

Anthrax toxin activator (AtxA) regulates the major virulence genes in Bacillus anthracis. The bacterium produces the anthrax toxins, and understanding the mechanism of toxin production may facilitate the development of therapeutics for B. anthracis infection. Since the discovery of AtxA 25 years ago, the mechanism by which it regulates its targets has largely remained a mystery. Here, we provide evidence that AtxA binds to the promoter region of the pagA gene encoding the main central protective antigen (PA) component of the anthrax toxin. These data suggest that AtxA binding plays a direct role in gene regulation. Our work also assists in clarifying the role of CO2 in AtxA’s gene regulation and provides more evidence for the role of AtxA phosphorylation in virulence gene regulation.
KEYWORDS: AtxA binding, Bacillus anthracis, pagA promoter regulation
ABSTRACT
Anthrax toxin activator (AtxA) is the master virulence gene regulator of Bacillus anthracis. It regulates genes on the chromosome as well as the pXO1 and pXO2 plasmids. It is not clear how AtxA regulates these genes, and direct binding of AtxA to its targets has not been shown. It has been previously suggested that AtxA and other proteins in the Mga/AtxA global transcriptional regulators family bind to the curvature of their DNA targets, although this has never been experimentally proven. Using electrophoretic mobility shift assays, we demonstrate that AtxA binds directly to the promoter region of pagA upstream of the RNA polymerase binding site. We also demonstrate that in vitro, CO2 appears to have no role in AtxA binding. However, phosphomimetic and phosphoablative substitutions in the phosphotransferase system (PTS) regulation domains (PRDs) do appear to influence AtxA binding and pagA regulation. In silico, in vitro, and in vivo analyses demonstrate that one of two hypothesized stem-loops located upstream of the RNA polymerase binding site in the pagA promoter region is important for AtxA binding in vitro and pagA regulation in vivo. Our study clarifies the mechanism by which AtxA interacts with one of its targets.
IMPORTANCE Anthrax toxin activator (AtxA) regulates the major virulence genes in Bacillus anthracis. The bacterium produces the anthrax toxins, and understanding the mechanism of toxin production may facilitate the development of therapeutics for B. anthracis infection. Since the discovery of AtxA 25 years ago, the mechanism by which it regulates its targets has largely remained a mystery. Here, we provide evidence that AtxA binds to the promoter region of the pagA gene encoding the main central protective antigen (PA) component of the anthrax toxin. These data suggest that AtxA binding plays a direct role in gene regulation. Our work also assists in clarifying the role of CO2 in AtxA’s gene regulation and provides more evidence for the role of AtxA phosphorylation in virulence gene regulation.
INTRODUCTION
Bacillus anthracis is a Gram-positive, spore-forming bacterium that is the causative agent of the disease anthrax. The bacteria produce the anthrax toxins lethal toxin and edema toxin, composed of protective antigen (PA) plus lethal factor (LF) and PA plus edema factor (EF), respectively. The pXO1 and pXO2 plasmids of B. anthracis carry genes essential for virulence (1, 2). The pXO2 plasmid contains genes encoding the poly-d-γ-glutamic acid capsule, and pXO1 contains the toxin genes pagA, lef, and, cya and encodes the master virulence regulator anthrax toxin activator (AtxA).
Since its discovery as a transactivator of anthrax toxin synthesis 25 years ago, AtxA has been found to regulate genes on the chromosome, pXO1, and pXO2 of B. anthracis, including genes that encode the anthrax toxins and capsule synthesis (3, 4). AtxA is a 55.6-kDa protein normally existing as a homodimer (5). It contains two helix-turn-helix (HTH) domains, the function of which is typically associated with DNA binding. In addition to the HTH domains, AtxA also has two phosphotransferase system (PTS) regulation domains (PRDs) and an EIIB-like domain (6). Virulence regulators containing PRDs and having similarities to AtxA have been found in other Gram-positive pathogens. These proteins are often referred to as PRD-containing virulence regulators (PCVRs) (7) and as members of the Mga/AtxA family (8). The two groups of proteins are defined by their similar domains and functions. It has been suggested that proteins in these classes may bind to the structure of the promoter rather than its sequence, but limited work has been done exploring this area (9, 10).
