

Abstract
CRISPR-Cas systems are able to acquire immunological memories (spacers) from bacteriophages and plasmids in order to survive infection, however this often occurs at low frequency within a population making it difficult to detect. Here we have developed CAPTURE (CRISPR Adaptation PCR Technique Using Re-amplification and Electrophoresis) a versatile and adaptable protocol to detect spacer acquisition events by electrophoresis imaging, with a sensitivity that can identify spacer acquisition in 1 in 105 cells. Our method harnesses two simple PCR steps, separated by automated electrophoresis and extraction of size-selected DNA amplicons, allowing the removal of unexpanded arrays from the sample pool, and enabling a 1000 times more sensitive detection of new spacers than existing PCR protocols. CAPTURE is a straightforward method requiring only one day to enable detection of spacer acquisition in all native CRISPR systems and facilitate studies aimed both at unravelling the mechanism of spacer integration and more sensitive tracing of integration events in natural ecosystems.
Keywords: CRISPR, adaptation, spacer acquisition, detection, CAPTURE, CRISPR array
Introduction
Microbes have the unique capacity to acquire resistance against bacteriophage and plasmids by the incorporation of small DNA fragments (spacers), derived from these invaders, into the genome of the infected cell1,2. Cells that successfully incorporate these spacers in their CRISPR array become immunized against further infection by this specific invader1,2. The inserted spacers are expressed and processed into CRISPR RNAs (crRNAs) and act as guides for Cas protein effector complexes which help find, bind and cleave matching invader nucleic acid sequences, resulting in invader elimination and host survival2,3.
To date a number of evolutionarily diverse CRISPR-Cas systems have been identified and classified into 2 main classes (I and II) and six main types (I – VI) based on their cas gene repertoire4. Due to the rarity of spacer acquisition events, which are estimated to occur in 1 in 107 cells in some CRISPR-Cas systems5,6, adaptation remains hard to observe. Spacer acquisition was first shown in the type II-A system of Streptococcus thermophilus1 and subsequently in a number of type I systems7,8, including the well characterized type I-E system9–11. Some type III systems were shown to acquire spacers by converting RNA into DNA through a reverse transcriptase mechanism12 but spacer acquisition in type III systems seems to be very rare.
Comparison to Existing Approaches
Current methods to detect CRISPR spacer acquisition utilize the conserved aspects of CRISPR array structure and the spacer acquisition mechanism. CRISPR loci are generally comprised of repeat-spacer arrays preceded by an AT-rich leader sequence, which acts as the transcriptional promoter and binding site for factors assisting with spacer acquisition13. Spacers are typically between 30 and 40 base pairs, and are inserted at the leader proximal end of the array duplicating the first repeat in the process2. The insertion of a spacer results in an increase in CRISPR array size which can be assessed by PCR and harnessed as an indicator for acquisition events5,7,9–11. In addition to these conventional PCR approaches, reporter systems have been developed that give a detectable signal upon spacer insertion due to a frameshift14,15. While these reporter systems provide a quantifiable and sensitive method of detection, their reliance upon a frameshift requires strain engineering and limits the number of CRISPR types and systems this technique is applicable to14,15. Conventional array amplifying PCR methods are simple and user friendly but often lack the sensitivity that is required to study many aspects of CRISPR adaptation.
Advances in the study of adaptation were made in parallel with the increasing availability of deep sequencing allowing detection of spacer acquisition in minor fractions of the bacteria present in the population7. However, massive sequencing efforts suffer from high levels of non-informative PCR amplicons, representing unexpanded CRIPSR arrays from cells that did not acquire new spacers. Recently published work, showed the development of a method, SENECA, to eliminate such unexpanded arrays from sequencing samples. This method requires the presence of an engineered restriction endonuclease site flanking the first repeat, which can be harnessed to prevent PCR amplification of non-expanded CRISPR arrays in a second PCR round16. Here we present a general strategy termed CAPTURE (CRISPR Adaptation PCR Technique Using Re-Amplification and Electrophoresis), which can be adapted to any native CRISPR array. The method has been applied in recently published work from our lab 17 and builds on, combines and improves existing tricks to selectively amplify expanded CRISPR arrays 5,7,9–11. Our method harnesses two PCR steps, separated by automated electrophoresis and extraction of size-selected DNA amplicons, allowing the removal of unexpanded arrays from the sample pool (Figure 1). Following this, PCR re-amplification of the captured DNA pool with spacer acquisition specific primers allows visible detection of spacer acquisition within just 1 in 105 cells (0.01%) of the population, and potentially up to 1 in 108 cells after deep sequencing. With this method we enable the direct study of CRISPR-Cas systems with low acquisition rates, which appears to be a common feature of a large number of bacteria and archaea5,6.
Figure 1. Overview of the CAPTURE protocol.

Outline of the major steps required in order to detect spacer acquisition in a population of cells with high sensitivity. Dashed lines indicate steps where choices have to be made before proceeding to the next step.
Potential Applications
The study of CRISPR adaptation reveals the physical interactions of hosts with their invaders, and can be utilized in computational methods to couple viral invaders to unknown hosts and to track spacer acquisition events over time, in sequenced strains18,19. Because each bacterial strain has a different history, differences in spacer content can be used to distinguish closely related pathogenic strains, such as Mycobacterium and Yersinia species20. Typically only a small fraction of spacers can be mapped back to known invaders suggesting a vast amount of unexplored invader diversity in nature19. Aside from strain typing, the sequence of new spacers provides mechanistic information for example helping to identify the protospacer adjacent motif (PAM), a critical sequence motif in the DNA that authenticates DNA for cleavage by Cas proteins in type I, II and V CRISPR systems and prevents self-targeting of the CRISPR array7–11,17. Here, we aimed to develop a highly sensitive, simple and generally applicable method that could be used to detect the occurrence of such rare spacer acquisition events and unravel further mechanistic details of uncharacterized CRISPR systems. The use of CAPTURE can provide a wealth of information of both uncharacterized, newly discovered and well known CRISPR systems. The method helps to elucidate the role of Cas proteins, the identity of the PAM, the preferred spacer substrates of Cas1-Cas2, and the minimal requirements for both adaptation and defense as exemplified by Kieper et al. where the role of the Cas4 protein in adaptation was recently uncovered17.
