

Abstract
Rationale:
Congenital glucose–galactose malabsorption (CGGM) is a rare, autosomal recessive, hereditary disease that usuallypresents in newborns. CGGM manifests as severe diarrhea, hyperosmolar dehydration, and malnutrition. It does not respond to routine treatment and often is life-threatening.
Patient concerns:
We described a Chinese infant girl with refractory diarrhea, who suffered from severe dehydration and malnutrition even if with fluid replacement therapy and fed with several special formulas.
Diagnoses:
The genetic analysis identified CGGM with SLC5A1 mutations. c.1436G > C (p.R479T) was a novel mutation.
Interventions:
The patient was managed by free-glucose and galactose formula, and then special low-carbohydrate dietary therapy.
Outcomes:
The patient improved immediately after starting a free-glucose and galactose formula, and kept healthy with special low-carbohydrate diet. She had been followed up with nutritional management for 20 months.
Lessons:
This report highlights the importance of differential diagnosis of congenital diarrhea and enteropathies. For CGGM, free-glucose and galactose milk powder was the most effective treatment. Low-carbohydrate diet gradually introduced was still a great challenge that requires continuing guidance from child nutritionists and dietitians. Long-term nutrition management was extremely important to ensure the normal growth and development of children.
Keywords: congenital glucose-galactose malabsorption, free-glucose and galactose formula, low-carbohydrate diet, nutrition management, SLC5A1
1. Introduction
Congenital glucose–galactose malabsorption (CGGM) is a genetic disease. Because the SLC5A1 mutation causes the structural and functional deletion of the sodium-dependent glucose cotransporter-1 (SGLT-1) in the intestinal mucosa, glucose and galactose are unable to be absorbed by the intestine, resulting in a series of clinical manifestations.[1] To date, >40 SLC5A1 mutations have been identified in CGGM patients.[2,3] Additionally, CGGM is extremely rare and only approximately 300 cases have been reported worldwide.[4] The incidence of CGGM varies in different populations, and consanguineous correlations are evident in some areas, which confirms the autosomal recessive heredity pattern of CGGM.[2,3,4,5] There is no specific therapy for CGGM, and the most effective treatment is long-term special dietary therapy that avoids foods containing glucose and galactose.[3,5,6] However, maintaining normal physical and neurological development of CGGM children is still very challenging, especially the long-term nutrition management. Thus far, few relevant studies have been reported.[7]
2. Methods and materials:
2.1. Ethics statement
The study was approved by the Children's Hospital of Zhejiang University Ethics Committee. The parent of the patient provided informed consent for publication of the case.
2.2. Case report
A 16-day-old female infant was admitted to our hospital because of repeated diarrhea for 14 days. She experienced diarrhea 8 to 10 times per day with voluminous, yellow, watery stools, without blood or mucus. She had no fever and vomiting. Diarrhea was not improved after the treatment with lactose-free cow's milk powder. Her weight was 370 g less than her birth weight. Her antenatal and natal histories were uneventful. She was breast-fed after birth. The parents were healthy and denied intermarriage and a family history of genetic diseases.
The physical examination findings were: weight, 3.58 kg (weight-for-age [WFA]: −0.10 SD); length, 53 cm (Length-for-age [LFA]: 0.63 SD); head circumference, 31 cm (head circumference-for-age [HCFA]: −3.84 SD) (Fig. 1).[8] Her fontanelle was slightly concave. Otherwise, the examination was normal. Laboratory finding were: the blood pH, 7.32; sodium, 164 mmol/L; chlorine, 140 mmol/L; HCO3-, 14 mmol/L; actual base excess, −10.3; blood urea nitrogen 10.8 mmol/L; others were normal. Admission diagnosis included persistent neonatal diarrhea, mild hypertonic dehydration, and metabolic acidosis.
Figure 1.
