

Abstract
Background
An on-going debate exists as to whether partial ventilatory support is lung protective in an acute phase of ARDS. So far, the effects of different respiratory efforts on the development of ventilator-associated lung injury (VALI) have been poorly understood.
To test the hypothesis whether respiratory effort itself promotes VALI, acute lung injury (ALI) was induced in 48 Sprague Dawley rats by hydrochloric acid aspiration model. Hemodynamics, gas-exchange, and respiratory mechanics were measured after 4 h of ventilation in pressure control (PC), assist-control (AC), or pressure support with 100% (PS100), 60% (PS60), or 20% (PS20) of the driving pressure during PC. VALI was assessed by histological analysis and biological markers.
Results
ALI was characterized by a decrease in PaO2/FiO2 from 447 ± 75 to 235 ± 90 mmHg (p < 0.001) and dynamic respiratory compliance from 0.53 ± 0.2 to 0.28 ± 0.1 ml/cmH2O (p < 0.001). There were no differences in hemodynamics or respiratory function among groups at baseline or after 4 h of ventilation. The reduction of mechanical pressure support was associated with a compensatory increase in an inspiratory effort such that peak inspiratory transpulmonary pressures were equal in all groups. The diffuse alveolar damage score showed significant lung injury but was similar among groups. Pro- and anti-inflammatory proteins in the bronchial fluid were comparable among groups.
Conclusions
In experimental ALI in rodents, the respiratory effort was increased by reducing the pressure support during partial ventilatory support. In the presence of a constant peak inspiratory transpulmonary pressure, an increased respiratory effort was not associated with worsening ventilator-associated lung injury measured by histologic score and biologic markers.
Keywords: Acute lung injury, ALI, Acute respiratory distress syndrome, ARDS, Ventilator-associated lung injury, VALI, Mechanical ventilation, Assisted spontaneous breathing
Background
Ventilator-associated lung injury (VALI) is a complication of mechanical ventilation. Tidal volume, airway pressure, and cyclic opening and closing of alveolar lung regions have been shown to impact the degree of VALI. A lung-protective ventilation strategy with reduced tidal volume [1], limited inspiratory plateau and driving pressure [2], and positive end-expiratory pressure (PEEP) [3] has been shown to improve outcomes in patients with acute respiratory distress syndrome (ARDS).
Controlled modes of mechanical ventilation that deliver full ventilatory support have been used to completely offload the work of breathing and thereby “rest the respiratory muscles.” In a recent clinical trial of patients with moderate to severe ARDS, abolishing spontaneous breathing activity with muscle paralysis during the first 48 h of treatment improved survival [4]. On the other hand, partial ventilatory support allows spontaneous breathing efforts during mechanical ventilation and preserves respiratory muscle function [5]. Partial ventilatory support, traditionally reserved for use in weaning, is now often used in all phases of mechanical ventilation [6, 7], although the potential of increased respiratory effort to the dependent lung to contribute to VILI is a matter of debate [8].
Preserved spontaneous breathing during partial ventilatory support is potentially protective because it may improve the homogeneity of ventilation and ventilation-perfusion distributions. Several experimental studies have investigated the role of spontaneous breathing on VALI. If spontaneous breathing was associated with a decrease in peak inspiratory transpulmonary pressure (PTPi), the development of VALI was attenuated with both higher [9] or decreased [10] inspiratory effort. On the other hand, the increased respiratory drive has been demonstrated to cause potentially injurious swings in transpulmonary pressure, so-called patient self-inflicted lung injury (P-SILI) [11] with a concomitant increase in PTPi that presumably augmented VALI [12].
Thus, it seems that the effect of spontaneous breathing on VALI is mainly related to peak PTPi. However, the role of inspiratory effort independent of PTPi was not investigated. We hypothesized that increased inspiratory effort, maintaining peak PTPi constant, is associated with VALI.
Materials and methods
Sixty-three male Sprague Dawley rats (456 ± 70 g, Charles River Laboratories, Wilmington, MA) were anesthetized with pentobarbital 55 mg/kg intraperitoneally and instrumented as described previously [13] (for a more detailed description see Additional file 1). Briefly, animals were tracheotomized with a 14 G tube and mechanically ventilated (EVITA4, Draeger Medical Canada Inc., Richmond, ON, Canada) with inspired oxygen fraction (FiO2) of 0.4. Twenty gauge catheters were inserted into the carotid artery and jugular vein for blood pressure monitoring and blood gas analysis (ABL510 + OSM3, Radiometer Copenhagen, Denmark). The FiO2 was set to 1.0 2 min before obtaining blood samples for gas analysis and set back to 0.4 after each measurement. The femoral artery was cannulated with a thermocouple probe (ADInstruments Inc., Colorado Springs, CO, USA) for cardiac output measurements. Sedation was maintained by infusion of ketamine 20 mcg/kg/min intravenously.
Measurements
Cardiac output was measured by transcardio-pulmonary temperature dilution of 0.5 ml of saline solution (LabChart 6.0, ADInstruments). Gas flow and airway pressures (PAW) were measured proximal to the endotracheal tube by a heated pneumotachograph (Hans Rudolph Inc., Shawnee, KS). Esophageal pressure (PES) was measured via a water-filled 20 G catheter with multiple perforations inserted into the esophagus. Dynamic compliance of the respiratory system was defined as tidal volume (VT) divided by the pressure difference between end-inspiratory and end-expiratory airway pressure. The maximum difference between end-inspiratory airway pressure and PES was defined as peak inspiratory transpulmonary pressure (PTPi). Respiratory effort was assessed by calculation of pressure-time-product (PTP) and work of breathing (WOB), which were calculated separately for total (WOBT), ventilator (WOBV), and respiratory muscles (WOBRM) from the recorded flow and PES tracings according to standard formulae (see Additional file 1). A data collection system was used (PowerLab, ADInstruments).