Limitations
The use of CAPTURE includes the requirement of a gel extraction machine or more laborious manual extraction methods7. In order to increase both the utility of this protocol as a tool for the detection of spacer acquisition and to remove non-expanded CRISPR arrays, multiple PCR amplification steps are required. Re-amplification in PCR 2 with primer sets 2, 3 or 4 may introduce PCR bias, which prohibits using absolute abundance levels of new spacers with high confidence. Typically, only unique spacer sequences are used for downstream analysis (Table 1). It is therefore important that all users take this into consideration when analysing sequencing results. The removal of duplicates during the processing of sequencing reads is recommended and ensures that re-amplification bias does not influence the prevalence of certain spacer sequences in the population. We advise users to consider using biological triplicates and to compare the results obtained from all unique and non-unique spacers. If the degenerate primers are used, it is possible to carry out normalization to correct for the bias introduced by the fixed 3’ nucleotides of the primer21. This allows the user to draw accurate conclusions about the prevalence of certain spacers in the population.
Table 1. Primer sets and their limitations.
| Step in protocol | Primer Set | Desired Outcome for Sample | Limitations | Solution |
|---|---|---|---|---|
| 4 | Set 1, Initial Primers | Detection | Does not select specifically for expanded arrays. Can only detect expanded arrays when adaptation occurs in at least 10% of the entire bacterial population | Try size selection and re-amplification |
| 17(A) | Set 2, Internal Primers | Detection | Does not specifically bind expanded arrays and thus does not provide an extra level of detection | Try primer set 3 or 4 for more specific binding of expanded arrays |
| 17(A) | Set 2, Internal Primers | Sequencing | Creates a PCR bias in deep sequencing results and could strongly influence the prevalence of certain spacer sequences | Remove duplicate sequences during analysis. Use biological replicates to confirm observed trends |
| 17(B) | Set 3, Degenerate Primers | Sequencing | Creates a PCR bias due to both stronger G-C annealing at the 3’ end and exclusion of the primer ending with the same 3’ nucleotide as the existing leader proximal spacer | Complete a normalization during analysis that accounts for the bias introduced by omitting a primer containing the same 3’ nucleotide as the first spacer21. |
| 17(C) | Set 4, Repeat Primers | Sequencing | Cannot be used for all systems due to the differing repeats sequences. The primers designed need to bind in opposite directions specifically within the repeat sequence. Subsequently these primers may be very short and if the repeat region for a system is AT-rich it may be difficult to design primers with an appropriate annealing temperature. | Try Set 3, degenerate primers |
| 17(C) | Set 4, Repeat Primers | Sequencing | Requires further optimization of annealing temperature to prevent PCR artefacts arising from other sections of the array incorrectly amplified in PCR 1 | The degenerate primers (Set 3) require less optimization but increase the PCR bias |
| 17(C) | Set 4 Repeat Primers | Sequencing | Creates PCR bias in deep sequencing results and could strongly influence the prevalence of certain spacer sequences | Remove duplicate sequences during analysis. Use biological replicates to confirm observed trends |
Experimental Design
This protocol contains a series of steps involving size selection and extraction in combination with two PCR amplifications, PCR 1 and PCR 2. The options provided for these steps can be used in various combinations both to detect if the CRISPR adaptation module in your strain is active, as well as to aid the preparation of a deep sequencing sample containing a majority of arrays that have acquired spacers. Not all steps of the protocol need to be completed if the desired outcome can be reached at an earlier step (Figures 1 and 2). Prior to use of this protocol, primer design and re-amplification options should be carefully considered, as these will differ depending on both the CRISPR array of the strain used and further actions to be taken with the final sample (Figure 2, Supplementary Figure 1 and Supplementary Table 1).
Figure 2. CAPTURE decision chart.

This chart indicates by following the arrows which steps should be taken and which primers should be designed for detection or preparing a sequencing sample.
PCR 1, Amplifying CRISPR arrays from a population (Step 4)
The described protocol is designed with the assumption some sequence knowledge or metagenomics data has previously been obtained for the bacterial strain or population of interest. Exact sequence information of the CRISPR array to be studied is required for the primer design step. Primers for PCR 1, the initial array amplification, must be specifically designed for each strain to bind within the leader sequence and where possible within the closest known spacer to the leader, as the sequence of both binding sites can differ greatly between species and subtypes of the CRISPR system. Careful design of primer Set 1 allows amplification of all expanded arrays present within the population and enables elimination of all repeats downstream from spacer 1 (Figure 3). Elimination of excess repeats is important for later steps of the protocol because if a second re-amplification is required, the primer set may bind the repeats specifically, resulting in amplification only when a new spacer is inserted and two repeats are present. For an example of the primers that can be designed, here for the Type I-E CRISPR system of Escherichia coli BW25113, see (Supplementary Figure 1 and Supplementary Table 1).
Figure 3. Primer design for amplifying the CRISPR arrays within a population.

Schematic of the CRISPR array consisting of a leader sequence (L) and repeats (R) interspaced by short DNA sequences termed spacers (coloured squares). When acquisition occurs an additional spacer (+1) is added at the leader proximal end (orange) of the array. Primers indicated by black arrows (numbered 1 and 2) are designed to allow PCR amplification of both expanded and unexpanded arrays within the population.
Size Selection and Extraction Parameters (Steps 8-14)
In addition to considering which part of the CRISPR array is amplified it is also important to consider the amplified array product size in relation to the following size selection and extraction steps. Smaller bands will allow a higher degree of size separation during use of the Blue Pippin cassette. The highest percentage of agarose available in a Blue Pippin cassette is 3% and allows selection within the range of 100 – 250 bp. This equally applies to users completing the size selection and extraction manually from an agarose gel. For manual extraction we recommend use of a 3% agarose gel to increase expanded and non-expanded array separation. Following this the expanded array band must be excised from the gel, extracted and purified. For optimal selection of only the expanded arrays we suggest both the gel electrophoresis separation and subsequent DNA extraction to be completed twice7.
Controls (All Steps)
We recommend the use of an unexpanded (native) array control sample throughout the entire procedure to allow assessment of size extraction success and proportion of unexpanded arrays in the final DNA sample. The control should be the same as the CRISPR array of the strain of interest, in which the reverse primer (for example primer 2) will bind in the most leader proximal spacer (Supplementary Figure 1 and Supplementary Table 1). Either the parental strain with no new spacers or a synthetic DNA construct with the sequence of the unexpanded array could be used as controls throughout this protocol. Use of the original strain of interest as a control can begin at Step 1 with the samples to be tested. A synthetic DNA control should be added as a separate sample during size extraction (Steps 8 -15). Such a control determines the carryover of the non-expanded arrays in the size selection and extraction step and allows optimization of the parameters for both use of the blue pippin and manual extraction (Figure 4). The control also aids in the identification of PCR amplification artefacts that can arise due to unspecific annealing temperatures for primer binding or, internal binding of the repeats creating larger PCR amplicons. These artefacts can appear as expanded arrays in the control after size selection and can be removed with small optimizations (see Table 2).
