

Abstract
This study is to analyze the functional genes and metabolic pathways of dexamethasone degradation in Burkholderia through genome sequencing.
A new Burkholderia sp. CQQ001 (B. CQ001) with dexamethasone degrading activity was isolated from the hospital wastewater and sequenced using Illumina Hiseq4000 combined with the third-generation sequencing technology. The genomes were assembled, annotated, and genomically mapped. Compared with six Burkholderia strains with typical features and four Burkholderia strains with special metabolic ability, the functional genes and metabolic pathways of dexamethasone degradation were analyzed and confirmed by RT-qPCR.
Genome of B. CQ001 was 7,660,596 bp long with 6 ring chromosomes. The genes related to material metabolism accounted for 80.15%. These metabolism related genes could participate in 117 metabolic pathways and cover various microbial metabolic pathways in different environments and decomposition pathways of secondary metabolites, especially the degradation of aromatic compounds. The steroidal metabolic pathway containing 1 ABC transporter and 9 key metabolic enzymes related genes were scattered in the genome. Among them, the ABC transporter, KshA, and KshB increased significantly under the culture conditions of dexamethasone sodium phosphate as carbon source.
B. CQ001 is a bacterium with strong metabolic function and rich metabolic pathways. It has the potential to degrade aromatics and other exogenous chemicals and contains genes for steroid metabolism. Our study enriches the genetic information of Burkholderia and provides information for the application of Burkholderia in bioremediation and steroid medicine production.
Keywords: Burkholderia, degradation, dexamethasone, genome
1. Introduction
Burkholderia was divided into an independent genus in 1993 from Palleroni rRNA homologous groups of Pseudomonas according to the 16S rRNA sequences, DNA / DNA homology, cell lipid and fat characteristics. Burkholderia is widely found in the natural environment.[1] It has been reported in recent years that Burkholderia has a very unique metabolic ability and can use more than 200 kinds of organic matter as carbon sources, nitrogen sources and energy for growth and reproduction. A small number of bacteria in the Burkholderia are the plant and human pathogens.[2,3] Some bacteria of Burkholderia can produce a variety of metabolites with antibacterial activity, which has been used in drug production, such as phenazine,[4] phenylphrrole,[5] etc. Some bacteria of Burkholderia can degrade toxic compounds such as trichlorethylene (TCE), polychlorinated biphenyls (PCBs), and phenanthrene in wastewater and have been used in biological control[6,7,8] However, the role of Burkholderia in degrading steroid hormones has not been reported.
Steroid compound is an important element of life and the basic material of sterols, steroids, bile acids, and vitamin D. Steroid drugs are widely used in medicine, and dexamethasone is a commonly used steroid in clinic. Due to the particular molecular structure of the steroid compounds, steroid drugs are mainly produced through converting sterols into useful steroid drug intermediates by specific microorganisms, such as Rhodococcus and Mycobacterium.[9,10] The mechanism of metabolism of steroidal compounds in microorganisms has been widely studied. The steroid metabolic gene clusters have been identified and the functions of some key enzymes have also been identified.[11,12,13] However, the degradation pathways and functional genes related to the degradation of steroid hormone still need further investigation.
The steroid hormone is stable and difficult to be degraded in the natural environment. Drug residues of steroid hormone can pollute the environment in various ways, especially in those in hospital and urban wastewater. Several papers have reported different degrees of dexamethasone contamination in factories, hospital wastewater, urban wastewater, and drinking water.[14,15,16,17] Some of them also suggest that environmental dexamethasone contamination can have adverse effects on organisms.[18] Thus, understanding the mechanisms by which microorganisms metabolize steroid compounds is also of great value in the prevention and control of environmental pollution by steroids.
In our previous study, a dexamethasone-degrading strain was isolated from the wastewater, which was identified as a Burkholderia strain via 16S rDNA sequencing.[18] This is the first reported Burkholderia strain that can degrade steroid hormone, which is named as Burkholderia sp. CQ001. In this paper, the whole genome sequencing analysis of Burkholderia CQ001 was carried out to explore its genetic and metabolic characteristics and the mechanism of dexamethasone degradation. Our results may provide valuable information for the construction of engineering bacteria that can degrade dexamethasone.
