

Abstract
Background
The DelIVery for Pulmonary Arterial Hypertension clinical trial was a multi-center, prospective, single arm, Investigational Device Exemption study utilizing a fully implantable, programmable intravascular delivery system consisting of a pump and a catheter for intravenous treprostinil. The study met its primary endpoint and demonstrated that the intravascular delivery system significantly reduced catheter related complications at 22,000 subject-days of follow-up compared with a predefined objective performance criterion. Here we summarize the results obtained during a 6.4-year follow-up period.
Methods
Throughout study follow-up, participants had clinic visits and medication refills at least every 12 weeks (dependent on the subjects’ dose). All adverse events and intravascular delivery system complications were evaluated and recorded.
Results
Sixty pulmonary arterial hypertension subjects were followed post device implantation for approximately 282 patient-years (range 87 days to 6.4 years). Of the 60 subjects, 14 died (1 related to intravascular delivery system pump failure), 2 withdrew after lung transplants, and 2 withdrew due to pump pocket infection. No catheter-related bloodstream infections, catheter thrombosis or occlusions, or catheter kinks occurred through 282 patient-years. Two participants had adverse events of abdominal pain, rash, due to subcutaneous treprostinil “leaks” after one catheter puncture and one catheter laceration during pump refill and replacement, respectively. Eight pump failure events occurred: seven pump motor stalls and one early replacement (faulty battery).
Conclusion
Delivery of treprostinil with an intravascular delivery system is a safe alternative to an external delivery system, while providing enhanced life experiences. To preserve the risk–benefit ratio, treatment at specialized pulmonary arterial hypertension centers is recommended until training is disseminated at other sites.
Keywords: pulmonary arterial hypertension, treprostinil, prostacyclin, internal device
Introduction
Current World Symposium Proceedings in Pulmonary Hypertension recommend treatment with parenteral prostacyclins for high-risk newly diagnosed or rapidly progressing pulmonary arterial hypertension (PAH) requiring escalation of therapy.1,2 However, risks and complexities associated with current external delivery systems limit the use of parenteral therapy.3,4 In addition, the use of prostacyclins may be limited in part as all PAH therapies are expensive, with use limited by individual political and geographic constraints. The DelIVery for Pulmonary Arterial Hypertension (PAH) clinical trial was a multi-center, prospective, single arm, Investigational Device Exemption study utilizing a fully implantable, programmable intravascular delivery system (IDS) consisting of a pump and a catheter for intravenous (IV) treprostinil. An exemption study allows the device, in this case the approved Medtronic SynchroMed II model 8637 pump with the novel 10642 catheter designed by Medtronic for treprostinil delivery at low flow rates, to be used in clinical study as a novel system to collect safety and outcome data. The study met its primary endpoint and demonstrated a reduced rate of catheter-related complications (systemic bloodstream infections, site infections, and complications from catheter thrombosis, mechanical dysfunction, or catheter dislocation) or procedure-related pneumothorax using the implantable system after 22,000 patient-days of implant compared with a predefined objective performance criterion (OPC).5 A completely implantable delivery system (IDS) for treprostinil infusion allowed PAH patients to return to normal daily activities (e.g. bathing without fear of infection and sleeping without worry of dislocating or kinking the infusion catheter), by reducing the risks of these specific complications associated with external delivery systems. This manuscript provides performance and safety information, and protocol and technical adjustments of the IDS over a 6.4-year follow-up period since the first patient was implanted.
Methods
The subject selection, design, and detailed methods of the DelIVery for PAH trial have been previously published including a description of the unique catheter design6 and system implant techniques utilized in the study (model 10642 Implantable Intravascular Catheter, model 8637 SynchroMed II Implantable Programmable Pump, and the model 8840 N’Vision Clinician Programmer (Medtronic, plc).5 A total of 10 US centers participated in this long-term observational study under an Investigational Device Exemption in accordance with the amended Declaration of Helsinki. The institutional review boards (IRBs) at each center approved the protocol, and written informed consent was obtained from all patients. The study was registered with clinicaltrials.gov under number NCT01321073. For full details of the participating IRBs, please see the online supplement.