Figure 4. Gel Electrophoresis images of CRISPR arrays before (Step 5) and after (Step 15) size selection.

The proportion of expanded CRISPR arrays (indicated by the black arrow labelled +1) in the sample (10-1) is greatly increased after size selection (right) compared to the control (C).
Table 2. Troubleshooting.
| Step | Problem | Possible Reason | Solution |
|---|---|---|---|
| 5 | Multiple bands seen after PCR amplification | Primers are binding to multiple regions | Annealing temperature of primers must be adjusted (increased) to be as specific as possible |
| 5 | No PCR products | Primers were not optimized to specific conditions | Consider a gradient PCR or decreased annealing temperature |
| 16 | Low DNA yield after size selection | Up to 50% of DNA can be lost during the extraction procedure | A Blue Pippin cassette in which your DNA fragment size is in the middle of the selection range should be chosen Programming a wider selection range should also increase your yield Increase the concentration of DNA loaded into the blue pippin cassette by pooling multiple PCRs |
| 15 | A large proportion of unexpanded arrays are present in the extracted sample | Selection range was too close to the unexpanded array fragment size | Increase the size of the collection starting point to as close to the expanded array size as possible To widen the overall range and increase the yield as mentioned previously you can instead increase the end collection size |
| 18 | A large proportion of unexpanded arrays are present after internal primer re-amplification | The acquisition rate may be very low | Try using the degenerate or repeat primers to re-amplify the extracted fragment pool |
| 18 | No PCR product can be visualized after re-amplification of size extracted fragments | Low DNA yield from the extraction step | Run the extracted fragment pool directly on a gel to ensure product was extracted |
| 18 | No or low PCR yield | Not enough template used for PCR Primers were not optimized to specific conditions |
The template DNA concentration as indicated in the protocol can be increased Consider a gradient PCR or lower annealing temperature |
| 18 | High levels of the expanded band can be seen in the control after re-amplification | There may be carryover of longer products amplified in PCR 1 from the entire array. There could be a contamination or an amplification artefact, caused by repeats binding to each other, present in the sample |
Increase the annealing temperature to increase the primer binding specificity in PCR 1 Increase the annealing temperature in PCR 2 Lower the number of cycles and template used for the re-amplification PCR and check control levels |
| 18 | High levels of the unexpanded array band can be seen in the control after re-amplification | A large proportion of unexpanded or control arrays are still present after size extraction | Lower the number of cycles and template used for the re-amplification PCR and check control levels Increase the annealing temperature to increase the primer binding specificity Consider changing primer set for the re-amplification If this does not work repeat the size extraction using the previously extracted sample pool Or alternatively use the size extracted DNA directly for deep sequencing |
| 18 | Bands are not clearly distinguishable enough on the agarose gel to determine expanded: unexpanded array ratio | Gel was not run long enough or DNA yield is low and hard to visualize | Increase the percentage of your gel and run the products for longer If this does not work consider staining with SYBR Gold for clearer bands |
PCR 2, Re-Amplification after Size Selection (Step 17)
Re-amplification is an important additional step to this protocol as often the initial amplification (PCR 1) is not sensitive enough to detect acquisition occurring at a low frequency in the population (Figure 4). In addition, when high frequency spacer acquisition can be detected in the PCR 1 there is often still a large amount of non-expanded arrays present in the sample, these non-expanded arrays can act as a mask to low frequency acquisition events which are revealed after size selection and re-amplification (Figure 5). In this protocol through size selection and re-amplification we aim to maximise both the detection limit and the number of expanded array sequences obtained from deep sequencing. There are three options, Step 17 A, B or C, using primer sets 2, 3 or 4 respectively, for the second amplification step (PCR 2) of this method. The three primer sets we refer to as; (Set2) internal primers (Set 3) degenerate primers and (Set 4) repeat primers, should be carefully designed based on the desired output, subsequent plans for the sample pool and sequence analyses (Figure 2, Figure 5, Supplementary Figure 1 and Supplementary Table 1). The internal primers (Set 2) allow re-amplification of the expanded arrays enabling confirmation of successful spacer acquisition taking place in just 1 in 103 cells in the population of interest (Figure 6a). The degenerate primers (Set 3) allow extremely sensitive detection of acquisition at an occurrence of just 1 in 105 cells (Figure 6b). This amplification is based on the use of 3 forward primers, these primers anneal their 3’ nucleotide only with new spacers beginning with a nucleotide different from the original spacer5 (Figure 5, Supplementary Figure 1). The repeat primer (Set 4) strategy ensures that the short 60 bp array products can only be amplified in the presence of 2 repeats (i.e. an expanded array), this process relies on the removal of a repeat during the first PCR amplification but allows high sensitivity detection of acquisition occurring in just 1 in 106 cells and a final sample population ready for further sequencing (Figure 5, Figure 6c).
Figure 5. Primer design options for re-amplification of expanded CRISPR arrays.

Schematic of the three possible primer options for the second PCR amplification step. The internal primers (left) bind in spacer 1 (primer #2) and the leader sequence (#3) internally of primer #1 used in the first PCR step before size selection. The degenerate primers (middle) consist of 3 forward primers (DP) that anneal their 3’ nucleotide only with new spacers (orange) starting with a nucleotide different from the original spacer (blue), in combination with #2. The repeat primers (right) bind within the repeat (#7 and #8) orientated such that a product is only amplified when two repeats are present i.e. the array is expanded.
Figure 6. Anticipated Detection Results after Re-amplification.

Populations containing between 1:10 (10-1) and 1:106 (10-6) expanded:unexpanded CRISPR arrays plus an unexpanded control (0) were compared and visualized on a 2% agarose gel after completion of Step 15, Re-amplification after size selection. a) Re-amplification using internal primers option A, b) re-amplification with degenerate primers option B, c) re-amplification with repeat binding primers option C. The black arrow labelled +1 indicates the expected fragments size when the array has expanded. Here it is shown that while the internal primers (a) have a low sensitivity to detect acquisition, occurring in at least 1% of the entire bacterial population, CAPTURE with the more specific degenerate primers (b) and repeat primers (c) is able to detect acquisition events that occurred in just 0.01% of the population.
Sequencing (Steps 20-31)
A user may choose to sequence new spacers to determine spacer length distribution (can be consistent or variable), spacer source (bacteriophage genome, plasmid or host genome), or to retrieve the PAM of the newly acquired spacers. Deep sequencing may be used to increase the sensitivity at which new spacers are detected to enable discovery of rare spacer acquisition events. When deep sequencing is carried out it is advisable to re-amplify expanded PCR amplicons using the CAPTURE method to minimize the presence of non-expanded CRISPR array amplicons. A user may choose not to sequence new spacers, by following the protocol up to and including step 15, when only general spacer acquisition activity is monitored over time, or for example when trying to answer questions about the general effect of cas gene mutants on the spacer acquisition activity.
