

Abstract
Purpose
Abnormalities in cerebrospinal fluid (CSF) have been reported in Hurler syndrome, a fatal neurodegenerative lysosomal disorder. While no biomarker has predicted neurocognitive response to treatment, one of these abnormalities, glycosaminoglycan nonreducing ends (NREs), holds promise to monitor therapeutic efficacy. A trial of intrathecal enzyme replacement therapy (ERT) added to standard treatment enabled tracking of CSF abnormalities, including NREs. We evaluated safety, biomarker response, and neurocognitive correlates of change.
Methods
In addition to intravenous ERT and hematopoietic cell transplantation, patients (N = 24) received intrathecal ERT at four peritransplant time points; CSF was evaluated at each point. Neurocognitive functioning was quantified at baseline, 1 year, and 2 years posttransplant. Changes in CSF biomarkers and neurocognitive function were evaluated for an association.
Results
Over treatment, there were significant decreases in CSF opening pressure, biomarkers of disease activity, and markers of inflammation. Percent decrease in NRE from pretreatment to final intrathecal dose posttransplant was positively associated with percent change in neurocognitive score from pretreatment to 2 years posttransplant.
Conclusion
Intrathecal ERT was safe and, in combination with standard treatment, was associated with reductions in CSF abnormalities. Critically, we report evidence of a link between a biomarker treatment response and neurocognitive outcome in Hurler syndrome.
Keywords: mucopolysaccharidosis, biomarkers, enzyme replacement therapy, neurocognitive decline, intrathecal therapy
INTRODUCTION
Neurocognitive decline, a distinguishing symptom of the severe form of mucopolysaccharidosis type I (MPS I), is quantified in very young affected children by successive losses in early IQ points. The pathogenesis of this decline is not completely understood but has long been an area of investigation and theory development.1–4 Besides neurocognitive decline, severe MPS I (Hurler syndrome, MPS IH, OMIM 607014) is distinguished from the attenuated phenotypes by its early lethality, as the natural history of severe MPS I involves death within the first decade of life.5,6 MPS I is an autosomal recessive disorder caused by pathogenic variants in the IDUA gene, resulting in insufficient production of α-L-iduronidase, a lysosomal enzyme critical to degrading long chains of sugar, or glycosaminoglycans (GAG).5 In all phenotypes of MPS I, including Hurler syndrome and the attenuated forms, accumulating GAG in nearly all organ systems both directly and indirectly2,7–9 causes worsening cardiac and pulmonary dysfunction, hepatosplenomegaly, bone and joint disease, ophthalmologic and auditory problems, and increased risk for central nervous system (CNS) complications such as hydrocephalus and cervical spinal cord compression.
Current recommended treatment for Hurler syndrome is hematopoietic cell transplantation (HCT), which arrests neurocognitive decline, halts accumulating somatic burden in many organ systems, and prevents early death by providing a stable source of enzyme via donor-derived cells of hematopoietic origin.10–13 Deficient enzyme can also be supplemented with intravenous recombinant iduronidase, i.e., enzyme replacement therapy (ERT). Intravenous ERT is presumed to be unable to penetrate the blood–brain barrier to treat the CNS manifestations of Hurler syndrome,8,14 but it is used as an adjunct to HCT to decrease morbidity and mortality.15–17 For most patients with Hurler syndrome, neurocognitive outcomes include IQ scores that are below the average range even following successful HCT.12,13,18 This persistent neurocognitive morbidity is often the target of innovative therapies for neurodegenerative MPS types, through increasing enzyme in the CNS.14,19–23
However, existing therapies and those in trial all suffer from a serious lack of precision in predicting neurocognitive outcomes. There has been considerable emphasis on identifying biomarker correlates of therapeutic outcomes, but to date no biomarker has shown accurate, reliable clinical correspondence with neurocognitive course.3,14 GAG levels in urine or serum may be different between severe and attenuated phenotypes, but not to a degree to distinguish phenotype reliably,8,24 nor does urine or serum GAG level predict neurocognition with any precision. Of the three major GAG chains, including heparan sulfate, keratan sulfate, and dermatan sulfate, accumulation of heparan sulfate has been implicated in neurocognitive abnormalities.2,3,5 Recently a focus on the predictable degradation of GAG starting with the nonreducing end (NRE) of the chains led to development of a methodology for quantifying disease-specific biomarkers from these NREs.25 It was proposed that this method be used for monitoring treatment efficacy. This proposal could be tested when our phase I trial of intrathecal (IT) administration of ERT for Hurler syndrome presented the unique opportunity to obtain serial samples of cerebrospinal fluid (CSF) to analyze for potential biomarker response, including elevated heparan sulfate–derived NREs. We examined whether observed NRE changes were associated with change in neurocognitive function in the first two years following treatment. This trial (NCT00638547) involved standard of care at the University of Minnesota (i.e., peritransplant intravenous ERT as an adjunct to HCT), with the addition of IT ERT as means of “bridge therapy” between diagnosis and the anticipated time to achieve donor-derived microglial engraftment in the brain. The present study evaluated safety of IT ERT, monitored alterations in CSF abnormalities at each time point that IT enzyme was delivered, and measured neurocognitive functioning prospectively to assess for clinical correlates of biochemical change.
MATERIALS AND METHODS
Patient eligibility and study design
This was part of a two-year, single center, open label phase I trial of IT ERT as adjunctive therapy to intravenous ERT and HCT in Hurler syndrome. Schedule of therapies, biomarker sampling, and neurocognitive evaluations is depicted in Figure S1. Patients were eligible for study enrollment if they (1) had a diagnosis of Hurler syndrome, (2) were aged >6 months and <4 years, (3) were under consideration for HCT at our center, and (4) had received ≤4 previous doses of intravenous ERT. Among the 39 patients with Hurler syndrome evaluated for HCT at the University of Minnesota during the study period (2008–2014), 12 did not meet inclusion criteria and 1 declined the addition of IT ERT to the standard HCT protocol. Thus 26 patients were enrolled to receive IT ERT in the peri-HCT period (Figure S2). One patient withdrew due to poor cardiac function detected prior to IT therapy, and a second received one dose of IT ERT and elected to undergo HCT at another institution and withdrew from the study. All patients also received concomitant intravenous ERT as described below. Lumbar puncture with CSF collection was performed at each IT infusion time point by one of the authors (J.T., W.P.M., P.J.O., T.C.L.). All therapies were administered by one of the authors (J.T., W.P.M., P.J.O., T.C.L.). This study was approved by the Committee on the Use of Human Subjects in Research at the University of Minnesota. This protocol was reviewed and approved by the FDA (IND 100782). This trial was registered at www.clinicaltrials.gov as NCT00638547. Informed consent was obtained from all legal guardians.