Implant and follow-up procedures
The implant procedure has been previously published in detail.7 Implanted subjects were seen at scheduled follow-up visits as part of routine clinical care for patients with advanced PAH and depending on dose at planned pump refill visits, and at unscheduled follow-up visits as clinically indicated. During follow-up visits, the implanted pump was refilled with treprostinil via percutaneous needle access to the pump reservoir at intervals dependent on patient dose (up to a maximum of 12 weeks). At the time of a refill, residual drug remaining in the pump reservoir was removed, quantified, and discarded, and the pump refilled with fresh drug (10 mg/mL concentration). In all cases the programmed dose versus actual dose delivered was assessed and any dose discrepancy was tracked for each subject. In the later part of the study, the concentrated treprostinil was chilled at approximately 4℃ for at least 30 min prior to use in refill procedure to limit any leakage of drug from the refill needle.
Adverse events (AE) and IDS complications
All AEs were reported throughout the study. This excluded documented pre-existing conditions unless the nature or severity of the condition worsened. Serious AEs were defined as those that were life-threatening that could result in death, hospitalization, or require intervention to prevent a serious outcome. Catheter failures were defined as events that required replacement of the catheter. Pump failure was defined as inability to restart the pump after unplanned cessation. Pump stall is a cessation of the pump. System modification is defined as replacement of the catheter and/or pump.
An independent Adverse Events Advisory Committee (AEAC) reviewed all AEs. The AEAC and the investigators worked with the biomedical engineers throughout the study to determine causation of any mechanical pump or catheter and, system related issues. All AEs were adjudicated by the AEAC and steering committee and appropriate changes to the study protocol were made regarding process and clinical care.
Statistical methods
Summary statistics were reported as mean ± standard deviation for continuous variables, and count (%) for categorical variables. Survivorship analysis was conducted using the Kaplan–Meier method, with events that included death, catheter failure, pump failure, pump replacement, and system modification. Subjects were censored at their last known follow-up. Survivorship point estimates and 95% confidence intervals are provided. Comparisons were made using Fishers exact test with p values below 0.05 considered statistically significant.
Results
Participants (n = 60) were implanted from June 2011 through November 2012 and have accumulated a total of 281.8 patient-years of follow-up (mean 4.7 ± 1.4 years; range 87 days to 6.4 years). Forty-two participants (70.0%) remain in the study, 14 (23.3%) died, and 4 (6.7%) withdrew (Fig. 1). The 42 participants still in the study had an average age of 47.6 years at implant, 85.7% are female, versus the entire study population which had an average age of 50.1 years and 80% female (Table 1). One of the 14 deaths was adjudicated by the AEAC to be related to the investigational system. In this case, a patient died due to disease progression after a pump failure confounded by a subsequent clinical care failure as adjudicated by the AEAC. The subject was transferred to their study center with symptoms of worsening dyspnea. Unfortunately, the recommended study protocol procedure for pump interrogation and assessment in the event of worsening symptoms, and if indicated, the subsequent initiation of treprostinil via an external pump, was not followed. Thus, the pump failure was not detected with subsequent death of the patient. Engineers analyzed the pump system post-mortem and confirmed pump failure.
Fig. 1.