Experimental protocol
Initially, sedated animals were paralyzed with additional pancuronium 0.5 mg/kg/h intravenously and ventilated using pressure-controlled ventilation. The level of inspiratory driving pressure (ΔPAW) was set to achieve VT of 8 ml/kg, and the mechanical respiratory rate was set to maintain a PaCO2 < 60 mmHg, while avoiding flow at end-expiration. The positive end-expiratory pressure (PEEP) was set to 5 cmH2O and inspiration-expiration ratio set to 1:1. Lung injury was induced by intra-tracheal instillation of 0.5 ml of 0.2 m HCl as described previously [13]. Physiologic measurements were performed at baseline (BL) and 1 h after induction of lung injury (ALI-BL) (Additional file 1: Figure S1). A recruitment maneuver was performed by increasing PEEP to 10 cmH2O for 2 min, and animals were afterwards randomized to one of the following 5 groups:
PC: Pressure control with neuromuscular blockade and unchanged ventilatory settings.
AC: Assist-control mode without paralysis and the pressure controlled mechanical breaths set as before.
PS100: Spontaneous breathing with pressure support (PS) set equal to 100% of the previous ΔPAW (value required to obtain a VT of 8 ml/kg during pressure control).
PS60: Spontaneous breathing with PS set equal to 60% of the previous ΔPAW.
PS20: Spontaneous breathing with PS set equal to 20% of the previous ΔPAW.
After randomization, neuromuscular blockers were discontinued in groups 2–5. Animals remained in these settings for 4 h. Only if needed, the ΔPAW was adjusted to keep the VT < 10 ml/kg in case of changing respiratory system compliance. In PS, the respiratory rate was self-adjusted by the animals. Physiologic measurements were taken hourly with the final measurement after 4 h or the last one before death (ALI-End) (Additional file 1: Figure S1). Only animals receiving at least 2 h of the study ventilation regimen were included in the analysis. Arterial and venous blood samples were taken for cytokine analysis, then the animals were killed and the lungs dissected for histologic analysis as described previously [13]. The right middle lobe was weighed and dried for 48 h at 37 °C for analysis of wet-to-dry ratio.
Histopathology and cytokine analysis
After formalin fixation, the lungs were embedded in paraffin, cut, and stained with hematoxylin eosin. A lung pathologist blinded to the experimental group (Z.X.) graded the degree of diffuse alveolar damage (DAD) [13] (Additional file 1: Table S2).
Blood samples were immediately centrifuged and supernatant plasma stored at − 80°. Broncho-alveolar lavage fluid (BALF), and plasma samples were analyzed by multiple enzyme-linked immune assay (Luminex® technology, Panomics Inc., Fremont, CA) for tumor necrosis factor-α (TNF-α); interleukin (IL)-1β, IL6, and IL10; intracellular adhesion molecule (ICAM1); macrophage inflammatory protein (MIP1α); and KC (CXCL1 chemokine). Regulated upon Activation Normal T-cell Expressed and Secreted (RANTES) and Monocyte Chemotactic Protein (MCP1). Standard dilution curves were constructed to calculate concentrations in pg/ml.
Statistical analysis
Data were tested for normality by Kolmogorov-Smirnov test and also empirically based on visual inspection of Q-Q plots. Parametric data are presented as mean ± SD, while non-parametric data are expressed as median (interquartile range). Since sample size calculation is difficult, we assumed an effect size similar to previously published experiments [10–13] giving 8–10 animals per group. The sample size was based on pilot studies and on our past experience with ventilator strategies in small animals. We tested the hypothesis that increased WOBRM would increase DAD during assisted ventilation. Using data from previous experiments and a calculated effect size of 0.531, a sample size of 10 animals per group would provide the appropriate power (1-β = 0.82) to identify significant (α = 0.05) differences (G*Power 3.1.9.3, Duesseldorf, Germany).
Included for analysis were only animals that had completed at least 2 h of study ventilation after ALI-BL measurement. Baseline measurements were not included in the analysis, but are shown for informative purpose only. For parametric data, physiologic measurements were analyzed by a mixed-model ANOVA for repeated measurements with Wilk’s lambda indicating significance for between-group and within-group differences. If significant, post hoc comparison using SNK correction was used to analyze between-group differences. Repeated measurements were analyzed by paired t test and correlations by Spearman’s rho. Cytokines and W/D ratios were analyzed by one-way ANOVA, followed by post hoc comparison using SNK correction. Non-parametric data was analyzed accordingly by Kruskal-Wallis followed by a Mann-Whitney U test as appropriate. Categorical data were analyzed by chi-square (Statistical Package for the Social Sciences 15.0, SPSS Inc., Chicago, Ill, USA).
Results
All data except DAD score were normally distributed. There were no differences in hemodynamics or respiratory mechanics among groups at baseline. Intra-tracheal HCl caused similar hemodynamic and respiratory function compromise. Fifteen experiments had to be terminated early for various reasons, i.e., inadvertent severe lung damage, problems with instrumentation, handling, malfunction of equipment, drug application, hemodynamic compromise of other reason, and protocol violation. These experiments were stopped, and no analyses of cytokines or histology were performed, leaving 48 animals for analysis. Averaging data for all groups, the mean arterial pressure decreased from 144 ± 23 at BL to 119 ± 27 mmHg at ALI-BL (p < 0.001), although cardiac output remained stable at 122 ± 30 and 121 ± 34 ml/min (p = 0.34), respectively. Lung injury was characterized by a decrease in PaO2/FiO2 from 447 ± 75 to 235 ± 90 mmHg (p < 0.001) and in dynamic compliance from 0.53 ± 0.2 to 0.28 ± 0.1 ml/cmH2O (p < 0.001). At ALI-BL, the respiratory mechanics’ data were comparable among groups (Table 1).