2. Material and methods
2.1. Ethics statement
This article does not involve materials from humans or animals. Thus, ethical approval is not necessary.
2.2. Whole genome sequencing and analysis of B. CQ001
B. CQ001 was cultured in medium containing dexamethasone sodium phosphate at 37°C, 180 r/minute for 18 hours.[18] Bacterial DNA was extracted using DNA preparation kit (TaKaRa). The purified DNA was sequenced using Illumina Hiseq4000 platform and the 3rd sequencing technology by Shanghai Meiji Biological Medicine Technology Co., Ltd. (Shanghai, China).
PE library (500 bp) and PacBio library (8–10 kb) were prepared. The resulted sequences were examined and analyzed via bioinformatic analysis, and the genome was sketched by DNAplotte. Barrnap 0.4.2 and tRNAscan-SE v1.3.1 were used to predict the rRNA and tRNA. Glimmer3.02 was used to predict the genes. The sequences were compared against NR, GENES, and STRING. The annotation of COG (Clusters of Orthologous Groups of proteins) (http://www.ncbi.nlm.nih.gov/COG/) and KEGG (Kyoto Encyclopedia of Genes and Genomes) (http://www.genome.jp/kegg/) were obtained.
2.3. Comparative genomics analysis
Totally, 6 typical Burkholderia strains were selected out from NCBI database (Burkholderia xenovorans LB400, Burkholderia vietnamiensis G4, Burkholderia mallei ATCC23344, Burkholderia cenocepacia J2315, Burkholderia pseudomallei K96243, and Burkholderia gladioli BSR3). The basic characteristics of B. CQ001 genome were analyzed and compared with these 6 strains, including length of genes, GC content, numbers of genes, tRNA, and rRNA.
According to the 16S rRNA, 4 strains with high homology and special metabolic ability were chosen (Burkholderia phenoliruptrix BR3459, Burkholderia xenovorans LB400, Burkholderia kuruiensis M130, and Burkholderia vietnamiensis G4). Their genomes were downloaded from NCBI database, and the MUSCLE software was used for multiple alignment. All the homologous genes were clustered at the genome level, and the genome - based phylogenetic tree was constructed according to the NJ method. OrthoMCL software was used to analyze the amino acid (or nucleotide) sequences of all the tested strains. The homologous genes between different strains and the specific genes in each strain were obtained after clustering.
2.4. Confirmation of the dexamethasone-degrading genes via RT-qPCR
The related genes of ABC transporters and the KSH enzyme (KshA, KshB1, and KshB2) were tested by RT-qPCR. Briefly, B. CQ001 was cultured in media containing dexamethasone sodium phosphate (induced group) and inorganic sucrose broth (non-induced group) at 37°C, 200 r/minute for 24 hours. The total RNA was extracted using RNAprep Pure Bacteria Kit (TIANGEN). cDNA was reverse-transcribed using RT reagent Kit with gDNA Eraser (TaKaRa). RT-PCR was carried out using 2-step method via SYBR Premix Ex Taq II (TaKaRa) and Applied Biosystems 7300 Real Time PCR System (Bio-Rad). The 16S rRNA that stably expressed in different hosts was used as the housekeeping gene. The primers used in this study were listed in Table 1, which were designed and synthesized in Sangon Biotech Co., Ltd., (Shanghai, China). The relative expression level was calculated as fold change using the 2-ΔΔCt method.
Table 1.
Primer sequences used in the RT-qPCR experiment.
2.5. Statistical analysis
All data are expressed as means ± standard deviation (SD). Values of P ≤ .05, .01, or .001 were considered to be statistically significant (∗), highly significant (∗∗), or extremely significant (∗∗∗), respectively. Data were analyzed using SPSS 12.0 (SPSS Inc., Chicago, IL) for Windows.