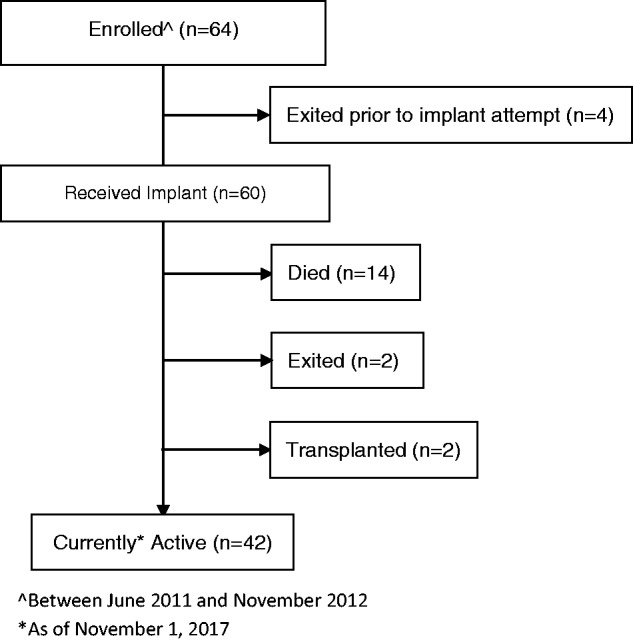
CONSORT flow diagram.
Table 1.
Baseline characteristics of implanted subjects.
| Baseline characteristics | |
|---|---|
| Enrolled subjects (n = 60) | |
| Age (years) | 50.1 ± 13.5 |
| Gender (female) | 48 (80.0%) |
| Race/Ethnic origin | |
| Asian | 2 (3.3%) |
| Black or African American | 3 (5.0%) |
| Hispanic or Latino | 8 (13.3%) |
| White or Caucasian | 47 (78.3%) |
| Classification of PAH | |
| Idiopathic | 35 (58.3%) |
| Heritable | 2 (3.3%) |
| Associated | 23 (38.3%) |
| Congenital heart disease | 4 (6.7%) |
| Dose of remodulin (ng/kg/min) | 71.4 ± 27.8 |
| I | 10 (17) |
| II | 30 (50%) |
| III | 20 (33%) |
| IV | 0 (0.0%) |
PAH: pulmonary arterial hypertension.
Continuous variables: mean ± SD. Categorical variables: count (%).
Two subjects withdrew from the study after lung transplantation and two withdrew due to infections following pump replacement out of the 17 total pump replacements. Figure 2 shows the Kaplan–Meier survival curve for the study using death from any cause as the event. The five-year post-implant survival estimate was 76.4% (95% CI: 63.3–85.3%). The rate of catheter-related complications throughout the study was 0.08/1000 patients-days versus the OPC of 2.5/1000 patients-days (p < 0.0001). This includes the events previously described (5) that contributed to the primary-endpoint occurring within the first 22,000 patient-days (three catheter dislocations, one venous stenosis, one punctured catheter, and one pneumothorax), plus one additional catheter puncture during pump refill, and one catheter laceration during pump replacement.
Fig. 2.

Kaplan–Meier survivorship plot with 95% confidence limit error bars. Five-year estimate from implant to death 76.4% (95% CI 63.3–85.3%).
The two catheter punctures seen during the study occurred in the same patient at different refills. For both cases, the patient experienced pain and a rash near the pump, and after infection was ruled out, fluoroscopy was conducted and demonstrated that the catheter body had migrated anterior to the refill port on the pump. The catheter was removed and inspected, and it was determined that the catheter had been punctured by the refill needle. Treprostinil was being administered subcutaneously through the hole instead of intravenously through the catheter tip and causing pain and erythema (known to occur with subcutaneous infusion of treprostinil with external infusion systems). The punctures did not result in symptoms of treprostinil over-/under-dosing because the patient continued to receive the correct dose (subcutaneously administered treprostinil is 100% bioavailable).8 Unfortunately, after replacement of the second punctured catheter, the patient developed an infection and the patient exited the study (mentioned above).
The catheter laceration was discovered after the patient had severe erythema anterior to the pump for approximately two months despite antibiotic treatments for possible infection. The pump and catheter were explanted and upon inspection it was determined that the catheter had been cut by the scalpel during a routine pump replacement two months earlier. Several months after explant, all symptoms resolved, and the patient received a new pump and catheter.