Intrathecal enzyme replacement administration
IT ERT was administered by standard lumbar puncture using sterile technique. At each time point, recombinant iduronidase (0.05 mg/kg) was delivered over 1–2 minutes in 4 mL of Elliott’s B® solution. Infusion of IT ERT was planned at four discrete time points with respect to HCT: 8–12 weeks before HCT, 2 weeks before HCT, 100 days after HCT, and 6 months after HCT (Figure S1). The timing coincided with scheduled sedation. Dosing every 3 months was based on the experience in the MPS I canine model.26 For both time points pre-HCT, IT ERT was given prior to commencing the transplant preparative regimen. In two instances, patients received intravenous ERT prior to the first IT ERT dose.
Allogeneic hematopoietic stem cell transplantation
Transplant allografts were selected according to institutional guidelines and based upon donor–recipient human leukocyte antigen matching characteristics. Stem cell sources included unrelated umbilical cord blood (n = 21), unrelated donor marrow (n = 2), and noncarrier sibling donor marrow (n = 1). All 24 patients underwent a myeloablative, busulfan-based preparative regimen. Of those, 20 were treated with alemtuzumab (0.9 mg/kg), cyclophosphamide (200 mg/kg), and busulfan (targeted total area-under-the-curve exposure [AUC] of 75 mg × hour/mL). The remaining four patients received thymoglobulin (10 mg/kg), fludarabine (160 mg/m2), and busulfan (targeted total AUC of 90 mg/hour/mL). Twenty-three patients successfully engrafted; one did not and was retransplanted 6 weeks later, with reduced intensity preparative regimen including fludarabine, busulfan, and total body irradiation. Both times this patient's stem cell sources were umbilical cord blood; engraftment was successful upon second HCT.
Peritransplant intravenous recombinant iduronidase administration
All patients were treated with peritransplant intravenous ERT (0.58 mg/kg, once weekly) per institutional guidelines; patients received an average of 12 doses pretransplant and 8 doses posttransplant.
Cerebrospinal fluid evaluation
During the initial evaluation and subsequent IT delivery time points, a lateral decubitus lumbar puncture was performed with an opening pressure measurement; CSF was obtained and analyzed for cell count, protein concentration, glucose concentration, and biomarker analysis. End tidal CO2 was monitored and maintained from 25–40 mm Hg to ensure opening pressure accuracy. Methodology for biomarker analysis is presented in the supplementary information (Table S1). For protein and glucose concentrations and biomarker analysis, comparison ranges were based on previously published findings for unaffected controls whose CSF was compared with that of patients with Hurler syndrome.27
Neurocognitive assessment
A standard neurocognitive evaluation protocol previously described18 was used for all patients at baseline (before any intervention, except for two patients who had received prior intravenous ERT) and after HCT at annual visits to the University of Minnesota. The neurocognitive evaluations included a test of developing intellectual function: the Mullen Scales of Early Learning28 or the Bayley Scales of Infant and Toddler Development, Third Edition.29 Both of these tests yield a norm-referenced score that reflects overall neurocognitive functioning (emerging IQ). Specifically, the IQ is represented by age-based standardized scores with a population mean of 100 and standard deviation of 15, with qualitative ranges of average (85–115), below average (70–84), and impaired (<70).
Statistics
Design and analyses followed an intention to treat approach as all study patients received all therapies. Continuous measures were summarized as mean (standard deviation) along with median (interquartile range [IQR]) and categorical measures as frequency (%). The change in mean biomarker from time point 1 to subsequent time points was evaluated using generalized estimating equations with exchangeable working correlation structure and robust variance estimation for confidence intervals and p value.30 A Wald test from nested models was also used to evaluate if a mean biomarker at any time point varied significantly from the other time points. Additionally, the pairwise comparisons between time point 1 and time points 2, 3, and 4 were adjusted for multiple comparisons using the Benjamini–Hochberg correction. Comparison of baseline IQ scores of patients with missing versus complete data was evaluated with generalized estimating equations assuming independence working correlation structure for baseline IQ. The association between biomarkers and percent change in IQ was evaluated using generalized estimating equations with independence working correlation structure and robust variance estimation for confidence intervals and p values. These analyses were conducted using R v3.4.3 (R Foundation for Statistical Computing, Vienna, Austria).
RESULTS
Patients and safety
Characteristics of the 24 patients who entered the trial are summarized in Table 1. There were five deaths, all following HCT: one due to myocardial infarction, one due to mycotic thrombus, one from influenza A, and two from respiratory failure (presumed viral pneumonia). None of the deaths were attributed to IT ERT. There were no adverse events related to IT ERT. An exact binomial test comparing survival within our study sample with the 84% survival rate in a large modern cohort31 indicated no difference in rates of survival (p = 0.574).
Table 1.
Patient characteristics: values expressed are mean (SD) and median (IQR) for continuous measures or N (%) for categorical measures
| Covariate (N) | Mean (SD) | Median (IQR) |
|---|---|---|
| (Total N = 24) | ||
| Male | 12 (50.0%) | |
| Age at transplant (months) (N = 24) | 16.3 (6.17) | 15.1 (12.8, 18.9) |
| IQ (M = 100 ± 15): | ||
| Baseline (N = 22) | 84.6 (13.4) | 86.0 (73.5, 97.0) |
| Year 1 (N = 18) | 76.7 (16.8) | 78.5 (65.5, 90.8) |
| Year 2 (N = 15) | 79.1 (18.5) | 79.0 (69.5, 90.5) |
IQR interquartile range.
Cerebrospinal fluid evaluation
The CSF opening pressure, total protein, glucose, red cell count, and white cell count were measured at the time of IT ERT infusion. The upper limit of normal opening pressure in a child is 20 cm H2O;32 the mean opening pressure in Hurler syndrome patients prior to ERT therapy was 24.2 cm H2O (SD = 7.4), which decreased over the treatment period (Fig. 1, Table 2). When changes in opening pressure were analyzed (Table 2), the reduction of 4.5 cm H2O was significant from time point 1 to 2, i.e., prior to any intervention to just before HCT, reflecting change based on a single dose of IT enzyme and concurrent intravenous enzyme (p = 0.0499). Overall there was clinically significant variability in opening pressure over the full treatment period (p = 0.0003). Mean opening pressure decreased from time point 1 to 3 (p = 0.0004), before increasing again at time point 4, such that it was not significantly different from time point 1 (p = 0.0861). Mean concentrations of CSF total protein did not significantly vary between any time point (p = 0.7760; Table 2), but mean concentrations of CSF glucose did vary significantly (p = 0.0150; Table 2).
