

Abstract
The conventional oxidative Heck reaction between aryl boronic acids and alkenes typically involved the PdII/Pd0/PdII catalytic cycle incorporating an external oxidant and often suffered C=C bond isomerization for internal alkyl-substituted alkenes via chain-walking. Herein, we demonstrate that the regioselectivity (γ-selectivity vs. δ-selectivity) and pathway selectivity (hydroarylation vs. oxidative Heck coupling) of a directed Ni-catalyzed alkene arylation can be controlled by judicious tuning of the coordination environment around the nickel catalyst via optimization of an appropriate phosphine ligand and directing group. In this way, the Ni(0)-catalyzed oxidative Heck arylation that relies on transfer hydrogenation of an acceptor olefin is developed with excellent E/Z selectivity and regioselectivity. Mechanistic investigations suggest that the addition of the acceptor is crucial for lowering the energy for carbometalation and for enabling catalytic turnover.
Subject terms: Catalytic mechanisms, Homogeneous catalysis, Synthetic chemistry methodology
Conventional palladium-catalyzed Heck reactions rely on the addition of oxidants and may suffer from low selectivity. Here, the authors report a nickel-catalyzed oxidative Heck arylation driven by transfer hydrogenation of an acceptor olefin leading to high E/Z selectivity and regioselectivity.
Introduction
Transition-metal-catalyzed cross-coupling reactions involving alkenes enable conversion of abundant feedstocks to value-added products through C–C bond formation1–16. Heck-type reactions, which can be carried out both in classical polarity and oxidative modes, are the archetypal method for arene–alkene coupling. Despite significant advances during the past several decades1–10, Heck-type reactions still suffer from significant limitations, including a difficulty controlling selectivity with internal alkyl-substituted alkenes due to the lack of steric and electronic differentiation in the key migratory insertion step and issues of C=C bond isomerization due to chain-walking17. Additionally, though notable exceptions exist18,19, oxidative Heck reactions with arylmetal species have traditionally required use of comparatively expensive palladium catalysts with specially tailored reoxidation systems20–26. Recently, Zhou reported a highly selective Ni-catalyzed hydroarylation of styrenes and 1,3-dienes with arylboron compounds, which was proposed to proceed via oxidation addition of Ni(O) to an alcohol O–H bond to form a Ni–H species, hydrometallation across the alkene, transmetalation with the organoboron reagents, and C–C reductive elimination (Fig. 1a, path A)27,28. In 2018, we extended this strategy to hydroarylation of non-conjugated alkenes by introducing a directing group on the substrate29–35. During the course of this study, we detected byproducts arising from hydrogenolysis of the Ar–B bond, indicating that transmetalation might precede migratory insertion. In this case, a Ni(H)(Ar) intermediate would be the active form of the catalyst that engages the alkene (Fig. 1a, path B). This catalytic paradigm offers exciting possibilities for controlling regioselectivity and pathway selectivity to access different products from a common set of reagents.
Fig. 1.
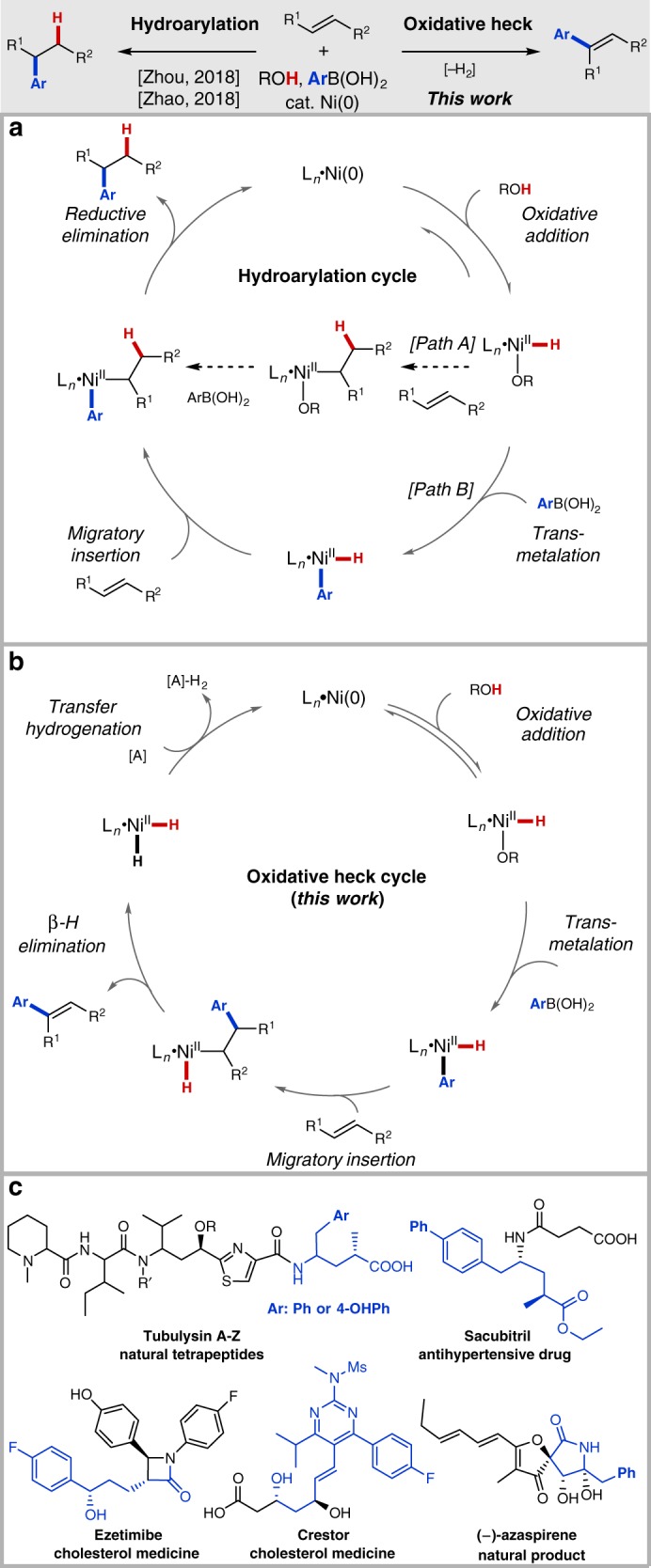
Inspiration and motivation for the present study. a Catalytic cycle for previously reported Ni-catalyzed hydroarylation reaction. b Envisioned oxidative Heck reaction. c Relevant biologically active compounds containing γ-aryl carboxylic acid substructure
We were attracted to the possibility of diverting this process to obtain non-chain-walking oxidative-Heck products by using internal alkyl-substituted alkenes and aryl boronates as the coupling partners. In the proposed cycle (Fig. 1b), migratory insertion of the aryl group in preference to the hydride group would first take place. Subsequently, β-hydride elimination would then furnish the arylated alkene product along with a Ni(H)2 species that would reduce a sacrificial alkene to close the catalytic cycle. If successful, this sequence of events would result in a Ni(O)-catalyzed oxidative.