Table 1.
Respiratory mechanics
| PC (n = 9) | AC (n = 10) | PS100 (n = 9) | PS60 (n = 10) | PS20 (n = 10) | Main effect | |||
|---|---|---|---|---|---|---|---|---|
| Within group | Between group | |||||||
| PAW [cmH2O] | BL | 15 ± 3 | 15 ± 4 | 13 ± 2 | 14 ± 3 | 13 ± 1 | ||
| ALI-BL | 22 ± 4 | 22 ± 4 | 18 ± 3 | 20 ± 4 | 21 ± 1 | |||
| ALI-End | 26 ± 6#* | 25 ± 7 | 19 ± 7 | 15 ± 5* | 9 ± 3#* | 0.060 | 0.000 | |
| PTPinsp [cmH2O] | BL | 16 ± 4 | 15 ± 4 | 13 ± 4 | 14 ± 4 | 13 ± 5 | ||
| ALI-BL | 22 ± 5 | 23 ± 4 | 20 ± 6 | 19 ± 6 | 19 ± 4 | |||
| ALI-End | 25 ± 7 | 26 ± 7 | 24 ± 9 | 23 ± 6 | 23 ± 8 | 0.006 | 0.987 | |
| PTPexp [cmH2O] | BL | 5 ± 3 | 4 ± 2 | 4 ± 3 | 6 ± 2 | 4 ± 4 | ||
| ALI-BL | 4 ± 3 | 5 ± 2 | 6 ± 5 | 4 ± 3 | 3 ± 3 | |||
| ALI-End | 5 ± 4 | 5 ± 4 | 6 ± 4 | 4 ± 2 | 3 ± 6 | 0.790 | 0.337 | |
| VT [ml/kg BW] | BL | 9.6 ± 1.2 | 8.7 ± 0.8 | 8.7 ± 0.6 | 8.8 ± 1 | 8.6 ± 0.9 | ||
| ALI-BL | 8.4 ± 1.6 | 9.4 ± 2.0 | 8.9 ± 2.8 | 11 ± 4 | 8.6 ± 1.9 | |||
| ALI-End | 9.0 ± 1.2 | 9.0 ± 1.2 | 9.1 ± 1.0 | 10 ± 3 | 8.9 ± 1.2 | 0.919 | 0.853 | |
| RR [min−1] | BL | 77 ± 18 | 81 ± 15 | 84 ± 8 | 87 ± 7 | 85 ± 9 | ||
| ALI-BL | 75 ± 17 | 82 ± 14 | 85 ± 11 | 92 ± 6 | 84 ± 11 | |||
| ALI-End | 86 ± 16# | 94 ± 17# | 71 ± 31 | 77 ± 17# | 80 ± 20 | 0.213 | 0.011 | |
| VE [ml/min] | BL | 334 ± 115 | 328 ± 68 | 348 ± 42 | 353 ± 56 | 339 ± 48 | ||
| ALI-BL | 323 ± 60 | 346 ± 86 | 364 ± 82 | 403 ± 91 | 352 ± 58 | |||
| ALI-End | 318 ± 70 | 411 ± 117 | 320 ± 115 | 396 ± 123 | 330 ± 130 | 0.662 | 0.345 | |
BL baseline measurement on pressure controlled ventilation, ALI-BL measurement on pressure-controlled ventilation after induction of lung injury, ALI-End last measurement after 2–4 h of experimental ventilation mode, PAW peak airway pressure, PTPi peak inspiratory transpulmonary pressure, VT tidal volume, RR respiratory rate, VE minute ventilation, *p < 0.05 vs. other groups #p < 0.05 vs. ALI-BL
After randomization, VT remained similar among groups (Table 1), but ΔPAW had to be increased in PC and decreased in PS100 to maintain the target VT. Minute ventilation remained constant despite the reduction of ventilatory support. PTPi was similar among groups, independent from the level of ventilatory support (Table 1, Additional file 1: Figure S2).
The level of ventilatory support did not significantly affect hemodynamics. The mean arterial pressure, but not cardiac output, decreased similarly in all groups over time (Table 2). However, animals in PS20 exhibited more episodes of hemodynamic destabilization, leading to the lowest rate of animals achieving 240 min of ventilation (PC 44%, AC 70%, PS100 55%, PS60 60%, and PS20 30%; p = 0.68). In prematurely deceased animals, the time between ALI-BL and death was on average 211 ± 35 min, which was not significantly different among groups (Additional file 1: Table S1).
Table 2.