Fig. 1.
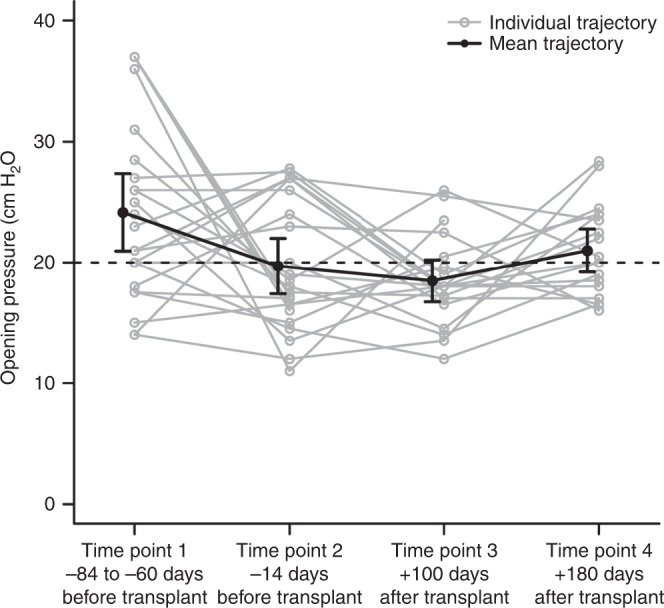
Change in cerebrospinal fluid opening pressure throughout treatment. Cerebrospinal fluid opening pressure on lumbar puncture decreased significantly from time point 1 to 2, i.e., prior to transplant (p = 0.0499). At time point 3, which is 100 days posttransplant, the opening pressure was also significantly lower than time point 1 (p = 0.0004). Opening pressure increased somewhat at time point 4, which is 6 months posttransplant, such that it was no longer significantly different from time point 1 (p = 0.0861).
Table 2.
Comparison of mean measures across all time points and pairwise comparisons for changes from time point 1 to follow-up time points
| Time point | Mean (SD) | Δ From time point 1 (95% CI) | p value |
|---|---|---|---|
| Opening pressure (cm H2O; ref range ≤ 20 cm H2O) (ref. 32) | |||
| 1 (N = 23) | 24.2 (7.4) | - | - |
| 2 (N = 23) | 19.7 (5.3) | −4.5 (−8.6, −0.4) | 0.0499 |
| 3 (N = 20) | 18.5 (3.7) | −5.7 (−8.6, −2.8) | 0.0004 |
| 4 (N = 19) | 21.0 (3.7) | −3.2 (−6.8, 0.5) | 0.0861 |
| Any time point | - | - | 0.0003 |
| CSF protein (mg/dL; non-MPS ref 15–60 mg/dL (ref. 27) | |||
| 1 (N = 23) | 31.0 (13.9) | - | - |
| 2 (N = 24) | 30.8 (10.7) | −0.3 (−5.4, 4.7) | 0.8951 |
| 3 (N = 20) | 33.9 (12.6) | 2.2 (−3.7, 8.1) | 0.8951 |
| 4 (N = 21) | 33.0 (15.9) | 1.1 (−6.7, 8.9) | 0.8951 |
| Any time point | - | - | 0.7760 |
| CSF glucose (mg/dL; non-MPS ref 40–70 mg/dL (ref. 27) | |||
| 1 (N = 23) | 48.8 (4.8) | - | - |
| 2 (N = 24) | 46.2 (5.3) | −2.6 (−4.3, −1.0) | 0.0058 |
| 3 (N = 21) | 48.7 (7.2) | −0.1 (−2.4, 2.3) | 0.9536 |
| 4 (N = 21) | 51.0 (8.8) | 2.3 (−1.6, 6.2) | 0.3834 |
| Any time point | - | - | 0.0150 |
| CSF antidrug antibodies | |||
| 1 (N = 23) | 0.012 (0.041) | - | - |
| 2 (N = 23) | 0.082 (0.085) | 0.069 (0.034, 0.104) | 0.0003 |
| 3 (N = 20) | 0.003 (0.003) | −0.009 (−0.024, 0.006) | 0.2534 |
| 4 (N = 19) | 0.002 (0.002) | −0.010 (−0.026, 0.005) | 0.2534 |
| Any time point | - | - | <0.0001 |
| I0S6 (ng/mL; non-MPS ref < 30 ng/mL (ref. 27) | |||
| 1 (N = 20) | 240.4 (100.4) | - | - |
| 2 (N = 20) | 157.9 (76.6) | −82.5 (−112.2, −52.8) | <0.0001 |
| 3 (N = 20) | 127.2 (55.5) | −113.2 (−145.9, −80.5) | <0.0001 |
| 4 (N = 20) | 110.1 (45.9) | −130.3 (−166.0, −94.6) | <0.0001 |
| Any time point | - | - | <0.0001 |
| I0S0 (ng/mL; non-MPS ref < 15 ng/mL (ref. 27) | |||
| 1 (N = 20) | 58.2 (32.0) | - | - |
| 2 (N = 20) | 29.9 (15.4) | −28.3 (−42.0, −14.6) | <0.0001 |
| 3 (N = 20) | 16.8 (12.8) | −41.5 (−56.9, −26.0) | <0.0001 |
| 4 (N = 20) | 11.2 (10.6) | −47.1 (−61.2, −32.9) | <0.0001 |
| Any time point | - | - | <0.0001 |
| Heparan sulfate (ng/mL; non-MPS ref < 120 ng/mL (ref. 27) | |||
| 1 (N = 20) | 283.7 (99.5) | - | - |
| 2 (N = 20) | 262.9 (80.9) | −20.8 (−60.1, 18.6) | 0.3008 |
| 3 (N = 20) | 212.9 (83.8) | −70.8 (−118.0, −23.5) | 0.0050 |
| 4 (N = 20) | 195.1 (60.2) | −88.6 (−131.2, −46.0) | 0.0001 |
| Any time point | - | - | 0.0001 |
| Heparin cofactor II–thrombin (ng/mL; non-MPS ref < 0.25 ng/mL (ref. 27) | |||
| 1 (N = 21) | 7.1 (3.1) | - | - |
| 2 (N = 23) | 4.8 (3.5) | −2.3 (−3.6, −1.0) | 0.0006 |
| 3 (N = 21) | 2.8 (1.9) | −4.4 (−5.7, −3.1) | <0.0001 |
| 4 (N = 21) | 3.3 (3.1) | −3.8 (−5.7, −2.0) | <0.0001 |
| Any time point | - | - | <0.0001 |
| MCP-1 (pg/mL; no reference range (ref. 27) | |||
| 1 (N = 10) | 976.7 (401.1) | - | - |
| 2 (N = 10) | 891.4 (526.7) | −85.3 (−257.5, 86.9) | 0.3317 |