Materials
Biological Materials
CAUTION Use gloves and a lab coat when working with biological samples.
-
-
Escherichia coli K12 BW25113 (Parental strain KEIO Collection, National BioResource Project (NBRP) E. coli Strain)22.
-
-
E. coli PIM5, a variant of the KEIO strain E. coli K12 JW12252 (ref. 10, Brouns lab collection). CRITICAL: The protocol described below permits the use of any bacterial strain in which sequence information of the CRISPR array is known or is suspected to be similar to a known array to allow the primer design.
Reagents
Lysogeny Broth (LB) Medium (Sigma Aldrich, cat. no. L3022)
GeneJET Genomic DNA Extraction Kit (Thermo Fisher Scientific, cat. no. K0721)
CAUTION Components of the kit are dangerous if swallowed and harmful to aquatic life if released into the environment.
OneTaq® Quick-Load® 2X Master Mix with Standard Buffer (Bioké, cat. no. M0486S)
Agarose (Promega, cat no. V3125)
40X TAE buffer stock (Promega, cat no. V4281)
SYBR Safe (Thermo Fisher Scientific, cat no. S33102)
SYBR Gold (Thermo Fisher Scientific, cat no. S11494)
SmartLadder SF (Eurogentec, cat no. MW-1800-04)
Quick-Load® Purple Low Molecular Weight DNA Ladder (NEB, cat no. N0557S)
Primer sets (Figure 3, Figure 5, Supplementary Figure 1 and Supplementary Table 1.) (Integrated DNA Technologies)
Gibco Distilled Water (Thermo Fisher Scientific, cat. No. 10977035)
Absolute Ethanol (Sigma Aldrich, cat. no. 46139) CAUTION Flammable, so keep away from heat sources.
GeneJET PCR Purification Kit (Thermo Fisher Scientific, cat. no. K0701)
CAUTION Components of the kit are dangerous if swallowed and harmful to aquatic life if released to the environment.
Isopropanol (Sigma Aldrich, cat no. 33539)
Qubit™ dsDNA HS Assay Kit (Thermo Fisher Scientific, cat no. Q32854)
Blue Pippin Cassette kit 3% agarose, including TAE Buffer and Internal Marker Q3 (Sopachem, cat. no. BDF3010)
CRITICAL Cassettes must be stored at room temperature (~20ºC). The TAE buffer and marker should be stored separately at 4ºC.
CRITICAL If the Blue Pippin machine is not available for this protocol the selection and extraction step can be carried out manually, additionally requiring:
GeneJET Gel Extraction Kit (Thermo Fisher Scientific, cat. no. K0691)
Next Generation Sequencing library preparation requires:
NEBNext® Ultra™ II DNA Library Prep Kit for Illumina® (Bioke, cat no. E7645S)
NEBNext® Multiplex Oligos for Illumina (Index Primers Set 1) (Bioke, cat no. E7335S)
Agencourt AMPure XP Magnetic Beads 5 mL (Thermo Fisher Scientific, cat no. NC9959336)
2100 Bioanalyzer Instrument (Agilent Technologies, cat no. G2939BA)
Equipment
PCR tubes (Sarstedt, cat no. 72.991.002)
Implen NanoPhotometer NP80
Incubators (New Brunswick, cat. no. M1335-0002)
Microcentrifuge tubes (Eppendorf, cat. no. Z606901)
Pipette tips (Thermo Fisher Scientific, cat. no. 7325)
Pipettes (Thermo Fisher Scientific, cat. no. 4642080)
Biometra TOne Thermocycler
Centrifuge
BioRad Gel Doc XR+
VortexBlue Pippin (Sopachem, cat. no. BLU0001)
CRITICAL If you do not have the blue pippin machine on offer in your facility, gel extraction of the expanded arrays can be done manually by first concentrating the DNA samples for each condition. This is followed by manual gel extraction; the entire sample including 1 μl loading dye per 6 μl of sample should be loaded in a 3% agarose gel and run at 100 V for ~ 1 hr to maximise band separation, the expanded array (larger) band should be excised from the gel as accurately as possible and DNA extracted using the GeneJET Gel Extraction Kit. The manual gel extraction can be repeated a second time to further remove remaining non-expanded arrays.
Qubit™ 4 Fluorometer (Thermo Fisher Scientific, cat no. Q33226)
Qubit™ Assay Tubes (Thermo Fisher Scientific, cat no. Q32856)
DynaMag™-96 Bottom Magnet (Magnetic Stand) (Thermo Fisher Scientific, cat no. 12332D)
Reagent Set Up
Fresh Lysogeny Broth (LB) Medium
To produce 1L of media weigh 5 g/L yeast extract, 10 g/L NaCl, 10 g/L tryptone and mix well with 800 mL distilled H2O. After mixing thoroughly add the final 200mL of distilled H2O. The media must then be sterilized by autoclaving at 121°C for 20 mins and can be stored short term at room temperature.
Degenerate Primer Master Mix
To create the degenerate primer master mix used in Step 16, mix the three forward degenerate primers containing differing 3’ nucleotides that do not match the existing spacer in your system (See Experimental Design and Supplementary Table 1) equally to give a final concentration of ~3.3 μM of each primer and total concentration of 10 μM. The primer master mix can be stored in the – 20ºC freezer indefinitely.
1X TAE Buffer
Dilute a 40X TAE Buffer stock using MilliQ H2O. 1X TAE buffer can be stored at room temperature for a recommended maximum of 3 years.
Agarose Gels
Make fresh when needed by combining agarose and 1X TAE buffer wt/vol. The most commonly used gels in this protocol are 2% i.e. 1 g agarose and 50 mL 1X TAE. Mix and heat these components for ~ 1 min. Then add 5 μL of Sybr safe (10,000X) to allow visualization of the DNA and pour into a gel mold with a comb. Leave to set for at least 10 mins.
Procedure
Preparation of samples for use as PCR template
TIMING ~12 hrs + 1 hr
1| Set up an overnight culture of your strain of interest at the optimal temperature for growth (here, 37ºC, 180 rpm).
2| (Optional) Extract the genomic DNA of your sample using the GeneJET Genomic DNA Extraction Kit (ThermoFisher Scientific) following the supplied protocol. Elute the DNA in 200 μl of molecular biology grade H2O or less where a low concentration is expected.
CRITICAL STEP If your sample is acquired from the environment or a different source, there may be a more appropriate kit to complete this step.
CRITICAL STEP Genomic extraction is recommended when your sample has a low cell density or may contain contaminants that will affect subsequent PCR reactions.