Throughout the study, there were 17 pump replacements, 9 were routine, as device was nearing expected end of service, 8 pumps have failed in 8 (13%) patients that involved 7 motor stalls and 1 battery issue. Seven of the patients with pump failures had no symptoms and the pump failures were identified after the pumps alarmed and / or during pump refill. As described above, there was one undetected pump failure, which was adjudicated by the AEAC to have contributed to their death. Six of these seven surviving subjects had their pump replaced and did not experience another pump failure event. One patient was considered by the investigator to be too sick to have pump replacement surgery and decided to have palliative-only care.
All failed pumps were returned to the manufacturer for a biomedical engineering investigation and destructive forensic analysis. Causes of each of the pump failures were identified, which involved high battery resistance, shaft wear, and holes in pump tubing. Design changes to the shaft wear and battery failure modes have been implemented to mitigate future issues. Investigation continues into the cause of the holes in the pump tubing, which has only occurred in pumps that were implanted for more than 51 months and had more than 33 refills. The seven patients with pump failures had the IDS implanted for 4–5 years and had between 18,000 and 25,000 motor turns (Fig. 3). Notably the majority of the patients with this range and follow-up duration and number of motor turns did not experience a pump failure. Of note, there is a correlation with longer duration of implant and flow rate (dose) with the number of motor turns.
Fig. 3.

Scatter plot of years post-implant (x-axis) vs. estimated pump motor turns (y-axis), with pump motor stalls identified with solid circles.
Kaplan–Meier survivorship plots using catheter failure, pump failure, pump replacement or pump explant without replacement, and system modification as the events are provided (Fig. 4(a) to (d)). Five-year survival from catheter failure was 94.3% and from pump failure was 87.7%. Five-year survival from pump replacement or pump explant without replacement was 74.9%, and survivorship from system modification was 69.8%.
Fig. 4.


Kaplan–Meier survivorship plots with 95% confidence limit error bars. (a) Five-year estimate from implant to catheter failure 94.3% (95% CI 83.1–98.2%). (b) Five-year estimate from implant to pump failure 87.7% (95% CI 72.9–94.7%). (c) Five-year estimate from implant to pump replacement or explanted without replacement 74.9% (95% CI 60.0–85.0%). (d) Five-year estimate from implant to system modification 69.8% (95% CI 54.9–80.7%).
Refill reactions
Throughout the study, patients had treprostinil-related reactions soon after their pump was refilled, including local reactions near the pump refill site such as pain, erythema, and swelling and / or systemic reactions including flushing, headache, nausea, and hypotension. Upon investigation of the refill procedure by the biomedical team, it was determined that when the needle was pulled out through the skin a small droplet of drug could be expelled from the needle opening causing a local site reaction. The team also determined that refilling the pump with chilled drug would reduce the pressure in the pump reservoir thus limiting residual drug from exiting the needle as it was withdrawn at the completion of the refill process.
The FDA approved the option to use chilled drug during refills in December 2013. Since, treprostinil refill site reactions have been significantly reduced from 4% (42/1054) when using non-chilled drug to 1.4% (20/1467) with chilled drug (p value < 0.0001). Serious refill reactions were also significantly reduced from 0.9% (10/1054) when using non-chilled treprostinil compared to 0.1% (2/1467) with chilled drug (p value = 0.006). All AEs due to refill reactions resolved within 24–48 h. No subject had a pocket fill, which is defined as an inadvertent injection of drug during refill directly into the subcutaneous tissue or the pump pocket instead of into the pump.
Adverse events
During the study follow-up period there were 340 AEs in the 60 subjects that were adjudicated as related to the system and/or treprostinil. Forty-seven of those (13.8%) were adjudicated as serious. The majority were events commonly seen after an invasive procedure (e.g. pain, swelling at procedure site) or associated with treprostinil infusion (i.e. refill reactions). Figure 5 shows the proportion of patients with an AE during every six-month interval post-implant for the five most common system and/or drug-related events. The rate of AEs reduced over time except in the post-refill reaction, as expected.
Fig. 5.

Adverse event proportion over time for the five most common therapy related adverse events.