| 3 (N = 10) | 760.9 (197.2) | −215.8 (−477.2, 45.6) | 0.1586 |
| 4 (N = 10) | 595.7 (169.5) | −381.0 (−641.5, −120.5) | 0.0124 |
| Any time point | - | - | 0.0006 |
| MIP-1b (pg/mL; no reference range27) | |||
| 1 (N = 10) | 15.0 (7.0) | - | - |
| 2 (N = 10) | 10.8 (3.6) | −4.2 (−7.5, −0.9) | 0.0205 |
| 3 (N = 10) | 14.1 (7.2) | −0.9 (−6.3, 4.5) | 0.7370 |
| 4 (N = 10) | 9.0 (4.8) | −6.0 (−10.2, −1.8) | 0.0162 |
| Any time point | - | - | 0.0070 |
| SDF1 (pg/mL; no reference range27) | |||
| 1 (N = 10) | 385.1 (94.7) | - | - |
| 2 (N = 10) | 351.7 (123.0) | −33.3 (−144.9, 78.2) | 0.8374 |
| 3 (N = 10) | 334.9 (168.7) | −50.1 (−179.4, 79.2) | 0.8374 |
| 4 (N = 10) | 387.7 (303.1) | 2.6 (−192.5, 197.8) | 0.9788 |
| Any time point | - | - | 0.5378 |
| IL-1Ra (pg/mL; no reference range27) | |||
| 1 (N = 10) | 18.7 (6.6) | - | - |
| 2 (N = 10) | 25.0 (32.7) | 6.3 (−12.8, 25.3) | 0.5203 |
| 3 (N = 10) | 34.1 (33.5) | 15.3 (−3.1, 33.8) | 0.2497 |
| 4 (N = 10) | 32.0 (31.8) | 13.2 (−5.5, 31.9) | 0.2497 |
| Any time point | - | - | 0.2759 |
CI confidence interval, CSF cerebrospinal fluid, MPS mucopolysaccharidosis.
Biochemical abnormalities within the CSF included biomarkers of disease activity (NREs, total heparan sulfate [HS], heparin cofactor II–thrombin complex [HCIIT]) and inflammatory factors (SDF1a, IL-1ra, MCP-1, and MIP-1b; Table S2), all of which we previously found to be elevated in treatment-naïve patients with Hurler syndrome.27 The heparan sulfate NREs I0S0 and I0S6 separately showed significant reductions at each time point and overall (p < 0.0001 for each; Table 2; Fig. 2). The reduction in mean NRE I0S0 was more than 80% from time point 1 to 4, while for NRE I0S6 it was approximately 54% for this same time span. Some patients had undetectable NRE I0S0 at time point 4 (100% reduction). These observations of significant decrements in each NREs I0S0 and I0S6 include the span prior to HCT, i.e., time points 1 and 2 (p < 0.0001 for both; Table 2). Evaluation of HS showed a significant drop, approximately 30%, between time points 1 and 4 (p = 0.0001; Table 2, Fig. 2), but only a decrease of 7% between time points 1 and 2 (p = 0.3008; Table 2). Baseline HCIIT values were elevated in the CSF, consistent with prior observations.27 There was a significant HCIIT decrement by about a third between time points 1 and 2, as well as an overall decrease by more than half between time points 1 and 4 (p < 0.0001, Table 2, Fig. 2). Although most patients did not have CSF antidrug antibodies (ADA) at time point 1, 87% (i.e., 20/23) of patients showed a transient increase in antibody detection at time point 2. Almost universally, the CSF had little to no detectable antibody at time points 3 and 4, i.e., post-HCT (Table 2).
Fig. 2.
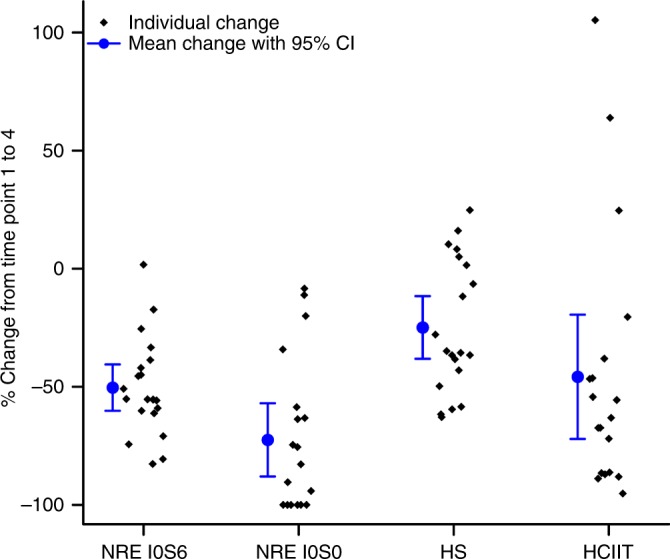
Decrease in biochemical abnormality from pretreatment to last follow-up. Percent change in biochemical abnormalities in the cerebrospinal fluid from time point 1 to 4, i.e., before any treatment to final intrathecal recombinant iduronidase infusion, 6 months posttransplant. All reductions were significant at p < 0.0001. Nonreducing ends (NREs) I0S0 and I0S6 (N = 20, N = 19, respectively) separately showed significant reductions, by more than 80% for I0S0 and 54% for I0S6. Heparan sulfate (HS, N = 20) decreased by 30%, and heparin cofactor II–thrombin complex (HCIIT, N = 19) decreased by more than half. CI confidence interval.