AtxA activity is regulated by several mechanisms. Two phosphorylation sites in the PRDs of AtxA have been suggested to regulate AtxA function. Phosphorylation at H199 is suggested to promote the DNA binding activity of AtxA, while phosphorylation at H379 has been shown to inhibit AtxA function by preventing dimerization of the protein and inhibiting AtxA activity (5, 6). In addition to phosphorylation, toxin gene expression is highly dependent on the presence of CO2 in the environment. Previous work in our lab found a number of genes that respond to CO2 through an AtxA-dependent mechanism (11). However, the relationship between AtxA and CO2 remains unclear.
Although regulation of AtxA expression has been well characterized (4, 12–14), it is not clear how AtxA regulates its targets. An AtxA-dependent start site called P1 was localized by primer extension at bp −59 relative to the pagA start codon (15), whereas our RNA sequencing (RNA-seq) analysis (11) found stable transcripts beginning at bp −25, which we refer to here as the transcriptional start site (TSS). Previous work suggested that high levels of intrinsic curvature exist in the three anthrax toxin promoters near their AtxA-dependent start sites (9), and more recently, another group provided indirect evidence that AtxA binds to the pagA promoter (16). However, further characterization of AtxA-dependent transcription is required to understand AtxA regulation of its targets. We hypothesized that AtxA binds directly to its targets. We used the promoter region of pagA to assess AtxA’s regulation effects. Using electrophoretic mobility shift assays and DNase I footprinting, we identify a region of the pagA promoter to which AtxA binds. Additionally, because phosphorylation was suggested to regulate DNA binding, we assessed how phosphomimetic and phosphoablative substitutions at the phosphorylation sites impacted DNA binding. We found that AtxA with the H199D substitution bound DNA more efficiently, consistent with previous reports, while the H199A substitution greatly decreased binding. We were unable to purify the AtxA H379D protein but were able to show in vivo that the H379D substitution drastically reduced the expression of PA. This work furthers our understanding of the mechanisms of B. anthracis gene regulation.
RESULTS
Purification and characterization of AtxA WT and mutant proteins.
The BL21(DE3)RIPL Escherichia coli strain containing pPROEx HTc-AtxA plasmids was used to express AtxA wild type (WT) and its phosphorylation site mutants. After purification, the proteins were cleaved with tobacco etch virus (TEV) protease to remove the N-terminal His6 tag. The purified proteins ran on SDS gels as 50-kDa proteins, consistent with the molecular weight of a monomer of AtxA (55.6 kDa) (Fig. 1A). N-terminal sequencing confirmed the identity of the purified proteins, with all proteins reporting the sequence GAMGIPMLTPIS. Electrospray-mass spectrometry (ES-MS) gave masses within 1 to 2 mass units of the masses calculated from the amino acid sequences. While 3 of the 4 desired proteins were obtained in good yields and purity, the AtxA H379D protein could not be obtained in a soluble form. Expression levels were good from plasmids having several different tags, but in every case, the protein was in inclusion bodies and could not be solubilized.
FIG 1.