3. Results
3.1. Whole genome sequencing and analysis of B. CQ001
To better understand the gene information of B. CQ001, we carried out whole genome sequencing via Illumina Hiseq4000 and 3rd generation sequencing technology. The resulted genome has been uploaded to the GenBank database. The number is PRJNA329146. B. CQ001 harbored 6 circular chromosomes. We speculate that the genome of B. CQ001 contains 2 chromosomes and 4 giant plasmids according to the genomic characteristics, chromosome size and reported gene information of Burkholderia.[19] The 6 circular chromosomes are shown in Figure 1, and the information of B. CQ001 genome is shown in Table 2. We found that chromosome 1 was a typical Burkholderia chromosome and was also the core chromosome. The main function of chromosome 1 was related to regulation of DNA replication and cellular physiological and biochemical activities. Most of the metabolic genes located on chromosome 2, which were associated with the transport of carbohydrates, the generation and transformation of energy, and the synthesis, transport, and decomposition of secondary metabolites, which determined the niche of the strain. The 4 giant plasmids were characteristic replicons. The existence of the 4 giant plasmids was critical for the specific metabolic pathways of B. CQ001. The whole genome of B. CQ001 reveals the high plasticity, specificity, and variability of the bacterium at the genetic level.
Figure 1.
Genome of Burkholderia sp. CQ001. The genome size, GC content, COG category, homologous genes were analyzed via GenomeViz and Circos softwares. The outermost circle represents CDs on the sense strand. Going inwards, the next circle represents CDs on the antisense strand. Different colors represent different COG categories. The third circle represents rRNA and tRNA. The next circle indicates GC content (below average GC content (blue) and above average GC content (yellow)). The innermost represents GC skew value (G-C/G+C). The value is positive when CDs locate on the positive strand, and negative when CDs locate on the negative strand. COG = Clusters of Orthologous Groups of proteins.
Table 2.
The information of B. CQ001 genome.

3.2. Functional annotation and classification of B. CQ001 genes
To better understand the function of the B. CQ001 genome, we compared the sequences against the STRING database (V9.05). Approximately 69.8% of the coding genes were classified (Fig. 2A), which included 408 genes associated with energy generation and transformation (C), 396 genes associated with carbohydrate transport and metabolism (G), 388 genes associated with cell wall and outer membrane synthesis (M), and 262 genes associated with secondary metabolic product synthesis, transport and decomposition (Q). These results indicate that this bacterium possesses abundant metabolic genes, and has powerful ability in energy generation and conversion. The genes associated with secondary metabolism synthesis are also abundant, resulting in a strong metabolic ability. Additionally, 429 genes were not classified (S), suggesting that there still are many genes to be investigated.
Figure 2.
Functional classification of Burkholderia sp. CQ001 genes. (A) Functional classification chart of COG; (B) secondary classification map of metabolic pathways in KEGG.
3.3. Analysis of metabolic pathways
To better understand the metabolic pathways of B. CQ001, we compared the genes against KEGG database. We found that B. CQ001 could participate in 117 metabolic pathways (Fig. 2B). About 80.15% of the genes were related to material metabolism, covering almost all the microbial metabolic pathways in different environments and the decomposition pathways of secondary metabolites, especially the degradation pathways of aromatic compounds. The number of genes involved in metabolism and degradation of exogenous chemicals (A: Metabolism/Xenobiotics biodegradation and metabolism) were relatively higher. We found that a pathway involved in steroidal metabolism, which contained 1 gene encoding the transporter of steroidal metabolism and 9 genes associated with the key enzymes of steroidal metabolism (Table 3). Therefore, B. CQ001 is a powerful bacterium with strong metabolic ability and various metabolic pathways, which can metabolize the steroids.
Table 3.
Key enzymes involved in steroidal degradation in Burkholderia sp.CQ001.
To further understand the distribution of dexamethasone-degrading related genes in B. CQ001, we found out the location of these genes according to relative position of the coding genes (Fig. 3). We found that ABC transporter (ORF03369_1), KshA (ORF05568_1), KshB1 (ORF05593_1), and KshB2 (ORF06355_1) showed dispersed distribution, which provides information for further study of the mechanism of steroidal degradation.