Pump flow rate accuracy: The flow rate accuracy (defined as the actual delivered volume / programmed delivery volume) of the IDS filled with treprostinil has been observed to decrease over time.5 The decline in flow rate accuracy was observed by noting more residual drug in the reservoir than expected at scheduled pump refills. The decline in flow rate accuracy correlated with the cumulative volume of drug delivered, which is dependent on the flow rate and implant time / refill number. Figure 6 shows the 95% tolerance interval of an exponential model of refill accuracy versus refill number in the DelIVery for PAH trial. If during a pump refill, the accuracy ratio was calculated to be outside the gray area of the graph in Fig. 6, clinicians were instructed to up-titrate the programmed dose if medically necessary. There were no serious clinical events related to flow rate accuracy. However, some patients clinically noted changes in their functional status and required up-titration of dose or change in their concomitant PAH therapies.
Fig. 6.

Implantable system for remodulin expected accuracy ratio for a 40-mL refill volume. The shaded area represents the DelIVery for PAH clinical trial accuracy data (95/95% tolerance interval of the exponential data model).
Discussion
Continuous IV prostacyclin infusion therapy for severe PAH is effective with significant clinical data supporting long-term use.1 It is often utilized late in patient care, and often at very advanced stages of disease due to under-recognition of disease severity and because of its complex delivery system and inherent safety concerns, particularly related to the constant risk of life-threatening blood stream infections (BSIs) associated with external pump delivery.9 Successful initiation and use of parenteral prostacyclins require strong family/social support, compliance with medication changes, and scrupulous catheter care. Infection, as with any long-term indwelling central venous catheter, can result in significant morbidity and mortality that has been reported in PAH patients treated with IV prostacyclin therapy.4 Because the IDS is internally implanted, the infection risks associated with external systems are eliminated. In fact, the approved labeling comparison for BSI rates has a 44-fold reduction for the fully implanted system compared to external systems (1 per 133 patient-years vs. 1 per 3 patient-years, respectively).8
The recent FDA approval 30 July 2018 (announced 31 July 2018 press release) of this IDS provides clinicians who manage severe PAH access to an effective therapy with a markedly reduced risk of infections, including serious BSIs, and offers them the opportunity to utilize this therapy, after initial stabilization of a patient on externally administered drug. Its use also offers the ability for a patient to live a near normal lifestyle without the limitations imparted by the need for an external pump and infusion system. This included showering, bathing, swimming and other related activities, and limited the need for daily medication preparation.
The risks associated with pump reservoir refills were mitigated by having refills performed at clinical trial centers with well trained, experienced clinicians who were prepared to manage the complications that could arise with inadvertent injection or leakage of treprostinil into the subcutaneous space surrounding the pump. Such device pocket injection did not occur in any experience. Refill reactions, manifested as both local and systemic adverse effects, were due to a small amount of treprostinil being extruded from the refill needle during withdrawal.
Injecting chilled drug into the pump reservoir causes the propellant surrounding the drug reservoir to condense to a liquid state, which transiently lowers the reservoir pressure below ambient. This causes any residual drug in the needle to stay in the needle when it is withdrawn from the reservoir and thereby decreases the frequency and severity of refill reactions. If the reservoir pressure is above ambient when the needle is withdrawn from the reservoir, any residual drug in the needle may be expelled from the needle and delivered into the subcutaneous tissue resulting in local and potential systemic reactions.