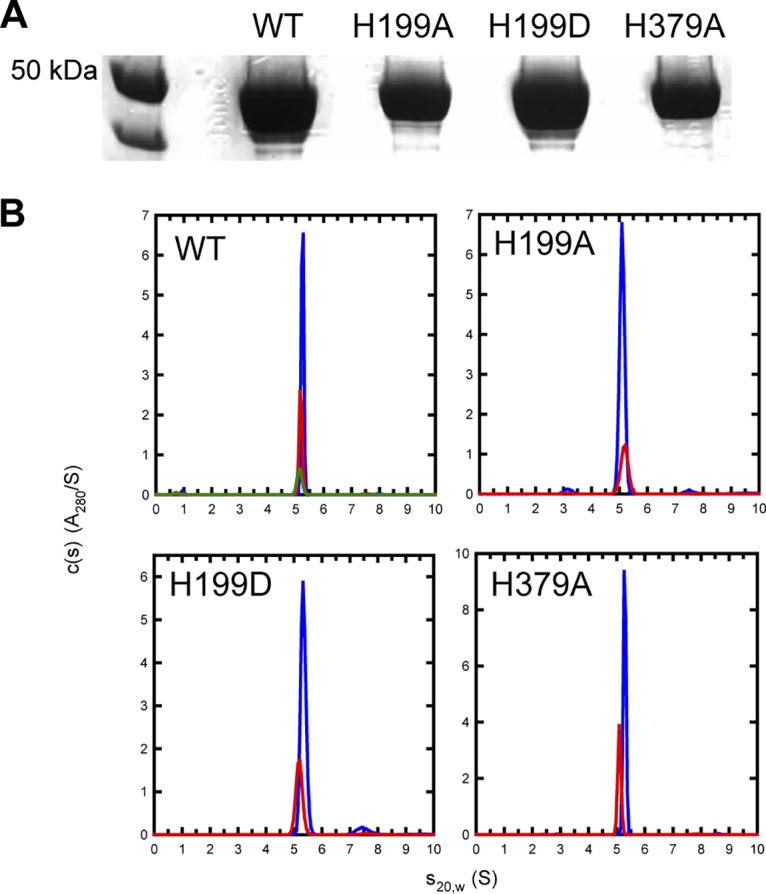
Purification and oligomeric state of AtxA WT and mutants. (A) SDS-PAGE of AtxA variants expressed from BL21(DE3)RIPL cells containing derivatives of the pProEx HTc-AtxA plasmids. Lane 1, PageRuler unstained protein ladder (Thermo Scientific); lane 2, AtxA; lane 3, AtxA H199A; lane 4 AtxA H199D; and lane 5, AtxA H379A. (B) Absorbance c(s) distributions for AtxA at 11.3 μM (blue), 5.3 μM (red), 2.0 μM (green) protein, AtxA H199A at 24 μM (blue) and 6 μM (red) protein, AtxA H199D at 20 μM (blue) and 7 μM (red) protein, and AtxA H379A at 17 μM (blue) and 8 μM (red) protein. All proteins were analyzed in 500 mM NaCl, 10 mM HEPES (pH 7.2), and 5 mM 2-mercaptoethanol. All data show a major species at 5.16S with an estimated molar mass of 107 kDa indicative of an AtxA dimer (monomer, 55.6 kDa).
Sedimentation velocity experiments done at concentrations of 2 to 24 μM confirmed previous reports that AtxA exists as a dimer (Fig. 1B). This was confirmed for the WT and H199A-, H199D-, and H379A-substituted proteins. The sedimentation velocity analyses demonstrated uniform size distributions of all four proteins assayed. No evidence for higher oligomeric states was found. As noted, the H379D protein was not available to be analyzed in this way.
AtxA specifically binds to the promoter region of pagA.
Protective antigen is a component of both anthrax toxins and is encoded by the gene pagA. We hypothesized that AtxA binds to the promoter region of the gene because the transcription of pagA is highly regulated by AtxA. The purified proteins described above were used in electrophoretic mobility shift assays (EMSAs) to assess their binding to pagA. Due to the high isoelectric point of the protein, a buffer and gel system (designated EABE) operating at elevated pH was created to facilitate entry of the protein into the gel matrix. We created a series of overlapping, infrared dye-labeled fragments encompassing an area approximately 1,200 bp upstream of the pagA open reading frame, chosen as the promoter region based on previous work (3) (Fig. 2A). Incubation of purified AtxA protein with one of these fragments distinctly decreased its mobility, demonstrating specific binding. The PAG4 fragment, encompassing a region immediately upstream of the transcriptional start site, was shown to bind to AtxA (Fig. 2A and B). The mobility shift was dependent on AtxA concentration, with half-maximal binding seen at 100 to 200 nM (Fig. 2C). The reaction mixture of PAG4 with AtxA also contained large excesses of poly(dI-dC) and serum albumin to eliminate nonspecific interactions, showing that AtxA is specifically recognizing the PAG4 DNA. To further confirm the specificity of AtxA binding to the PAG4 fragment, we added unlabeled PAG4 in excess and found that this blocked binding to the dye-labeled PAG4 DNA (Fig. 2D).