3| (Optional) Measure the DNA concentration in ng/μl with a NanoPhotometer.
PAUSE POINT Can be stored at 4ºC overnight or frozen at -20ºC for long term storage.
Amplification of CRISPR arrays present in sample population
TIMING ~3 hrs
4| Using either the overnight culture from Step 1 directly, or the sample prepared in optional Steps 2 and 3 as a template, prepare a PCR reaction as follows:
| Reagent | Volume Added | Final Concentration |
|---|---|---|
| OneTaq® Quick-Load® 2X Master Mix with Standard Buffer | 25 μl | 1X |
| FWD Primer Set 1 (10μM) | 2 μl | 0.4 μM |
| REV Primer Set 1 (10μM) | 2 μl | 0.4 μM |
| Template DNA or Overnight Culture of your strain of interest | x μl | ~ 100 ng |
| Molecular biology grade H2O | Up to 50 μl |
Then perform a PCR amplification under the following conditions:
Conditions must be adapted to your specific primers and or product length
CRITICAL STEP If you are using an overnight culture as template for the initial PCR increasing your denaturation time to 5 mins at 98 ºC can help aid cell lysis making more genomic DNA available as a template in PCR the reaction tube.
5| Run the PCR products (5-10 μl) on a 2% (wt/vol) agarose gel containing SYBR safe (1 μl / 10 ml) at 100 V for ~30min to check for the presence of a single band of your desired size (gel extraction or PCR optimization is needed if more bands are present).
CRITICAL STEP The gel percentage can be adapted for your expected fragment size. A higher percentage of agarose allows larger separation of smaller bands.
?TROUBLESHOOTING
6| Clean and concentrate the PCR(s) using the ThermoFisher Scientific GeneJET PCR Purification kit, following the manufacturer’s instructions. Elute in 30 μl molecular biology grade H2O.
CRITICAL STEP Add isopropanol in equal proportion to the DNA binding buffer when purifying fragments smaller than 500 bp.
CRITICAL STEP The PCR reaction must be purified and H2O must be used during the elution of samples from kits, as salts in the PCR reaction or elution buffer can interfere in future steps using the Blue Pippin Machine.
7| Measure the concentration of DNA (ng/μl) on the NanoPhotometer. In the next steps large quantities of DNA can be lost so a concentration of at least 30 ng/μl in 30 μl is advised; run additional PCRs and pool if more DNA is needed.
CRITICAL STEP Between 20 and 50% of the DNA sample can be lost during the Blue Pippin process so it is better to load as much as possible without exceeding the maximum of 5 μg of DNA
PAUSE POINT DNA can be stored at 4ºC overnight or frozen at -20ºC for long term storage.
Size Selection and Extraction of Expanded Arrays
TIMING ~2 hrs 45 mins
CRITICAL In the absence of the Blue Pippin, manually extracting the expanded arrays from a gel will take ~ 4 hrs
8| Based on the size (or sizes if multiple spacers were acquired) of your arrays visualized in Step 5 choose an appropriate Blue Pippin Cassette. For arrays between 100 – 250 bp the 3% Gel Cassette with Internal Standards Marker Q3 (Cat. no. BDF3010) is advised.
9| Calibrate the Blue Pippin machine as described in the Sage Science Blue Pippin Operations Manual.
10| Load your cassette of choice into the machine, exchange the elution buffer and check buffer levels as described in the Sage Science Blue Pippin Operations Manual.
CRITICAL STEP The loading buffer must be at room temperature (~20ºC) before use in the Blue Pippin gel cassette
11| Prepare your samples for loading into the Blue Pippin (final volume 40 μl) by adding 10 μl of assigned marker or loading solution. CRITICAL STEP: This step is cassette dependant.
CRITICAL STEP Samples should be vortexed well and spun down before loading. The loading solution contains a densifying agent that will allow the sample to sink below the electrophoresis buffer layer in the well, increasing the size selection accuracy.
CRITICAL STEP The fluorescein labels (on the DNA marker) will degrade at room temperature, minimize time spent on the bench or keep on ice.
12| Program the selection range as outlined in the Sage Science Blue Pippin Operations Manual to collect the size range in which all expanded arrays are included.
CRITICAL STEP Programming a wider selection range can improve DNA yield (For example ~20 bp either side of your desired fragment size).
13| Load samples slowly into appropriate lanes and run the program as described in the Sage Science Blue Pippin Operations manual.
14| Collect the DNA extracted for the defined size range from the elution chamber.
15| (Optional) Visualize DNA by gel electrophoresis (Figure 4). Run the extracted DNA products (5-10 μl) on a 2% (wt/vol) agarose gel containing SYBR safe (1 μl/10 ml) at 100 V for ~30 min and image to check for the presence of expanded arrays.
CRITICAL STEP This step should be carried out if the user simply desires to check if their CRISPR system actively incorporates new spacers or not. Visualization of the extracted products allows confirmation of the ability to acquire spacers, and can be followed by optional deep sequencing.
CRITICAL STEP If the user desires to carry out deep sequencing without further re-amplification this step should be carried out. Visualization of the extracted products allows assessment of the success of the extraction and verification of the presence of expanded arrays in the sample before sequencing.
?TROUBLESHOOTING
16| Measure the concentration of DNA from Step 14 using the Nanophotometer to assess which step needs to be taken next (See Figure 2 and Experimental Design).
?TROUBLESHOOTING
PAUSE POINT DNA can be stored at 4ºC overnight or frozen at -20ºC for long term storage.
CRITICAL STEP If your DNA concentration is high enough immediately after this step to be used as input DNA for next generation sequencing library preparation with your kit of choice it is possible to proceed directly to sequencing (Step 20). However, it is important to note that higher starting concentrations are often recommended for library preparation kits as DNA can be lost when using magnetic beads.
Re-Amplification of the size selected arrays to increase detection sensitivity
TIMING ~2 hrs
17| Use the size selected DNA fragments as template DNA for a PCR amplification with one of the primer sets outlined in the Experimental Design section ‘Re-Amplification after Size Selection’ (Figure 5, Supplementary Figure 1 and Supplementary Table 1). Follow option A for re-amplification with internal primers, option B for re-amplification with degenerate primers, or option C for re-amplification with repeat primers. Option A should be used when a high acquisition rate is observed for your strain (Figure 6a). Options B and C (degenerate and repeat primers respectively) are capable of detecting rare events, enabling sensitive detection on gel, further detection through deep sequencing and removing the unexpanded background (Figure 6b and c). However, only the option C maintains full spacer diversity (Table 1).
CRITICAL STEP Primer set must be system specific and chosen carefully see details in Experimental Design and recommendations below.