Hemodynamics and gas exchange
| PC (n = 9) | AC (n = 10) | PS100 (n = 9) | PS60 (n = 10) | PS20 (n = 10) | Main effect | |||
|---|---|---|---|---|---|---|---|---|
| Within group | Between group | |||||||
| HR [min−1] | BL | 420 ± 33 | 394 ± 43 | 388 ± 90 | 405 ± 32 | 370 ± 36 | ||
| ALI-BL | 377 ± 65 | 382 ± 45 | 374 ± 53 | 375 ± 65 | 410 ± 41 | |||
| ALI-End | 357 ± 96 | 324 ± 67# | 353 ± 64 | 342 ± 39 | 375 ± 42# | 0.005 | 0.830 | |
| MAP [mmHg] | BL | 139 ± 26 | 134 ± 32 | 152 ± 15 | 150 ± 16 | 147 ± 23 | ||
| ALI-BL | 101 ± 37 | 120 ± 24 | 124 ± 22 | 125 ± 23 | 124 ± 24 | |||
| ALI-End | 47 ± 40# | 74 ± 44# | 80 ± 16# | 78 ± 26# | 72 ± 35# | 0.000 | 0.943 | |
| CO [ml/min] | BL | 116 ± 44 | 107 ± 27 | 128 ± 24 | 130 ± 27 | 124 ± 26 | ||
| ALI-BL | 92 ± 29 | 103 ± 17 | 147 ± 30 | 129 ± 18 | 146 ± 41 | |||
| ALI-End | 128 ± 47 | 91 ± 52 | 130 ± 29 | 132 ± 29 | 113 ± 42 | 0.575 | 0.139 | |
| pHa | BL | 7.26 ± 0.09 | 7.26 ± 0.06 | 7.25 ± 0.08 | 7.25 ± 0.07 | 7.28 ± 0.05 | ||
| ALI-BL | 7.22 ± 0.11 | 7.23 ± 0.07 | 7.22 ± 0.06 | 7.24 ± 0.07 | 7.22 ± 0.05 | |||
| ALI-End | 7.07 ± 0.12# | 7.08 ± 0.15# | 7.04 ± 0.19# | 7.13 ± 0.15# | 7.04 ± 0.09# | 0.000 | 0.763 | |
| PaO2 [torr] | BL | 445 ± 78 | 440 ± 76 | 451 ± 85 | 448 ± 84 | 450 ± 67 | ||
| ALI-BL | 204 ± 119 | 234 ± 105 | 271 ± 91 | 243 ± 73 | 240 ± 62 | |||
| ALI-End | 92 ± 50# | 118 ± 108# | 166 ± 126# | 106 ± 67# | 83 ± 64# | 0.000 | 0.873 | |
| PaCO2 [torr] | BL | 59 ± 19 | 59 ± 10 | 59 ± 14 | 59 ± 10 | 57 ± 11 | ||
| ALI-BL | 60 ± 14 | 57 ± 14 | 61 ± 13 | 61 ± 14 | 65 ± 11 | |||
| ALI-End | 74 ± 27 | 68 ± 24 | 78 ± 19# | 79 ± 26# | 87 ± 20# | 0.000 | 0.734 | |
BL baseline measurement on pressure controlled ventilation, ALI-BL measurement on pressure-controlled ventilation after induction of lung injury. Blood gas analyses performed with FiO2 = 1.0. ALI-End last measurement after 2–4 h of experimental ventilation mode, HR heart rate, MAP mean arterial pressure, CO cardiac output, *p < 0.05 vs. other groups #p < 0.05 vs. ALI-BL
After the establishment of lung injury, oxygenation continued to deteriorate similarly in all groups from ALI-BL to ALI-End (Table 2). PaCO2 inversely correlated to reduced ventilatory support and tended to be highest in PS20 (n.s.). The increase in PaCO2 was significant only for pressure support groups.
As expected, the establishment of ALI was associated with an increase in the average WOBT from 0.46 ± 0.2 J/l at BL to 0.95 ± 0.47 J/l at ALI-BL (p < 0.001). The calculated WOBV was highest in PC and decreased with ventilatory support to a minimum with PS20. The WOBRM was lowest in PC and increased with reduced ventilatory support (Fig. 1, Additional file 1: Table S3). The pressure-time product behaved similar to WOBRM; both parameters correlated significantly (r = 0.742, p < 0.001). Across all groups, the higher the work performed by the ventilator (WOBV), the lower the mean arterial pressure (r = − 0.389, p = 0.007) (Additional file 1: Figure S3).
Fig. 1.

Work of breathing exerted by the animal (WOBRM) at baseline and after 4 h (ALI-End) of ventilation with pressure control (PC), assist control (AC), or pressure support ventilation with 100%(PS100), 60%(PS60), or 20%(PS20) of previous pressure control level after establishment of ALI.*p < 0.05 vs. indicator line; #p < 0.05 vs. all other groups
There were no significant differences in wet-to-dry ratios among groups (Additional file 1: Figure S3). The wet-to-dry ratio decreased with the higher pressure-time product (r = − 0.305, p = 0.037). Lung damage was mainly related to the epithelium, neutrophil infiltration, and edema, but not hemorrhage. The level of ventilatory support did not affect the total alveolar damage score (Fig. 2) or its components (Additional file 1: Table S4). The alveolar damage score correlated inversely with pressure-time product (r = − 0.469, p = 0.049) (Additional file 1: Figure S6).
Fig. 2.
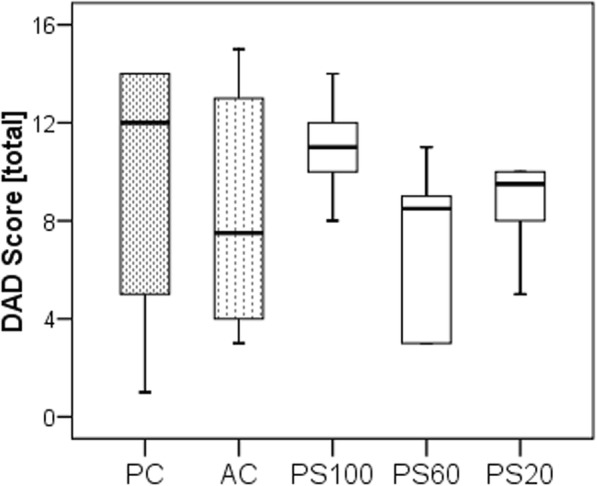
Diffuse alveolar damage (DAD) total score after 4 h of ventilation in pressure control (PC), assist control (AC), or pressure support ventilation with 100%(PS100), 60%(PS60), or 20%(PS20) of previous pressure control level. Differences among groups were not statistically significant (p = 0.097)
The concentrations of inflammatory markers were not different between arterial and venous blood samples and did not differ among the variated levels of ventilatory support (Additional file 1: Tables S5, S6, S7). IL-6 (Fig. 3a), KC, MIP1a, and MCP1 were higher in the BAL compared to plasma; IL1β, ICAM, and TNF-α (Fig. 3b) were equal in the BAL and plasma. RANTES was higher in plasma compared to BAL in all groups and IL-10 only in PS60 and PS20 groups.