FIG 2.
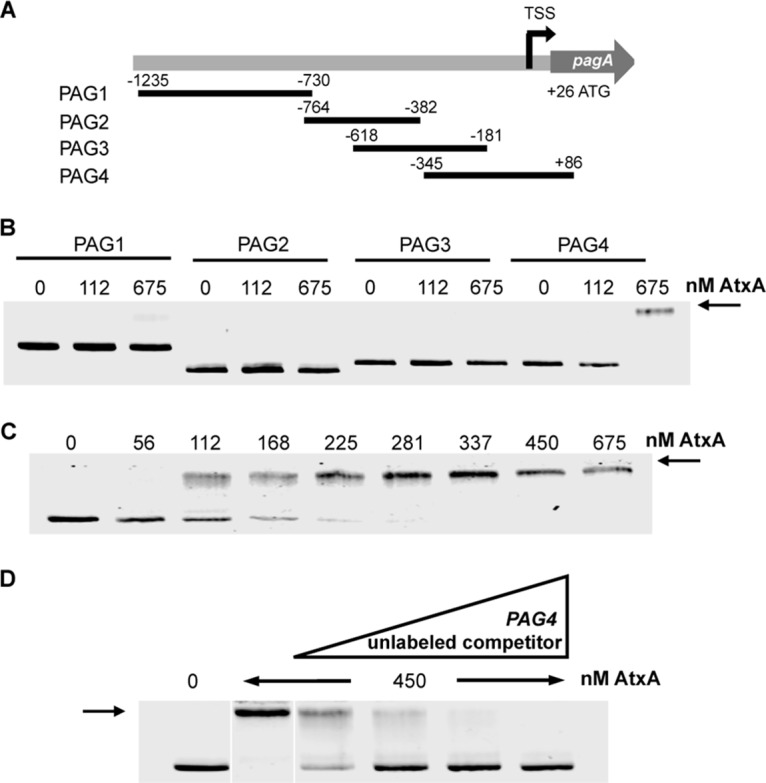
AtxA binds the pagA promoter region. (A) Map depicting overlapping fragments of pagA promoter region synthesized with IR700 primers using PCR. Numbers represent distance in base pairs from transcriptional start site as indicated by “TSS.” TSS was determined previously from RNA-seq data (11). (B to D) Electrophoretic mobility shift assays using infrared labeled fragments of the pagA promoter region. Fragments were generated via PCR and end labeled with IR700 dye. The labeled probe (14.2 ng) was added to each reaction mixture and incubated with the indicated concentrations of protein. Samples were run on 2% agarose in EABE buffer. (B) EMSA of overlapping fragments depicted in panel A with various concentrations of AtxA. (C) Labeled PAG4 fragment incubated with increasing concentrations of AtxA. (D) Labeled PAG4 fragment incubated with 10×, 30×, 60×, and 100× unlabeled PAG4 generated via PCR and 450 nM AtxA. Arrows indicate shifted fragments.
Sodium bicarbonate does not impact binding of AtxA.
Although it has been previously established that AtxA and CO2 work synergistically to regulate their targets, we still do not understand how they interact. To see if CO2-HCO3− affects the binding activity of AtxA, we added various amounts of NaHCO3 to the EMSAs. For low levels of HCO3−, like the 20 to 30 mM concentrations found in animal hosts that can be infected by B. anthracis, we saw that HCO3− had no impact on binding (Fig. 3A). Larger amounts of sodium bicarbonate (100 to 200 mM) reduced the binding of AtxA significantly. To assess whether this response was specific to sodium bicarbonate or was an effect of the higher ionic strength, parallel tests were done with potassium nitrate. The addition of potassium nitrate at 100 mM and 200 mM also reduced binding (Fig. 3B). These experiments do not provide evidence that bicarbonate directly affects AtxA binding to DNA but instead suggest that high salt concentrations disrupt the AtxA-DNA complexes.
