.gov/contentfiles/lvguidelines/adultandadolescentgl.pdf. Accessed October 29, 2018. [Google Scholar]
- 7.Anderson MS, Gilmartin J, Cilissen C, et al. Safety, tolerability and pharmacokinetics of doravirine, a novel HIV non-nucleoside reverse transcriptase inhibitor, after single and multiple doses in healthy subjects. Antivir Ther (Lond). 2015;20:397–405. [DOI] [PubMed] [Google Scholar]
- 8.Colombier MA, Molina JM. Doravirine: a review. Curr Opin HIV AIDS. 2018;13:308–314. [DOI] [PubMed] [Google Scholar]
- 9.Feng M, Sachs NA, Xu M, et al. Doravirine suppresses common nonnucleoside reverse transcriptase inhibitor-associated mutants at clinically relevant concentrations. Antimicrob Agents Chemother. 2016;60:2241–2247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Feng M, Wang D, Grobler JA, et al. In vitro resistance selection with doravirine (MK-1439), a novel nonnucleoside reverse transcriptase inhibitor with distinct mutation development pathways. Antimicrob Agents Chemother. 2015;59:590–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Smith SJ, Pauly GT, Akram A, et al. Rilpivirine and doravirine have complementary efficacies against NNRTI-resistant HIV-1 mutants. J Acquir Immune Defic Syndr. 2016;72:485–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.U.S. Department of Health and Human Services, National Institutes of Health. Division of AIDS (DAIDS) Table for Grading the Severity of Adult and Pediatric Adverse Events, Version 2.0. [November 2014]. Available at: https://rsc.niaid.nih.gov/sites/default/files/daids-ae-grading-table-v2-nov2014.pdf. Accessed October 29, 2018. [Google Scholar]
- 13.Clopper CJ, Pearson ES. The use of confidence or fiducial limits illustrated in the case of binomial. Biometrika. 1934;26:404–413. [Google Scholar]
- 14.Johnson M, Kumar P, Molina JM, et al. Switching to Doravirine/Lamivudine/Tenofovir disoproxil fumarate (DOR/3TC/TDF) maintains HIV-1 virologic suppression through 48 weeks: results of the DRIVE-SHIFT trial. J Acquir Immune Defic Syndr 2019;81:463–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.