

Abstract
The present study aimed to evaluate the cellular and molecular effects of low concentrations of the flavonoid, fisetin, on K562 human chronic myeloid leukemia cells, in the context of both potential anti-proliferative and anti-metastatic effects. Thiazolyl blue tetrazolium bromide assay, Trypan blue exclusion assay, Annexin V/propidium iodide test, cell cycle analysis, Transwell migration and invasion assays, the fluorescence staining of β-catenin and F-actin as well as reverse transcription-quantitative polymerase chain reaction were performed to achieve the research goal. Furthermore, the nature of the interaction between fisetin and arsenic trioxide in the K562 cells was analyzed according to the Chou-Talalay median-effect method. We found that low concentrations of fisetin had not only a negligible effect on the viability and apoptosis of the K562 cells, but also modulated the mRNA levels of selected metastatic-related markers, accompanied by an increase in the migratory and invasive properties of these cancer cells. Although some markers of cell death were significantly elevated in response to fisetin treatment, these were counterbalanced through anti-apoptotic and pro-survival signals. With decreasing concentrations of fisetin and arsenic trioxide, the antagonistic interactions between the 2 agents increased. On the whole, the findings of this study suggest that careful consideration should be taken when advising cancer patients to take fisetin as a dietary supplement and when considering fisetin as a potential candidate for the treatment of chronic myeloid leukemia. Further more detailed studies are required to confirm our findings.
Keywords: fisetin, dietary flavonoids, chronic myeloid leukemia, cell death, cell migration and invasion, arsenic trioxide, combination therapy
Introduction
In recent years, dietary polyphenols have attracted increasing scientific attention as potential chemopreventive and chemotherapeutic agents, due to their putative low toxicity profiles, their common availability, low cost and a wide range of biological activities (1-4). Despite the large variety of structural variants of dietary polyphenols, not all of these share the same antitumor activity; hence, a considerable focus of researchers has been concentrated on selected groups, including flavonoids. One of the most intensely studied compound of this class has become fisetin (3,3′,4′,7-tetrahydroxyflavone; FIS), whose anticancer properties have relatively recently been discovered, and earlier mainly its antimicrobial, as well as its neuroprotective activities had been known. FIS belongs to a flavonol subgroup of flavonoids and constitutes common ingredient of the human diet, as it can be found in several fruits, vegetables and nuts, including strawberries, apples, kiwi fruit, grapes, persimmons, onions and cucumbers (5). There is accumulating evidence that FIS may exert anti-proliferative, anti-inflammatory, pro-death, anti-invasive, anti-migratory and anti-angiogenic effects in a large panel of human cancer cell lines and several animal models (6-8). At the cellular and molecular level, these anti-carcinogenic effects of FIS have been shown to be associated with the modulation of multiple signal transduction pathways, including the phosphatidylinositol 3-kinase/AKT/mammalian target of rapamycin (PI3K/AKT/mTOR), cyclooxygenase 2 (COX-2), Janus kinase/signal transducer and activator of transcription (JAK/STAT), extracellular-signal regulated kinase 1/2 (ERK1/2), c-Jun N-terminal kinase (JNK) and p38 mitogen-activated protein kinase (p38 MAPK) pathways, AMP-activated protein kinase (AMPK) signaling, KIT receptor signaling, growth hormone receptor signaling, as well as Wnt/β-catenin, epidermal growth factor receptor (EGFR) and nuclear factor κB (NF-κB) pathways. Furthermore, FIS has been found to bind and inhibit the activity of cyclin-dependent kinases (CDKs) 2, 4, 6, decrease the levels of cyclin B1, D1 and cyclin E, suppress retinoblastoma protein (pRb) phosphorylation, increase the levels of p21, p27 and p53, as well as to function as an antagonist of DNA topoisomerases I, II and as a ligand for the androgen receptor (AR) (5,7,9). At the cellular level, FIS possesses a microtubule stabilizing property and binds to β-tubulin, leading to the disruption of microtubule dynamics (10). It has been demonstrated that FIS-induced cancer cell death occurs predominantly through apoptosis, although its ability to trigger autophagy and mitotic catastrophe has also been documented (11-13). FIS-induced apoptosis has been found to be associated with endoplasmic reticulum (ER) stress (14,15), the activation of the caspase cascade (16-19), the inhibition of heat shock factor 1 (HSF1) (20), the induction (19) or the suppression (17) of reactive oxygen species (ROS) production, the downregulation of anti-apoptotic Bcl-2, Bcl-XL, Mcl-1L, cIAP-1/2, XIAP, survivin and TRAF1 proteins, and the upregulation of pro-apoptotic Bcl-2 family members, such as Bax, Bim and Bad (16,19,21,22). The occurrence of apoptosis following treatment with FIS has been demonstrated in a wide variety of cancer types, including chronic myelogenous leukemia (23,24), promyelocytic leukemia (25), melanoma (26), hepatocellular carcinoma (18), multiple myeloma (19), breast (27), bladder (22), liver (6), pancreatic (28), cervical (29), prostate (16), colon (20,30,31), epidermoid (32), laryngeal (33) and oral squamous cell carcinoma (34,35). However, the therapeutic application of FIS, similar to other flavonoids, has been hampered due to insufficient oral bioavailability as a result of its poor aqueous solubility, poor absorption, as well as extensive and rapid metabolism. Therefore, considerable efforts have been made to improve the bioavailability of FIS through e.g., co-crystallization with caffeine, nicotinamide and isonicotinamide, complexation with cyclodextrins and encapsulation with nanoparticles (36-39).
Although numerous in vitro studies have been devoted to investigating the antitumor efficacy, as well as the mechanisms of action of FIS, only few of these have used low, in vivo achievable concentrations of this agent. To limit a lack of reproducibility of the in vitro studies in clinical trials, the use of clinically relevant concentrations in the in vitro testing of agents is currently strongly recommended (40). Therefore, in this study, we aimed to investigate the cellular and molecular effects of in vivo attainable concentrations of FIS on K562 human chronic myeloid leukemia (CML) cells. Furthermore, since we, as well as others have previously reported that FIS can act synergistically with certain anticancer drugs (27,41-43), thereby establishing its potential as a possible candidate for combination therapy, herein we also aimed to assess whether this flavonoid may enhance the anticancer effects exerted by arsenic trioxide (ATO) against K562 leukemic cells.
Materials and methods
Cell culture and treatment
The K562 human chronic myeloid leukemia cells (ATCC) were maintained in Roswell Park Memorial 1640 medium (RPMI-1640; BioWhittaker, Lonza) supplemented with 10% fetal bovine serum (FBS, Sigma-Aldrich) and 50 µg/ml gentamycin (Sigma-Aldrich). A humidified atmosphere of 95% air and 5% CO2 at a constant temperature of 37°C was provided by the CO2 incubator. After reaching approximately 70-80% confluency, the cells were passaged and seeded on 12- or 6-well plates (BD Falcon) for further assays. Concentrated stock solutions of FIS (100 mM; Abcam) were prepared in dimethyl sulfoxide (DMSO; Sigma-Aldrich), stored at -25°C and serially diluted in complete growth medium right before use. ATO (Sigma-Aldrich) was dissolved in 1 M of NaOH to generate 100 mM stock solution, which was then stored at 4°C and serially diluted in complete growth medium immediately prior to cell treatment. The control cells were cultured under identical conditions, although without the addition of the test agents. The final concentrations of DMSO or NaOH (0.02 and 0.0001%, respectively) did not influence the experimental results (data not shown). The cells were evaluated for mycoplasma contamination by DAPI staining methods. Cells at low passage were used for all the experiments (≤5). The range of concentrations for cell viability assays (10-50 µM) were selected to enable the construction of the dose-response curves for the estimation of the half-maximal inhibitory concentration values and drug-drug interaction analysis. Since in vivo achievable levels of FIS are only up to 20 µM (44), 10 and 20 µM concentrations of this flavonoid were employed for all subsequent experiments. Likewise, clinically-relevant (1.5-2.5 µM) and sub-therapeutic (0.5-1.0 µM) concentrations of ATO were used in drug-drug interaction experiments, as such an approach has previously been recommended to limit its toxicity and to reduce unwanted side-effects (45,46). Given the stability and cellular uptake of several flavonoid compounds (47,48), the incubation time was 24 and/or 48 h, unless otherwise stated.
Cell viability assays
The cytotoxic effects of the test agents on the viability of the K562 cells was evaluated by the thiazolyl blue tetrazolium bromide (MTT) colorimetric assay, as well as Trypan blue dye exclusion test. For both experiments, the cells were seeded into 12-well plates and treated with FIS at concentrations of 10, 20, 30, 40, 50 µM for 24 or 48 h. After the indicated treatment period, the cells were incubated in the MTT working solution, which was prepared by diluting the MTT stock solution [made by dissolving 5 mg of MTT (Sigma-Aldrich) in 1 ml of PBS] with the culture medium without phenol red (HyClone, Thermo Fisher Scientific) at a ratio of 1:9. Following 3 h of incubation in the CO2 incubator at a standard condition, the resulting formazan crystals were dissolved in isopropanol (POCH, Poland) and the absorbance was read at 570 nm using a spectrophotometer (Spectra Academy, K-MAC). Following Trypan blue staining (3 min at room temperature), at least 100 cells per data point were counted under an Eclipse E800 light microscope (Nikon Corp.) and blue stained cells were considered dead. The cell viability was calculated assuming the absorbance of control cells as 100%.
Drug interaction analysis
To determine the combined effects of FIS and ATO on K562 cells, the data of MTT assay were analyzed with CompuSyn software (ComboSyn Inc.) based on the combination index method of Chou and Talalay (49). This method is entirely based on the physical, chemical and mathematical principles of the mass-action law, i.e., unified median-effect equation and the combination index equation, and it is the most cited and the broadest cited method for drug combination studies (50). For MTT assays, ATO (at concentrations of 0.5, 1.0, 1.5, 2.0, 2.5 µM) or FIS (10, 20, 30, 40, 50 µM) were added to the cells for 24 h as either single or combined agents at a fixed concentration ratio of 1:20. The combination index (CI) values for each concentration and the corresponding effect level, referred to as the fraction affected (fa; the fraction of cells inhibited after the drug exposure, e.g., 0.5 when cell growth is inhibited by 50%), were determined. The resulting combination index offers a quantitative determination of drug interaction, where CI <1, = 1 and >1 indicates synergism, an additive effect and antagonism, respectively. For a graphical illustration of drug interaction, the Fa-CI plot was constructed by computerized simulation of serial CI values over a range of fa levels from 0.001 (IC0.1) to 0.90 (IC90). The dose reduction index (DRI) at fa=0.5 was also calculated to indicate the fold of dose reduction allowed for each agent in the combination treatment compared to the single-agent treatment that is required to achieve the same effect level. DRI = 1 signifies no dose reduction, whereas DRI >1 and <1 represent favorable and unfavorable dose-reduction, respectively (51).