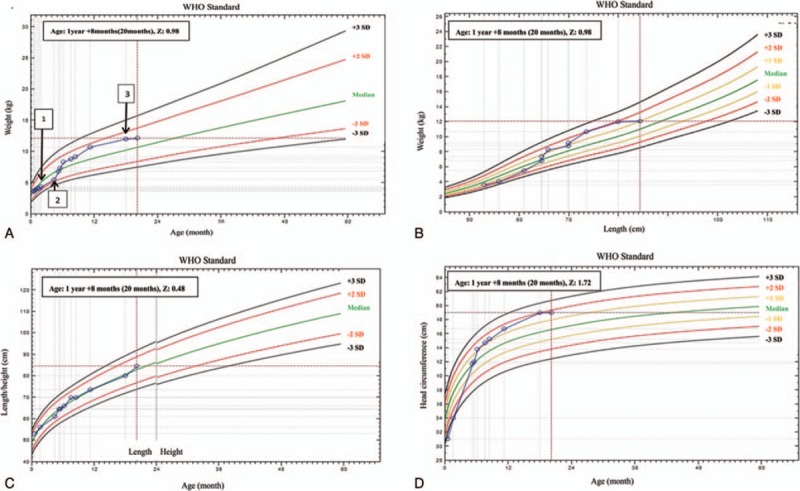
The patient's growth curve (calculated using WHO software www.who.int/childgrowth/software). (A) Weight-for-age (WFA) curve: 1. at discharge; 2. at the beginning of taking Galatomin 19 formula; 3. at the age of 18 months. (B) Weight-for-length (WFL) curve. (C) Length-for-age (LFA) curve. (D) Head circumference-for-age (HCFA) curve.
After admission treatment included acidosis correction and fluid replacement, but watery diarrhea persisted. The infant was fed with a variety of formulas including lactose-free cow's milk formula (Similac Lactose Free, Abbott), amino acid formula (Neocate, Nutricia, the Netherlands), and extensively hydrolyzed formula (Pepti Junior, Nutricia, the Netherlands), but the stool weight was still 350 to 600 g/day. Blood Na+ was 161 to 178 mmol/L. Her weight gained slowly despite adequate intake. At 30 days after admission, the nutrition assessment indicated moderate malnutrition. Owing to the poor gastrointestinal absorption, temporary fasting was recommended and total parenteral nutrition was administered. The goals were: calories 75 kcal/kg/day[9] and protein 2.5 g/kg/day.[10] After fasting, the stool decreased significantly (0–30 g/day). As the intake of enteral nutrition (EN) of Pepti Junior formula (Nutricia, the Netherlands) increased, the stool weight increased. According to the clinical manifestations,[11,12] galactose malabsorption could not be excluded. Then, she was fed with soy-based lactose-free formula (Similac Soy Isomil, Abbott). Fortunately, the diarrhea symptoms did not worsen significantly, and no hyperosmolar dehydration occurred. The volume of the formula was increased to 780 mL/day (calories provided by EN reached 130 kcal/kg/day), but the stool was only 50 to 100 g/day and the weight gain increased from 4.03 to 4.33 kg (Fig. 1). She was discharged 44 days after admission and was followed in the Clinical Nutrition Outpatient every 2 to 4 weeks. At about 1 month after discharge, the genetic tests (completed by Medical Testing Center of My Genostics) showed that the patient had heterozygous mutations at 2 loci of the SLC5A1 gene on chromosome 22, exos 11, and exos 12. They were, c.1135C > T (p.R379X) and c.1436G > C (p.R479T) (Fig. 2), which indicates congenital glucose–galactose malabsorption (OMIM 606824). c.1436G > C (p.R479T) was a novel mutation.
Figure 2.