After subjects experienced pump failure, the investigative team sought to determine the etiology of these events. After destructive forensic evaluation by the manufacturer, pump design changes have been implemented that address many of the observed pump failures and motor stalls. Recommended management of an implanted pump includes periodic demonstration to the patient of the audible pump alarm to assure prompt recognition of the alarm; telemetered interrogation and review of pump logs at all patient encounters, including whenever a patient presents with an alarm or significant change in their symptoms. The risks of early prophylactic pump replacement need to be balanced against the risks of mechanical pump failure. The risks of pump replacement surgery include the inherent risk of anesthesia in patients with severe PAH and the risk of peri-operative infection. The risk of infection associated with this abdominal wall surgical replacement of the pump during the clinical trial was 12% (2/17), similar to Medtronic’s unpublished data available from their product surveillance registry of the SynchroMed II 40 mL pump used for intrathecal infusion applications, which estimates a pump replacement infection rate of 7%.10
One safety measure that is recommended is programming the pump to minimum rate mode and placing barriers between exposed junctions during pump replacement. This will limit the risk of drug leakage into the subcutaneous tissues during pump replacement. The interruption of drug delivery during pump replacement was for a short duration so that external line delivery was not required but should be quickly available if the procedure time is extended due to unusual issues.
Collaborative teamwork with clinicians, patients, and industry helped improve study outcomes during all phases of the study. The investigative team: (1) developed better surgical procedures to reduce the risk of catheter and pump displacement and mitigate catheter migration over the pump refill port, (2) refined the technique for pump reservoir refills to minimize the risk of local and systemic refill reactions, (3) quantified the reduction in pump flow rate that occurs over time based on the number of refills and dose adjust, and (4) developed protocols to manage catheter, pump, total system replacement, and peri-lung transplant procedures. These processes and guidance required a collaborative relationship with academia and industry and will help with the larger scale distribution of the system. The IDS will require not just an expert PAH team but a coordinated approach for each aspect of the care: evaluation, implantation, recovery, refills, emergency management, and replacement. The pump improved outcomes but is not a simple trade-off for the external system.
Who is best suited for an implantable system? As in this phase 1 study, the best suited patients are not WHO functional class 4, are able to tolerate the procedure itself, are able to return to the treatment center for follow-up refills, have prior experience with treprostinil, and did not require frequent escalation of prostacyclin therapy. New risk stratification guidelines1 aim for near clinical perfection such as, patients are often WHO functional class 4 at the initiation of parenteral prostacyclin therapy, which may be too late.9 Perhaps the availability of the IDS will encourage patients and physicians to utilize IV therapy when, in the past, it was not encouraged or wanted due to the permanent indwelling IV and infection risk. The observed survival statistics are similar to reported survival with prostacyclin therapy in historical PAH patients.
Limitations
A quality of life assessment tool was collected for each patient, but only 12-months post-implant. The results of this study cannot necessarily be extrapolated to real world clinical practice as the study was performed in carefully selected and highly experienced PAH centers. Some patients may need adjustment of their dose based on the IDS pump flow rate changes experienced over time if associated with changes in symptoms or other objective assessments of an individual patient’s status, such as measured exercise capacity. A coordinated approach between the patient and PAH expert team can minimize symptoms based on these mechanistic changes. In addition, due to the pharmacologic properties of other prostacyclin analogs (primarily lack of long-term thermal stability) the system can currently only be used with treprostinil for continuous IV infusion of prostacyclin therapy.
Conclusion
After more than 280 patient-years of experience, the fully implanted treprostinil IV delivery system has been shown to result in a significant decrease in the risk of BSIs and catheter dislocations, and prevented catheter occlusions in PAH patients, while providing patients enhanced day-to-day living experience (swimming and bathing). As with all implantable devices/systems, state-of-the art equipment, and extensive medical and surgical experience are required to minimize the risks of peri-operative infection, pocket fills and refill reactions and system malfunction. Our reported experience will facilitate the implementation of this novel, implantable IV delivery system into the PAH patient community with continued guidance and oversight.
Supplemental Material
Supplemental material, PUL878615 Supplemetal Material for Long-term results of the DelIVery for Pulmonary Arterial Hypertension trial by Mardi Gomberg-Maitland, Robert C. Bourge, Shelley M. Shapiro, James H.Tarver III Dianne L. Zwicke, Jeremy P. Feldman, Murali M. Chakinala, Robert P. Frantz, Fernando Torres, Remzi Bag, Jeffrey A. Murphy, Amy A. Lautenbach, Marty Morris, Leigh Peterson and Aaron B. Waxman in Pulmonary Circulation
Acknowledgments
We would like to thank all our subjects, study coordinators, and research teams at each respective center.