MPS disease has been shown to have an inflammatory component, in both the brain and bones.1–3 This includes previous findings of elevated CSF cytokines in this patient cohort when still treatment naïve.27 In the current study, cytokines were able to be assayed in ten patients. The levels of SDF1a, IL-1ra, MCP-1, and MIP-1b (Table S2) were taken at serial time points, and the mean concentrations of SDF1a and IL-1ra were not significantly different over time (p = 0.5378 and p = 0.2759, respectively; Table 2). However, MCP-1 and MIP-1b varied significantly over time (p = 0.0006 and p = 0.0070, respectively) with their mean values decreasing from time point 1 to 4 (p = 0.0124 and p = 0.0161, respectively; Table 2).
To identify whether a strong response for one biomarker (e.g., I0S6) was also reflected in responsiveness of other biomarkers within the same patient, we conducted Pearson correlations for percent decrease for each measure in Table 2. Within patient, percent decrease of NRE I0S6 was strongly correlated with percent decrease of I0S0 and heparan sulfate, as expected given that both I0S6 and I0S0 are derived from this GAG (Table S3). Within patient, percent decrease of I0S6 was negatively correlated with percent decrease of MCP-1 and SDF1, suggesting they increase relative to the decrease in I0S6. However, many correlations were smaller and suggest that a good response (in % decrease) may not necessarily reflect a linear change in other measures.
Neurocognitive outcomes
Neurocognitive outcomes, henceforth referred to as IQ scores, at 1 and 2 years after HCT were compared with baseline (i.e., pretreatment) scores. Of the 24 patients treated with all four doses of IT therapy, 22 had baseline neurocognitive assessment data and 15 had neurocognitive assessment data at all three time points (Table 1). There was no difference in baseline IQ scores between the 15 who had complete data at all time points and the 7 who had missing data (p = 0.1428). The mean baseline IQ score was 84.6 (SD = 13.4; N = 22), which is on the boundary between average and below average (normative mean ± SD = 100 ± 15). At 2 years post-HCT, the mean IQ was below average at 79.1 (SD = 18.5; N = 15). Neurocognitive outcomes at 2 years post-HCT were examined for a possible association with attenuating NRE; inflammatory markers were subject to exploratory evaluation. Loss of IQ points is generally reported following HCT;13,18,33 therefore, results that are closer to no loss (quantified as zero percent change), or even gain (quantified as a positive percent change), are considered to reflect a favorable neurocognitive trajectory of development. More specifically, percent change in IQ is a continuous variable that is considered more favorable when values approach a positive number, and less favorable when values approach a negative number. Percent decrease in NRE I0S6 from time point 1 to 4 was positively associated with more favorable neurocognitive development (i.e., percent change in IQ) from baseline to 2 years post-HCT (Fig. 3). Decrease in I0S6 over the four time points is depicted in the supplementary information (Figure S3). A more favorable percent change in IQ by 2 years post-HCT was also associated with lower levels of NREs I0S0 and I0S6 at time point 3 (p < 0.0001 and p = 0.0078, respectively) and NRE I0S6 at time point 4 (p = 0.0054). Decrease in NRE I0S0 from time point 1 to 4 was not associated with IQ change. CSF MCP-1 at time point 4 was plotted with change in IQ, and the resulting scatterplot showed a rather clear slope suggestive of a relationship. However, this was a small subset of the sample, as only seven participants had both MCP-1 and IQ assessed and therefore a meaningful statistical evaluation could not be performed. Statistical evaluations were not performed on the relationship between IQ change and any inflammatory markers for this reason.
Fig. 3.
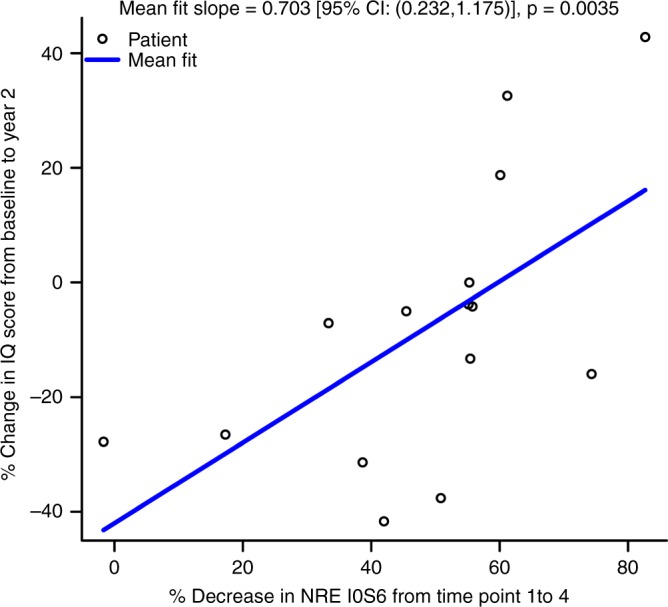
Correspondence of biochemical change with IQ outcome. Percent change in IQ from baseline (i.e., pretreatment) to 2-year posttransplant anniversary is plotted against percent reduction in nonreducing end (NRE) I0S6 from time point 1 to 4, i.e., pretreatment through final intrathecal recombinant iduronidase infusion occurring 6 months posttransplant (N = 15). A significant association was found, such that percent decrease in I0S6 was significantly associated with more favorable change in IQ (p = 0.0035). CI confidence interval.
DISCUSSION
This study systematically tracked changes in CSF characteristics during this novel treatment with IT ERT, which was added to standard dual therapy of HCT with peritransplant intravenous ERT. This therapy was safe and well tolerated, and deaths on this study resulted from HCT-related morbidity and not IT enzyme infusion. Although mortality was slightly higher than reported in recent trials,6,31 this difference was not statistically meaningful. Causes of death matched previous reports of HCT-related mortality,31 including infection in four of the five cases, suggesting tragic chance was to blame rather than the therapy protocol. Of import, the method of therapy delivery in this protocol enabled the unusual opportunity to evaluate serial samples of CSF to track potential treatment response. This tracking revealed significant decreases in CSF opening pressure as well as biomarkers including heparan sulfate NREs, HCIIT, and several inflammatory cytokines across all time points. Among these reduced biomarkers were NREs, recently proposed as tools for monitoring treatment efficacy.25 In an urgent hunt for more precise methods of predicting treatment outcomes, the present study’s findings that greater reduction in an NRE was associated with a more favorable neurocognitive trajectory has important implications. This potential link between a biochemical response during treatment and a longer-term neurocognitive outcome in Hurler syndrome may support understanding disease pathophysiology and optimizing patient management.