Figure 3.
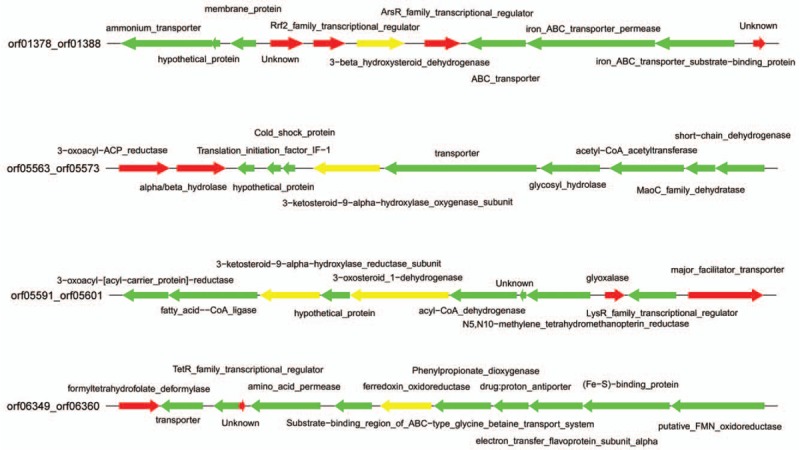
Location of key genes in Burkholderia sp. CQ001 for degradation of dexamethasone. Each arrow represents a gene. The direction of the arrow indicates the transcriptional direction. The length represents the size of each gene.
3.4. Analysis of basic characteristics and homology of B. CQ001 genomes
We further compared the B. CQ001 genome with 6 other typical Burkholderia genomes (Table 4). To better understand the metabolic characteristic of B. CQ001, we compared this bacterium with 4 strains with high homology and specific metabolic ability using OrthoMCL. Our results showed that 2856 homologous genes and 2758 specific genes in B. CQ001 (accounting for 33.2% of the coding genes), which were mainly related to the metabolisms of different exogenous substances (Fig. 4). The phylogenetic tree showed that B. CQ001 was close to B. vietnamiensis G4 at the evolutionary level (Fig. 5). It has been shown that B. vietnamiensis G4 can degrade biotoxic and carcinogenic TCE in surface water and groundwater, indicating the similar ability of B. CQ001.
Table 4.
Comparison of B. CQ001 with other Burkholderia genomes.
Figure 4.
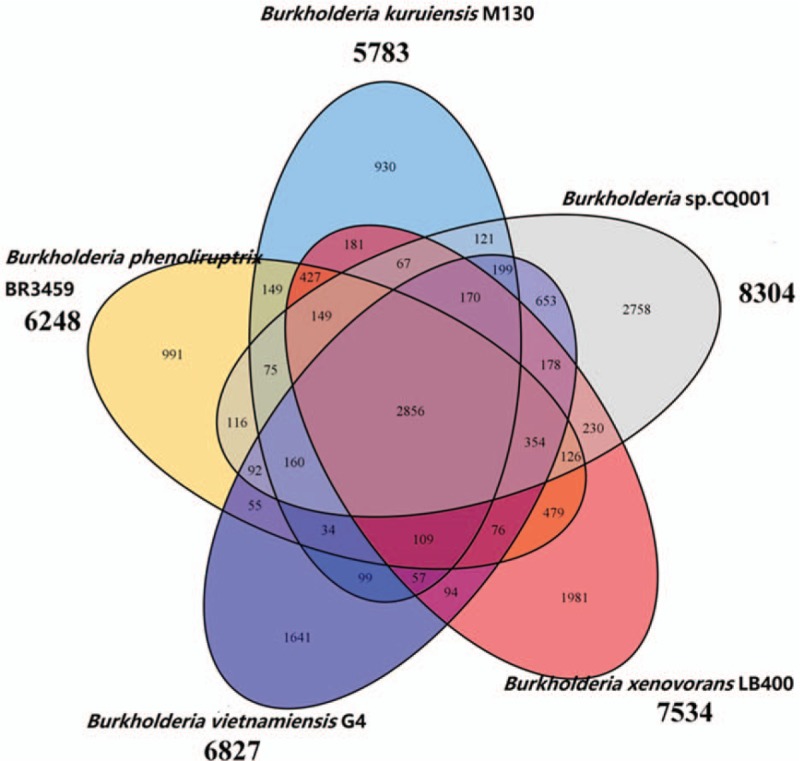
Homology analysis of Burkholderia sp. CQ001 and other Burkholderia strains (Burkholderia phenoliruptrix BR3459, Burkholderia xenovorans LB400, Burkholderia kuruiensis M130, and Burkholderia vietnamiensis G4).