Heck reaction coupled to a transfer hydrogenation process36–44. We envisioned that selectivity for this pathway over other alternatives could be achieved by tuning the coordination environment around nickel with an appropriate phosphine ligand and directing group45–52. Herein we describe the realization of this goal. In particular, we report complementary transformations for both γ-selective hydroarylation and oxidative Heck coupling. These methods are efficient and scalable, providing a modular means of assembling δ-aryl olefinic acids, which are versatile building blocks for synthesizing drugs and other biologically active compounds (Fig. 1c)53–57.
Results
Condition screening
In a pilot experiment, 4-pentenoic acid masked with the 8-aminoquinoline (AQ) directing group (1a, 0.2 mmol) was subjected to our reported conditions for hydroarylation of 3-butenoic acid derivatives29,58,59, which gave a complicated mixture of hydoarylation and Heck-type products (Table 1, entry 1). Next, various phosphine ligands were evaluated, and the effects were pronounced (entries 2–14). With PCy3 or PnBu3 as the ligand, the δ-selective Heck-type product 5a was the major product, albeit in low yield (entries 2-3). Surprisingly, use of bulkier phosphine ligands resulted in exclusive formation of the δ-selective hydroarylation product 3a (entries 4–5). In contrast, when PMe3 was used as the ligand, only γ-selective hydroarylation product 4a was observed in 85% yield. Notably, the γ-selectivity of this directed hydroarylation protocol is opposite to that of Loh’s Pd-catalyzed reductive Heck system, which is δ-selective for this type of substrate60. Using PCy3 or PnBu3 as ligand, we found that adjusting the molar ratio of 1a and 2a to 1.5:1 dramatically increased the yield of oxidative Heck product 5a (entries 15–16). Notably, the reduced form of alkene 1a was detected in all the reactions, suggesting that 1a was serving as an H2 acceptor. Based on this insight, we systematically examined various hydrogen acceptors so that alkene 1a could be used as the limiting reagent. Pleasingly, after an exhaustive screen of alkene acceptors, we identified cinnamaldehyde derivative A18 (1.0 equiv) as an efficient sacrificial alkene, and with this additive, 1 equiv 1a could be converted to 5a in 94% isolated yield.
Table 1.
Reaction optimization
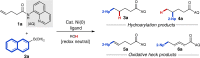
| |||||
|---|---|---|---|---|---|
| Entry | Ligand | Yield[%]a (3a:4a:5a:6a) | Entry | Ligand | Yield[%]a (3a:4a:5a:6a) |
| 1 | PPh3 | 47 (0/17/25/5) | 9 | PPhCy2 | 43 (0/10/29/4) |
| 2 | PCy3 | 41 (0/0/35/6) | 10 | P(OMe)3 | 30 (0/14/16/0) |
| 3 | PnBu3 | 39 (0/9/30/0) | 11 | PnPr3 | 29 (0/13/16/0) |
| 4 | PtBu3 | 25 (25/0/0/0) | 12 | PMe2Ph | 32 (0/25/7/0) |
| 5 | XPhos | 35 (35/0/0/0) | 13 | PtBuCy2 | 32 (0/15/17/0) |
| 6 | PMe3 | 85 (0/85/0/0) | 14 | Xantphos | 38 (0/16/19/3) |
| 7 | PPh2Cy | 44 (0/17/22/5) | 15b | PCy3 | 79 (0/0/79/0) |
| 8 | DCYPE | 0 | 16b | PnBu3 | 92 (0/0/92/0) |
| |||||
Reactions conditions: Ni(cod)2 (10 mol%), ligand (20 mol%), CsOPiv (1.5 equiv), 1a (0.2 mmol), 2a (0.4 mmol), t-AmylOH (1 mL), 70 °C, 48 h
a1H NMR yields with C2H2Br4 as internal standard
bMolar ratio of 1a:2a = 1.5:1
cPnBu3 (20 mol%), H2 scavenger (2.0 equiv). The 1H NMR yield of 5a is given
d1.0 equiv H2 acceptor
eThe isolated yield is given in parenthesis
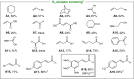
Substrate scope of hydroarylation
Given the opposite selectivity of our Ni-catalytic system compared to Loh’s Pd-catalyzed reductive Heck system for hydroarylation of 4-pentenoic acids60, we next briefly probed the substrate scope of the γ-selective hydroarylation (Table 2). A diverse collection of arylboronic acids could be coupled to 1a, furnishing the corresponding γ-arylated products in good to excellent yields. Cyclic alkenes and δ,δ-disubstituted alkenes also underwent hydroarylation, though in these cases the reactions proceeded with δ-selectivity. Notably, these substrates are unreactive in Loh’s Pd-catalyzed system.