(A) Re-Amplification with Internal Primers
(i) Prepare a PCR reaction as follows, using the fragments collected in Step 14 as a template:
| Reagent | Volume Added | Final Concentration |
|---|---|---|
| OneTaq® Quick-Load® 2X Master Mix with Standard Buffer | 25 μl | 1X |
| FWD Internal Primer Set 2 (10μM) | 2 μl | 0.4 μM |
| REV Internal Primer Set 2 (10μM) | 2 μl | 0.4 μM |
| Size Extracted Template DNA | x μl | |
| Molecular biology grade H2O | Up to 50 μl |
Perform PCR amplification under the following conditions:
Conditions must be adapted to your specific primers and or product length
(B) Re-Amplification with Degenerate Primers
(i) Prepare a PCR reaction as follows, using the fragments collected in Step 14 as a template:
| Reagent | Volume Added | Final Concentration |
|---|---|---|
| OneTaq® Quick-Load® 2X Master Mix with Standard Buffer | 25 μl | 1X |
| FWD Degenerate Primer Master Mix, Set 3 (10μM) | 2 μl | 0.4 μM |
| REV Degenerate Primer Set 3 (10μM) | 2 μl | 0.4 μM |
| Size Extracted Template DNA | x μl | |
| Molecular biology grade H20 | Up to 50 μl |
Perform PCR amplification under the following conditions:
Conditions must be adapted to your specific primers and or product length
(C) Re-Amplification with Repeat Primers
(i) Prepare a PCR reaction as follows, using the fragments collected in Step 14 as a template:
| Reagent | Volume Added | Final Concentration |
|---|---|---|
| OneTaq® Quick-Load® 2X Master Mix with Standard Buffer | 25 μl | 1X |
| FWD Repeat Primer Set 4 (10μM) | 1 μl | 0.2 μM |
| REV Repeat Primer Set 4 (10μM) | 1 μl | 0.2 μM |
| DMSO | 1.5 μl | 3% |
| Size Extracted Template DNA | 0.5 μl | |
| Molecular biology grade H2O | Up to 50 μl |
Perform PCR amplification under the following conditions:
Conditions must be adapted to your specific primers and or product length
Visualizing spacer acquisition by Gel Electrophoresis
TIMING ~45mins
18| Run the PCR products (5-10 μl) on a 2% (wt/vol) agarose gel containing SYBR safe (1 μl/10 ml) at 100 V for ~30 min to check for the presence of a single band of your desired size (gel extraction or PCR optimization is needed if more bands are present).
?TROUBLESHOOTING
19| Assess whether adaptation has occurred (presence of a band with the expected size) and when next generation sequencing is planned assess if your sample pool has a greater proportion of expanded arrays compared to the unexpanded control array (Figure 6).
Next generation sequencing to investigate spacer content (Optional)
TIMING ~ 1 day sample library preparation, 1-2 days sequencing
CRITICAL: Sequencing of samples with a next generation sequencing (NGS) platform allows even greater detection and more importantly sequence specific information for the spacers acquired within the population. There are many kits and sequencing platforms that could be used to sequence expanded array PCR amplicons, below we provide the preferences from our lab.
20| Measure the concentration of DNA (ng/μl) in your samples from Step 17 using the Flurometric Qubit machine for higher accuracy. Prepare samples with the dsDNA HS kit (10 pg/μL to 100 ng/μL) in a thin walled tube to allow accurate measurement.
21| Choose the appropriate library preparation kit for your samples input concentrations and barcoding required. Here we use the NEBNext® Ultra™ II DNA Library Prep Kit for Illumina® in combination with NEBNext® Multiplex Oligos for Illumina.
22| Prepare your samples with the chosen kit to add flow cell binding adapters and where applicable individual barcodes to each sample. This involves end preparation (A-tailing), adaptor ligation, size selection using magnetic beads, PCR amplification to add barcodes and a final clean up with magnetic beads, following the manufacturer’s instructions.
CRITICAL STEP Beads need to be at room temperature before use. Remove from 4 °C 30 mins before use.
23| Assess the quality of your sample using the Qubit (DNA concentration in ng/μl) and where available Bioanalyzer (fragment size and molar concentration).
24| If you will be sequencing your sample in house: Based on the guidelines for your sequencing instrument (here Miseq) pool all barcoded sample libraries at the desired proportions to the recommended final concentration here (2 nM). Denature your sample with an equal amount of freshly diluted 0.2 N NaOH and dilute to the appropriate loading concentration and volume for the flow cell following the Illumina guidelines for the specific platform. CRITICAL: The entire sample is not required for sequencing and it is therefore recommended to store a portion of your sample library before dilution and denaturation at -20 °C for long term storage.
CRITICAL STEP Use equimolar amounts of all barcoded libraries to maintain equal depth among all samples sequenced.
CRITICAL STEP The recommended loading concentration will differ for each sequencing machine. The recommended concentration for the MiSeq is 6 – 10 pM, here we use 4 pM, as we load very small fragments that cluster more efficiently than the recommended minimum 250 bp fragments.
PAUSE POINT Prepared sample library can be stored long term at 20 ºC until sequencing will be carried out.
25| Following the specific instructions for the sequencing instrument load the appropriately diluted sample into the reagent cartridge. Next clean and load the flow cell into the machine, check for any additional solutions required and start the run. Alternatively frozen libraries prepared above can be sent for custom sequencing.
26| In order to analyse sequencing data for each sample, the sequences must first be quality assessed, normalized, merged where necessary and de-multiplexed (separated by unique barcodes into samples). This can be carried out at a basic level by the sequencing instrument itself, or a software package or more detailed pipeline can be used7,17,21.
27| Once de-multiplexed, for each sample all spacer-containing sequences can be extracted by identifying conserved CRISPR array regions such as the 3′ end of the degenerate primer and the 5′ end of the repeat. The sequences flanked by these regions can be extracted as spacer sequences23.
28| In order to assess spacer content extracted sequences are aligned to potential spacer sources such as the host genome, other known factors in the cell such as plasmids or when the potential sources are unknown, sequences can be screened using BLAST (Basic Local Alignment Search Tool) or other sequence databases24.
29| After extraction of all spacer sequences duplicate spacers should be removed to determine the number of unique spacers acquired and to remove the duplication bias created by PCR amplification. In addition, normalization should be applied to the sequences if any form of degenerate primer was used for re-amplification in PCR 2 (See Table 1)21.
30| The orientation of the extracted spacers then needs to be determined to enable extraction of the PAM containing nucleotides adjacent to the target sequence7,17,25. Web tools such as WebLogo can then be used to further aid determination of the strongest PAM sequence for the system of interest9,26.