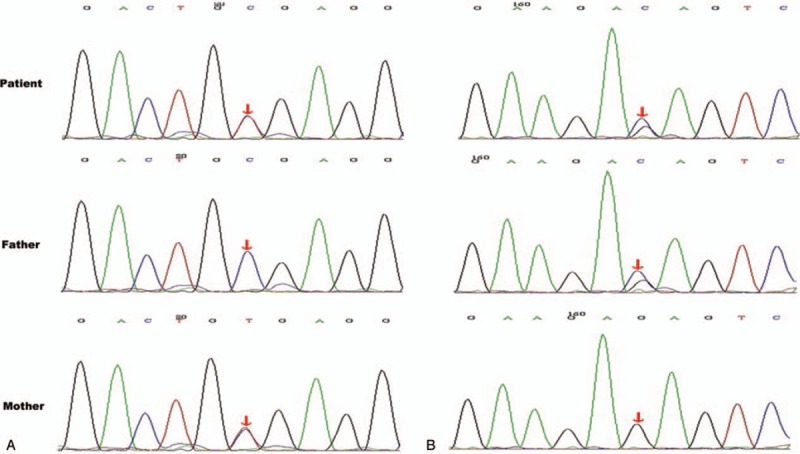
Genetic test report and mutation analysis was performed for the patient and her parents. The patient had heterozygous mutations at 2 loci of the SLC5A1 gene. (A) The mutation was located on chromosome 22, SLC5A1, exon11, c.1135C > T. It was from maternal heterozygous mutation. (B) The mutation was located on chromosome 22, SLC5A1, exon12, c.1436G > C. It was from paternal heterozygous mutation.
Owing to glucose and galactose malabsorption, the optimal formula is carbohydrate-free formula. Galactomin19, (Nutricia International Ltd., Oslo, Norway), which has a caloric density of 20 kcal/oz (fructose 6.3 g/100 mL, protein 1.9 g/100 mL, and fat 4 g/100 mL), was purchased from a foreign country[3]; it contained no glucose and lactose, and fructose served as the sole source of carbohydrates. After she began consuming Galactomin19, the stools immediately became formed with once daily. The weight gain indicated catch-up growth and reached a normal weight at 5 M15D old (the age of 5 months and 15 days). The laboratory examination showed: the serum levels of zinc (57.4 mmol/L) and 25(OH) VitD2 (42.5 nmol/L) were slightly low, whereas the results of liver and kidney function tests, blood lipid, electrolyte, and iron metabolism assays were normal. Subsequently, she was given zinc gluconate granules (2.5 mg, TID) and VitD drops (800 IU, QD). At 6 months of age, a low-carbohydrate diet such as vegetables and fruits was introduced first. Galactomin 19 was still the main source of nutrition. Each new food was tried for 5 to 7 days, and stools were closely monitored. She was found to tolerate apple, purple cabbage, carrot, zucchini, asparagus but not pear. Later, fruits and vegetables with a slightly higher sucrose content were introduced, including banana, peach, potato, small sweet green peas, and pumpkin, but fruit juice was restricted. The patient tolerated all these foods except pumpkin. At the age of 8 months and 10 months, pork and chicken and egg and plant oil were added respectively; after the age of 1 year, fish and shrimp were added. The introduction of these foods was well tolerated. The intake of Galactomin 19 was decreased to 500 mL/day, and the high-carbohydrate foods were added gradually, including rice paste and noodles. Since the age of 15 months, she could tolerate pumpkin but not corn. In addition, when she spontaneously chose to eat unsupervised foods with refined sugar, such as chocolate, biscuits, and cake, she immediately developed diarrhea. The results of several serum electrolyte assays after discharge were normal. At the age of 18 months, the nutritional assessment (Fig. 1) showed she was overweight (WFA: 1.27 SD). The results of laboratory examination indicated: elevated blood triglyceride 3.1 mmol/L. The results of 72-hour dietary assessment demonstrated: energy intake 1000 kcal/day, fat 50.7%, carbohydrates 25%, and protein 24.3%. Compared with the Chinese Dietary Reference Intakes, the patient had the high intake of fat but insufficient of carbohydrates. The nutritional plan was: reduce the intake of high-fat foods and saturated fatty acids; increase the consumption of complex carbohydrates, including rice, noodles, potato, and pumpkin; use honey as sweetener. The follow-up evaluation at 20 months of age showed: the nutritional assessment result was normal. She demonstrated age-appropriate developmental skills (timeline was in Fig. 3).