Table 2.
Substrate scope of hydroarylation
|
Reaction conditions: Ni(cod)2 (10 mol%), PMe3 (20 mol%), CsOPiv (1.5 equiv), alkene 1 (0.2 mmol), arylboronic acid 2 (0.4 mmol), t-AmylOH (1 mL), 70 °C, 48 h. Values represent isolated yields
aThe isolated yield in parenthesis was obtained by use of Ni(cod)2 (5 mol%) and PMe3 (10 mol%)
b120 °C
cPnBu3 as ligand
Substrate scope of oxidative Heck reaction
Next, we investigated the substrate scope and functional group tolerance of the oxidative Heck reaction with respect to the organoboron reagent using 1a as the model alkene substrate (Table 3). To our delight, a wide variety of para- and meta-substituted arylboronic acids with different electronic properties afforded the desired products in good to excellent yields and with high E-selectivity (5ae–ao). ortho-Substituted arylboronic acids were slightly lower yielding, presumably due to steric encumbrance (5ac−ad). Multi-substituted arylboronic acids (5ap−ar) and heteroarylboronic acids (5as−av) also provided the corresponding Heck products in high yields. Additionally, cyclic and acyclic alkenyl boronic acids were competent coupling partners, enabling access to conjugated dienoic acid derivatives (5aw−ax). Importantly, this method was found to be compatible with an array of functional groups, including halo, acetyl, alkoxycarbonyl, cyano, and methoxy substituents, and could be carried out on gram scale (1.14 g of 5ab, 84%). The structure of 5a was confirmed by single-crystal X-ray diffraction.
Table 3.
Scope of organoboronic acids and alkenes in oxidative Heck arylation

|
Reaction conditions: Ni(cod)2 (10 mol%), ligand (20 mol%), CsOPiv (1.5 equiv), alkene (0.2 mmol), arylboronic acid (0.4 mmol), t-AmylOH (1 mL), 70 °C, 48 h. Values correspond to isolated yields. In cases where the E/Z ratio is not specified, only the E-alkene product was observed
aThe values parentheses represent the isolated yield using Ni(cod)2 (5 mol%) and ligand (10 mol%)
bPMe3 (20 mol%), 120 °C
ct-AmylOH (1.5 mL)
dZ/E ratios were determined by 1H NMR
eThe Z/E ratio of substrate 1i was 91:9
fNi(cod)2 (20 mol%), PnBu3 (40 mol%), 120 °C, 72 h
We then evaluated various γ,δ-unsaturated carboxamides (Table 3). Terminal alkenes bearing a single substituent at the α- or β-position reacted smoothly to afford the desired products with the complete E-selectivity (5ba−ea). The β,β-disubstituted terminal alkene was also reactive, selectively furnishing desired product 5fa. In contrast, the sterically more congested α,α-disubstituted terminal alkene only gave 36% yield, even after brief optimization of the conditions (5ga). (E)-Internal alkenes bearing an alkyl group at the δ-position could also be efficiently converted into the corresponding E- configured products (5ha–ka). However, aryl substitution at the δ-position led to clear attenuation of reactivity (5la). Notably, when (Z)-alkene 1i was used, the reaction stereospecifically generated (Z)-product 5ia. In addition, the reaction was also compatible with β-substituted internal alkenes (5ma–na), a γ-substituted terminal alkene (5oa), and an α,δ-disubstituted alkene (5pa), though in the latter two cases Z/E selectivity was low. In addition to γ,δ-unsaturated substrates, δ,ε-unsaturated AQ carboxamides were also examined. Surprisingly, with this substrate class, the connectivity of the products was the same as with the (E)-γ,δ-unsaturated internal alkenes (5qa–qd) suggesting potential alkene isomerization. A representative internal δ,ε-unsaturated alkene was unreactive in this isomerization/oxidative Heck cascade reaction.
Synthetic utility
To demonstrate the synthetic utility of our method, we first sought to prepare carboxylic acid 4j, a key intermediate in the synthesis of the natural products aristelegone A, heritol, and xanthorrhizol (Fig. 2, upper panel)61,62. This intermediate has previously been synthesized through a sequence involving condensation followed by Michael addition and decarboxylation. As a second example, unsaturated carboxylic ester 7 was also prepared via δ-selective oxidative Heck arylation. Compound 7 has been employed as a key intermediate in the synthesis of the natural product curcudiol (Fig. 2, lower panel)63,64. The literature method to access 7 involves a multi-step sequence including Grignard addition and Johnson–Claisen rearrangement.
Fig. 2.