Figure 5.
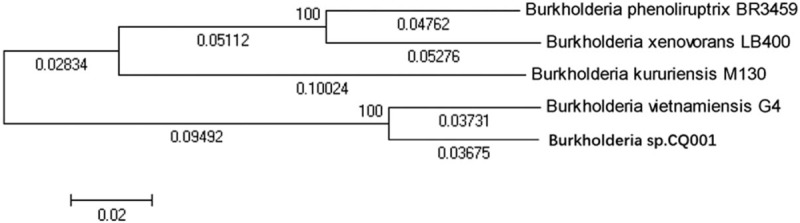
Phylogenetic trees according to the whole genome sequences of Burkholderia sp. CQ001.
3.5. Functional confirmation of dexamethasone-degrading related genes
According to the mechanisms of multi-step degradation of steroid,[20,21] the chemical structure of dexamethasone and the genomic sequences of B. CQ001, we speculate that ABC transporters and the KSH enzyme may be involved in the degradation of dexamethasone. To confirm this speculation, RT-qPCR was carried out to validate the expression of the related genes. We found that the levels of these genes in the presence of dexamethasone sodium phosphate were higher than those in the presence of sucrose (Fig. 6). The change of the ABC transporter was the highest (increased by 20.53 folds). These results confirm that the genes studied are related to the degradation of dexamethasone. We also speculate that KshA and KshB1 are oxygenase and reductase that are strongly induced by dexamethasone, and KshB2 is not as sensitive to dexamethasone as KshB1, which may be due to the regulatory mechanism of KstR2.[11,22]
Figure 6.
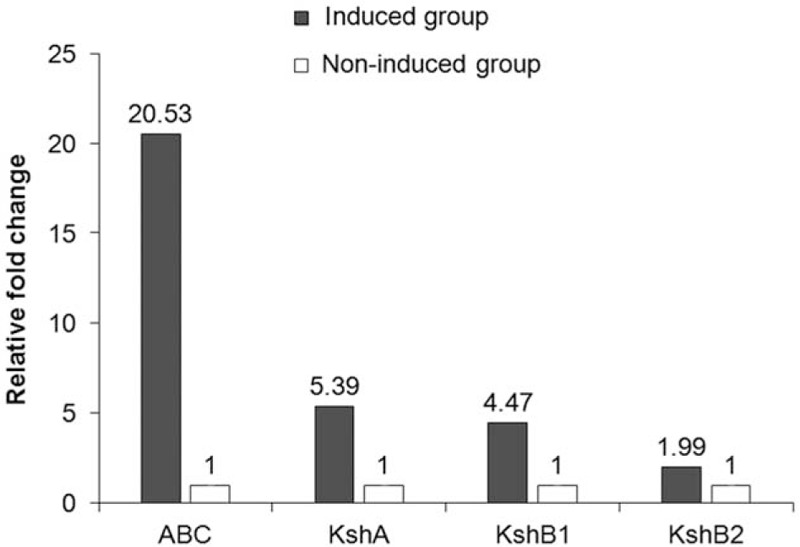
The relative expression of genes in Burkholderia sp. CQ001 in the presence of 2 carbon sources (induced group: dexamethasone sodium phosphate; non-induced group: sucrose). Gene expression was detected with RT-qPCR. All data are expressed as means ± standard deviation (SD).