Fig. 3.

Measurement of cytokines in the bronchoalveolar fluid (BALF) and arterial (Art) and venous (Ven) samples. Data are presented as mean ± SD [pg/ml], upper panel (a): IL6. Measured levels were higher in the BALF than the plasma, but differences among groups were not statistically significant; lower panel (b): TNF-a. No significant differences were found between BALF and plasma or among groups
Discussion
In a rodent acid aspiration model of mild to moderate acute lung injury, we found that increased respiratory effort when generated such as to produce identical peak inspiratory transpulmonary pressure and tidal volume had no effect on gas exchange, histologic, and biologic markers of lung injury.
We tried to control for most known factors associated with the development of VALI. The lung injury model and dosage of HCl was chosen in order to achieve severe lung damage and keep the model stable during the experiment time. Protective ventilation was maintained both during controlled and partial ventilatory support ventilation, with a PEEP sufficient to prevent major regional lung collapse in small animals [13]. Sedation was administered continuously and equally in all groups. The duration of ventilation after induction of lung injury was at least 3 h making it reasonable to investigate the production of inflammatory proteins. Supposedly, previous studies investigating the influence of transpulmonary pressure on VALI resulted in different levels of PTPI. The innovative approach of our study was to investigate the effect of different levels of inspiratory effort (while keeping PTPi constant) on VALI and the production of inflammatory proteins.
Despite varying levels of partial ventilatory support, the animals maintained constant tidal volumes and minute ventilation. The reduction of pressure support caused a compensatory increase in the respiratory effort to match previous PTPi and preserve tidal volume. To achieve similar PTPi, the distribution of forces across the lungs must be different during partial ventilatory support: the alveolar pressure is lower and the pleural pressure more negative with progressive reduction in pressure support, which could impact hemodynamics. Consequently, the decrease of partial ventilatory support was associated with an increase of WOBRM and pressure-time product.
Our data suggest that, during partial ventilatory support, PTPi is more important than respiratory effort “per se” in generating VALI, suggesting that the major factor causing volutrauma is the degree of lung stretch [14, 15] .
Previous experimental studies showed a possible protective effect of hypercapnia on the development of VALI [16], although severe hypercapnia is associated with worse outcome in patients with ARDS if used as an indicator of the inability to sufficiently ventilate the failing lung [17]. In our study, all animals developed hypercapnia, but the increase was significant only for those ventilated in pressure support. IL-10 was the only mediator increasing in PS60 and PS20 groups, the modes with the highest PaCO2, demonstrating a possible, anti-inflammatory effect.
VALI is characterized by structural injury (alveolar damage score) and biochemical mediators. While cytokine mRNA expression is a sensitive measure, it is not necessarily followed by biologic injury. Conversely, the detection of active protein has the advantage of a definite effect, although proof of origin, i.e., the lung, may be problematic. The fact that some, but not all inflammatory markers (IL1β, TNF-α) had concentrations similar in BALF and plasma implies differences in alveolar secretion of biomarkers. Conversely, those cytokines that were increased in BALF, but not plasma (IL6, KC, MCP1), likely originate in the lung before spilling into the blood. The absence, presence, or amount of inspiratory effort did not affect lung-derived pro-inflammatory substances under the condition of a constant PTPi.
Previous work by Yoshida et al. [10, 12] seems conflicting with our results. There are substantial differences in our study design that may explain some of the different findings. First, most important, we have controlled for the transpulmonary pressure which was equal in all groups rather than PTP being an outcome of the ventilation strategy. The amount of alveolar damage was closely correlated with increased PTP. We have also used a more physiologic calculation of the total alveolar stretching pressure, as we calculated the pressures across the lung (PTPi) from real-time tracings. Yoshida et al. have used a fixed sum of PPLAT + ΔPes, regardless whether it was becoming effective to the lungs. Second, we have used a more realistic ventilation strategy. Yoshida et al. have used chemical agents to increase respiratory drive and thereby added spontaneous breathing on top, increasing minute ventilation and PEEPi, a condition neither physiological nor clinically applied [12]. In another study Yoshida et al. used assist-control ventilation; however, from the printed curves, there is evidence of severe subject-ventilator dyssynchrony, another important mechanism of VALI [10]. In our study, pressure support was used to gradually increase respiratory effort while letting the subject maintain minute ventilation and controlling PTPi. Third, we used the most physiologic model of acid installation to induce lung injury [13]. The lavage model is prone to spontaneous improvement and very minimal influence on alveolar damage, while the extent of the actual lung injury is mainly depending on the ventilation settings. In contrast, the acid aspiration model creates characteristic inflammation and lung injury very similar to clinical situations. We did not specifically design our study to investigate the importance of spontaneous breathing on VALI. Although previous investigations suggested that strong spontaneous breathing in severe ARDS might augment lung injury [10], a large observational clinical study established an association of assisted ventilation with better outcome compared controlled mechanical ventilation and paralysis in patients from mild to severe ARDS [18].