Synthesis of key intermediates towards natural products. a The application of hydroarylation to approach the key intermediates in the synthesis of the natural products. b The application of oxidative Heck coupling toward the key intermediate in the synthesis of the natural product curcudiol
Discussion
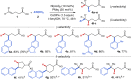
Mechanism investigation
We then carried out a series of experiments to elucidate the mechanism of the directed oxidative Heck reaction. As an initial control experiments, we first subjected two representative substrates lacking the AQ directing group, pent-4-enoic acid and N-phenylpent-4-enamide, to the standard reaction conditions using 2-naphthalenylboronic acid as the coupling partner (Fig. 3a). In these experiments, no reaction took place, indicating that the AQ directing group on the olefin substrate is indispensable for reactivity. Next, to probe the formation of putative Ni–H species, we performed a stoichiometric reaction between t-amylOH (1.0 equiv.) and Ni(COD)2 (1.0 equiv.) in C6D6 at r.t. and analyzed the crude reaction mixture by 1H NMR. We observed a new peak at −23.24 ppm, which is consistent with previously described Ni–H species in the literature65–67. Several additional experiments were then performed under catalytic conditions. First, we found that the addition of hydrogen acceptor A18 (1.0 equiv) under conditions that were otherwise identical to the optimal conditions for γ-selective hydroarylation resulted in a significant increase in the yield of Heck product 5a (Fig. 3b). This result indicates that compound A18 may play a role beyond serving as a hydrogen acceptor. In particular, this additive could be bound to nickel as a ligand during the key migratory insertion event and could lower the activation energies for carbometalation compared to hydrometalation (see below). Second, we exposed alkene 1a to the reaction conditions in t-BuOD and were unable to detect deuterium incorporation (Fig. 3c). The result is consistent with a scenario in which [Ni(H)(OR)] is inactive in alkene insertion until it undergoes transmetalation with arylboronic acid to give an active [Ni(H)(Ar)] species. Third we performed the reaction with δ,δ-di-deuterated alkene 1a-d2. The product, 5ab-d1 (89% yield), contained a single deuterium atom at the δ position (Fig. 3d). In addition, it was found that the reduced acceptor R18 was partially deuterated at both the α- and β-positions (Fig. 3d). These results are consistent with an irreversible β-H elimination step followed by transfer hydrogenation to acceptor A18. Last, we performed the standard reaction in t-BuOD (Fig. 3e). There was no deuterium incorporation in the Heck product 5ab. Instead, the deuterium was transfered to the α- and β-positions of reduced acceptor R18 implying that: (1) hydrometalation of alkene 1a does not happen under the oxidative Heck reaction conditions; (2) hydrogen is transferred from t-BuOH to compound A18 as part of catalyst turnover.
Fig. 3.

Experiments to elucidate Heck coupling mechanism. a Control experiments establishing the need for the AQ-directing group in both hydroarylation and Heck coupling. b The role of the compound A18. c The importance of aryl boronic acid in alkene insertion. d The reaction with δ,δ-di-deuterated alkene 1a-d2. e Deuteration experiments in t-BuOD
To gain further insight into mechanism and the turnover-limiting step, we examined reaction kinetics at various initial concentrations of alkene 1a, aryl boronic acid 2o, acceptor A18, and Ni(COD)2 (See Supplementary Figs. 12–19). Based on initial rates, the reaction is first-order with respect to the Ni0 catalyst and zeroth-order with respect to the alkene 1a, aryl boronic acid 2o, and the acceptor A18. These results are consistent with turnover-limiting migratory insertion from a complex of the general form, [Ni(Ar)(1a)(A18)], as discussed more below. Additionally, we also performed a brief series of experiments to study the mechanism of the γ-selective hydroarylation (Fig. 4). First, we found that the alkene 1a is unreactive in the absence of arylboronic acid under the hydroarylation conditions (Fig. 4a). However, in the presence of arylboronic acid 2a, most of the terminal alkene 1a is isomerized to internal alkene 1a’ after 5 h under the hydroarylation conditions (Fig. 4b). In addition, we exposed alkene 1a to hydroarylation conditions in t-BuOD for 5 h. Deuterium incorporation was detected at all four positions of isomerized alkene 1a’ (Fig. 4c). Several conclusions can be drawn from these results: (1) [Ni(H)(OR)] is unreactive in alkene insertion in the absence of arylboronic acid; (2) [Ni(H)(Ar)] species promotes isomerization of alkene 1a; (3) internal alkene 1a’ is likely an intermediate for this γ-selective hydroarylation; and (4) isomerization of alkene 1a is reversible. Furthermore, we find that the addition of the hydrogen acceptor A18 in the hydroarylation reaction for 5 h partially inhibits the isomerization of alkene 1a and initiates the oxidative Heck arylation (Fig. 4d). The isomerization of alkene 1a does not occur under the Heck reaction conditions (Fig. 4e).
Fig. 4.

A series of experiments to study the mechanism of the γ-selective hydroarylation. a The control experiment in the absence of arylboronic acid. b The monitoring of the alkene substrate in hydroarylation for 5 h. c Deuterium incorporation into isomerized alkene. d The hydroarylation reaction in the presence of the compound A18 for 5 h. e The reaction by use of PnBu3 as the ligand for 5 h
DFT calculations
On the basis of literature reports and our own mechanistic experiments, we propose a catalytic cycle that begins with oxidation addition of Ni(O) to an alcohol O–H bond to form intermediate A. This is followed by transmetalation of the arylboronic acid, which affords the key [Ni(Ar)(H)] species (Fig. 5). An alternative sequence involving initial coordination of Ni(O) to the AQ group and subsequent O–H oxidation addition was also considered; however, this pathway was found to be much higher in energy (see Supplementary Fig. 21). To understand the underlying origin of the divergent reactivity, DFT calculations were performed using alkene 1a and phenylboronic acid (2b) as model reactants (Fig. 5). First, starting from [Ni(Ar)(H)] species B, ligand exchange of one PMe3 for 1a in the presence of CsOPiv delivers complex C. Then, the reaction can proceed along two divergent pathways: Pathway I (hydroarylation; left) or Pathway II (oxidative Heck coupling; right). In Pathway I, C undergoes γ-selective hydrometalation via TS1A. The resulting six-membered nickelacycle C1-1 either directly loses a second PMe3 ligand upon coordination of the quinoline group to form a more stable species C2 or undergoes β-hydride elimination to form C1-3 via TS1D. The other pathway to form C2 via TS1B is also possible, though it requires higher energy. Next, C2 undergoes reductive elimination via transition state TS1C to produce complex C3. Finally, C3 releases the hydroarylated product C4 by binding a PMe3 ligand to regenerate the Ni(O) catalyst, thereby closing the catalytic cycle. In contrast, the oxidative Heck coupling cycle proceeds in the presence of a hydrogen acceptor and involves the following steps. First, 1,4-addition of the Ni–H moiety from [Ni(Ar)(H)] species C to the hydrogen acceptor and concomitant release of PMe3 forms intermediate C'1-1. Subsequent carbometalation across the C=C bond via TS2B leads to formation of linear intermediate C'2. β-Hydride elimination via TS2C results in olefinic complex C'3, and subsequent transfer hydrogenation furnishes the product C'4 and regenerates the Ni0 catalyst. DFT calculations indicate that: (1) the energy of direct carbometalation of C via TS2A in the absence of hydrogen acceptor is much higher than that of hydrometalation step via TS1A (37.6 vs. 21.0); and (2) the addition and coordination of hydrogen acceptor significantly lowers the energy of the carbometalation step (37.6 vs. 20.8). Therefore, the presence of a hydrogen acceptor is expected to accelerate the oxidative Heck coupling, which is consistent with the experimental results. Transition states TS2B and TS1A are close in energy (20.8 vs. 21.7), meaning that hydroarylation and Heck coupling are competitive.