Cell apoptosis assay
To quantify cell apoptosis, the Annexin V-Alexa Fluor 488 and Propidium Iodide kit (Thermo Fisher Scientific) was used according to the manufacturer's instructions with some modifications. In brief, following 24 and 48 h of incubation with 10 and 20 µM FIS, the cells were collected from 6-well plates, centrifuged (300 × g, 5 min, room temperature), resuspended in Annexin V Binding Buffer (ABB) and incubated with Annexin V-Alexa Fluor 488 at room temperature in the dark. Following 20 min of incubation, the cells were once more centrifuged (300 × g, 5 min, room temperature) and resuspended in ABB. Finally, 1 µl of propidium iodide (PI) was added for 5 min at room temperature in the dark. The data were acquired with the Tali image-based cytometer (Thermo Fisher Scientific) and analyzed using the FCS express Research Edition Software (version 4.03; De Novo Software). The sum of the early (Annexin V+/PI−) and late (Annexin V+/PI+) apoptotic cells represented the total apoptosis.
Cell cycle assay
To examine cell populations in various phases of the cell cycle, the measurement of the DNA content was performed using the Guava Cell Cycle reagent (Merck KGaA) according to the manufacturer's protocol. Briefly, the cells treated with 10 and 20 µM FIS for 24 and 48 h were harvested from 6-well plates, centrifuged (300 × g, 5 min, room temperature) and then fixed in ice-cold 70% ethanol at 4°C followed by overnight incubation at −25°C. The cells were then centrifuged at 650 × g for 5 min at room temperature and washed with PBS. Following centrifugation at 500 × g for 7 min at room temperature, the cells were resuspended in Guava Cell Cycle reagent, and incubated for 30 min at room temperature in the dark. The cells were then analyzed with the Guava easyCyte 6HT-2L Benchtop Flow Cytometer (Merck KGaA), and the percentage of cells in each phase of the cell cycle was determined using InCyte software (version 4.03; De Novo Software).
Transwell invasion and migration assays
The invasion and migration of the K562 cells was assessed using the Transwell chamber system. The upper side of 24-well polycarbonate filter inserts (8 µm pore size; Corning, Inc.) were pre-coated for 24 h at 37°C with 40 µl of Matrigel (2 mg/ml in serum-free medium). The cells treated or untreated with the flavonoid were placed in the upper compartment of the chamber at a density of 3.0×105. Transwell inserts were then placed in the lower chamber filled with 750 µl of medium containing 10% FBS as a chemoattractant and allowed to invade for 40 h under standard culture conditions provided by a CO2 incubator. After the indicated period of time, invaded or migrated cells recovered from the lower chamber were counted with the Tali image-based cytometer (Thermo Fisher Scientific), and the results were expressed relative to control cells, set as 1. Transwell migration assay was performed in a manner similar to the above-described invasion assay with the exception that Matrigel was omitted and the cells were counted following 20 h of incubation.
Fluorescence staining of β-catenin and F-actin
β-catenin and filamentous actin (F-actin) fluorescence staining was performed according to a previously described protocol (52). Briefly, control and FIS-treated K562 cells were collected from 12-well plates and centrifuged onto glass coverslips (768 × g, 10 min, room temperature), and then fixed with 4% paraformaldehyde (Serva), as well as permeabilized with 0.25% Triton X-100. F-actin was labeled with Alexa Fluor 488-conjugated phalloidin (dilution 1:40 in PBS, 20 min; Life Technologies). Non-specific background was blocked with 1% bovine serum albumin (BSA). Rabbit polyclonal anti-β-catenin antibody was used as a primary antibody to label β-catenin (dilution 1:150 in BSA, 60 min; cat. no. SAB4500541, Sigma-Aldrich), followed by incubation with a secondary goat anti-rabbit antibody conjugated to Alexa Fluor 594 (dilution 1:200 in BSA, 60 min; cat. no. R37117, Thermo Fisher Scientific). Cell nuclei (DNA) were stained with 4′,6-diamidino-2-phenylindole (DAPI, dilution 1:20,000, 10 min; Sigma-Aldrich). All incubations were carried out at room temperature, whereas fluorescence staining was performed in the dark. Finally, the slides were mounted in Aqua-Poly/Mount (Polysciences) and analyzed using the C1 laser-scanning confocal microscope (with a 100X oil immersion objective; Nikon) and Nikon EZ-C1 software version 3.80 (both from Nikon). The acquisition parameters, including laser power, pixel dwell time and gains, were maintained at the same level for all fluorescence images captured with the confocal microscope. The fluorescence intensity of β-catenin was normalized to DAPI signal and measured using ImageJ software (version 1.52k; National Institutes of Health).
Reverse transcription-quantitative polymerase chain reaction (RT-qPCR)
Total RNA was isolated from the K562 cells using the Total RNA Mini Plus kit (A&A Biotechnology) according to the provided manufacturer's instructions, followed by the spectrophotometric determination of RNA concentration and purity (BioSpectrometer basic; Eppendorf). A one-step RT-qPCR was carried out with LightCycler RNA Master SYBR-Green I kit (Roche Applied Science) on a LightCycler 2.0 Instrument (Roche Applied Science). The total reaction mixture (20 µl per single LightCycler capillary) contained 100 ng of RNA and 0.2 µM of each primer (oligo.pl) in addition to the LightCycler RNA Master SYBR-Green I kit components. The following thermocycling conditions were used: One cycle of reverse transcription for 20 min at 61°C, one cycle of denaturation for 1 min at 95°C, and 45 cycles of denaturation for 5 sec at 95°C, followed by annealing and extension for 20 sec at 55-60°C (depending on the melting temperature of the primers) and 5 sec at 72°C, respectively. The results obtained from at least 3 independent experiments were analyzed with LightCycler Software version 4.0. The expression of each target mRNA relative to the glyceraldehyde-3-phosphate dehydrogenase (GAPDH) internal control was calculated based on the ΔΔCq method (2−ΔΔCq method) (53). The primers used for the PCR reactions are listed in Table I.
Table I.
Details of the investigated and reference genes.
| Gene | Primer sequence |
|---|---|
| AIF | Forward: 5′-CTGAAAGACGGCAGGAAGGTAG-3′ |
| Reverse: 5′-CTCCAGCCAATCTTCCACTCAC-3′ | |
| AKT | Forward: 5′-GGCTATTGTGAAGGAGGGTTG-3′ |
| Reverse 5′-TCCTTGTAGCCAATGAAGGTG-3′ | |
| BAX | Forward: 5′-AGATGTGGTCTATAATGCGTTTTCC-3′ |
| Reverse: 5′-CAGAAGGCACTAATCAAGTCAAGGT-3′ | |
| BCL2 | Forward: 5′-AACATCGCCCTGTGGATGAC-3′, |
| Reverse: 5′-AGAGTCTTCAGAGACAGCCAGGAG-3 | |
| BECN1 | Forward: 5′-AGCTGCCGTTATACTGTTCTG-3′ |
| Reverse: 5′-ACTGCCTCCTGTGTCTTCAATCTT-3′ | |
| CASP3 | Forward: 5-TGGTTCATCCAGTCGCTTTG-3′ |
| Reverse: 5′-CATTCTGTTGCCACCTTTCG-3′ | |
| CD44 | Forward: 5′-ACCCCAACTCCATCTGTGC-3′ |
| Reverse: 5′-TTCTGGACATAGCGGGTG-3′ | |
| GAPDH | Forward: 5′-ACAACTTTGGTATCGTGGAAGG-3′ |
| Reverse: 5′-GCCATCACGCCACAGTTTC-3′ | |
| MAP1LC3B | Forward: 5′-CGGTGATAATAGAACGATACAAGG-3′ |
| Reverse: 5′-CTGAGATTGGTGTGGAGACG-3′ | |
| MMP2 | Forward: 5′-GATACCCCTTTGACGGTAAGGA-3′ |
| Reverse: 5′-CCTTCTCCCAAGGTCCATAGC-3′ | |
| MMP9 | Forward: 5′-CCCTGGAGACCTGAGAACCA-3′ |
| Reverse: 5′-CCCGAGTGTAACCATAGCGG-3′ | |
| MTOR | Forward: 5′-TCACATTACCCCCTTCACCA-3′ |
| Reverse: 5′-TCAGCGAGTTCTTGCTATTCC-3′ | |
| PI3K | Forward: 5′-AGTAGGCAACCGTGAAGAAAAG-3′ |
| Reverse: 5′-GAGGTGAATTGAGGTCCCTAAGA-3′ | |
| PYK2 | Forward: 5′-GAGACCTACCGCTGTGAAC-3′ |
| Reverse: 5′-CTGCTAGGGATGAGGTTTTG-3′ | |
| RHOA | Forward: 5′-CCAAATGTGCCCATCATCCTAGTTG-3′ |
| Reverse: 5′-TCCGTCTTTGGTCTTTGCTGAACAC-3′ | |
| RIP3 | Forward: 5′-AATTCGTGCTGCGCCTAGAAG-3′ |
| Reverse: 5′-TCGTGCAGGTAAAACATCCCA-3′ | |
| ROCK1 | Forward: 5′-GGTGCTGGTAAGAGGGCATT-3′ |
| Reverse: 5′-AGCATCCAATCCATCCAGCAA-3′ | |
| SNAI1 | Forward: 5′-TTCAACTGCAAATACTGCAACAAG-3′ |
| Reverse: 5′-CGTGTGGCTTCGGATGTG-3′ | |
| SNAI2 | Forward: 5′-TGTGACAAGGAATATGTGAGCC-3′ |
| Reverse: 5′-TGAGCCCTCAGATTTGACCTG-3′ | |
| SQSTM1/P62 | Forward: 5′-GGGGACTTGGTTGCCTTTT-3′ |
| Reverse: 5′-CAGCCATCGCAGATCACATT-3′ | |
| TWIST | Forward: 5′-GTCCGCAGTCTTACGAGGAG-3′ |
| reverse: 5′-GCTTGAGGGTCTGAATCTTGCT-3′ | |
| VIM | Forward: 5′- AGTCCACTGAGTACCGGAGAC-3′ |
| Reverse: 5′-CATTTCACGCATCTGGCGTTC-3′ |
Statistical analysis
Statistical analysis was performed with the Prism software package (version 7.01, GraphPad Software). The evaluation of data normality was carried out with the Shapiro-Wilk test. One-way or two-way ANOVA with Tukey's or Sidak's post hoc comparisons, Kruskal-Wallis with Dunn's post hoc tests or unpaired t-test with Welch's correction were used for data comparison where appropriate. All values are expressed as the means ± standard deviation and all statistical assessments were two-sided. Probability (P)-values <0.05 was considered to indicate statistically significant differences.