4. Discussion
In this study, we revealed the whole genomic sequences of a previous identified dexamethasone-degrading Burkholderia strain CQ001. We found that B. CQ001 had the basic characteristics of Burkholderia strains and the largest chromosomes among known Burkholderia strains. The bacterium possessed abundant metabolic genes, which participated in various metabolic pathways, and had a strong potential of metabolizing exogenous chemical.
Rhodococcus and Mycobacteria [23,24] are the main steroid-degrading bacteria being studied currently. However, the functional genes involved are still unclear. In this study, we sequenced and analyzed genome of B. CQ001 and confirmed the key genes involved in dexamethasone degradation. We found a metabolic pathway of steroidal metabolism and genes responsible for degradation of steroid hormone in B. CQ001. ABC transporters encoded by Mce family may be involved in the active uptake of dexamethasone.[25] The gene encoding cholesterol oxidase in the process of steroidal open-loop reaction was not found,[26] while the gene encoding 3β-hydroxy steroid dehydrogenase/isomerase (3β-HSD) was found.[27] We speculate that 3β-HSD is the first key enzyme of steroidal degradation. This process does not need the dehydrogenation and isomerization of 3-sterone-Δ1-dehydrogenase due to the fact that dexamethasone hormones are derivatives of steroidal metabolism. In the process of dexamethasone degradation, the hydroxylation on B ring of dexamethasone caused by 3-sterone-9α-hydroxylation enzyme (KSH) is the most critical step in the open loop of steroidal nucleus. The generated 9α-hydroxy-1,4-diene steroid is extremely unstable and the high molecular energy can cause the automatical pyrolysis of B ring on C9.[26] The steroid after the open loop is catalyzed by Hsa family protein and finally decomposed into ATP, small molecule organic salt and the coenzyme.[28]
KSH consists of the terminal oxygenase (KshA) and the iron-sulfur protein reductase (KshB). Two isoenzymes, KshB1 and KshB2, were found in B. CQ00. KshB1 is a reductase that could be strongly induced by dexamethasone, while KshB2 is a negative regulator that is the key factor for the induction of steroidal substrate, which is less sensitive to dexamethasone than B1.[29] The regulatory mechanism of the open loop and the branch catabolism of steroidal nucleus are not yet clear, and the pathways after the open loop remain to be further explored.
Previous studies found that the genes associated with steroidal metabolism tend to cluster together in most of microorganism and the expression of many genes in the gene cluster increased significantly under the induction of steroidal substrates.[30,31] However, other studies found that nearly 60% important genes were not clustered and located outside the gene clusters.[11,32] The results in this study also confirmed that the genes associated with steroidal metabolism were not clustered. In addition, unspecified aggregation of steroid-related genes, the widespread presence of isoenzyme coding sequences, and genes with unknown functions have increased the difficulty to understand the degradation mechanism of steroids. Thus, it is of great value to investigate the upstream and downstream of the steroid-related genes, which needs to be further studied.
In conclusion, we successfully isolated an effective dexamethasone sodium and dexamethasone-degrading strain from the hospital wastewater before. To better understand the pathways and mechanisms of dexamethasone degradation, we further sequenced and analyzed its genome in the present study. The genetic features of B. CQ001 disclosed herein not only enriched the genetic information of Burkholderia but also provided further information on the degradation mechanism of steroid hormones and degradation-related enzymes. Our results lay the foundation for further study on the mechanism underlying bacterial steroid degradation and the development of valuable engineering bacteria for bioremediation and steroid production.
Author contributions
Conceptualization: Yi Wang.
Data curation: Dan Si, Zhibang Yang.
Methodology: Dan Si, Yuxia Xiong, Jin Zhang, Lianju Ma, Jinyang Li.
Writing – original draft: Dan Si.
Writing – review & editing: Yi Wang.
Footnotes
Abbreviations: 3β-HSD = 3β-hydroxy steroid dehydrogenase/isomerase, PCBs = polychlorinated biphenyls, TCE = trichlorethylene.