Fig. 5.

DFT calculations for reaction mechanism. a Proposed catalytic cycle with free energies computed by DFT (kcal mol−1). b Structures of transition states TS1A, TS1B, TS2A, and TS2B
In summary, we describe a transfer-hydrogenation-coupled oxidative Heck reaction catalyzed by a versatile Ni(O)/PR3/alcohol catalytic system. With the aid of a cleavable AQ directing group, a broad range of γ,δ- and δ,ε-unsaturated carboxamides, including traditionally challenging non-conjugated internal alkenes, are tolerated. The arylated alkene products are typically obtained with high E/Z selectivity and as single positional isomers. Mechanistic studies are consistent with a transfer hydrogenation process involving an active [Ni(Ar)(H)] species. The hydrogen acceptor promotes the carbometalation step and formally accepts dihydrogen from the metal. To our knowledge, intermolecular Ni(O)-catalyzed oxidative boron Heck reaction has not been reported before.
Methods
General procedure for Heck arylation
In an argon-filled glovebox, an oven-dried 25-mL Schlenk tube equipped with a Teflon-coated magnetic stir bar was charged successively with alkene 1 (0.2 mmol, 1.0 equiv.), arylboronic acid 2 (0.4 mmol, 2.0 equiv.), Ni(cod)2 (0.02 mmol, 0.0055 g, 0.1 equiv.), CsOPiv (0.3 mmol, 0.0702 g, 1.5 equiv.), (E)-3-(3,4,5-trimethoxyphenyl)acrylaldehyde A18 (0.2 mmol, 0.0444 g, 1.0 equiv.), anhydrous t-AmylOH (1 mL), and PnBu3 (0.04 mmol, 0.0081 g, 0.2 equiv.). The tube was sealed with a Teflon screw cap, moved out of the glovebox, and placed on a hotplate pre-heated to 70 °C with vigorous stirring. After 48 h, the reaction mixture was cooled to room temperature and diluted with EtOAc. The organic layer was washed with brine solution and was then dried over anhydrous MgSO4. The reaction mixture was filtered through a short pad of Celite, and the solvent was evaporated under vacuum to give the crude product. The resulting residue was purified by silica gel flash column chromatography (hexane/EtOAc = 10/1) to afford the corresponding product. For details of the synthetic procedures and NMR spectra of these compounds in this manuscript see Supplementary information.
Supplementary information
Description of Additional Supplementary Files
Acknowledgements
We are grateful for the financial support from the National Key Research and Development Program of China (2019YFA0210500), the National Natural Science Foundation of China (21871146, 21602115), 1000-Talent Youth Program (020/BF180181), the Natural Science Foundation of Tianjin (18JCYBJC20400), the Fundamental Research Funds for the Central Universities and Nankai University.
Author contributions
H.L. performed the experiments. H.K., B.Z., and X.X. performed the DFT calculation. K.E. and D.Z. conceived the concept, directed the project and wrote the paper.
Data availability
The authors declare that the data supporting the findings of this study are available within the article and its Supplementary Information Files.
Competing interests
The authors declare no competing interests.
Footnotes
Peer review information Nature Communications thanks Troels Skrydstrup and the other, anonymous, reviewer(s) for their contribution to the peer review of this work
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Xiaosong Xue, Email: xuexs@nankai.edu.cn.
Keary M. Engle, Email: keary@scripps.edu
Dongbing Zhao, Email: dongbing.chem@nankai.edu.cn.
Supplementary information
Supplementary information is available for this paper at 10.1038/s41467-019-12949-1.
References
- 1.Heck RF. Palladium-catalyzed reactions of organic halides with olefins. Acc. Chem. Res. 1979;12:146–151. doi: 10.1021/ar50136a006. [DOI] [Google Scholar]
- 2.Beletskaya IP, Cheprakov AV. The Heck reaction as a sharpening stone of palladium catalysis. Chem. Rev. 2000;100:3009–3066. doi: 10.1021/cr9903048. [DOI] [PubMed] [Google Scholar]
- 3.Oestreich, M. The Mizoroki–Heck Reaction (Wiley, Chichester, 2009).