We cannot exclude the possibility that increased effort during PS is associated with vascular injury, since DAD did not differentiate alveolar and perivascular hemorrhage. Previous studies have reported increased hemorrhage with the addition of respiratory effort, when rabbits were ventilated with tidal volumes of 7–9 ml/kg12. Additionally, DAD and hemorrhage were higher in AC with spontaneous breathing in severe lung injury and higher PTPi [10]. However, in these studies, the work of breathing and pressure-time product were not measured. We found no difference in DAD and hemorrhage with increased respiratory effort.
Recent studies during volume control ventilation reported possible effects of increased mechanical power on lung injury [19, 20]. However, during assisted ventilation, the mechanical power to the lungs was delivered partly by the ventilator and partly by the inspiratory effort of the subject. In our study, the total mechanical power was similar at different ventilatory settings, suggesting that during assisted ventilation, it is mandatory to partition subject inspiratory effort.
Our study does have limitations that must be considered. First, we used an aspiration model in rats; this is a clinically relevant model of lung injury, but we cannot exclude the possibility that different injury models in different species may produce different results. We have used a model of lung injury, which may exhibit changes over time, but is relatively constant in these changes and the produced biological effect making it advantageous over other models [13]. Second, we investigated only pressure support and not other partial ventilatory support modalities. Third, those animals ventilated with a lower level of support had hypercapnic acidosis (HA) suggesting a higher grade of ventilation/perfusion mismatch. We cannot rule out the possibility that HA attenuated possible higher lung injury at lower levels of pressure support. Fourth, since we did not measure lung volume and recruitment, different values for strain and atelectrauma [21] could have influenced our results. However, if pressure across the lung and tidal inflation are constant, increasing activation of the respiratory muscles is not associated with increased lung damage. Fifth, our approach was a more dynamic and physiological one, rather than analyzing the spatial distribution of gas across the lungs. Even CT scanning is not able to differentiate between the collapsed lung and edematous lung in non-aerated areas. From histology in similar experiments [10], it is evident that non-aerated lung was mainly due to alveolar/interstitial edema and infiltrates. The selected level of PEEP (5 cmH2O) in small animals physiologically exerts equal effects to the lungs as higher levels in larger animals (i.e., likely equaling PEEP 10 cmH2O in rabbits and PEEP 15 cmH2O in dogs) or humans and thus appears appropriate to assure sufficient aeration. Further, the influence of edematous lung on Va/Q mismatch and atelectasis formation in the dependent lung is much less in rodents with lungs weighing a few grams only as compared to larger animals or humans. Sixth, we did not monitor neuromuscular blockage but assessed the absence or presence of spontaneous breathing by the esophageal tracings, a method that has been used by other investigators as well [10, 12]. Seventh, not all animals completed the planned 240 min of the experiment. Most experimental data in small animals come from shorter experiments, commonly being ≤ 2 h of mechanical ventilation [22, 23]. Considering the kinetics of cytokine development after injury, the measured plasma levels depend on the experimental setup. The lower rate of finishing animals in PC and PS20 might have been underestimated by a type II error. Although not powered to detect a difference in mortality among groups, our data suggest that the type of ventilation mode independently from the inspiratory effort might have an effect on survival.
Conclusions
This study provides experimental evidence that in a clinically relevant animal model of acute lung injury, the degree of VALI is not correlated with increased respiratory effort per se, when tidal volume and end-inspiratory transpulmonary pressures are relatively constant across differing respiratory efforts. Our results suggest that in setting the ventilator during partial ventilatory support, PTPi is more important than inspiratory effort to minimize VALI. In application to patient care, it has to be considered that these results were obtained with lung-protective settings and might not be true for different ventilator conditions. As a clinical consequence, monitoring of esophageal pressure may be useful to optimize ventilatory setting [24].
Supplementary information
Additional file 1: Supplemental digital content
Acknowledgements
We thank Nancy McGrath and Raymond Chankalal for their invaluable help in the lab and for preparing the histology slides. We thank Sara Whynot, MLT, and Juan Zou, MD PhD, for their support and help in conducting the experiments. We thank Julie Kang at St. Michael’s Hospital, Toronto, for her help in the cytokine analyses. We owe special thanks to Lorenzo Ball MD, Genoa, for statistical review of the manuscript.
Abbreviations
- ALI
Acute lung injury
- ARDS
Acute respiratory distress syndrome
- BALF
Bronco-alveolar lavage fluid
- CDYN
Dynamic compliance respiratory system
- ICAM
Intracellular adhesion molecule
- IL1β, -6, -10
Interleukin
- IP
Intraperitoneal
- IV
Intravenous
- KC
CXCL1 chemokine
- MCP1
Monocyte chemotactic protein
- MIP1α
Macrophage inflammatory protein
- PAW
Airway pressure
- PC
Pressure control
- PES
Esophageal pressure
- PS
Pressure support
- PTP
Pressure time product
- PTPi
Maximum inspiratory transpulmonary pressure
- RANTES
Regulated upon activation normal T cell expressed and secreted
- RR
Respiratory rate
- TNF-α
Tumor necrosis factor α
- VE
Minute ventilation
- VT
Tidal volume
- WOBRM
Work of breathing performed by respiratory muscles
- WOBT
Total work of breathing
- WOBV
Work of breathing performed by the ventilator
Authors’ contributions
DH conceived the study, participated in the experiments, the analysis of data, and writing of the manuscript. AS helped analyze the data. ZX performed the histological scoring. NI participated in the experiments and interpretation of the data. HZ participated in the cytokine analysis. ASS helped in the design of the study, interpretation of the data, and writing of the manuscript. PP participated in conceiving the study, interpretation of data, and writing of the manuscript.