The authors have no conflicts of interests to disclose.
References
- [1]. Lemaire B, Chimphango SB, Stirton C, et al. Biogeographical patterns of legume-nodulating Burkholderia spp.: from African fynbos to continental scales. Appl Environ Microbiol 2016;82:5099–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2]. Francis F, Kim J, Ramaraj T, et al. Comparative genomic analysis of two Burkholderia glumae strains from different geographic origins reveals a high degree of plasticity in genome structure associated with genomic islands. Mol Genet Genomics 2013;288:195–203. [DOI] [PubMed] [Google Scholar]
- [3]. Johnson MM, Ainslie KM. Vaccines for the prevention of melioidosis and glanders. Curr Trop Med Rep 2017;4:136–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4]. Xu N, Ahuja EG, Janning P, et al. Trapped intermediates in crystals of the FMN-dependent oxidase PhzG provide insight into the final steps of phenazine biosynthesis. Acta Crystallogr D Biol Crystallogr 2013;69:1403–13. [DOI] [PubMed] [Google Scholar]
- [5]. el-Banna N, Winkelmann G. Pyrrolnitrin from Burkholderia cepacia: antibiotic activity against fungi and novel activities against streptomycetes. J Appl Microbiol 1998;85:69–78. [DOI] [PubMed] [Google Scholar]
- [6]. Hamid S, Bae W, Kim S, et al. Enhancing co-metabolic degradation of trichloroethylene with toluene using Burkholderia vietnamiensis G4 encapsulated in polyethylene glycol polymer. Environ Technol 2014;35:1470–7. [DOI] [PubMed] [Google Scholar]
- [7]. Chen WT, Shen SM, Shu CM. Application of ethylene diamine tetra acetic acid degrading bacterium Burkholderia cepacia on biotreatment process. Bioresour Technol 2015;193:357–62. [DOI] [PubMed] [Google Scholar]
- [8]. Festa S, Coppotelli BM, Madueno L, et al. Assigning ecological roles to the populations belonging to a phenanthrene-degrading bacterial consortium using omic approaches. PloS One 2017;12:e0184505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9]. Wu K, Li W, Song J, et al. Production, purification, and identification of cholest-4-en-3-one produced by cholesterol oxidase from Rhodococcus sp. in aqueous/organic biphasic system. Biochem Insights 2015;8:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10]. Xiong LB, Liu HH, Xu LQ, et al. Role identification and application of SigD in the transformation of soybean phytosterol to 9alpha-hydroxy-4-androstene-3,17-dione in mycobacterium neoaurum 2017;65:626–31. [DOI] [PubMed] [Google Scholar]
- [11]. Fernandez-Cabezon L, Garcia-Fernandez E, Galan B, et al. Molecular characterization of a new gene cluster for steroid degradation in Mycobacterium smegmatis. Environ Microbiol 2017;19:2546–63. [DOI] [PubMed] [Google Scholar]
- [12]. Mohn WW, Wilbrink MH, Casabon I, et al. Gene cluster encoding cholate catabolism in Rhodococcus spp. J Bacteriol 2012;194:6712–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13]. Van der Geize R, Yam K, Heuser T, et al. A gene cluster encoding cholesterol catabolism in a soil actinomycete provides insight into Mycobacterium tuberculosis survival in macrophages. Proc Natl Acad Sci U S A 2007;104:1947–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14]. Chang H, Hu J, Shao B. Occurrence of natural and synthetic glucocorticoids in sewage treatment plants and receiving river waters. Environ Sci Technol 2007;41:3462–8. [DOI] [PubMed] [Google Scholar]
- [15]. Van der Linden SC, Heringa MB, Man HY, et al. Detection of multiple hormonal activities in wastewater effluents and surface water, using a panel of steroid receptor CALUX bioassays. Environ Sci Technol 2008;42:5814–20. [DOI] [PubMed] [Google Scholar]