- 4.McCartney D, Guiry PJ. The asymmetric Heck and related reactions. Chem. Soc. Rev. 2011;40:5122–5150. doi: 10.1039/c1cs15101k. [DOI] [PubMed] [Google Scholar]
- 5.Johansson Seechurn CCC, Kitching MO, Colacot TJ, Snieckus V. Palladium‐catalyzed cross‐coupling: a historical contextual perspective to the 2010 Nobel Prize. Angew. Chem. Int. Ed. 2012;51:5062–5085. doi: 10.1002/anie.201107017. [DOI] [PubMed] [Google Scholar]
- 6.Le Bras J, Muzart J. Intermolecular dehydrogenative Heck reactions. Chem. Rev. 2011;111:1170–1214. doi: 10.1021/cr100209d. [DOI] [PubMed] [Google Scholar]
- 7.Yeung CS, Dong VM. Catalytic dehydrogenative cross-coupling: forming carbon−carbon bonds by oxidizing two carbon−hydrogen bonds. Chem. Rev. 2011;111:1215–1292. doi: 10.1021/cr100280d. [DOI] [PubMed] [Google Scholar]
- 8.Liu C, et al. Oxidative coupling between two hydrocarbons: an update of recent C–H functionalizations. Chem. Rev. 2015;115:12138–12204. doi: 10.1021/cr500431s. [DOI] [PubMed] [Google Scholar]
- 9.Karimi, B. et al. Transition-metal-catalyzed oxidative Heck reactions. Synthesis, 9, 1399–1427 (2010).
- 10.Lee A-L. Enantioselective oxidative boron Heck reactions. Org. Biomol. Chem. 2016;14:5357–5366. doi: 10.1039/C5OB01984B. [DOI] [PubMed] [Google Scholar]
- 11.Kakiuchi F, Murai S. Catalytic C−H/Olefin Coupling. Acc. Chem. Res. 2002;35:826–834. doi: 10.1021/ar960318p. [DOI] [PubMed] [Google Scholar]
- 12.Pan S, Shibata T. Recent advances in iridium-catalyzed alkylation of C–H and N–H bonds. ACS Catal. 2013;3:704–712. doi: 10.1021/cs400066q. [DOI] [Google Scholar]
- 13.Zeng X. Recent advances in catalytic sequential reactions involving hydroelement addition to carbon–carbon multiple bonds. Chem. Rev. 2013;113:6864–6900. doi: 10.1021/cr400082n. [DOI] [PubMed] [Google Scholar]
- 14.Yang L, Huang H. Transition-metal-catalyzed direct addition of unactivated C–H bonds to polar unsaturated bonds. Chem. Rev. 2015;115:3468–3517. doi: 10.1021/cr500610p. [DOI] [PubMed] [Google Scholar]
- 15.Crisenza GEM, Bower JF. Branch selective Murai-type alkene hydroarylation reactions. Chem. Lett. 2016;45:2–9. doi: 10.1246/cl.150913. [DOI] [Google Scholar]
- 16.Dong Z, Ren Z, Thompson SJ, Xu Y, Dong G. Transition-metal-catalyzed C–H alkylation using alkenes. Chem. Rev. 2017;117:9333–9403. doi: 10.1021/acs.chemrev.6b00574. [DOI] [PubMed] [Google Scholar]
- 17.Eberhardt NA, Guan H. Nickel hydride complexes. Chem. Rev. 2016;116:8373–8426. doi: 10.1021/acs.chemrev.6b00259. [DOI] [PubMed] [Google Scholar]
- 18.Lv L, Zhu D, Li C-J. Direct dehydrogenative alkyl Heck-couplings of vinylarenes with umpolung aldehydes catalyzed by nickel. Nat. Commun. 2019;10:715. doi: 10.1038/s41467-019-08631-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ilies L, Okabe J, Yoshikai N, Nakamura E. Iron-catalyzed, directed oxidative arylation of olefins with organozinc and Grignard reagents. Org. Lett. 2010;12:2838–2840. doi: 10.1021/ol1009448. [DOI] [PubMed] [Google Scholar]
- 20.Delcamp JH, Brucks AP, White CM. A general and highly selective chelate-controlled intermolecular oxidative Heck reaction. J. Am. Chem. Soc. 2008;130:11270–11271. doi: 10.1021/ja804120r. [DOI] [PubMed] [Google Scholar]
- 21.Werner EW, Sigman MS. A highly selective and general palladium catalyst for the oxidative Heck reaction of electronically nonbiased olefins. J. Am. Chem. Soc. 2010;132:13981–13983. doi: 10.1021/ja1060998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang C, Santiago CB, Kou L, Sigman MS. Alkenyl carbonyl derivatives in enantioselective redox relay Heck reactions: accessing α,β-unsaturated systems. J. Am. Chem. Soc. 2015;137:7290–7293. doi: 10.1021/jacs.5b04289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nallasivam JL, Fernandes RA. Pd-catalyzed site-selective mono-allylic substitution and bisarylation by directed allylic C−H activation: synthesis of anti-γ-(Aryl,Styryl)-β-hydroxy acids and highly substituted tetrahydrofurans. J. Am. Chem. Soc. 2016;138:13238–13245. doi: 10.1021/jacs.6b06438. [DOI] [PubMed] [Google Scholar]
- 24.Chen Z-M, Hilton MJ, Sigman MS. Palladium-catalyzed enantioselective redox-relay Heck arylation of 1,1-disubstituted homoallylic alcohols. J. Am. Chem. Soc. 2016;138:11461–11464. doi: 10.1021/jacs.6b06994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yuan Q, Sigman MS. Palladium-catalyzed enantioselective relay Heck arylation of enelactams: accessing α,β-unsaturated δ-lactams. J. Am. Chem. Soc. 2018;140:6527–6530. doi: 10.1021/jacs.8b02752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Romine AM, Yang KS, Karunananda MK, Chen JS, Engle KM. Synthetic and mechanistic studies of the a versatile heteroaryl thioether directing group for Pd(II) catalysis. ACS Catal. 2019;9:7626–7640. doi: 10.1021/acscatal.9b01471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xiao L-J, et al. Nickel(0)-catalyzed hydroarylation of styrenes and 1,3-dienes with organoboron compounds. Angew. Chem. Int. Ed. 2018;57:461–464. doi: 10.1002/anie.201710735. [DOI] [PubMed] [Google Scholar]