FIG 3.
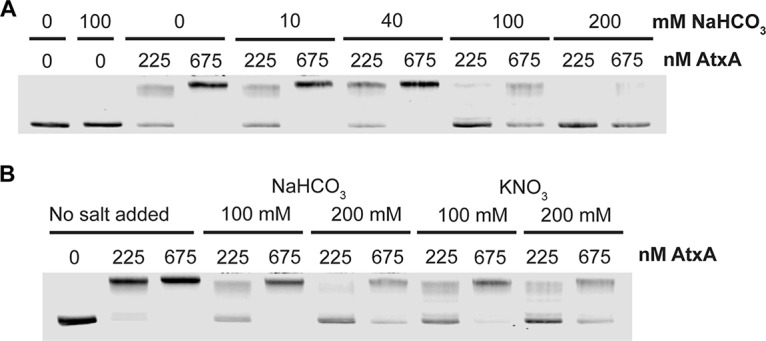
High salt inhibits binding of AtxA to PAG4. Electrophoretic mobility shift assay using infrared labeled PAG4 fragment of pagA promoter region. PAG4 (14.2 ng) was added to each reaction mixture and incubated with the indicated concentrations of protein. Samples were run on 2% agarose in EABE buffer. (A) Fragment PAG4 incubated with various amounts of NaHCO3 and AtxA. (B) Fragment PAG4 incubated with increasing amounts of AtxA and either no salt, NaHCO3, or KNO3.
Phosphomimetic substitutions alter the binding capacity and function of AtxA.
We next examined the effects on AtxA binding of phosphomimetic and phosphoablative substitutions at the two histidine residues previously shown to be phosphorylated (6). Phosphomimetic substitutions were created by changing the histidine residue to an aspartic acid, and phosphoablative substitutions were created by changing the residue to an alanine. We found that the H199D protein showed a higher affinity for PAG4 than did WT AtxA, consistent with previous work on the H199D substitution (5, 6). The phosphoablative substitution at residue 199 (H199A) clearly reduced the affinity of AtxA to the probe (Fig. 4A). The phosphoablative H379A substitution did not affect the ability of AtxA to bind DNA (Fig. 4B). Because we could not produce soluble AtxA H379D protein, we were unable to assess its behavior in this EMSA.
FIG 4.
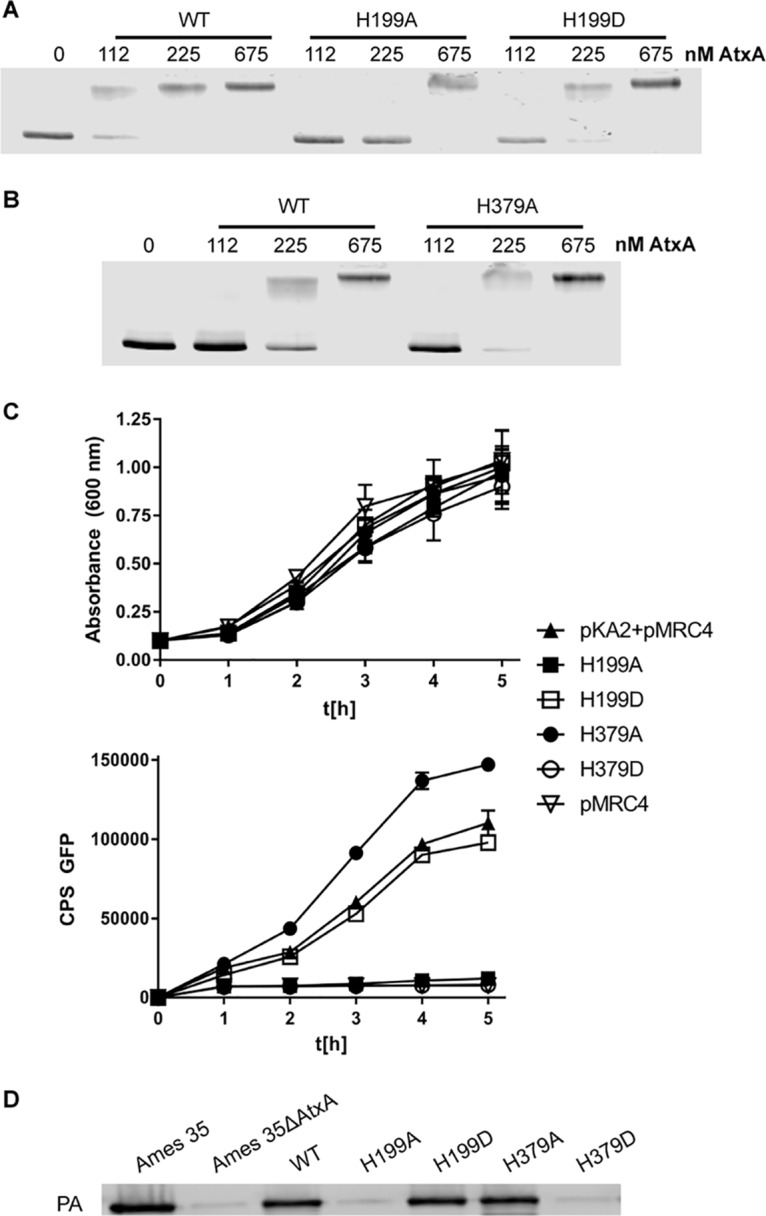
Phosphomimetic and phosphoablative substitutions of AtxA. (A and B) EMSAs with PAG4 infrared labeled fragment generated via PCR run on a 2% agarose EABE gel with various amounts of protein. (A) EMSA with AtxA, AtxA H199A, and AtxA H199D. (B) EMSA with AtxA and AtxA H379A. (C) Growth and fluorescence measured over 5 h in BH500 