Results
Effects of FIS treatment on the viability of the K562 human chronic myeloid leukemia cells
To examine the cytotoxic effects of FIS towards the K562 cells, we first performed an MTT assay, which is a sensitive and reliable indicator of metabolically active mitochondria. As depicted in Fig. 1A, FIS at concentrations ranging from 10 to 50 µM was not highly toxic to the K562 cells, as approximately 80% of the cells managed to survive following treatment with the highest concentration of this compound, regardless of the incubation time. At low concentrations (10 and 20 µM), FIS did not cause any apparent changes in the viability of the K562 cells; however, at higher concentrations (30-50 µM), it led to a statistically significant, although slight decrease in the survival rate of these cells. Furthermore, all tested concentrations resulted in no significant differences in cell viability between the two incubation periods (24 vs. 48 h). The results of MTT assays were confirmed by Trypan blue exclusion assay (Fig. 1B).
Figure 1.
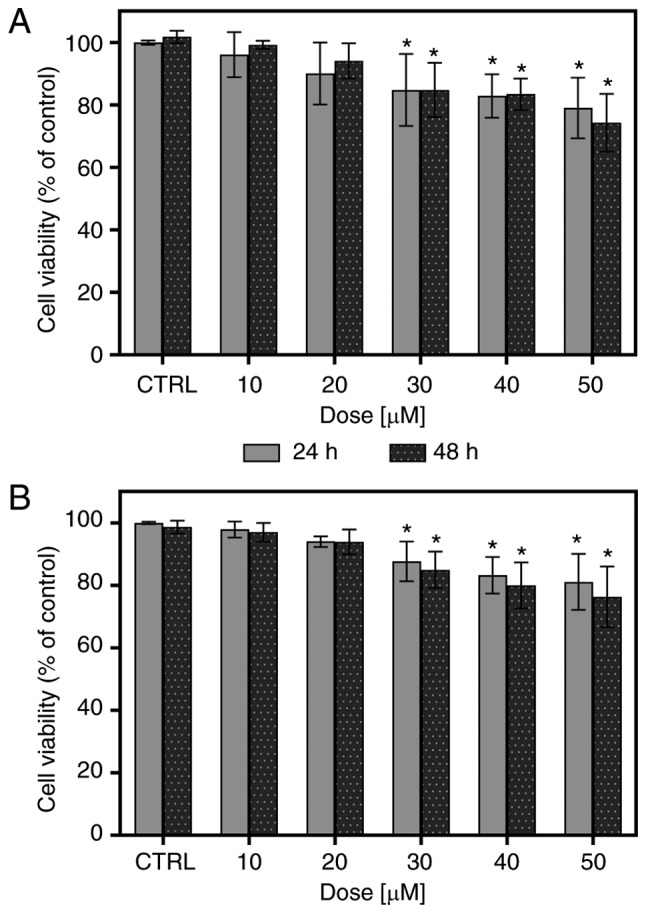
Determination of cell viability by (A) MTT assay and (B) Trypan blue dye exclusion assay. The K562 cells were treated with increasing concentrations of fisetin for 24 or 48 h or left untreated (CTRL, which is represented as 100%). Statistically significant differences between the fisetin treatment groups and the control group were marked with an asterisk (*P<0.05; two-way ANOVA with Tukey's post hoc test). There was no signifi-cant difference between 24 and 48 h treatment (P>0.05; two-way ANOVA with Sidak's post hoc test). Data represent the means ± standard deviation of 5 (MTT assay) or 4 (Trypan blue assay) independent replicates.
Effects of FIS on apoptosis induction and cell cycle distribution in the K562 human chronic myeloid leukemia cells
To determine whether FIS at low concentrations can induce the apoptosis and cell cycle arrest of the K562 cells, Annexin V/PI double staining and PI single staining were performed, respectively, following 24 and 48 h of exposure to the 10 and 20 µM concentrations of this flavonoid. As shown in Fig. 2A, FIS was not found to be a potent inducer of apoptosis and triggered only low cell death in the K562 cells. At the 10 µM concentration of the agent, the increase in the number of apoptotic cells was significant only following the longer exposure time; however, simultaneously, there were no statistically significant differences between the two incubation times (24 vs. 48 h). At the 20 µM concentration of FIS, the increase in apoptosis was found to be significant in response to both exposure time periods in comparison to the control; however, in this case as well, the longer incubation time did not result in a marked change in apoptosis when compared to the shorter incubation time. As depicted in Fig. 2B, treatment of the K562 cells with FIS at both concentrations did not cause any significant changes in cell cycle distribution when compared to the untreated cells.
Figure 2.
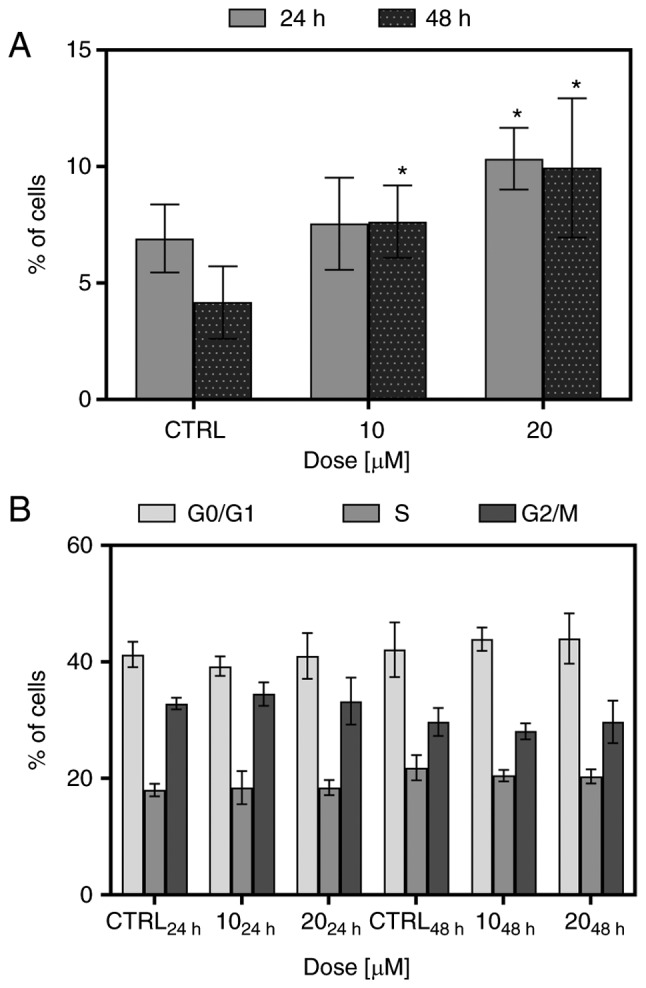
Determination of (A) apoptosis by Annexin V/PI assay and (B) cell cycle distribution by PI staining. The K562 cells were treated with 10 and 20 µM fisetin for 24 and 48 h or left untreated. (A) The sum of the early (Annexin V+/PI−) and late (Annexin V+/PI+) apoptotic cells represents the total apoptosis. An asterisk indicates statistically significant differences between the control and fisetin treatment groups (*P<0.05; two-way ANOVA with Tukey's post hoc test). Data represent the means ± standard deviation of (A) 3 or (B) 5 independent experiments.
Effects of FIS on the migratory and invasive behaviors of the K562 human chronic myeloid leukemia cells
To examine whether FIS at low concentrations can affect the invasive and migratory ability of the K562 cells, Transwell invasion and migration assays were conducted. As shown in Fig. 3A, the FIS-treated K562 cells exhibited a significantly greater capacity to invade through the Matrigel matrix than the untreated ones. Likewise, treatment of the K562 cells with both tested concentrations of this flavonoid visibly increased the migratory capability through the Transwell membranes compared with that of the control cells; however, statistical significance was reached only at the concentration of 20 µM (Fig. 3B).
Figure 3.
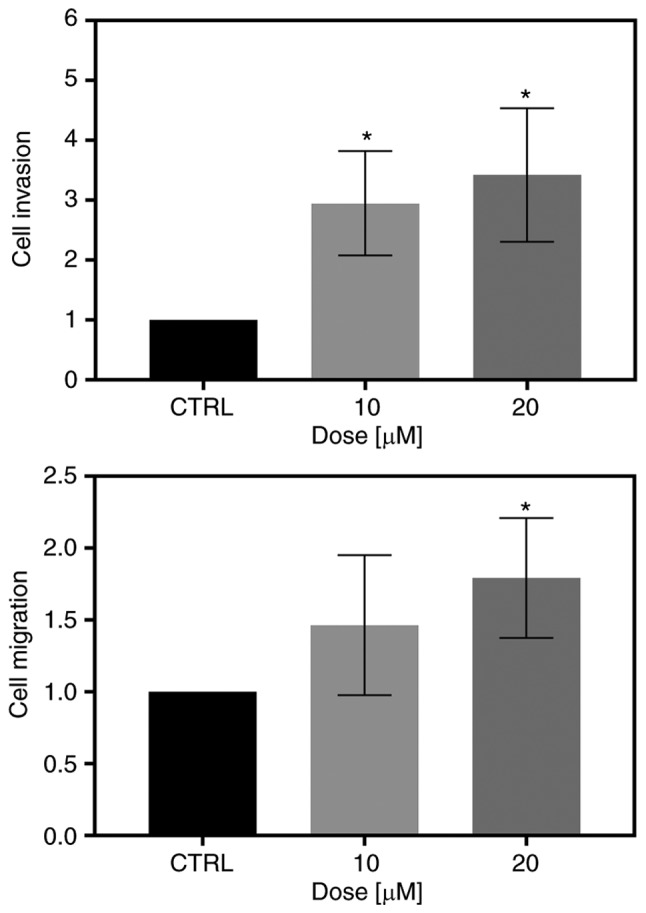
Evaluation of invasion and migration using a Transwell assay. The K562 cells were treated with 10 or 20 µM fisetin or left untreated (CTRL). The results are expressed relative to control cells (set as 1) and statistical significance between control and treated groups is labeled with an asterisk (*P<0.05; one-way ANOVA with Tukey's post hoc test). Data represent the means ± standard deviation of 2 (migration assay) or 3 (invasion assay) independent experiments, each in duplicate.
Effects of FIS on F-actin and β-catenin in the K562 human chronic myeloid leukemia cells
As shown in Fig. 4A, treatment of the K562 cells with FIS resulted in an increase in F-actin staining and its more visible redistribution toward the plasma membrane when compared to the untreated cells. Furthermore, FIS treatment enhanced the nuclear localization of β-catenin (Fig. 4).
Figure 4.

Assessment of F-actin and β-catenin staining under a confocal microscope. The K562 cells were treated with 10 or 20 µM fisetin for 24 h or left untreated (CTRL). (A) F-actin (green), β-catenin (red) and cell nuclei (DNA, blue) were stained as described in the Materials and methods. (B) The fluorescence intensity of nuclear β-catenin was normalized to the DAPI signal and measured using ImageJ software (****P<0.0001; ns, not significant; Kruskal-Wallis with Dunn's post hoc test). The representative images of 2 independent experiments are shown; scale bar, 100 µm.