- 28.Chen Y-G, et al. Nickel-catalyzed enantioselective hydroarylation and hydroalkenylation of styrenes. J. Am. Chem. Soc. 2019;141:3395–3399. doi: 10.1021/jacs.8b13524. [DOI] [PubMed] [Google Scholar]
- 29.Lv H, Xiao L-J, Zhao D, Zhou Q-L. Nickel(0)-catalyzed linear-selective hydroarylation of unactivated alkenes and styrenes with aryl boronic acids. Chem. Sci. 2018;9:6839–6843. doi: 10.1039/C8SC02101E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Derosa J, Tran VT, Boulous MN, Chen JS, Engle KM. Nickel-catalyzed β,γ-dicarbofunctionalization of alkenyl carbonyl compounds via conjunctive cross-coupling. J. Am. Chem. Soc. 2017;139:10657–10660. doi: 10.1021/jacs.7b06567. [DOI] [PubMed] [Google Scholar]
- 31.Shrestha B, et al. Ni-catalyzed regioselective 1,2-dicarbofunctionalization of olefins by intercepting Heck intermediates as imine-stabilized transient metallacycles. J. Am. Chem. Soc. 2017;139:10653–10656. doi: 10.1021/jacs.7b06340. [DOI] [PubMed] [Google Scholar]
- 32.Li W, Boon JK, Zhao Y. Nickel-catalyzed difunctionalization of allyl moieties using organoboronic acids and halides with divergent regioselectivities. Chem. Sci. 2018;9:600–607. doi: 10.1039/C7SC03149A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Thapa S, et al. Ni-catalysed regioselective 1,2-diarylation of unactivated olefins by stabilized heck intermediates as pyridylsilyl-coordinated transient metallacycles. Chem. Sci. 2018;9:904–909. doi: 10.1039/C7SC04351A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Basnet P, et al. Synergistic bimetallic Ni/Ag and Ni/Cu catalysis for regioselective γ,δ-diarylation of alkenyl ketimines: addressing β-H elimination by in situ generation of cationic Ni(II) catalysts. J. Am. Chem. Soc. 2018;140:15586–15590. doi: 10.1021/jacs.8b09401. [DOI] [PubMed] [Google Scholar]
- 35.Derosa J, et al. Nickel-catalyzed 1,2-diarylation of simple alkenyl amides. J. Am. Chem. Soc. 2018;140:17878–17883. doi: 10.1021/jacs.8b11942. [DOI] [PubMed] [Google Scholar]
- 36.Ruan J, Li X, Saidi O, Xiao J. Oxygen and base-free oxidative Heck reactions of arylboronic acids with olefins. J. Am. Chem. Soc. 2008;130:2424–2425. doi: 10.1021/ja0782955. [DOI] [PubMed] [Google Scholar]
- 37.Cooper P, Crisenza GEM, Feron LJ, Bower JF. Iridium-catalyzed α-selective arylation of styrenes by dual C−H functionalization. Angew. Chem. Int. Ed. 2018;57:14198–14202. doi: 10.1002/anie.201808299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Takeuchi R, Yasue H. Cationic rhodium complex-catalyzed highly selective dehydrogenative silylation of styrene. Organometallics. 1996;15:2098–2102. doi: 10.1021/om9508849. [DOI] [Google Scholar]
- 39.Beller M, Eichberger M, Trauthwein H. Anti-Markovnikov functionalization of olefins: rhodium-catalyzed oxidative aminations of styrenes. Angew. Chem. Int. Ed. 1997;36:2225–2227. doi: 10.1002/anie.199722251. [DOI] [Google Scholar]
- 40.Wang D, Astruc D. The golden age of transfer hydrogenation. Chem. Rev. 2015;115:6621–6686. doi: 10.1021/acs.chemrev.5b00203. [DOI] [PubMed] [Google Scholar]
- 41.Nakai K, Yoshida Y, Kurahashi T, Matsubara S. Nickel-catalyzed redox-economical coupling of alcohols and alkynes to form allylic alcohols. J. Am. Chem. Soc. 2014;136:7797–7800. doi: 10.1021/ja500666h. [DOI] [PubMed] [Google Scholar]
- 42.Whittaker AM, Dong VM. Nickel-catalyzed dehydrogenative cross-coupling: direct transformation of aldehydes into esters and amides. Angew. Chem. Int. Ed. 2015;54:1312–1315. doi: 10.1002/anie.201410322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yang P, Xu H, Zhou J. Nickel-catalyzed asymmetric transfer hydrogenation of olefins for the synthesis of α- and β-amino acids. Angew. Chem. Int. Ed. 2014;53:12210–12213. doi: 10.1002/anie.201407744. [DOI] [PubMed] [Google Scholar]
- 44.Shiota H, Ano Y, Aihara Y, Fukumoto Y, Chatani N. Nickel-catalyzed chelation-assisted transformations involving ortho C−H bond activation: regioselective oxidative cycloaddition of aromatic amides to alkynes. J. Am. Chem. Soc. 2011;133:14952–14955. doi: 10.1021/ja206850s. [DOI] [PubMed] [Google Scholar]
- 45.Rosen BM, et al. Nickel-catalyzed cross-couplings involving carbon−oxygen bonds. Chem. Rev. 2011;111:1346–1416. doi: 10.1021/cr100259t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Montgomery, J. Organonickel Chemistry. In Organometallics in Synthesis; Lipshutz, B. H., Ed.; John Wiley & Sons, Inc.: Hoboken, 2013; pp 319−428.