Figure 3.
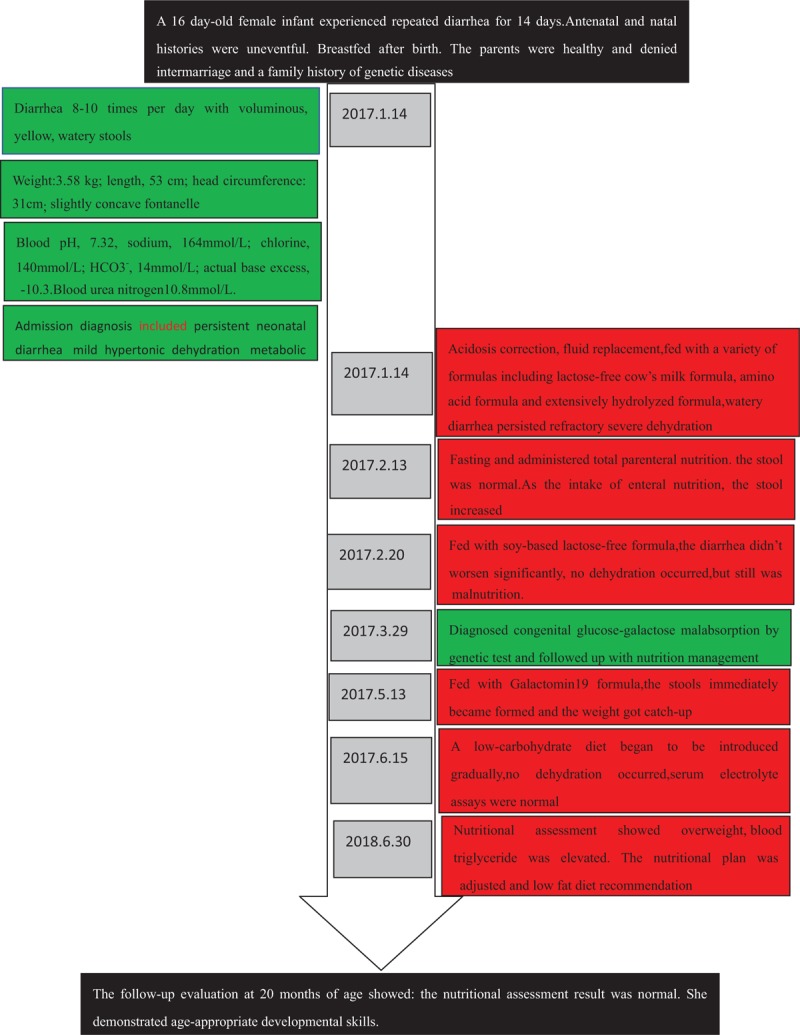
Timeline picture of the patient.
3. Discussion
CGGM is an autosomal, recessive hereditary disease caused by an abnormal SGLT-1 in the human intestinal mucosa. Its pathogenic gene, SLC5A1, is located on chromosome 22.[13] The secretion of water and electrolytes in the jejuna can be stimulated by the intake of osmotic substances, such as glucose, galactose. Normally, water enters the intestinal mucosa along with glucose and galactose via the sodium-coupled transporter.[14] However, because glucose and galactose cannot be absorbed in CGGM children, the large amount of unabsorbed water, electrolytes, and glucose that passes through the small intestine into the colon exceeds the absorption capacity of the intestinal tract and thus results in severe watery diarrhea.[4,15] The feces of the CGGM patients are acidic and contain a large amount of glucose, resulting in malnutrition and even death. However, the symptoms of abdominal distention, vomiting, and anorexia are usually uncommon.[1,7] Owing to long-term repeated dehydration, some CGGM children develop renal calculi, polyuria, renal tubular acidosis, rachitis, and gross hematuria.[1,5]
The most direct and effective treatment for CGGM is avoidance of foods containing glucose and galactose.[5] The currently available special carbohydrate-free formulas without glucose and galactose mainly include Ross carbohydrate-free formula (Abbott) and 3232A (Mead Johnson).[7] To meet the demand for energy and carbohydrates, fructose must be added to these 2 types of special formula. Because fructose enters into the blood via passive absorption and is not dependent on the SGLT-1, it can be used to replace glucose and galactose in foods. In addition, Galactomin 19 (Nutricia, Europe) formula powder contains fructose as a direct and sole source of carbohydrates. At present, such formula powder products are not marketed in China, so they need to be purchased from foreign countries.