Authors’ information
The work was performed at Dalhousie University, Halifax NS, Canada, Departments of Anesthesiology, Critical Care and Physiology & Biophysics, 1276 South Park St, 10 West Victoria, Halifax NS, B3H 2Y9, Canada.
Funding
This study was supported by Dalhousie University, Clinical Research Scholar, and the Lung Association of Nova Scotia, Halifax, Canada.
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Ethics approval and consent to participate
The Dalhousie University Committee on Laboratory Animals approved the protocol (UCLA 08-132), and the care and handling of the animals were in accordance with the National Institutes of Health Guidelines for ethical animal treatment.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary information accompanies this paper at 10.1186/s40635-019-0272-z.
References
- 1.Putensen C, Theuerkauf N, Zinserling J, Wrigge H, Pelosi P. Meta-analysis: ventilation strategies and outcomes of the acute respiratory distress syndrome and acute lung injury. Ann Intern Med. 2009;151:566–576. doi: 10.7326/0003-4819-151-8-200910200-00011. [DOI] [PubMed] [Google Scholar]
- 2.Amato MB, Meade MO, Slutsky AS, Brochard L, Costa EL, Schoenfeld DA, Stewart TE, Briel M, Talmor D, Mercat A, Richard JC, Carvalho CR, Brower RG. Driving pressure and survival in the acute respiratory distress syndrome. N Engl J Med. 2015;372:747–755. doi: 10.1056/NEJMsa1410639. [DOI] [PubMed] [Google Scholar]
- 3.Briel M, Meade M, Mercat A, Brower RG, Talmor D, Walter SD, Slutsky AS, Pullenayegum E, Zhou Q, Cook D, Brochard L, Richard JC, Lamontagne F, Bhatnagar N, Stewart TE, Guyatt G. Higher vs lower positive end-expiratory pressure in patients with acute lung injury and acute respiratory distress syndrome: systematic review and meta-analysis. JAMA. 2010;303:865–873. doi: 10.1001/jama.2010.218. [DOI] [PubMed] [Google Scholar]
- 4.Papazian L, Forel JM, Gacouin A, Penot-Ragon C, Perrin G, Loundou A, Jaber S, Arnal JM, Perez D, Seghboyan JM, Constantin JM, Courant P, Lefrant JY, Guerin C, Prat G, Morange S, Roch A. Neuromuscular blockers in early acute respiratory distress syndrome. N Engl J Med. 2010;363:1107–1116. doi: 10.1056/NEJMoa1005372. [DOI] [PubMed] [Google Scholar]
- 5.Levine S, Nguyen T, Taylor N, Friscia ME, Budak MT, Rothenberg P, Zhu J, Sachdeva R, Sonnad S, Kaiser LR, Rubinstein NA, Powers SK, Shrager JB. Rapid disuse atrophy of diaphragm fibers in mechanically ventilated humans. N.Engl.J Med. 2008;358:1327–1335. doi: 10.1056/NEJMoa070447. [DOI] [PubMed] [Google Scholar]
- 6.Gonzalez M, Arroliga AC, Frutos-Vivar F, Raymondos K, Esteban A, Putensen C, Apezteguia C, Hurtado J, Desmery P, Tomicic V, Elizalde J, Abroug F, Arabi Y, Moreno R, Anzueto A, Ferguson ND. Airway pressure release ventilation versus assist-control ventilation: a comparative propensity score and international cohort study. Intensive Care Med. 2010;36:817–827. doi: 10.1007/s00134-010-1837-1. [DOI] [PubMed] [Google Scholar]
- 7.Esteban A, Frutos-Vivar F, Muriel A, Ferguson ND, Penuelas O, Abraira V, Raymondos K, Rios F, Nin N, Apezteguia C, Violi DA, Thille AW, Brochard L, Gonzalez M, Villagomez AJ, Hurtado J, Davies AR, Du B, Maggiore SM, Pelosi P, Soto L, Tomicic V, D’ Empaire G, Matamis D, Abroug F, Moreno RP, Soares MA, Arabi Y, Sandi F, Jibaja M, Amin P, Koh Y, Kuiper MA, Bulow HH, Zeggwagh AA, Anzueto A. Evolution of mortality over time in patients receiving mechanical ventilation. Am J Respir Crit Care Med. 2013;188:220–230. doi: 10.1164/rccm.201212-2169OC. [DOI] [PubMed] [Google Scholar]
- 8.Yoshida Takeshi, Engelberts Doreen, Otulakowski Gail, Katira Bhushan, Post Martin, Ferguson Niall D., Brochard Laurent, Amato Marcelo B. P., Kavanagh Brian P. Continuous Negative Abdominal Pressure Recruits Lungs at Lower Distending Pressures. American Journal of Respiratory and Critical Care Medicine. 2018;197(4):534–537. doi: 10.1164/rccm.201705-0979LE. [DOI] [PubMed] [Google Scholar]
- 9.Carvalho NC, Guldner A, Beda A, Rentzsch I, Uhlig C, Dittrich S, Spieth PM, Wiedemann B, Kasper M, Koch T, Richter T, Rocco PR, Pelosi P, de Abreu MG. Higher levels of spontaneous breathing reduce lung injury in experimental moderate acute respiratory distress syndrome. Crit Care Med. 2014;42:e702–e715. doi: 10.1097/CCM.0000000000000605. [DOI] [PubMed] [Google Scholar]
- 10.Yoshida T, Uchiyama A, Matsuura N, Mashimo T, Fujino Y. The comparison of spontaneous breathing and muscle paralysis in two different severities of experimental lung injury. Crit Care Med. 2013;41:536–545. doi: 10.1097/CCM.0b013e3182711972. [DOI] [PubMed] [Google Scholar]