Effects of FIS on the expression of pro-survival, cell death-related and metastasis-related markers in the K562 human chronic myeloid leukemia cells
To assess the effects of FIS at low concentrations on the K562 cells at a molecular level, the mRNA expression of a range of cell death-related and metastasis-related markers was investigated by RT-qPCR. As depicted in Fig. 5A-C, the flavonoid modulated the transcript level of apoptosis-associated genes. It upregulated the expression of BAX and BCL2, resulting in the ratio of BCL2 to BAX being marginally higher than 1 (1.06 and 1.19 for 10 and 20 µM FIS, respectively; Fig. 4A). Furthermore, this compound increased the mRNA levels of caspase-3 and AIF; however, in the case of the former, statistical significance was reached only at the concentration of 20 µM of the agent (similar to the results obtained for BCL2; Fig. 5A-C). In addition to apoptosis, the expression of two markers of regulated necrosis, RIP3 and PARP1, was also markedly upregulated by FIS (Fig. 5D and E). Furthermore, since the autophagy-associated gene, BECN1 was significantly overexpressed upon treatment of the K562 cells with 20 µM FIS (Fig. 5F), the mRNA levels of two other autophagy markers, LC3B and p62, were also measured in these cells. However, the expression levels of these markers did not indicate the activation of autophagy in response to FIS exposure, as the expression of the former was unaltered and that of the latter was markedly elevated (Fig. 5G and H). Likewise, the mRNA level of the master negative regulator of autophagy, MTOR, increased upon FIS treatment (Fig. 5I). The agent also upregulated the expression of two other critical nodes in the pro-survival PI3K/AKT and mTOR signaling pathways, whereby PI3K mRNA was significantly increased only at the higher concentration of FIS, and AKT mRNA only at the lower one (Fig. 5J and K).
Figure 5.

Measurement of the expression level of pro-survival and death-associated markers by RT-qPCR. The K562 cells were treated for 24 h with 10 and 20 µM FIS or left untreated (CTRL). RT-qPCR measurement of the (A) BCL2/BAX, (B) CASP3, (C) AIF, (D) RIP3, (E) PARP1, (F) BECN1, (G) MAP1LC3B, (H) SQSTM1/P62, (I) MTOR, (J) PI3K and (K) AKT mRNA expression level. Relative gene expression was normalized to the GAPDH housekeeping gene and expressed as a fold difference relative to a calibrator sample (untreated cells; assumed as 1). An asterisk denotes statistically significant differences in comparison to the control (*P<0.05; one-way ANOVA with Tukey's post hoc test). Data represent the means ± standard deviation of at least 3 independent experiments. BCL2, B-cell lymphoma 2; BAX, BCL2 associated X; CASP3, caspase-3; AIF, apoptosis-inducing factor; RIP3, receptor interacting serine/threonine-protein kinase 3; PARP1, poly(ADP-ribose) polymerase 1; BECN1, Beclin 1; MAP1LC3B, microtubule associated protein 1 light chain 3 beta; SQSTM1/P62, sequestosome 1/PP62; MTOR, mammalian target of rapamycin; PI3K, phosphatidylinositol 3-kinase; AKT, protein kinase B.
FIS also influenced the expression of metastasis-related genes in the K562 cells. At both the tested concentrations, it significantly increased the CD44, MMP2, SNAIL, SLUG, VIM and ROCK1 mRNA levels; however, it simultaneously had no effect on RHOA expression (Fig. 6A-G). Furthermore, there was a significant upregulation of MMP9, TWIST, PYK2 and CTNNB1 expression following treatment with 20 µM FIS. The 10-fold lower concentration of the flavonoid also upregulated the mRNA level of MMP9, TWIST and PYK2; however, in these cases, statistical significance was not reached (Fig. 6H-K).
Figure 6.

Measurement of the expression level of metastasis-related markers by RT-qPCR. The K562 cells were treated for 24 h with 10 and 20 µM FIS or left untreated (CTRL). RT-qPCR measurement of the (A) CD44 (B) MMP2, (C) SNAI1, (D) SNAI2, (E) VIM, (F) ROCK1, (G) RHOA, (H) MMP9, (I) TWIST, (J) PYK2, (K) CTNNB1 mRNA expression level. Relative gene expression was normalized to the GAPDH housekeeping gene and expressed as a fold difference relative to a calibrator sample (untreated cells; assumed as 1). An asterisk denotes statistically significant differences in comparison to control (*P<0.05; one-way ANOVA with Tukey's post hoc test). Data represent the means ± standard deviation of at least 3 independent experiments. CD44, cluster of differentiation 44; MMP2, matrix metalloproteinase 2; SNAI1, snail family transcriptional repressor 1; SNAI2, snail family transcriptional repressor 2; VIM, vimentin; ROCK1, Rho associated coiled-coil containing protein kinase 1; RHOA, Ras homolog family member A; MMP9, matrix metalloproteinase 9; TWIST, Twist family BHLH transcription factor 1; PYK2, proline-rich tyrosine kinase 2; CTNNB1, catenin beta 1.
Combined effects of FIS and ATO on the K562 human chronic myeloid leukemia cells
The interaction between FIS and ATO was analyzed based on the results of MTT assays and the median-effect principle of Chou and Talalay (49). As shown in Fig. 7A, a single treatment with ATO, at concentrations ranging from 0.5 to 2.5 µM, had little if any effect on the viability of the K562 cells, the results of which are consistent with those of previous studies on this cell line (54,55). In turn, the combination of FIS and ATO produced a highly variable effect on cell survival in dependence of the concentrations of both agents. Co-treatment with 10 µM FIS and 0.5 µM ATO resulted in a slight increase in the viability of the K562 cells; however, the results did not achieve statistical significance compared to the control or either agent alone (Fig. 7A). The calculated CI value indicated a very potent antagonism between FIS and ATO at this experimental point (dose combination) (Table II). However, when the cells were simultaneously treated with higher concentrations of FIS (20 or 30 µM) and ATO (1 or 1.5 µM), a significant decrease in cell viability compared to the control cell population and ATO-treated cells, but not to the FIS-treated cells was observed (Fig. 7A). The computed CI values indicated the occurrence of a slight antagonism and synergism between the former (20 µM FIS + 1 µM ATO) and the latter (30 µM FIS + 1.5 µM ATO) dose-pair, respectively (Table II). At the two highest concentrations (40 µM FIS + 2 µM ATO and 50 µM FIS + 2.5 µM ATO), the combined cytotoxic effects of FIS and ATO against the K562 cells were greater than those induced by each agent individually (Fig. 7A). At these experimental points, the interactions of the two agents were moderately synergistic, as indicated by calculating the CI values (Table II). Apart from the determination of the CI values for actual experimental points, the CI values for a wide range of effect levels were generated by the computerized simulation, and depicted graphically as combination index curve (Fa-CI plot). As demonstrated in Fig. 7B, this analysis revealed that the interactions between FIS and ATO varied from antagonism at lower concentrations (i.e., the lowest fa values) to synergism at higher ones (i.e., higher fa values). The dose requirement to achieve 50% growth inhibition (fa=0.5) decreased to 48.3 and 2.4 µM for FIS, respectively, thus reducing the dose by 3.1- and 11.5-fold (Fig. 7B). Importantly, according to the previous reports (44), this concentration of the flavonoid is still outside physiologically attainable levels (≤20 µM). Likewise, the therapeutic concentrations of ATO have been reported to be approximately 1-2 µM (56).
Figure 7.
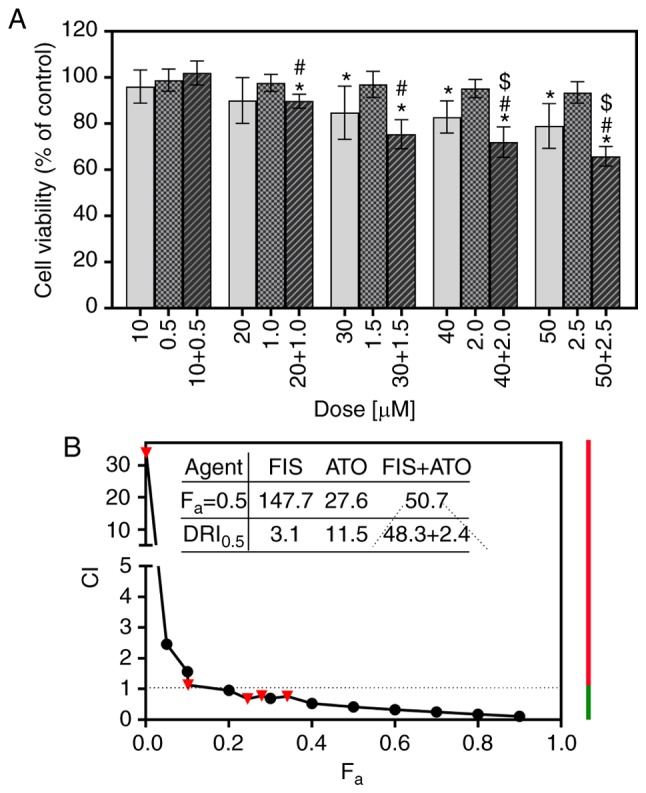
Combined effects of fisetin (FIS) and arsenic trioxide (ATO) on the K562 cells. The cells were treated with various concentrations of ATO (0.5, 1.0, 1.5, 2.0, 2.5 µM) and FIS (10, 20, 30, 40, 50 µM), either alone or in a fixed ratio of 1:20, for 24 h. (A) Cell viability was determined by MTT colorimetric assay. Data are expressed as a percentage of the control. The symbols $ and # indicate statistically significant differences compared with FIS or ATO treatment alone, respectively (P<0.05; unpaired t-test with Welch's correction). The asterisk (*) indicates statistically significant differences compared with the control. All values represent the means ± standard deviation of 5 independent experiments. (B) Combination index plot (Fa-CI plot) for FIS and ATO co-treatment in K562 cells. CI values are plotted as a function of the fractional inhibition (fa) of cell viability by computer simulation (CompuSyn software) from 0.001 to 0.90. Triangles represent CI values derived from the actual data points. CI <1 indicates synergism (denoted by green line), CI=1 designates additivity (denoted by a dashed line), and CI >1 represents antagonism (denoted by red line). The dose reduction index (DRI) =1 signifies no dose reduction, whereas DRI >1 and <1 represent a favorable and unfavorable dose-reduction, respectively.
Table II.