31| Finally, spacer content (nucleotide sequence) and length of the extracted sequences can be analysed and compared between conditions17.
Troubleshooting
Troubleshooting advice can be found in Table 2.
Timing
Step 1, Overnight growth ~12 hrs
Steps 2 and 3 (optional), Genomic DNA extraction 1 hr
Step 4, PCR amplification 1 2 hrs
Steps 5-7, Gel electrophoresis and purification of sample 1 hr
Steps 8-14, Extraction of expanded arrays 2 hrs
Step 15 (optional), Visualization of extracted DNA 45 mins
Steps 16, Determination of DNA concentration 10 mins
Step 17, PCR amplification 2 ~2 hrs
Steps 18 and 19, Gel electrophoresis to visualize expanded arrays and determine if sequencing is necessary 45 mins
Steps 20-24 (optional), Sample preparation for next generation sequencing ~1 day
Step 25-31 (optional), Next generation sequencing sequencing and analysis of prepared sample ~1-2 days
Anticipated Results
The outlined protocol provides a method enabling detection of spacer acquired by active CRISPR systems even at extremely low levels. To test the sensitivity of CAPTURE we mimicked a population in which only a small fraction of the cells had acquired one new spacer. To achieve this we used both the wild type strain Escherichia coli K12 BW25113 containing a type I-E CRISPR system27 and a variant of this strain, known to contain an extra 32 bp spacer10, referred to as unexpanded and expanded respectively (Figure. 3). The low prevalence of acquisition was simulated by the serial dilution of genomic DNA extracted from the expanded strain to 10-6 in DNA extracted from the unexpanded strain. Such a dilution series allowed us to test the true sensitivity of our method by simulating acquisition rates as low as 1 in 106 expanded arrays (0.0001% of the population) with an upper limit of 1 in 10 expanded arrays (10% of the population) acquiring a spacer (Figure 6). In our experience if all steps are followed this procedure can detect spacers acquired in just 1 in 105 cells of a population (Figure 6), a sensitivity which can be increased even further with the subsequent use of deep sequencing on the prepared sample17.
Supplementary Material
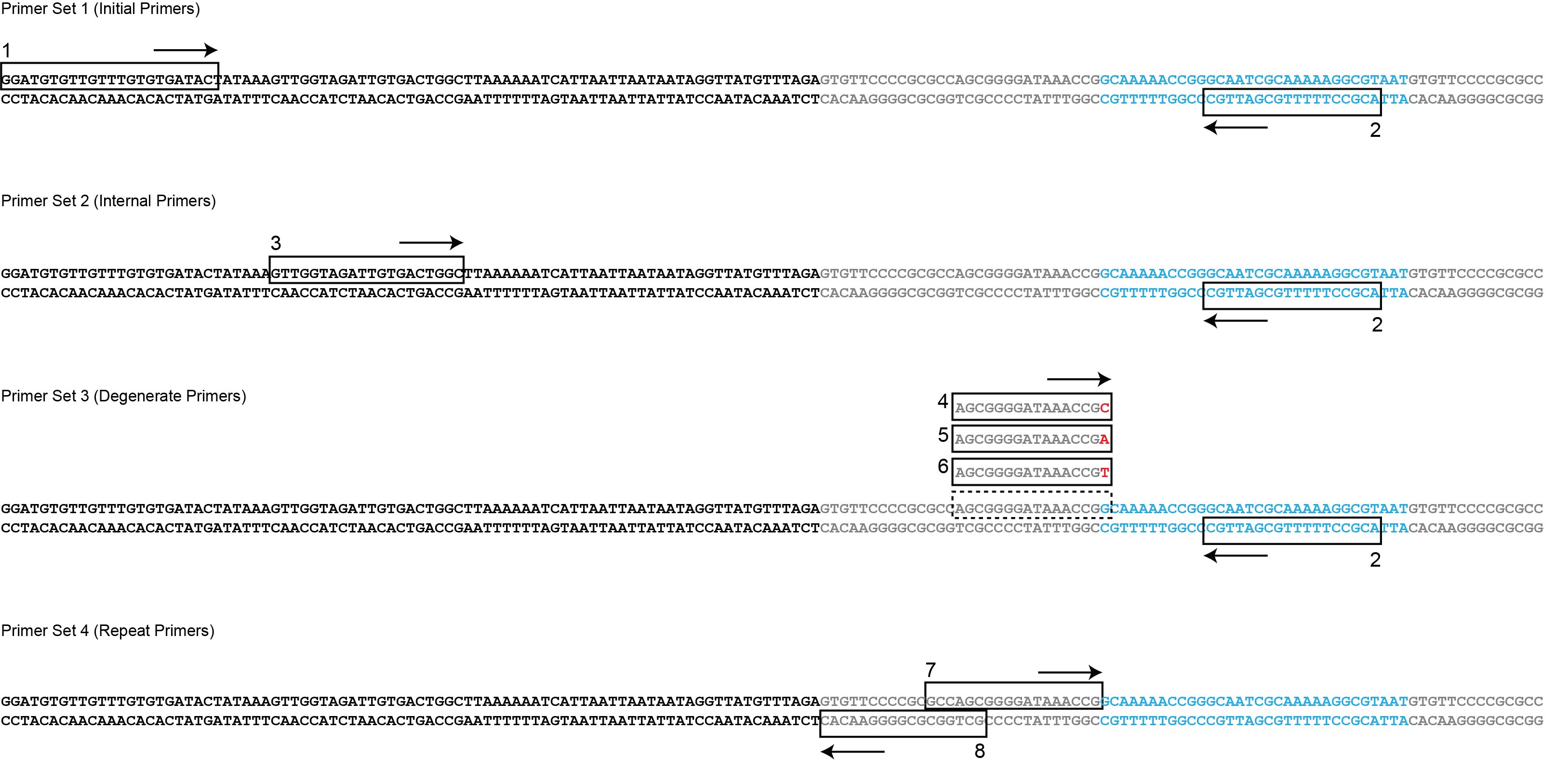
The double stranded DNA sequence of the array is shown here including the leader sequence (black) repeat sequences (grey) and the sequence of spacer 1 (blue). Primer sets designed for the type I-E CRISPR system of E. coli K12 are indicated with black boxes and directional arrows.
{kind=link}
Editorial Summary.
Here the authors describe CAPTURE, a versatile and sensitive protocol to detect spacer acquisition events in native CRISPR arrays using nested PCR amplification and amplicon size selection.
Acknowledgements
SJJB is supported by European Research Council (ERC) StG grant 639707, NWO Vidi grant 864.11.005, and a TU Delft start-up grant. This work was supported by the Netherlands Organisation for Scientific Research (NWO/OCW), as part of the Frontiers of Nanoscience (NanoFront) program. The authors would like to thank members of the Brouns Lab for discussions on the manuscript and feedback.