Neurocognitive decline in patients with Hurler syndrome continues for a period after HCT, then stabilizes.13,18,33 Earlier age at treatment predicts a significantly better neurodevelopmental course.8,12,13,34,35 Presumably, treatment at an earlier age reduces brain injury resulting from pathophysiological processes such as inflammation, oxidative stress, and other disruptions to intracellular and extracellular homeostasis.2,3 However, predicting neurocognitive status based on timing of treatment is unreliable due to numerous factors that can influence outcomes. In the current study, several patients showed an increase in IQ scores after therapy, demonstrating gains not only relative to their own development but also relative to the normative population. This is a dramatic outcome given the natural history of Hurler syndrome, which involves steep neurocognitive decline to a profoundly impaired state by age 4, if not sooner.36 All of the patients who demonstrated increases in IQ two years after treatment had at least 50% reduction in CSF NRE I0S6 across the four time points. This finding aligns with the idea that the focus on the nonreducing end of the GAG may be a more sensitive and specific biomarker than simply measuring the total extent of GAG accumulation,25 as NRE may have some association with key clinical manifestations.
Why was there a significant relationship between reduction in NRE I0S6 and percent change in IQ, but not between NRE I0S0 and IQ? The different statistical properties of these two biomarkers are likely a major reason. First, a plot of both NREs by age shows that I0S6 is more plentiful than I0S0, even though both decrease over time (reflected by age) after initiation of treatment (Figure S4). Thus, a larger sample size would likely be needed to detect a relationship between I0S0 and IQ, as the slope for this variable is smaller than I0S6. Second, there are differences in variability between I0S0 and I0S6. The observed data showed a greater range in change in IQ scores among those with 100% I0S0 reduction, leading to a lower slope relative to the relationship between I0S6 and IQ.
Intravenous ERT is presumed not to cross the blood–brain barrier to treat the neurologic pathology of Hurler syndrome.8,14 In line with this theory, there is clinical evidence of greater cumulative incidence of classic CNS structural manifestations of Hurler syndrome when IV ERT is the sole treatment, instead of HCT.6 Notably, despite the evidence that HCT has better ability to halt CNS disease progression than ERT, in the present study many CSF abnormalities showed improvement even before HCT. Not only did opening pressure decrease, but also there were significant decrements in NRE as well as HCIIT between time points 1 and 2, which are prior to HCT, raising the possibility that these improvements were facilitated with IT therapy. They also suggest that IT therapy may be associated with decreased CSF GAG. Further, as HCIIT has been shown to be a biomarker in several MPS diseases,37–39 these findings suggest HCIIT may be a useful surrogate marker of GAG accumulation.
The reductions in biochemical abnormalities in the CSF prior to transplant, i.e., during only IT and intravenous ERT therapy, align with reports of some benefit of IT ERT in humans with the attenuated phenotypes of MPS I.19,21 In one report, CSF GAG was reduced from slightly above normal: 13.3 mg/L (NV < 12 mg/L) to normal levels (10.3 mg/L) after therapy.19 That patient did not have elevations in CSF opening pressure at any point. In another report, the patient showed significantly improved memory and adaptive functioning, and mildly improved attention and IQ following treatment.21
The most notable limitation of this study is a lack of a comparison CSF from a group that received only intravenous ERT and HCT. Such a comparison would possibly allow for increased clarification of what changes resulted from IT therapy, but the frequency of CSF collection that would be needed for such a trial is not justified in patients who are not receiving intrathecal treatment. However, we emphasize that the unique access to serial CSF samples in this study enabled a separate investigation of a potential relationship between biomarker change and clinical outcome.
In terms of other limitations, enrollment bias may have influenced these results, as data were collected at a single institution, which has the potential to exclude patients whose health status or family resources prohibit travel. Also, previous studies of HCT for pediatric neurodegenerative metabolic diseases have noted that undergoing retransplantation for failed engraftment involves both repeated exposure to preparative regimens known to affect the CNS and extended delay in the arrest of the disease process, which can exacerbate neurocognitive decline.18,40 However, all results remained largely the same when the patient who underwent a second transplant was excluded from analyses. While age was not a significant predictor of IQ change in this group, it may have been challenging to detect age-related differences given the small sample and the fact that this entire group was generally young upon initiation of treatment (Table S4).
The findings of this study indicate that CSF biomarkers are responsive to therapy and that a possible relationship exists between real-time biomarker response and patients’ longer-term neurocognitive outcomes. This latter finding is important in light of the proliferation of new therapies aimed at reducing the neurocognitive and somatic disease burden that persists even when all treatment guidelines are followed. While there is extensive evidence that earlier treatment yields better clinical outcomes in Hurler syndrome8,12,13,34 predictions of neurocognitive outcomes based on timing of treatment are unreliable due to the multitude of factors, known and unknown, affecting the clinical course. Identifying biomarkers that correlate with neurocognitive function may expand the possibilities for disease monitoring, predicting the outcomes, and developing targeted therapies to yield improved clinical results.
Supplementary information
Acknowledgements
We are indebted to the patients who enabled this research. We thank Elsa Shapiro for neuropsychological supervision and consultation. The Lysosomal Disease Network (U54NS065768) is a part of the Rare Diseases Clinical Research Network (RDCRN), an initiative of the Office of Rare Diseases Research (ORDR), and the National Center for Advancing Translational Sciences (NCATS). This consortium is funded through a collaboration between NCATS, the National Institute of Neurological Disorders and Stroke (NINDS), and the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK). Translational research support and statistical consultation and analysis was provided by National Institutes of Health (NIH) UL1TR002494. Clinical trial support and drug was provided by Sanofi Genzyme Corporation, who played no role in data analysis, interpretation, manuscript preparation, or submission. Cytokine testing was supported by the Children's Cancer Research Fund. J.B.E., K.D.R., and K.E.K. were supported in part by NIH U54NS065768. A.M.K. and K.D.R. were supported in part by NIH UL1TR002494.