Once the CGGM infants are older than 4 months, infant foods with a low content of carbohydrates can be added gradually to ensure their gastrointestinal tolerance.
The foods that should be avoided in the children diagnosed with CGGM include[5]: all kinds of milk, butter, yogurt, cheese, and other dairy products; glucose, maltodextrin, corn syrup, lactose, and stevia; sugar (sucrose), ice cream, all desserts, candies, chocolate. The foods that are permitted include: special formula without galactose and glucose; fructose, honey; all vegetables; all fruits; all meat, fish, and eggs; all legumes; all fat and oil; small amounts of rice, potatoes, bread, unsweetened cereal, wheat, quinoa.
The parents of CGGM children should monitor food labels and identify the foods containing glucose and lactose. The parents of CGGM children should monitor food labels and identify the foods containing glucose and lactose. At first, low-carbohydrate vegetables and fruits can be added with caution. Then, protein foods and a careful amount of carbohydrates can be gradually added to the diet with increasing age. The specialized carbohydrate-free formula plus fructose is still the main source of nutrition. The restriction of carbohydrate intake is adjusted on the basis of stool weight. Their weight and height should be closely monitored. Most CGGM children can achieve normal growth as well as the normal development of nervous system if they are provided careful nutritional management.[5]
A child newly diagnosed with CGGM is a challenge for primary physicians, pediatric nutritionists, and specialists. The child nutritionists play an extremely important role in providing the healthcare institutions’ and pediatricians’ recommendations for milk-substitute powders and special dietary supplements for CGGM children. The nutritionists can assist the treatment team of children with CGGM with the choice of appropriate liquid formula, fructose, and solid foods. Additionally, they can provide the parents or caregivers with the following guidance: how to add fructose to the special sugar-free formula powder to meet the recommended energy requirements, selection of vitamins and minerals, how to purchase the special formula powder, and how to introduce solid foods into the diet. The nutritional management of CGGM children is a long-term, difficult process. Because intolerance to glucose and galactose is expected to improve with age, patients with CGGM exhibit better prognosis if they are diagnosed and treated in the neonatal period.[5,7] However, their ongoing nutritional management is still a great challenge that requires continuing guidance from child nutritionists and dietitians.
Author contributions
Formal analysis: Ting Zhang, Qi Long, Mengshan Lu
Methodology: Ming Ma, Weiyan Wang, Fei Chen
Conceptualization: Ming Ma, Weiyan Wang.
Formal analysis: Qi Long, Fei Chen, Ting Zhang, Mengshan Lu.
Funding acquisition: Ming Ma.
Investigation: Qi Long, Fei Chen, Ting Zhang, Mengshan Lu.
Methodology: Ming Ma, Weiyan Wang.
Project administration: Ming Ma.
Supervision: Weiyan Wang, Lihua Chen.
Writing – original draft: Ming Ma, Qi Long.
Writing – review & editing: Ming Ma, Fei Chen, Lihua Chen.
Footnotes
Abbreviations: CGGM = congenital glucose-galactose malabsorption, EN = enteral nutrition, HCFA = head circumference-for-age, LFA = length-for-age, SGLT-1 = Sodium dependent glucose cotransporter-1, WFA = weight-for-age, WFL = weight-for-length.
Statement of Ethics approval and patient consent: The study was approved by the Children's Hospital of Zhejiang University ethics committee. The parent of the patient provided informed consent for publication of the case.
Statement of Non-duplication: No part of this article has been published or submitted elsewhere in any language, including the abstract.