Footnotes
Author Contributions:
Conceived and designed the experiments: REM, CA, JV and SJJB. Performed the experiments: REM, CA and JV. Analyzed the data: REM, CA, JV and SJJB. Wrote the paper: REM and SJJB with input from all authors.
Competing Financial Interests:
The authors declare that no competing financial interests exist.
References
- 1.Barrangou R, et al. CRISPR provides acquired resistance against viruses in prokaryotes. Science. 2007;315:1709–12. doi: 10.1126/science.1138140. [DOI] [PubMed] [Google Scholar]
- 2.Jackson SA, et al. CRISPR-Cas: Adapting to change. Science. 2017;356 doi: 10.1126/science.aal5056. eaal5056. [DOI] [PubMed] [Google Scholar]
- 3.Brouns SJJ, et al. Small CRISPR RNAs guide antiviral defense in prokaryotes. Science. 2008;321:960–4. doi: 10.1126/science.1159689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Koonin EV, Makarova KS, Zhang F. Diversity, classification and evolution of CRISPR-Cas systems. Current Opinion in Microbiology. 2017;37:67–78. doi: 10.1016/j.mib.2017.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Heler R, et al. Cas9 specifies functional viral targets during CRISPR–Cas adaptation. Nature. 2015;519:199–202. doi: 10.1038/nature14245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hynes AP, Villion M, Moineau S. Adaptation in bacterial CRISPR-Cas immunity can be driven by defective phages. Nat Commun. 2014;5 doi: 10.1038/ncomms5399. 4399. [DOI] [PubMed] [Google Scholar]
- 7.Staals RHJ, et al. Interference-driven spacer acquisition is dominant over naive and primed adaptation in a native CRISPR–Cas system. Nat Commun. 2016;7 doi: 10.1038/ncomms12853. 12853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Datsenko KA, et al. Molecular memory of prior infections activates the CRISPR/Cas adaptive bacterial immunity system. Nat Commun. 2012;3 doi: 10.1038/ncomms1937. 945. [DOI] [PubMed] [Google Scholar]
- 9.Yosef I, Goren MG, Qimron U. Proteins and DNA elements essential for the CRISPR adaptation process in Escherichia coli. Nucleic Acids Res. 2012;40:5569–5576. doi: 10.1093/nar/gks216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Swarts DC, Mosterd C, van Passel MWJ, Brouns SJJ. CRISPR Interference Directs Strand Specific Spacer Acquisition. PLoS One. 2012;7:e35888. doi: 10.1371/journal.pone.0035888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yosef I, et al. DNA motifs determining the efficiency of adaptation into the Escherichia coli CRISPR array. Proc Natl Acad Sci USA. 2013;110:14396–401. doi: 10.1073/pnas.1300108110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Silas S, et al. Direct CRISPR spacer acquisition from RNA by a natural reverse transcriptase-Cas1 fusion protein. Science (80-.) 2016;351:aad4234–aad4234. doi: 10.1126/science.aad4234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nuñez JK, Bai L, Harrington LB, Hinder TL, Doudna JA. CRISPR Immunological Memory Requires a Host Factor for Specificity. Mol Cell. 2016;62:824–833. doi: 10.1016/j.molcel.2016.04.027. [DOI] [PubMed] [Google Scholar]
- 14.Amlinger L, Hoekzema M, Wagner EGH, Koskiniemi S, Lundgren M. Fluorescent CRISPR Adaptation Reporter for rapid quantification of spacer acquisition. Sci Rep. 2017;7 doi: 10.1038/s41598-017-10876-z. 10392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Díez-Villaseñor C, Guzmán NM, Almendros C, García-Martínez J, Mojica FJM. CRISPR-spacer integration reporter plasmids reveal distinct genuine acquisition specificities among CRISPR-Cas I-E variants of Escherichia coli. RNA Biol. 2013;10:792–802. doi: 10.4161/rna.24023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schmidt F, Cherepkova MY, Platt RJ. Transcriptional recording by CRISPR spacer acquisition from RNA. Nature. 2018;562:380–385. doi: 10.1038/s41586-018-0569-1. [DOI] [PubMed] [Google Scholar]
- 17.Kieper SN, et al. Cas4 Facilitates PAM-Compatible Spacer Selection during CRISPR Adaptation. Cell Rep. 2018;22:3377–3384. doi: 10.1016/j.celrep.2018.02.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Edwards RA, McNair K, Faust K, Raes J, Dutilh BE. Computational approaches to predict bacteriophage–host relationships. FEMS Microbiol Rev. 2016;40:258–272. doi: 10.1093/femsre/fuv048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Snyder JC, Bateson MM, Lavin M, Young MJ. Use of Cellular CRISPR (Clusters of Regularly Interspaced Short Palindromic Repeats) Spacer-Based Microarrays for Detection of Viruses in Environmental Samples. Appl Environ Microbiol. 2010;76:7251–7258. doi: 10.1128/AEM.01109-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shariat N, Dudley EG. CRISPRs: Molecular Signatures Used for Pathogen Subtyping. Appl Environ Microbiol. 2014;80:430–439. doi: 10.1128/AEM.02790-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Modell JW, Jiang W, Marraffini LA. CRISPR–Cas systems exploit viral DNA injection to establish and maintain adaptive immunity. Nature. 2017;544:101–104. doi: 10.1038/nature21719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Baba T, et al. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol Syst Biol. 2006;2 doi: 10.1038/msb4100050. 2006.0008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Biswas A, Gagnon JN, Brouns SJJ, Fineran PC, Brown CM. CRISPRTarget: bioinformatic prediction and analysis of crRNA targets. RNA Biol. 2013;10:817–27. doi: 10.4161/rna.24046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 25.Biswas A, Staals RHJ, Morales SE, Fineran PC, Brown CM. CRISPRDetect: A flexible algorithm to define CRISPR arrays. BMC Genomics. 2016;17:356. doi: 10.1186/s12864-016-2627-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Crooks GE, Hon G, Chandonia J-M, Brenner SE. WebLogo: a sequence logo generator. Genome Res. 2004;14:1188–90. doi: 10.1101/gr.849004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Díez-Villaseñor C, Almendros C, García-Martínez J, Mojica FJM. Diversity of CRISPR loci in Escherichia coli. Microbiology. 2010;156:1351–61. doi: 10.1099/mic.0.036046-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The double stranded DNA sequence of the array is shown here including the leader sequence (black) repeat sequences (grey) and the sequence of spacer 1 (blue). Primer sets designed for the type I-E CRISPR system of E. coli K12 are indicated with black boxes and directional arrows.