- [16]. Schriks M, van Leerdam JA, van der Linden SC, et al. High-resolution mass spectrometric identification and quantification of glucocorticoid compounds in various wastewaters in the Netherlands. Environ Sci Technol 2010;44:4766–74. [DOI] [PubMed] [Google Scholar]
- [17]. Zhongquan S, Yuzhen Z, Zhibang Y. Discussion on contamination of dexamethasone in wastewater. Chin Med Guid 2012;10:319–21. [Google Scholar]
- [18]. Xi Y, Li X, Si D. Effects of dexamethasone-contaminated water on mouse growth and intestinal microflora composition. Int J Clin Exp Med 2017;10:2165–72. [Google Scholar]
- [19]. Depoorter E, Bull MJ, Peeters C, et al. Burkholderia: an update on taxonomy and biotechnological potential as antibiotic producers. Appl Microbiol Biotechnol 2016;100:5215–29. [DOI] [PubMed] [Google Scholar]
- [20]. Yam KC, D’Angelo I, Kalscheuer R, et al. Studies of a ring-cleaving dioxygenase illuminate the role of cholesterol metabolism in the pathogenesis of Mycobacterium tuberculosis. PLoS Pathog 2009;5:e1000344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21]. Ouellet H, Johnston JB, de Montellano PR. Cholesterol catabolism as a therapeutic target in Mycobacterium tuberculosis. Trends Microbiol 2011;19:530–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22]. Garcia-Fernandez J, Galan B, Medrano FJ, et al. Characterization of the KstR2 regulator responsible of the lower cholesterol degradative pathway in Mycobacterium smegmatis. Environ Microbiol Rep 2015;7:155–63. [DOI] [PubMed] [Google Scholar]
- [23]. Malaviya A, Gomes J. Androstenedione production by biotransformation of phytosterols. Bioresour Technol 2008;99:6725–37. [DOI] [PubMed] [Google Scholar]
- [24]. Vasilevskaya AV, Yantsevich AV, Sergeev GV, et al. Identification of Mycobacterium tuberculosis enzyme involved in vitamin D and 7-dehydrocholesterol metabolism. J Steroid Biochem Mol Biol 2017;169:202–9. [DOI] [PubMed] [Google Scholar]
- [25]. García JL, Uhía I, García E, et al. Bacterial degradation of cholesterol and other contaminant steroids. Microbial Bioremediation of Nonmetals: Current Research 2011;23–43. [Google Scholar]
- [26]. Donova MV, Egorova OV. Microbial steroid transformations: current state and prospects. Appl Microbiol Biotechnol 2012;94:1423–47. [DOI] [PubMed] [Google Scholar]
- [27]. Uhia I, Galan B, Morales V, et al. Initial step in the catabolism of cholesterol by Mycobacterium smegmatis mc2 155. Environ Microbiol 2011;13:943–59. [DOI] [PubMed] [Google Scholar]
- [28]. Shen Y, Zhao H, Liu Y, et al. Effect of attapulgite on cell activity of steroid-transforming arthrobacter simplex. Adv Appl Biotechnol 2015;289–95. [Google Scholar]
- [29]. Lack NA, Yam KC, Lowe ED, et al. Characterization of a carbon-carbon hydrolase from Mycobacterium tuberculosis involved in cholesterol metabolism. J Biol Chem 2010;285:434–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30]. Bergstrand LH, Cardenas E, Holert J, et al. Delineation of steroid-degrading microorganisms through comparative genomic analysis. mBio 2016;7:e00166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31]. Horinouchi M, Kurita T, Yamamoto T, et al. Steroid degradation gene cluster of Comamonas testosteroni consisting of 18 putative genes from meta-cleavage enzyme gene tesB to regulator gene tesR. Biochem Biophys Res Commun 2004;324:597–604. [DOI] [PubMed] [Google Scholar]
- [32]. Griffin JE, Gawronski JD, Dejesus MA, et al. High-resolution phenotypic profiling defines genes essential for mycobacterial growth and cholesterol catabolism. PLoS Pathog 2011;7:e1002251. [DOI] [PMC free article] [PubMed] [Google Scholar]