Disclosure
J.B.E. received honoraria, consulting fees, and/or research support from ArmaGen, Gene Spotlight, Inc., Sangamo, and Sanofi Genzyme, and has done contract work for Shapiro Neuropsychology Consulting LLC. K.E.K. is a consultant for Shire Plc; has received research support from Shire, Sanofi Genzyme, and Alexion Pharmaceuticals, Inc.; and has done previous contract work for Shapiro Neuropsychology Consulting. M.P. is a consultant for Moderna Tx, Inc. and BioMarin Pharmaceuticals, Inc. and has received travel support from BioMarin Pharmaceuticals, Inc. L.E.P. has received honoraria, consulting fees, and/or research support from Sanofi Genzyme, Shire, and BioMarin. P.I.D. receives research support from Biomarin and Genzyme. W.P.M. is an employee of Sangamo Therapeutics, Inc.. P.J.O. has received honoraria, consulting fees, and/or research support from Sanofi Genzyme, Bluebird Bio, and Horizon. T.C.L. has received speaker fees and research support from Sanofi Genzyme. The other authors declare no conflicts of interest.
Footnotes
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
The online version of this article (10.1038/s41436-019-0522-1) contains supplementary material, which is available to authorized users.
References
- 1.Pereira VG, Martins AM, Micheletti C, D'Almeida V. Mutational and oxidative stress analysis in patients with mucopolysaccharidosis type I undergoing enzyme replacement therapy. Clin Chim Acta. 2008;387:75–79. doi: 10.1016/j.cca.2007.09.008. [DOI] [PubMed] [Google Scholar]
- 2.Campos D, Monaga M. Mucopolysaccharidosis type I: current knowledge on its pathophysiological mechanisms. Metab Brain Dis. 2012;27:121–129. doi: 10.1007/s11011-012-9302-1. [DOI] [PubMed] [Google Scholar]
- 3.Bigger BW, Begley DJ, Virgintino D, Pshezhetsky AV. Anatomical changes and pathophysiology of the brain in mucopolysaccharidosis disorders. Mol Genet Metab. 2018;125:322–331. doi: 10.1016/j.ymgme.2018.08.003. [DOI] [PubMed] [Google Scholar]
- 4.Ohmi K, Greenberg DS, Rajavel KS, Ryazantsev S, Li HH, Neufeld EF. Activated microglia in cortex of mouse models of mucopolysaccharidoses I and IIIB. Proc Natl Acad Sci U S A. 2003;100:1902–1907. doi: 10.1073/pnas.252784899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Neufeld E, Muenzer J. The mucopolysaccharidoses. In: Scriver CR, editor. The metabolic and molecular bases of inherited disease. New York: McGraw-Hill; 2001. pp. 3421–3452. [Google Scholar]
- 6.Eisengart JB, Rudser KD, Xue Y, et al. Long-term outcomes of systemic therapies for Hurler syndrome: an international multicenter comparison. Genet Med. 2018;20:1423–1429. doi: 10.1038/gim.2018.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Clarke LA. Pathogenesis of skeletal and connective tissue involvement in the mucopolysaccharidoses: glycosaminoglycan storage is merely the instigator. Rheumatology. 2011;50:v13–v8. doi: 10.1093/rheumatology/ker395. [DOI] [PubMed] [Google Scholar]
- 8.Muenzer J, Wraith JE, Clarke LA. Mucopolysaccharidosis I: management and treatment guidelines. Pediatrics. 2009;123:19–29. doi: 10.1542/peds.2008-0416. [DOI] [PubMed] [Google Scholar]
- 9.Woloszynek JC, Coleman T, Semenkovich CF, Sands MS. Lysosomal dysfunction results in altered energy balance. J Biol Chem. 2007;282:35765–35771. doi: 10.1074/jbc.M705124200. [DOI] [PubMed] [Google Scholar]
- 10.Krivit W, Aubourg P, Shapiro E, Peters C. Bone marrow transplantation for globoid cell leukodystrophy, adrenoleukodystrophy, metachromatic leukodystrophy, and Hurler syndrome. Curr Opin Hematol. 1999;6:377–382. doi: 10.1097/00062752-199911000-00004. [DOI] [PubMed] [Google Scholar]
- 11.Hobbs JR, Barrett AJ, Chambers D, et al. Reversal of clinical features of Hurler's disease and biochemical improvement after treatment by bone-marrow transplantation. Lancet. 1981;318:709–712. doi: 10.1016/S0140-6736(81)91046-1. [DOI] [PubMed] [Google Scholar]
- 12.Aldenhoven M, Wynn RF, Orchard PJ, et al. Long-term outcome of Hurler syndrome patients after hematopoietic cell transplantation: an international multicenter study. Blood. 2015;125:2164–2172. doi: 10.1182/blood-2014-11-608075. [DOI] [PubMed] [Google Scholar]
- 13.Shapiro EG, Nestrasil I, Rudser K, et al. Neurocognition across the spectrum of mucopolysaccharidosis type I: age, severity, and treatment. Mol Genet Metab. 2015;116:61–68. doi: 10.1016/j.ymgme.2015.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Scarpa M, Orchard PJ, Schulz A, et al. Treatment of brain disease in the mucopolysaccharidoses. Mol Genet Metab. 2017;122:25–34. doi: 10.1016/j.ymgme.2017.10.007. [DOI] [PubMed] [Google Scholar]
- 15.Ghosh A, Miller W, Orchard PJ, et al. Enzyme replacement therapy prior to haematopoietic stem cell transplantation in mucopolysaccharidosis type I: 10 year combined experience of 2 centres. Mol Genet Metab. 2016;117:373–377. doi: 10.1016/j.ymgme.2016.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tolar J, Grewal S, Bjoraker K, et al. Combination of enzyme replacement and hematopoietic stem cell transplantation as therapy for Hurler syndrome. Bone Marrow Transplant. 2007;41:531–535. doi: 10.1038/sj.bmt.1705934. [DOI] [PubMed] [Google Scholar]
- 17.Wynn R, Mercer J, Page J, Carr T, Jones S, Wraith J. Use of enzyme replacement therapy (laronidase) before hematopoietic stem cell transplantation for mucopolysaccharidosis I: experience in 18 patients. J Pediatr. 2009;154:135–139. doi: 10.1016/j.jpeds.2008.07.004. [DOI] [PubMed] [Google Scholar]