strains containing the pMRC4 fluorescence reporter for pagA and the pKA2 plasmid with AtxA phosphomimetic and phosphoablative substitutions. The sample denoted pMRC4 is a no-AtxA control and does not contain the pKA2 plasmid. CPS, counts per second. (D) Western blot of Ames 35 ΔAtxA strains transformed with pKA2 plasmids as listed. Samples were grown in NBY with 0.8% NaHCO3 and incubated in 15% CO2 at 37°C. The blot was assayed with an antibody against PA.
To evaluate how the native and histidine-modified AtxA proteins influence pagA promoter activity in B. anthracis, we constructed a 2-plasmid reporter system, with pKA2 encoding AtxA proteins and pMRC4 encoding a pagA promoter-driven green fluorescent protein (GFP) (see Fig. S1 in the supplemental material). These plasmids were transformed into the atxA-negative (and pXO1-negative) BH500 strain (17), which was then grown in nutrient broth-yeast extract (NBY) with NaHCO3, a condition which induces toxin production (18). Figure 4C shows growth curves and fluorescence over time for BH500 strains containing pMRC4 along with pKA2 plasmids expressing the four His substitution mutants. The highest GFP expression was observed for the strain producing AtxA H379A. The expression of GFP induced by native AtxA was comparable with induction by AtxA H199D, whereas AtxA H199A and AtxA H379D behaved similarly to the atxA-negative strain BH500(pMRC4) and did not activate the production of GFP. We also analyzed GFP expression in the same strains in NBY medium without added bicarbonate. No expression of GFP was induced in any of the strains, with the exception that low expression was seen in the AtxA H199D strain (data not shown). This activity was 3-fold less than the activity of the same strain in the presence of carbon dioxide.