- 47.Tasker SZ, Standley EA, Jamison TF. Recent advances in homogeneous nickel catalysis. Nature. 2014;509:299–309. doi: 10.1038/nature13274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Misal Castro LC, Chatani N. Nickel catalysts/N,N’-bidentate directing groups: an excellent partnership in directed C–H activation reactions. Chem. Lett. 2015;44:410–421. doi: 10.1246/cl.150024. [DOI] [Google Scholar]
- 49.Ananikov VP. Nickel: the “Spirited Horse” of Transition Metal Catalysis. ACS Catal. 2015;5:1964–1971. doi: 10.1021/acscatal.5b00072. [DOI] [Google Scholar]
- 50.Zhan B-B, Liu B, Shi B-F. Recent progress on nickel-catalyzed direct functionalization of unactivated C–H bonds. Chin. Sci. Bull. 2015;60:2907–2917. doi: 10.1360/N972015-00389. [DOI] [Google Scholar]
- 51.Xiao L-J, Ye M-C, Zhou Q-L. Nickel-catalyzed highly atom-economical C–C coupling reactions with π components. Synlett. 2019;30:361–369. doi: 10.1055/s-0037-1610410. [DOI] [Google Scholar]
- 52.Arun V, Mahanty K, Sarkar SD. Nickel-catalyzed dehydrogenative couplings. ChemCatChem. 2019;11:2243–2259. doi: 10.1002/cctc.201900254. [DOI] [Google Scholar]
- 53.Singh FV, Wirth T. Selenium-catalyzed regioselective cyclization of unsaturated carboxylic acids using hypervalent iodine oxidants. Org. Lett. 2011;13:6504–6507. doi: 10.1021/ol202800k. [DOI] [PubMed] [Google Scholar]
- 54.Takenaka K, Akita M, Tanigaki Y, Takizawa S, Sasai H. Enantioselective cyclization of 4-alkenoic acids via an oxidative allylic C−H esterification. Org. Lett. 2011;13:3506–3509. doi: 10.1021/ol201314m. [DOI] [PubMed] [Google Scholar]
- 55.Brücher O, Hartung J. Vanadium(V)-catalyzed oxidative bromination of acid labile alkenols and alkenes in alkyl carbonates. ACS Catal. 2011;1:1448–1454. doi: 10.1021/cs200349c. [DOI] [Google Scholar]
- 56.Kim J, et al. Identification of selective ERR inverse agonists. Molecules. 2016;21:80–96. doi: 10.3390/molecules21010080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nishikawa Y, et al. Enantioselective bromolactonization of trisubstituted olefinic acids catalyzed by chiral pyridyl phosphoramides. Chem. Eur. J. 2018;24:18880–18885. doi: 10.1002/chem.201804630. [DOI] [PubMed] [Google Scholar]
- 58.Rouquet G, Chatani N. Catalytic functionalization of C(sp2)−H and C(sp3)−H bonds by using bidentate directing groups. Angew. Chem. Int. Ed. 2013;52:11726–11743. doi: 10.1002/anie.201301451. [DOI] [PubMed] [Google Scholar]
- 59.Daugulis O, Roane J, Tran LD. Bidentate, monoanionic auxiliary-directed functionalization of carbon−hydrogen bonds. Acc. Chem. Res. 2015;48:1053–1064. doi: 10.1021/ar5004626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wang C, et al. Palladium-catalyzed regiocontrollable reductive Heck reaction of unactivated aliphatic alkenes. J. Am. Chem. Soc. 2018;140:9332–9336. doi: 10.1021/jacs.8b03619. [DOI] [PubMed] [Google Scholar]
- 61.Song S, Zhu S-F, Yang S, Li S, Zhou Q-L. Enantioselective iridium-catalyzed hydrogenation of β,γ-unsaturated carboxylic acids: an efficient approach to chiral 4-alkyl-4-arylbutanoic acids. Angew. Chem. Int. Ed. 2012;51:2708–2711. doi: 10.1002/anie.201107802. [DOI] [PubMed] [Google Scholar]
- 62.Chavan SP, Thakkar M, Kalkote UR. The first enantiospecific synthesis of (–)-Heritol: absolute configuration determination. Tetrahedron Lett. 2007;48:643–646. doi: 10.1016/j.tetlet.2006.11.123. [DOI] [Google Scholar]
- 63.Du ZT, Yu HR, Xu Y, Li Y, Li AP. Synthesis of bisabolane sesquiterpenes: a Johnson–Claisen rearrangement approach. Chin. Chem. Lett. 2010;21:813–815. doi: 10.1016/j.cclet.2010.02.024. [DOI] [Google Scholar]
- 64.Green JC, Jiménez-Alonso S, Brown ER, Pettus TRR. Total synthesis and repudiation of the Helianane family. Org. Lett. 2011;13:5500–5503. doi: 10.1021/ol2022214. [DOI] [PubMed] [Google Scholar]
- 65.Miiller U, Keim W, Kriiger C, Betz P. [{Ph2PCH2C(CF3)2O}NiH(PCy3)]: support for a nickel hydride mechanism in ethene oligomerization. Angew. Chem. Int. Ed. 1989;28:1011–1013. doi: 10.1002/anie.198910111. [DOI] [Google Scholar]
- 66.González-Sebastián L, Flores-Alamo M, García JJ. Nickel-catalyzed reductive hydroesterification of styrenes using CO2 and MeOH. Organometallics. 2012;31:8200–8207. doi: 10.1021/om300819d. [DOI] [Google Scholar]
- 67.Gaydou M, Moragas T, Juliá-Hernández F, Martin R. Site-selective catalytic carboxylation of unsaturated hydrocarbons with CO2 and Water. J. Am. Chem. Soc. 2017;139:12161–12164. doi: 10.1021/jacs.7b07637. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Description of Additional Supplementary Files
Data Availability Statement
The authors declare that the data supporting the findings of this study are available within the article and its Supplementary Information Files.