The combination index values at actual experimental points for the combination of fisetin and arsenic trioxide in K562 cells.
| Drug combination
|
CI | fa | Interaction type | |
|---|---|---|---|---|
| FIS (µM) | ATO (µM) | |||
| 10 | 0.5 | 33.91 | 0.001 | Very potent antagonism |
| 20 | 1.0 | 1.13 | 0.102 | Slight antagonism |
| 30 | 1.5 | 0.68 | 0.245 | Synergism |
| 40 | 2.0 | 0.78 | 0.279 | Moderate synergism |
| 50 | 2.5 | 0.76 | 0.341 | Moderate synergism |
The combination index (CI) values were calculated according to the method of Chou-Talalay using CompuSyn software. CI <1 represents synergism, with CI of 0.1-0.3 indicating strong synergism, 0.3-0.7 indicating synergism, CI of 0.7-0.85 indicating moderate synergism, CI of 0.85-0.90 indicating slight synergism, CI of 0.90-1.10 indicating a nearly additive effect, CI of 1.10-1.20 indicating a slight antagonism, CI of 1.20-1.45 indicating a moderate antagonism, CI of 1.45-3.3 indicating antagonism, CI of 3.3-10 indicating strong antagonism, and CI above 10 indicating very potent antagonism. fa, fraction affected by dose; FIS, fisetin; ATO, arsenic trioxide.
Discussion
Advances achieved over the past two decades in the treatment of CML, associated mainly with the development of BCR/ABL1 tyrosine kinase inhibitors (TKIs), have markedly improved the prognosis and survival of Philadelphia chromosome-positive (Ph+) CML patients (57). However, resistance to TKIs and their toxicity have become an increasingly important cause of treatment failure (58). Therefore, the identification and development of novel agents with therapeutic potential against CML is still an important subject for research. With this aim, in the present study, we investigated whether the flavonoid, FIS, at achievable serum concentrations, exhibits anticancer activity against K562 human chronic myeloid leukemia cells, in the context of both potential anti-proliferative and anti-metastatic effects. We decided to focus mainly on these concentrations of the flavonoid that have previously been reported to correspond with its in vivo attainable levels (≤20 µM) (44). The concentration issue is of particular importance in the field of both in vitro and flavonoid research since there is currently the conception that to limit a lack of reproducibility of the cell line studies in clinical trials, it is essential to employ only low, clinically relevant concentrations of tested agents. In the case of dietary flavonoids, this issue is mostly related to the fact that an increasing number of studies have been demonstrating a dual, dose-dependent functional effect of these compounds on cancer cell behavior, with a desired anticancer effect at high doses (usually >60 µM) and no or unfavorable response at lower ones (59-61). Our previous studies (41,42) and the current study seem to confirm such observations.
In this study, we found that low concentrations (10 and 20 µM) of FIS only negligibly affected the viability of the K562 cells through the induction of apoptosis, accompanied by the increase in the migratory and invasive properties of these leukemia cells. Some markers of cell death were significantly elevated in response to FIS treatment; however, they were counterbalanced through anti-apoptotic and pro-survival signals. These results are consistent with the current understanding that in response to either death or non-lethal stress from the treatment, apoptotic signals may have functions other than cell death, including the promotion of a rapid proliferation of neighboring surviving tumor cells, and the increase in their metastatic capacity. This process is referred to as apoptosis-induced proliferation (AiP) and its direct crosstalk with signaling networks linked to migration and invasion has recently been proposed (62-67). Diverse mechanisms of AiP, such as the 'Phoenix Rising' (PR) pathway have been more recently identified and the role of the latter in tissue regeneration, as well as tumor repopulation and resistance following cytotoxic therapies (either ionizing radiation or chemotherapy) has also been reported (68-70). Therefore, it is possible that pro-apoptotic proteins, mostly caspase 3 confer a pro-survival and pro-invasive phenotype to FIS-treated K562 cells through the PR pathway. However, the assessment of other key molecular players of the PR pathway [e.g., caspase-7, prostaglandin E2 (PGE2), protein kinase Cδ (PKCδ), calcium-independent phospholipase A2 (iPLA2), COX-2, NF-κB] should be performed, among other assays (71-72), to confirm this assumption.
There are currently a few studies on the effects of FIS on human CML cells and their authors suggest that this flavonoid has a promising activity against these cells (23,24). Indeed, in K562 cells, FIS has been shown to arrest the cell cycle at both the S and G2/M phases with the subsequent induction of mitochondrial- and caspase-dependent apoptosis. Simultaneously, wide genome microarray analysis has revealed that this flavonoid dose-dependently modulates hundreds of genes implicated in proliferation, differentiation, cell cycle, growth arrest, apoptosis, DNA repair, invasion and metastasis, including those being well-recognized oncogenes and tumor suppressor genes, e.g., NFKBIA (also known as IκBα), metallothionin (MT) family genes, PMAIP1/NOXA, CDKN1A (p21/WAF1/Cip1), GADD45B, MYC (v-myc avian myelocytomatosis viral oncogenehomolog), MYB (v-myb avian myeloblastosis viral oncogene homolog), C-KIT (v-kit Hardy-Zuckerman 4 feline sarcoma viral oncogene homolog), tubulin family members, such as TUBA1A and TUBA1C, serpin family members, STAT proteins and the downstream targets of JAK/STAT pathway, such as PIM1/2 kinases (24). The above-mentioned effects required the concentrations of FIS far outside those reported as physiologically achievable levels (up to 200 µM) as well as the prolonged incubation time (IC50 values at 48 and 72 h were estimated as 163 and 120 µM, respectively) (24). Simultaneously, it should be emphasized that the IC50 values reported by the cited authors for FIS-treated K562 cells were similar to those obtained by us, whereas in the studies presented by another research group, 50-µM FIS promoted a robust cell death of nearly 50% of K562 cells after 24 h of treatment (23). Following the same concentration and incubation period, we found that FIS induced apoptosis in nearly 14% of the K562 cells (data not shown). Such discrepancies between the in vitro drug effects can be quite often found in the literature and usually result from differences in culture conditions, which are rather difficult to discuss, when there is a lack of methodological details provided in the study. Furthermore, the cited authors have demonstrated that the FIS-induced apoptosis of CML cells was mediated by nitric oxide (NO), although the exact underlying mechanism was investigated in THP-1 acute monocytic leukemia cells, which were much more sensitive to this flavonoid than K562 cells (23). Therefore, even if, in contrast to our results, 50 µM of FIS is able to induce a high level of apoptosis of K562 CML cells, it is currently considered that the concentrations of FIS (as well as most other dietary polyphenols) >20 µM are unachievable in vivo, and some researchers set this limit even at half lower dose (10 µM) (73-75).
Our efforts towards assessing the effects of FIS on some markers of both cancer cell migration/invasion and cancer cell growth/death were related to emerging evidence demonstrating that some chemotherapeutic drugs, alongside their cytotoxic activity, may promote invasive behaviors of cancer cells, resulting in the induction or acceleration of metastasis formation (76,77). With regard to CML, a recent example of such action has been derived from imatinib studies. For instance, Ovcharenko et al reported an increase in the adhesion, migratory and invasive capabilities of K562 cells following imatinib exposure and identified proline-rich tyrosine kinase 2 (PYK2) as one of the major mediator of these processes (78). Herein, we found that of treatment the K562 cells with FIS also resulted in the upregulation of the PYK2 transcript level, in addition to its ability to increase the expression of ROCK1, although not the expression of upstream RHOA, both of which have been implicated in the proliferation, migration and invasion of CML cells and are currently considered as therapeutic targets for CML patients (79,80). Moreover, the PI3K/AKT/mTOR signaling pathway enhances not only the growth and survival of cancer cells, including CML, but also cell invasion, migration and drug resistance (81,82). Likewise, a high expression of CD44 has been linked to tumor invasion, metastasis, proliferation, recurrence and chemoresistance (83), and with regard to the K562 cell line, it has been suggested that the proliferation of these cells depends to a greater extent on CD44 (84). Therefore, it is not without significance that FIS led to the increase in the transcript levels of these above-mentioned markers in CML cells. Furthermore, although epithelial-mesenchymal transition (EMT)-related proteins are well recognized for playing a pivotal role in the invasiveness, migration capability, stemness, and drug resistance of epithelial tumors, evidence of their similar importance in the hematological malignancies (despite their mostly mesenchymal status, as blood cells originate from the embryonic mesoderm) has been increasingly provided in recent years (85-87). EMT-related transcription factors, including TWIST, SLUG and SNAIL have been linked with the enhanced aggressiveness of CML cells (88-90), and we found that FIS at in vivo relevant concentrations increased their mRNA level in K562 CML cells. Likewise, this flavonoid promoted the overexpression of the EMT-associated markers, vimentin and MMP2/9, as well as the nuclear localization of β-catenin. Furthermore, FIS treatment resulted in enriched F-actin staining and its visible remodeling toward the plasma membrane. These results were in agreement with our in vitro migration and invasion assays, and therefore, it can be hypothesized that all these cellular and molecular changes underlie the increased migration and invasion of K562 cells in the presence of this flavonoid and constitute a clear undesirable outcome of its in vitro testing in this cancer model. Although further studies are required, particularly those evaluating the effects of low-dose FIS on a larger panel of cancer-related markers at both mRNA and protein level, as well as the phosphorylation status of selected proteins, it can be concluded that a particular caution should be taken when considering FIS as a potential candidate for CML treatment.
Given the lack of utility of FIS in the monotherapy of CML, as well as our (41,42) and other (27,43,91) recent findings on its considerable efficacy in the combination with standard chemotherapeutic drugs in some cancer models, herein we also decided to assess whether this flavonoid may synergize with ATO in K562 cells. ATO is known for its high in vitro and clinical activity against acute promyelocytic leukemia (APL), and numerous preclinical data have also supported its role as a therapy for CML (92,93). ATO treatment has been shown to present similar cellular effects on CML cell lines as on APL ones, although higher and clinically unachievable concentrations are required to promote the cell cycle arrest and apoptosis of the former leukemia cells (54,55,94,95). Therefore, there has been a rationale for the development of optimized ATO-based combination regimens to maintain or potentiate a treatment response, while reducing its dosage to sub-pharmacological, non-toxic levels (95-98). In this study, we found that the interactions between FIS and ATO varied from potent antagonism at lower concentrations (i.e., the lowest fa values) to potent synergism at higher ones (i.e., higher fa values). This raises two important concerns. Firstly, the dose-dependence of the in vitro interaction patterns between FIS and ATO make it difficult to predict the nature of their interaction in vivo, where the drug concentrations vary depending on their pharmacokinetic profile and intra-tumoral distribution. Secondly, it was the low, physiologically achievable concentrations of FIS, which acted antagonistically with clinically relevant doses of ATO, what again implies that the utility of FIS in the combination of ATO as a chemotherapy for CML is rather limited. This also suggests that the supplementation of FIS during the treatment of CML patients with ATO may detrimentally affect therapeutic response. This seems of particular importance when taking into consideration that FIS is widely available in the form of dietary supplements (at the recommended dose of 100-200 mg per day), which are usually taken without medical supervision. However, since the interaction patterns between FIS and ATO were examined only based on the results of MTT assays and at a particular concentration range, it is of importance to confirm our findings using various concentration ratios of both agents and additional assays, e.g., Annexin V/PI assay.