- 18.Eisengart JB, Rudser KD, Tolar J, et al. Enzyme replacement is associated with better cognitive outcomes after transplant in Hurler syndrome. J Pediatr. 2013;162:375–380. doi: 10.1016/j.jpeds.2012.07.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Munoz-Rojas MV, Vieira T, Costa R, et al. Intrathecal enzyme replacement therapy in a patient with mucopolysaccharidosis type I and symptomatic spinal cord compression. Am J Med Genet A. 2008;146A:2538–2544. doi: 10.1002/ajmg.a.32294. [DOI] [PubMed] [Google Scholar]
- 20.Jones SA, Breen C, Heap F, et al. A phase 1/2 study of intrathecal heparan-N-sulfatase in patients with mucopolysaccharidosis IIIA. Mol Genet Metab. 2016;118:198–205. doi: 10.1016/j.ymgme.2016.05.006. [DOI] [PubMed] [Google Scholar]
- 21.Nestrasil I, Shapiro E, Svatkova A, et al. Intrathecal enzyme replacement therapy reverses cognitive decline in mucopolysaccharidosis type I. Am J Med Genet A. 2017;173:780–783. doi: 10.1002/ajmg.a.38073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Muenzer J, Burton BK, Harmatz P, et al. Efficacy and safety of intrathecal idursulfase in pediatric patients with mucopolysaccharidosis type II and early cognitive impairment: design and methods of a controlled, randomized, phase II/III multicenter study. Mol Genet Metab. 2018;123:S99–S100. doi: 10.1016/j.ymgme.2017.12.262. [DOI] [Google Scholar]
- 23.Giugliani R, Giugliani L, de Oliveira Poswar F, et al. Neurocognitive and somatic stabilization in pediatric patients with severe mucopolysaccharidosis type I after 52 weeks of intravenous brain-penetrating insulin receptor antibody-iduronidase fusion protein (valanafusp alpha): an open label phase 1–2 trial. Orphanet J Rare Dis. 2018;13:110. doi: 10.1186/s13023-018-0849-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Clarke LA, Atherton AM, Burton BK, et al. Mucopolysaccharidosis type I newborn screening: best practices for diagnosis and management. J Pediatr. 2017;182:363–370. doi: 10.1016/j.jpeds.2016.11.036. [DOI] [PubMed] [Google Scholar]
- 25.Lawrence R, Brown JR, Al-Mafraji K, et al. Disease-specific non-reducing end carbohydrate biomarkers for mucopolysaccharidoses. Nat Chem Biol. 2012;8:197–204. doi: 10.1038/nchembio.766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dickson P, McEntee M, Vogler C, et al. Intrathecal enzyme replacement therapy: successful treatment of brain disease via the cerebrospinal fluid. Mol Genet Metab. 2007;91:61–68. doi: 10.1016/j.ymgme.2006.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Raymond GV, Pasquali M, Polgreen LE, et al. Elevated cerebral spinal fluid biomarkers in children with mucopolysaccharidosis IH. Sci Rep. 2016;6:38305. doi: 10.1038/srep38305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mullen E. Mullen scales of early learning. Circle Pines, MN: American Guidance Service; 1995. [Google Scholar]
- 29.Bayley N. Bayley scales of infant and toddler development. PsychCorp, Pearson; 2006.
- 30.Halekoh U, Højsgaard S, Yan J. The R package geepack for generalized estimating equations. J Stat Softw. 2006;15:1–11. doi: 10.18637/jss.v015.i02. [DOI] [Google Scholar]
- 31.Rodgers NJ, Kaizer AM, Miller WP, Rudser KD, Orchard PJ, Braunlin EA. Mortality after hematopoietic stem cell transplantation for severe mucopolysaccharidosis type I: the 30-year University of Minnesota experience. J Inherit Metab Dis. 2017;40:271–280. doi: 10.1007/s10545-016-0006-2. [DOI] [PubMed] [Google Scholar]
- 32.Custer JW, Rau RE. The Harriet Lane handbook: a manual for pediatric house officers. 18th ed. Philadelphia, PA: Mosby/Elsevier; 2009. Johns Hopkins Hospital. Children's Medical and Surgical Center. [Google Scholar]
- 33.Souillet G, Guffon N, Maire I, et al. Outcome of 27 patients with Hurler's syndrome transplanted from either related or unrelated haematopoietic stem cell sources. Bone Marrow Transplant. 2003;31:1105. doi: 10.1038/sj.bmt.1704105. [DOI] [PubMed] [Google Scholar]
- 34.Poe MD, Chagnon SL, Escolar ML. Early treatment is associated with improved cognition in Hurler syndrome. Ann Neurol. 2014;76:747–753. doi: 10.1002/ana.24246. [DOI] [PubMed] [Google Scholar]
- 35.Peters C, Shapiro E, Anderson J, et al. Hurler syndrome: II. Outcome of HLA-genotypically identical sibling and HLA-haploidentical related donor bone marrow transplantation in fifty-four children. Blood. 1998;91:2601. doi: 10.1182/blood.V91.7.2601. [DOI] [PubMed] [Google Scholar]
- 36.Shapiro EG, Whitley CB, Eisengart JB. Beneath the floor: re-analysis of neurodevelopmental outcomes in untreated Hurler syndrome. Orphanet J Rare Dis. 2018;13:76. doi: 10.1186/s13023-018-0817-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Randall DR, Sinclair GB, Colobong KE, Hetty E, Clarke LA. Heparin cofactor II-thrombin complex in MPS I: a biomarker of MPS disease. Mol Genet Metab. 2006;88:235–243. doi: 10.1016/j.ymgme.2006.01.005. [DOI] [PubMed] [Google Scholar]
- 38.Langford-Smith K, Arasaradnam M, Wraith JE, Wynn R, Bigger BW. Evaluation of heparin cofactor II-thrombin complex as a biomarker on blood spots from mucopolysaccharidosis I, IIIA and IIIB mice. Mol Genet Metab. 2010;99:269–274. doi: 10.1016/j.ymgme.2009.10.175. [DOI] [PubMed] [Google Scholar]
- 39.Clarke LA, Hemmelgarn H, Colobong K, et al. Longitudinal observations of serum heparin cofactor II-thrombin complex in treated mucopolysaccharidosis I and II patients. J Inherit Metab Dis. 2012;35:355–362. doi: 10.1007/s10545-011-9369-6. [DOI] [PubMed] [Google Scholar]
- 40.Pierpont EI, Eisengart JB, Shanley R, et al. Neurocognitive trajectory of boys who received a hematopoietic stem cell transplant at an early stage of childhood cerebral adrenoleukodystrophy. JAMA Neurol. 2017;74:710–717. doi: 10.1001/jamaneurol.2017.0013. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.