- 11.Brochard L, Slutsky A, Pesenti A. Mechanical ventilation to minimize progression of lung injury in acute respiratory failure. Am J Respir Crit Care Med. 2017;195:438–442. doi: 10.1164/rccm.201605-1081CP. [DOI] [PubMed] [Google Scholar]
- 12.Yoshida T, Uchiyama A, Matsuura N, Mashimo T, Fujino Y. Spontaneous breathing during lung-protective ventilation in an experimental acute lung injury model: high transpulmonary pressure associated with strong spontaneous breathing effort may worsen lung injury. Crit Care Med. 2012;40:1578–1585. doi: 10.1097/CCM.0b013e3182451c40. [DOI] [PubMed] [Google Scholar]
- 13.Henzler D, Hochhausen N, Chankalal R, Xu Z, Whynot SC, Slutsky AS, Zhang H. Physiologic and biologic characteristics of three experimental models of acute lung injury in rats. Anesth Analg. 2011;112:1139–1146. doi: 10.1213/ANE.0b013e3182104dac. [DOI] [PubMed] [Google Scholar]
- 14.Engelberts D, Malhotra A, Butler JP, Topulos GP, Loring SH, Kavanagh BP. Relative effects of negative versus positive pressure ventilation depend on applied conditions. Intensive Care Med. 2012;38:879–885. doi: 10.1007/s00134-012-2512-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Slutsky AS, Ranieri VM. Ventilator-induced lung injury. N Engl J Med. 2013;369:2126–2136. doi: 10.1056/NEJMra1208707. [DOI] [PubMed] [Google Scholar]
- 16.O'Croinin D, Ni CM, Higgins B, Laffey JG. Bench-to-bedside review: permissive hypercapnia. Crit Care. 2005;9:51–59. doi: 10.1186/cc2918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nin N, Muriel A, Penuelas O, Brochard L, Lorente JA, Ferguson ND, Raymondos K, Rios F, Violi DA, Thille AW, Gonzalez M, Villagomez AJ, Hurtado J, Davies A, Du B, Maggiore SM, Soto L, D’ Empaire G, Matamis D, Abroug F, Moreno R, Soares MA, Arabi Y, Sandi F, Jibaja M, Amin P, Koh Y, Kuiper MA, Buelow HH, Zeggwagh AA, Anzueto A, Sznajder JI, Esteban A, VENTILA Group Severe hypercapnia and outcome of mechanically ventilated patients with moderate or severe acute respiratory distress syndrome. Intensive Care Med. 2017;43:200–208. doi: 10.1007/s00134-016-4611-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.van Haren F, Pham T, Brochard L, Bellani G, Laffey J, Dres M, Fan E, Goligher EC, Heunks L, Lynch J, Wrigge H, McAuley D. Spontaneous breathing in early acute respiratory distress syndrome: insights from the large observational study to understand the global impact of severe acute respiratory failure study. Crit Care Med. 2019;47:229–238. doi: 10.1097/CCM.0000000000003519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Silva PL, Ball L, Rocco PRM, Pelosi P. Power to mechanical power to minimize ventilator-induced lung injury? Intensive Care Med Exp. 2019;7(Suppl 1):38. doi: 10.1186/s40635-019-0243-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Santos RS, Maia LA, Oliveira MV, Santos CL, Moraes L, Pinto EF, CDS S, Machado JA, Carvalho AC, MVS F, Martins V, Capelozzi VL, Morales MM, Koch T, Gama de Abreu M, Pelosi P, Silva PL, Rocco PRM. Biologic impact of mechanical power at high and low tidal volumes in experimental mild acute respiratory distress syndrome. Anesthesiology. 2018;128:1193–1206. doi: 10.1097/ALN.0000000000002143. [DOI] [PubMed] [Google Scholar]
- 21.Chiumello D, Carlesso E, Cadringher P, Caironi P, Valenza F, Polli F, Tallarini F, Cozzi P, Cressoni M, Colombo A, Marini JJ, Gattinoni L. Lung stress and strain during mechanical ventilation for acute respiratory distress syndrome. Am J Respir Crit Care Med. 2008;178:346–355. doi: 10.1164/rccm.200710-1589OC. [DOI] [PubMed] [Google Scholar]
- 22.Saddy F, Oliveira GP, Garcia CS, Nardelli LM, Rzezinski AF, Ornellas DS, Morales MM, Capelozzi VL, Pelosi P, Rocco PR. Assisted ventilation modes reduce the expression of lung inflammatory and fibrogenic mediators in a model of mild acute lung injury. Intensive Care Med. 2010;36:1417–1426. doi: 10.1007/s00134-010-1808-6. [DOI] [PubMed] [Google Scholar]
- 23.Chiumello D, Pristine G, Slutsky AS. Mechanical ventilation affects local and systemic cytokines in an animal model of acute respiratory distress syndrome. Am J Respir Crit Care Med. 1999;160:109–116. doi: 10.1164/ajrccm.160.1.9803046. [DOI] [PubMed] [Google Scholar]
- 24.Akoumianaki E, Maggiore SM, Valenza F, Bellani G, Jubran A, Loring SH, Pelosi P, Talmor D, Grasso S, Chiumello D, Guerin C, Patroniti N, Ranieri VM, Gattinoni L, Nava S, Terragni PP, Pesenti A, Tobin M, Mancebo J, Brochard L. The application of esophageal pressure measurement in patients with respiratory failure. Am J Respir Crit Care Med. 2014;189:520–531. doi: 10.1164/rccm.201312-2193CI. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1: Supplemental digital content
Data Availability Statement
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.