In conclusion, the present study revealed that low-dose FIS had not only negligible effect on the viability and apoptosis of K562 cells, but also modulated the mRNA level of selected metastatic-related markers, accompanied by the increase in the migratory and invasive properties of these cancer cells. Although the levels of some markers of cell death were significantly elevated in response to FIS treatment, they were counterbalanced through anti-apoptotic and pro-survival signals. FIS dose-dependently interacts with ATO; however, it was the low, in vivo achievable concentrations of the flavonoid, which acted antagonistically with clinically relevant doses of cytostatic drug. All these results suggest that careful consideration should be taken when advising FIS as dietary supplement in cancer patients and when considering this flavonoid as a potential candidate for the treatment of CML. However, it should be emphasized that our results were obtained under specific in vitro culture conditions, hence their relevance to human health is currently limited, and only in vivo and particularly clinical studies will be able to verify the potential advantages and disadvantages of using FIS in patients with CML.
Acknowledgments
Not applicable.
Funding
The study was supported by research tasks no. 142 within the frame-work of basal research activity (Nicolaus Copernicus University in Toruń, Faculty of Medicine, Collegium Medicum in Bydgoszcz).
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Authors' contributions
AKW and DG conceived and designed the experiments, performed some of the experiments, analyzed the data, performed the statistical analysis and wrote the manuscript; PC, MHW, JD and PA performed experiments and helped to draft the manuscript; MG performed some of the experiments; AG revised the manuscript and was involved in the conception of the study. All authors have read and approved the final manuscript.
Ethics approval and consent to participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have not competing interests.
References
- 1.Thomasset SC, Berry DP, Garcea G, Marczylo T, Steward WP, Gescher AJ. Dietary polyphenolic phytochemicals-promising cancer chemopreventive agents in humans? A review of their clinical properties. Int J Cancer. 2007;120:451–458. doi: 10.1002/ijc.22419. [DOI] [PubMed] [Google Scholar]
- 2.Ramos S. Cancer chemoprevention and chemotherapy: Dietary polyphenols and signalling pathways. Mol Nutr Food Res. 2008;52:507–526. doi: 10.1002/mnfr.200700326. [DOI] [PubMed] [Google Scholar]
- 3.Sak K. Chemotherapy and dietary phytochemical agents. Chemother Res Pract. 2012;2012:282570. doi: 10.1155/2012/282570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Surh YJ. Cancer chemoprevention with dietary phytochemicals. Nat Rev Cancer. 2003;3:768–780. doi: 10.1038/nrc1189. [DOI] [PubMed] [Google Scholar]
- 5.Jash SK, Mondal S. Bioactive flavonoid fisetin-a molecule of pharmacological interest. J Org Biomol Chem. 2014;2:89–128. [Google Scholar]
- 6.Liu XF, Long HJ, Miao XY, Liu GL, Yao HL. Fisetin inhibits liver cancer growth in a mouse model: Relation to dopamine receptor. Oncol Rep. 2017;38:53–62. doi: 10.3892/or.2017.5676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Khan N, Asim M, Afaq F, Abu Zaid M, Mukhtar H. A novel dietary flavonoid fisetin inhibits androgen receptor signaling and tumor growth in athymic nude mice. Cancer Res. 2008;68:8555–8563. doi: 10.1158/0008-5472.CAN-08-0240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Khan N, Syed DN, Ahmad N, Mukhtar H. Fisetin: A dietary antioxidant for health promotion. Antioxid Redox Signal. 2013;19:151–162. doi: 10.1089/ars.2012.4901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Syed DN, Adhami VM, Khan N, Khan MI, Mukhtar H. Exploring the molecular targets of dietary flavonoid fisetin in cancer. Semin Cancer Biol. 2016;40-41:130–140. doi: 10.1016/j.semcancer.2016.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mukhtar E, Adhami VM, Sechi M, Mukhtar H. Dietary flavonoid fisetin binds to β-tubulin and disrupts microtubule dynamics in prostate cancer cells. Cancer Lett. 2015;367:173–183. doi: 10.1016/j.canlet.2015.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Suh Y, Afaq F, Khan N, Johnson JJ, Khusro FH, Mukhtar H. Fisetin induces autophagic cell death through suppression of mTOR signaling pathway in prostate cancer cells. Carcinogenesis. 2010;31:1424–1433. doi: 10.1093/carcin/bgq115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jia S, Xu X, Zhou S, Chen Y, Ding G, Cao L. Fisetin induces autophagy in pancreatic cancer cells via endoplasmic reticulum stress-and mitochondrial stress-dependent pathways. Cell Death Dis. 2019;10:142. doi: 10.1038/s41419-019-1366-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Salmela AL, Pouwels J, Varis A, Kukkonen AM, Toivonen P, Halonen PK, Perälä M, Kallioniemi O, Gorbsky GJ, Kallio MJ. Dietary flavonoid fisetin induces a forced exit from mitosis by targeting the mitotic spindle checkpoint. Carcinogenesis. 2009;30:1032–1040. doi: 10.1093/carcin/bgp101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kang KA, Piao MJ, Madduma Hewage SR, Ryu YS, Oh MC, Kwon TK, Chae S, Hyun JW. Fisetin induces apoptosis and endoplasmic reticulum stress in human non-small cell lung cancer through inhibition of the MAPK signaling pathway. Tumor Biol. 2016;37:9615–9624. doi: 10.1007/s13277-016-4864-x. [DOI] [PubMed] [Google Scholar]
- 15.Su CH, Kuo CL, Lu KW, Yu FS, Ma YS, Yang JL, Chu YL, Chueh FS, Liu KC, Chung JG. Fisetin-induced apoptosis of human oral cancer SCC-4 cells through reactive oxygen species production, endoplasmic reticulum stress, caspase-, and mitochondria-dependent signaling pathways. Environ Toxicol. 2017;32:1725–1741. doi: 10.1002/tox.22396. [DOI] [PubMed] [Google Scholar]
- 16.Khan N, Afaq F, Syed DN, Mukhtar H. Fisetin, a novel dietary flavonoid, causes apoptosis and cell cycle arrest in human prostate cancer LNCaP cells. Carcinogenesis. 2008;29:1049–1056. doi: 10.1093/carcin/bgn078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee WR, Shen SC, Lin HY, Hou WC, Yang LL, Chen YC. Wogonin and fisetin induce apoptosis in human promyelo-leukemic cells, accompanied by a decrease of reactive oxygen species, and activation of caspase 3 and Ca(2+)-dependent endo-nuclease. Biochem Pharmacol. 2002;63:225–236. doi: 10.1016/S0006-2952(01)00876-0. [DOI] [PubMed] [Google Scholar]
- 18.Chen YC, Shen SC, Lee WR, Lin HY, Ko CH, Shih CM, Yang LL. Wogonin and fisetin induction of apoptosis through activation of caspase 3 cascade and alternative expression of p21 protein in hepatocellular carcinoma cells SK-HEP-1. Arch Toxicol. 2002;76:351–359. doi: 10.1007/s00204-002-0346-6. [DOI] [PubMed] [Google Scholar]
- 19.Jang KY, Jeong SJ, Kim SH, Jung JH, Kim JH, Koh W, Chen CY, Kim SH. Activation of reactive oxygen species/AMP activated protein kinase signaling mediates fisetin-induced apoptosis in multiple myeloma U266 cells. Cancer Lett. 2012;319:197–202. doi: 10.1016/j.canlet.2012.01.008. [DOI] [PubMed] [Google Scholar]
- 20.Kim JA, Lee S, Kim DE, Kim M, Kwon BM, Han DC. Fisetin, a dietary flavonoid, induces apoptosis of cancer cells by inhibiting HSF1 activity through blocking its binding to the hsp70 promoter. Carcinogenesis. 2015;36:696–706. doi: 10.1093/carcin/bgv045. [DOI] [PubMed] [Google Scholar]
- 21.Sung B, Pandey MK, Aggarwal BB. Fisetin, an inhibitor of cyclin-dependent kinase 6, down-regulates nuclear factor-kappaB-regulated cell proliferation, antiapoptotic and metastatic gene products through the suppression of TAK-1 and receptor-interacting protein-regulated IkappaBalpha kinase activation. Mol Pharmacol. 2007;71:1703–1714. doi: 10.1124/mol.107.034512. [DOI] [PubMed] [Google Scholar]
- 22.Li J, Cheng Y, Qu W, Sun Y, Wang Z, Wang H, Tian B. Fisetin, a dietary flavonoid, induces cell cycle arrest and apoptosis through activation of p53 and inhibition of NF-kappa B pathways in bladder cancer cells. Basic Clin Pharmacol Toxicol. 2011;108:84–93. doi: 10.1111/j.1742-7843.2010.00613.x. [DOI] [PubMed] [Google Scholar]
- 23.Ash D, Subramanian M, Surolia A, Shaha C. Nitric oxide is the key mediator of death induced by fisetin in human acute monocytic leukemia cells. Am J Cancer Res. 2015;5:481–497. [PMC free article] [PubMed] [Google Scholar]
- 24.Adan A, Baran Y. Fisetin and hesperetin induced apoptosis and cell cycle arrest in chronic myeloid leukemia cells accompanied by modulation of cellular signaling. Tumor Biol. 2016;37:5781–5795. doi: 10.1007/s13277-015-4118-3. [DOI] [PubMed] [Google Scholar]
- 25.Adan A, Baran Y. The pleiotropic effects of fisetin and hesperetin on human acute promyelocytic leukemia cells are mediated through apoptosis, cell cycle arrest, and alterations in signaling networks. Tumor Biol. 2015;36:8973–8984. doi: 10.1007/s13277-015-3597-6. [DOI] [PubMed] [Google Scholar]
- 26.Syed DN, Lall RK, Chamcheu JC, Haidar O, Mukhtar H. Involvement of ER stress and activation of apoptotic pathways in fisetin induced cytotoxicity in human melanoma. Arch Biochem Biophys. 2014;563:108–117. doi: 10.1016/j.abb.2014.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Smith ML, Murphy K, Doucette CD, Greenshields AL, Hoskin DW. The dietary flavonoid fisetin causes cell cycle arrest, caspase-dependent apoptosis, and enhanced cytotoxicity of chemotherapeutic drugs in triple-negative breast cancer cells. J Cell Biochem. 2016;117:1913–1925. doi: 10.1002/jcb.25490. [DOI] [PubMed] [Google Scholar]