Author contributions
RCB and MGM had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis, including and especially any adverse effects. RCB, ABW, MG-M, AAL, MM, JAM, and LP contributed substantially to the study design, data analysis and interpretation, and the writing of the manuscript. SMS, JHT III, DLZ, RB, JPF, MMC, RPF, and FT contributed substantially to the data acquisition and interpretation, and the writing of the manuscript.
Conflict of interest
JAM, AAL, and MM are employees of Medtronic and LP is an employee of United Therapeutics, the funders of this study. MGM and BB consult for United Therapeutics.
Funding
Representatives and scientists from the Sponsor (Medtronic) and the Company Funding the study (United Therapeutics), participated in the study including device development, study design, data collation from study institutions, data analysis, and critical review of the manuscript.
Supplemental Material
Supplemental material for this article is available online.
References
- 1.Galie N, Channick RN, Frantz RP, et al. Risk stratification and medical therapy of pulmonary arterial hypertension. Eur Respir J 2019; 53(1). [DOI] [PMC free article] [PubMed]
- 2.Galie N, Corris PA, Frost A, et al. Updated treatment algorithm of pulmonary arterial hypertension. J Am Coll Cardiol 2013; 62: D60–72. [DOI] [PubMed] [Google Scholar]
- 3.Simonneau G, Barst RJ, Galie N, et al. Continuous subcutaneous infusion of treprostinil, a prostacyclin analogue, in patients with pulmonary arterial hypertension: a double-blind, randomized, placebo-controlled trial. Am J Respir Crit Care Med 2002; 165: 800–804. [DOI] [PubMed] [Google Scholar]
- 4.Kallen AJ, Lederman E, Balaji A, et al. Bloodstream infections in patients given treatment with intravenous prostanoids. Infect Control Hosp Epidemiol 2008; 29: 342–349. [DOI] [PubMed] [Google Scholar]
- 5.Bourge RC, Waxman AB, Gomberg-Maitland M, et al. Treprostinil administered to treat pulmonary arterial hypertension using a fully implantable programmable intravascular delivery system: results of the DelIVery for PAH Trial. Chest 2016; 150: 27–34. [DOI] [PubMed] [Google Scholar]
- 6.Morris M, Phares K, Zaccardelli D, et al. A novel catheter system for totally implantable intravenous drug therapy: assessment of catheter function and patency with trepostinil therapy. J Vasc Access 2008; 9: 20–27. [PubMed] [Google Scholar]
- 7.Waxman AB, McElderry HT, Gomberg-Maitland M, et al. Totally implantable IV treprostinil therapy in pulmonary hypertension assessment of the implantation procedure. Chest 2017; 152: 1128–1134. [DOI] [PubMed] [Google Scholar]
- 8.Therapeutics U. Remodulin (package insert), Research Triangle Park, NC: Therapeutics U, 2018. [Google Scholar]
- 9.Farber HW, Miller DP, Meltzer LA, et al. Treatment of patients with pulmonary arterial hypertension at the time of death or deterioration to functional class IV: insights from the REVEAL Registry. J Heart Lung Transplant 2013; 32: 1114–1122. [DOI] [PubMed] [Google Scholar]
- 10.Medtronic. Meditronic Product Surveillance Registry on Pump replacement infection (MDT2361484), 2019.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental material, PUL878615 Supplemetal Material for Long-term results of the DelIVery for Pulmonary Arterial Hypertension trial by Mardi Gomberg-Maitland, Robert C. Bourge, Shelley M. Shapiro, James H.Tarver III Dianne L. Zwicke, Jeremy P. Feldman, Murali M. Chakinala, Robert P. Frantz, Fernando Torres, Remzi Bag, Jeffrey A. Murphy, Amy A. Lautenbach, Marty Morris, Leigh Peterson and Aaron B. Waxman in Pulmonary Circulation