De-identification: Patient information had been de-identified.
Consent for publication: Consent for publication was obtained from the participants.
This study was supported by funded by the national key research and development programme of China (2016YFC1305301) and Education Department Fund of Zhejiang Province, China (Y201635973).
The authors report no conflicts of interest.
References
- [1]. Assiri A, Saeed A, Alnimri A, et al. Five Arab children with glucose-galactose malabsorption. Paediatr Intern Child Health 2013;33:108–10. [DOI] [PubMed] [Google Scholar]
- [2]. Al-Suyufi Y, ALSaleem K, Al-Mehaidib A, et al. SLC5A1 mutations in saudi arabian patients with congenital glucose-galactose malabsorption. J Pediatr Gastroenterol Nutr 2018;66:250–2. [DOI] [PubMed] [Google Scholar]
- [3]. Saadah OI, Alghamdi SA, Sindi HH, et al. Congenital glucose-galactose malabsorption: a descriptive study of clinical characteristics and outcome from Western Saudi Arabia. Arab J Gastroenterol 2014;15:21–3. [DOI] [PubMed] [Google Scholar]
- [4]. Pode-Shakked B, Reish O, Aktuglu-Zeybek C, et al. Bitterness of glucose/galactose: novel mutations in the SLC5A1 gene. J Pediatr Gastroenterol Nutr 2014;58:57–60. [DOI] [PubMed] [Google Scholar]
- [5]. Xin B, Wang H. Multiple sequence variations in SLC5A1 gene are associated with glucose-galactose malabsorption in a large cohort of Old Order Amish. Clin Genet 2011;79:86–91. [DOI] [PubMed] [Google Scholar]
- [6]. Berni Canani R, Pezzella V, Amoroso A, et al. Diagnosing and treating intolerance to carbohydrates in children. Nutrients 2016;8:157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7]. Abad-Sinden ANA, Borowitz S, Meyers R, et al. Nutrition management of congenital glucose-galactose malabsorption. J Am Diet Assoc 1997;97:1417–21. [DOI] [PubMed] [Google Scholar]
- [8]. Borghi E, de Onis M, Garza C, et al. Construction of the World Health Organization child growth standards: selection of methods for attained growth curves. Stat Med 2006;25:247–65. [DOI] [PubMed] [Google Scholar]
- [9]. Joosten K, Embleton N, Yan W, et al. nutrition EEECwgopp. ESPGHAN/ESPEN/ESPR/CSPEN guidelines on pediatric parenteral nutrition: energy. Clin Nutr 2018;37 (6 pt B):2309–14. [DOI] [PubMed] [Google Scholar]
- [10]. van Goudoever JB, Carnielli V, Darmaun D, et al. nutrition EEECwgopp. ESPGHAN/ESPEN/ESPR guidelines on pediatric parenteral nutrition: amino acids. Clin Nutr 2018;37 (6 pt B):2315–23. [DOI] [PubMed] [Google Scholar]
- [11]. Posovszky C. Congenital intestinal diarrhoeal diseases: a diagnostic and therapeutic challenge. Best Pract Res Clin Gastroenterol 2016;30:187–211. [DOI] [PubMed] [Google Scholar]
- [12]. Thiagarajah JR, Kamin DS, Acra S, et al. Advances in evaluation of chronic diarrhea in infants. Gastroenterology 2018;154:2045–59. e2046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13]. Wright EM, Loo DDF, Hirayama BA. Biology of human sodium glucose transporters. Physiol Rev 2011;91:733–94. [DOI] [PubMed] [Google Scholar]
- [14]. Hamilton KL, Butt AG. Glucose transport into everted sacs of the small intestine of mice. Adv Physiol Educ 2013;37:415–26. [DOI] [PubMed] [Google Scholar]
- [15]. Sabino-Silva R, Mori RC, David-Silva A, et al. The Na+/glucose cotransporters: from genes to therapy. Braz J Med Biol Res 2010;43:1019–26. [DOI] [PubMed] [Google Scholar]