- 28.Murtaza I, Adhami VM, Hafeez BB, Saleem M, Mukhtar H. Fisetin, a natural flavonoid, targets chemoresistant human pancreatic cancer AsPC-1 cells through DR3-mediated inhibition of NF-kappaB. Int J Cancer. 2009;125:2465–2473. doi: 10.1002/ijc.24628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ying TH, Yang SF, Tsai SJ, Hsieh SC, Huang YC, Bau DT, Hsieh YH. Fisetin induces apoptosis in human cervical cancer HeLa cells through ERK1/2-mediated activation of caspase-8-/caspase-3-dependent pathway. Arch Toxicol. 2012;86:263–273. doi: 10.1007/s00204-011-0754-6. [DOI] [PubMed] [Google Scholar]
- 30.Lim DY, Park JH. Induction of p53 contributes to apoptosis of HCT-116 human colon cancer cells induced by the dietary compound fisetin. Am J Physiol Gastrointest Liver Physiol. 2009;296:G1060–G1068. doi: 10.1152/ajpgi.90490.2008. [DOI] [PubMed] [Google Scholar]
- 31.Suh Y, Afaq F, Johnson JJ, Mukhtar H. A plant flavonoid fisetin induces apoptosis in colon cancer cells by inhibition of COX2 and Wnt/EGFR/NF-kappaB-signaling pathways. Carcinogenesis. 2009;30:300–307. doi: 10.1093/carcin/bgn269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pal HC, Sharma S, Elmets CA, Athar M, Afaq F. Fisetin inhibits growth, induces G2/M arrest and apoptosis of human epidermoid carcinoma A431 cells: Role of mitochondrial membrane potential disruption and consequent caspases activation. Exp Dermatol. 2013;22:470–475. doi: 10.1111/exd.12181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang XJ, Jia SS. Fisetin inhibits laryngeal carcinoma through regulation of AKT/NF-κB/mTOR and ERK1/2 signaling pathways. Biomed Pharmacother. 2016;83:1164–1174. doi: 10.1016/j.biopha.2016.08.035. [DOI] [PubMed] [Google Scholar]
- 34.Li YS, Qin XJ, Dai W. Fisetin suppresses malignant proliferation in human oral squamous cell carcinoma through inhibition of Met/Src signaling pathways. Am J Transl Res. 2017;9:5678–5683. [PMC free article] [PubMed] [Google Scholar]
- 35.Shih YL, Hung FM, Lee CH, Yeh MY, Lee MH, Lu HF, Chen YL, Liu JY, Chung JG. Fisetin induces apoptosis of HSC3 human oral cancer cells through endoplasmic reticulum stress and dysfunction of mitochondria-mediated signaling pathways. In Vivo. 2017;31:1103–1114. doi: 10.21873/invivo.11176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sowa M, Slepokura K, Matczak-Jon E. A 1:2 cocrystal of genistein with isonicotinamide: Crystal structure and Hirshfeld surface analysis. Acta Crystallogr C. 2013;69:1267–1272. doi: 10.1107/S0108270113029545. [DOI] [PubMed] [Google Scholar]
- 37.Sowa M, Slepokura K, Matczak-Jon E. Improving solubility of fisetin by cocrystallization. Cryst Eng Comm. 2014;16:10592. doi: 10.1039/C4CE01713G. [DOI] [Google Scholar]
- 38.Kadari A, Gudem S, Kulhari H, Bhandi MM, Borkar RM, Kolapalli VR, Sistla R. Enhanced oral bioavailability and anticancer efficacy of fisetin by encapsulating as inclusion complex with HPβCD in polymeric nanoparticles. Drug Deliv. 2017;24:224–232. doi: 10.1080/10717544.2016.1245366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ragelle H, Crauste-Manciet S, Seguin J, Brossard D, Scherman D, Arnaud P, Chabot GG. Nanoemulsion formulation of fisetin improves bioavailability and antitumour activity in mice. Int J Pharm. 2012;427:452–459. doi: 10.1016/j.ijpharm.2012.02.025. [DOI] [PubMed] [Google Scholar]
- 40.Smith MA, Houghton P. A proposal regarding reporting of in vitro testing results. Clin Cancer Res. 2013;19:2828–2833. doi: 10.1158/1078-0432.CCR-13-0043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Klimaszewska-Wisniewska A, Halas-Wisniewska M, Tadrowski T, Gagat M, Grzanka D, Grzanka A. Paclitaxel and the dietary flavonoid fisetin: A synergistic combination that induces mitotic catastrophe and autophagic cell death in A549 non-small cell lung cancer cells. Cancer Cell Int. 2016;16:10. doi: 10.1186/s12935-016-0288-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Klimaszewska-Wiśniewska A, Hałas-Wiśniewska M, Grzanka A, Grzanka D. Evaluation of anti-metastatic potential of the combination of fisetin with paclitaxel on A549 non-small cell lung cancer cells. Int J Mol Sci. 2018;19:E661. doi: 10.3390/ijms19030661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tripathi R, Samadder T, Gupta S, Surolia A, Shaha C. Anticancer activity of a combination of cisplatin and fisetin in embryonal carcinoma cells and xenograft tumors. Mol Cancer Ther. 2011;10:255–268. doi: 10.1158/1535-7163.MCT-10-0606. [DOI] [PubMed] [Google Scholar]
- 44.Khan N, Afaq F, Khusro FH, Mustafa Adhami V, Suh Y, Mukhtar H. Dual inhibition of phosphatidylinositol 3-kinase/Akt and mammalian target of rapamycin signaling in human nonsmall cell lung cancer cells by a dietary flavonoid fisetin. Int J Cancer. 2012;130:1695–1705. doi: 10.1002/ijc.26178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Atashrazm F, Lowenthal RM, Dickinson JL, Holloway AF, Woods GM. Fucoidan enhances the therapeutic potential of arsenic trioxide and all-trans retinoic acid in acute promyelocytic leukemia, in vitro and in vivo. Oncotarget. 2016;19:46028–46041. doi: 10.18632/oncotarget.10016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Moloudi K, Neshasteriz A, Hosseini A, Eyvazzadeh N, Shomali M, Eynali S, Mirzaei E, Azarnezhad A. Synergistic effects of arsenic trioxide and radiation: Triggering the intrinsic pathway of apoptosis. Iran Biomed J. 2017;21:330–337. doi: 10.18869/acadpub.ibj.21.5.330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bandaruk Y, Mukai R, Terao J. Cellular uptake of quercetin and luteolin and their effects on monoamine oxidase-A in human neuroblastoma SH-SY5Y cells. Toxicol Rep. 2014;1:639–649. doi: 10.1016/j.toxrep.2014.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shia CS, Tsai SY, Kuo SC, Hou YC, Chao PD. Metabolism and pharmacokinetics of 3,3′,4′,7-tetrahydroxyflavone (fisetin), 5-hydroxyflavone, and 7-hydroxyflavone and antihemolysis effects of fisetin and its serum metabolites. J Agric Food Chem. 2009;57:83–89. doi: 10.1021/jf802378q. [DOI] [PubMed] [Google Scholar]
- 49.Chou TC, Talalay P. Quantitative analysis of dose-effect relationships: The combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul. 1984;22:27–55. doi: 10.1016/0065-2571(84)90007-4. [DOI] [PubMed] [Google Scholar]
- 50.Chou TC. Theoretical basis, experimental design, and comput-erized simulation of synergism and antagonism in drug combination studies. Pharmacol Rev. 2006;58:621–681. doi: 10.1124/pr.58.3.10. [DOI] [PubMed] [Google Scholar]
- 51.Chou TC. Preclinical versus clinical drug combination studies. Leuk Lymphoma. 2008;49:2059–2080. doi: 10.1080/10428190802353591. [DOI] [PubMed] [Google Scholar]
- 52.Gagat M, Grzanka D, Izdebska M, Grzanka A. Effect of L-homocysteine on endothelial cell-cell junctions following F-actin stabilization through tropomyosin-1 overexpression. Int J Mol Med. 2013;32:115–129. doi: 10.3892/ijmm.2013.1357. [DOI] [PubMed] [Google Scholar]
- 53.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 54.Shim MJ, Kim HJ, Yang SJ, Lee IS, Choi HI, Kim T. Arsenic trioxide induces apoptosis in chronic myelogenous leukemia K562 cells: Possible involvement of p38 MAP kinase. J Biochem Mol Biol. 2011;35:377–383. doi: 10.5483/bmbrep.2002.35.4.377. [DOI] [PubMed] [Google Scholar]
- 55.Song LL, Tu YY, Xia L, Wang WW, Wei W, Ma CM, Wen DH, Lei H, Xu HZ, Wu YL. Targeting catalase but not peroxiredoxins enhances arsenic trioxide-induced apoptosis in K562 cells. PLoS One. 2014;9:e104985. doi: 10.1371/journal.pone.0104985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhu XH, Shen YL, Jing YK, Cai X, Jia PM, Huang Y, Tang W, Shi GY, Sun YP, Dai J, et al. Apoptosis and growth inhibition in malignant lymphocytes after treatment with arsenic trioxide at clinically achievable concentrations. J Natl Cancer Inst. 1999;91:772–778. doi: 10.1093/jnci/91.9.772. [DOI] [PubMed] [Google Scholar]
- 57.Larson RA. Is there a best TKI for chronic phase CML? Blood. 2015;126:2370–2375. doi: 10.1182/blood-2015-06-641043. [DOI] [PubMed] [Google Scholar]
- 58.Santos FP, Ravandi F. Advances in treatment of chronic myelogenous leukemia new treatment options with tyrosine kinase inhibitors. Leuk Lymphoma. 2009;50(Suppl 2):S16–S26. doi: 10.3109/10428190903383427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cui S, Wang J, Wu Q, Qian J, Yang C, Bo P. Genistein inhibits the growth and regulates the migration and invasion abilities of melanoma cells via the FAK/paxillin and MAPK pathways. Oncotarget. 2017;8:21674–21691. doi: 10.18632/oncotarget.15535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chen HH, Chen SP, Zheng QL, Nie SP, Li WJ, Hu XJ, Xie MY. Genistein promotes proliferation of human cervical cancer cells through estrogen receptor-mediated PI3K/Akt-NF-κB pathway. J Cancer. 2018;9:288–295. doi: 10.7150/jca.20499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Guo JM, Xiao BX, Liu DH, Grant M, Zhang S, Lai YF, Guo YB, Liu Q. Biphasic effect of daidzein on cell growth of human colon cancer cells. Food Chem Toxicol. 2004;42:1641–1646. doi: 10.1016/j.fct.2004.06.001. [DOI] [PubMed] [Google Scholar]
- 62.Fogarty CE, Bergmann A. Killers creating new life: Caspases drive apoptosis-induced proliferation in tissue repair and disease. Cell Death Differ. 2017;24:1390–1400. doi: 10.1038/cdd.2017.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Rudrapatna VA, Bangi E, Cagan RL. Caspase signalling in the absence of apoptosis drives JNK-dependent invasion. EMBO Rep. 2013;14:172–177. doi: 10.1038/embor.2012.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ryoo HD, Bergmann A. The role of apoptosis-induced proliferation for regeneration and cancer. Cold Spring Harb Perspect Biol. 2012;4:a008797. doi: 10.1101/cshperspect.a008797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gdynia G, Grund K, Eckert A, Böck BC, Funke B, Macher-Goeppinger S, Sieber S, Herold-Mende C, Wiestler B, Wiestler OD, Roth W. Basal caspase activity promotes migration and invasiveness in glioblastomacells. Mol Cancer Res. 2007;5:1232–1240. doi: 10.1158/1541-7786.MCR-07-0343. [DOI] [PubMed] [Google Scholar]
- 66.Mukai M, Kusama T, Hamanaka Y, Koga T, Endo H, Tatsuta M, Inoue M. Cross talk between apoptosis and invasion signaling in cancer cells through caspase-3 activation. Cancer Res. 2005;65:9121–9125. doi: 10.1158/0008-5472.CAN-04-4344. [DOI] [PubMed] [Google Scholar]
- 67.Portela M, Richardson HE. Death takes a holiday-non-apoptotic role for caspases in cell migration and invasion. EMBO Rep. 2013;14:107–108. doi: 10.1038/embor.2012.224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Li F, Huang Q, Chen J, Peng Y, Roop DR, Bedford JS, Li CY. Apoptotic cells activate the 'phoenix rising' pathway to promote wound healing and tissue regeneration. Sci Signal. 2010;3:ra13. doi: 10.1126/scisignal.2000634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Huang Q, Li F, Liu X, Li W, Shi W, Liu F, O'Sullivan B, He Z, Peng Y, Tan AC, et al. Caspase 3-mediated stimulation of tumor cell repopulation during cancer radiotherapy. Nat Med. 2011;17:860–866. doi: 10.1038/nm.2385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Donato AL, Huang Q, Liu X, Li F, Zimmerman MA, Li CY. Caspase 3 promotes surviving melanoma tumor cell growth after cytotoxic therapy. J Invest Dermatol. 2014;134:1686–1692. doi: 10.1038/jid.2014.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Cheng J, Tian L, Ma J, Gong Y, Zhang Z, Chen Z, Xu B, Xiong H, Li C, Huang Q. Dying tumor cells stimulate proliferation of living tumor cells via caspase-dependent protein kinase Cδ activation in pancreatic ductal adenocarcinoma. Mol Oncol. 2015;9:105–114. doi: 10.1016/j.molonc.2014.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Cheng J, He S, Wang M, Zhou L, Zhang Z, Feng X, Yu Y, Ma J, Dai C, Zhang S, et al. The caspase-3/PKCδ/Akt/VEGF-A signaling pathway mediates tumor repopulation during radiotherapy. Clin Cancer Res. 2019;25:3732–3743. doi: 10.1158/1078-0432.CCR-18-3001. [DOI] [PubMed] [Google Scholar]
- 73.Stevenson DE, Cooney JM, Jensen DJ, Wibisono R, Adaim A, Skinner MA, Zhang J. Comparison of enzymically gluc-uronidated flavonoids with flavonoid aglycones in an in vitro cellular model of oxidative stress protection. In Vitro Cell Dev Biol Anim. 2008;44:73–80. doi: 10.1007/s11626-007-9072-y. [DOI] [PubMed] [Google Scholar]
- 74.Zeng M, Sun R, Basu S, Ma Y, Ge S, Yin T, Gao S, Zhang J, Hu M. Disposition of flavonoids via recycling: Direct biliary excretion of enterically or extrahepatically derived flavonoid glucuronides. Mol Nutr Food Res. 2016;60:1006–1019. doi: 10.1002/mnfr.201500692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sak K. In vitro cytotoxic activity of flavonoids on human ovarian cancer cell lines. Cancer Sci Res. 2015;2:1–13. doi: 10.15226/csroa.2015.00112. [DOI] [Google Scholar]
- 76.Volk-Draper L, Hall K, Griggs C, Rajput S, Kohio P, DeNardo D, Ran S. Paclitaxel therapy promotes breast cancer metastasis in a TLR4-dependent manner. Cancer Res. 2014;74:5421–5434. doi: 10.1158/0008-5472.CAN-14-0067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Daenen LG, Roodhart JM, van Amersfoort M, Dehnad M, Roessingh W, Ulfman LH, Derksen PW, Voest EE. Chemotherapy enhances metastasis formation via VEGFR-1-expressing endothelial cells. Cancer Res. 2011;71:6976–6985. doi: 10.1158/0008-5472.CAN-11-0627. [DOI] [PubMed] [Google Scholar]
- 78.Ovcharenko A, Granot G, Rokah OH, Park J, Shpilberg O, Raanani P. Enhanced adhesion/migration and induction of Pyk2 expression in K562 cells following imatinib exposure. Leuk Res. 2013;37:1729–1736. doi: 10.1016/j.leukres.2013.10.005. [DOI] [PubMed] [Google Scholar]
- 79.Molli PR, Pradhan MB, Advani SH, Naik NR. RhoA: A therapeutic target for chronic myeloid leukemia. Mol Cancer. 2012;11:16. doi: 10.1186/1476-4598-11-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Qi P, Tang R, Liu F, Zhou J. MicroRNA-186 regulates cell malignancy by targeting ROCK1 in chronic myeloid leukemia. Int J Clin Exp Pathol. 2017;10:6290–6298. [Google Scholar]
- 81.Martini M, DeSantis MC, Braccini L, Gulluni F, Hirsch E. PI3K/AKT signaling pathway and cancer: An updated review. Ann Med. 2014;46:372–383. doi: 10.3109/07853890.2014.912836. [DOI] [PubMed] [Google Scholar]
- 82.Mirabilii S, Ricciardi MR, Piedimonte M, Gianfelici V, Bianchi MP, Tafuri A. Biological aspects of mTOR in leukemia. Int J Mol Sci. 2018;19:E2396. doi: 10.3390/ijms19082396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Xu H, Tian Y, Yuan X, Wu H, Liu Q, Pestell RG, Wu K. The role of CD44 in epithelial-mesenchymal transition and cancer development. Onco Targets Ther. 2015;8:3783–3792. doi: 10.2147/OTT.S95470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Chang G, Zhang H, Wang J, Zhang Y, Xu H, Wang C, Zhang H, Ma L, Li Q, Pang T. CD44 targets Wnt/β-catenin pathway to mediate the proliferation of K562 cells. Cancer Cell Int. 2013;13:117. doi: 10.1186/1475-2867-13-117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Chou YS, Yang MH. Epithelial-mesenchymal transition-related factors in solid tumor and hematological malignancy. J Chinese Med Assoc. 2015;78:438–445. doi: 10.1016/j.jcma.2015.05.002. [DOI] [PubMed] [Google Scholar]
- 86.Chen SC, Liao TT, Yang MH. Emerging roles of epithelial-mesenchymal transition in hematological malignancies. J Biomed Sci. 2018;25:37. doi: 10.1186/s12929-018-0440-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kahlert UD, Joseph JV, Kruyt FAE. EMT- and MET-related processes in nonepithelial tumors: Importance for disease progression, prognosis, and therapeutic opportunities. Mol Oncol. 2017;11:860–877. doi: 10.1002/1878-0261.12085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Wang N, Guo D, Zhao YY, Dong CY, Liu XY, Yang BX, Wang SW, Wang L, Liu QG, Ren Q, et al. TWIST-1 promotes cell growth, drug resistance and progenitor clonogenic capacities in myeloid leukemia and is a novel poor prognostic factor in acute myeloid leukemia. Oncotarget. 2015;6:20977–20992. doi: 10.18632/oncotarget.4007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Cosset E, Hamdan G, Jeanpierre S, Voeltzel T, Sagorny K, Hayette S, Mahon FX, Dumontet C, Puisieux A, Nicolini FE, Maguer-Satta V. Deregulation of TWIST-1 in the CD34+ compartment represents a novel prognostic factor in chronic myeloid leukemia. Blood. 2011;117:1673–1676. doi: 10.1182/blood-2009-11-254680. [DOI] [PubMed] [Google Scholar]
- 90.Kidan N, Khamaisie H, Ruimi N, Roitman S, Eshel E, Dally N, Ruthardt M, Mahajna J. Ectopic expression of Snail and Twist in Ph+ Leukemia cells upregulates CD44 expression and alters their differentiation potential. J Cancer. 2017;8:3952–3968. doi: 10.7150/jca.19633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Touil YS, Seguin J, Scherman D, Chabot GG. Improved antiangiogenic and antitumour activity of the combination of the natural flavonoid fisetin and cyclophosphamide in Lewis lung carcinoma-bearing mice. Cancer Chemother Pharmacol. 2011;68:445–455. doi: 10.1007/s00280-010-1505-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.O'Dwyer ME, La Rosée P, Nimmanapalli R, Bhalla KN, Druker BJ. Recent advances in Philadelphia chromosome-positive malignancies: The potential role of arsenic trioxide. Semin Hematol. 2002;39(2 Suppl):S18–S21. doi: 10.1053/shem.2002.33612. [DOI] [PubMed] [Google Scholar]
- 93.O'Dwyer M. Multifaceted approach to the treatment of bcrabl-positive leukemias. Oncologist. 2002;7(Suppl 1):S30–S38. doi: 10.1634/theoncologist.7-suppl_1-30. [DOI] [PubMed] [Google Scholar]
- 94.Potin S, Bertoglio J, Bréard J. Involvement of a Rho-ROCK-JNK pathway in arsenic trioxide-induced apoptosis in chronic myelogenous leukemia cells. FEBS Lett. 2007;581:118–124. doi: 10.1016/j.febslet.2006.12.016. [DOI] [PubMed] [Google Scholar]
- 95.Yan H, Wang YC, Li D, Wang Y, Liu W, Wu YL, Chen GQ. Arsenic trioxide and proteasome inhibitor bortezomib synergis-tically induce apoptosis in leukemic cells: The role of protein kinase Cdelta. Leukemia. 2007;27:1488–1495. doi: 10.1038/sj.leu.2404735. [DOI] [PubMed] [Google Scholar]
- 96.El Eit RM, Iskandarani AN, Saliba JL, Jabbour MN, Mahfouz RA, Bitar NM, Ayoubi HR, Zaatari GS, Mahon FX, De Thé HB, et al. Effective targeting of chronic myeloid leukemia initiating activity with the combination of arsenic trioxide and interferon alpha. Int J Cancer. 2014;134:988–996. doi: 10.1002/ijc.28427. [DOI] [PubMed] [Google Scholar]
- 97.Xu W, Wei W, Yu Q, Wu C, Ye C, Wu Y, Yan H. Arsenic trioxide and bortezomib interact synergistically to induce apoptosis in chronic myelogenous leukemia cells resistant to imatinibmesylate through Bcr/Abl-dependent mechanisms. Mol Med Rep. 2014;10:1519–1524. doi: 10.3892/mmr.2014.2333. [DOI] [PubMed] [Google Scholar]
- 98.La Rosée P, Johnson K, O'Dwyer ME, Druker BJ. In vitro studies of the combination of imatinib mesylate (Gleevec) and arsenic trioxide (Trisenox) in chronic myelogenous leukemia. Exp Hematol. 2002;30:729–737. doi: 10.1016/S0301-472X(02)00836-6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.