We also assessed the behavior of the altered AtxA proteins in a more native situation, in the Ames 35(pXO1+) strain having the AtxA gene deleted. The altered AtxA proteins were expressed from pKA2 plasmids in bacteria grown in NBY medium with NaHCO3, and PA secretion was assessed by Western blotting. Consistent with the data from the GFP reporter system, the H199D and H379A proteins supported PA production at normal levels, while the H199A and H379D proteins failed to support any PA production (Fig. 4D).
Examining the role of AtxA H379.
As noted in this and in previous studies (5, 6), the phosphomimetic substitution of Asp at the H379 site of AtxA results in decreased activity. Introduction of a modification (phosphorylation) to this residue or a negatively charged side chain (Asp) in the place of the histidine is suggested to prevent AtxA dimerization and, therefore, activity (5). We sought to assess the role of this residue by performing a selection for second-site suppressor mutations that restore activity, and possibly dimerization, even in the continued presence of H379D. This was conveniently done because of the relative ease of detecting a rare fluorescent colony in a background of nonfluorescent colonies in the BH500 strain containing pKA2-AtxA H379D and pMRC4. The AtxA H379D structural gene was subjected to error-prone PCR, and the resulting library was cloned back into pKA2. Sequencing of a random set of transformants showed that an average of 1.5 bases were altered per gene sequenced. By examining >5,000 colonies on plates, a single fluorescent clone was identified, and the atxA gene in this clone was found to code for an H379G mutation. This AtxA protein produced fluorescence at levels comparable to that of AtxA WT when it was used in our fluorescent reporter system to induce the expression of a gfp gene under the control of a pagA promoter. This result validated the effectiveness of the selection process, identified a new and previously uncharacterized change different from H379A that supports AtxA activity, and argues that second-site suppressor mutations of H379D are rare.
AtxA selectively binds to a region of the pagA promoter.
DNase I footprinting assayed using 6-carboxyfluorescein (6-FAM) fluorescently labeled DNA of the PAG4 fragment was done with the AtxA WT and AtxA H199D proteins. Samples were digested, purified, and run on capillary electrophoresis by the Plant-Microbe Genetics Facility at The Ohio State University. We were unable to find a single area of strong protection and instead saw small areas of protection in AT-rich regions upstream of the RNA polymerase binding site, as defined by Dai et al. (15) (Fig. S2). However, the EMSA data described above appear to be inconsistent with these putative areas of protection, as these areas lie within the PAG3 fragment, which did not bind AtxA in the EMSA. AtxA WT and AtxA H199D demonstrated protection in similar areas.
Analysis of the PAG4 fragment on the mfold Web server demonstrated the possible presence of two stem-loops, which we termed stem-loop I (SLI) and stem-loop II (SLII), located upstream of the RNA polymerase (RNAP) binding site (Fig. S3). To assess the role of this region in AtxA binding, we performed competitive EMSAs using two fragments differing only in the presence of the hypothesized SLI and the downstream 5 bp that extend into SLII (Fig. 5B). We found that the fragment PAG4-2 containing both intact stem-loop regions blocked binding of the dye-labeled probe DNA, while the shorter fragment PAG4-1 did not, providing strong evidence that this region, including the suspected SLI and the first 2 bp of SLII, is an important feature of the DNA to which AtxA binds (Fig. 5C). To further analyze the importance of each of the identified hypothesized stem-loops, we conducted further analysis on fragments generated using the pRSP plasmid, a form of pMRC4 with a shortened pagA promoter region which was shown to have activity similar to that of the full pagA promoter region present in pMRC4 (Fig. 6A). We performed competitive EMSAs with an IRDye 700-labeled RPAG fragment that contains the full 175-bp pagA promoter region of pRSP. We created two unlabeled competitor DNA fragments, one with the full pRSP pagA promoter but excluding just the region containing the hypothesized SLI, and the other with the same pRSP promoter area but excluding the region containing the potential SLII (Fig. 5D). We found that the unlabeled competitor, RPAGΔSLI, which contains the potential SLII, blocked the binding of AtxA to the IRDye-labeled probe, while the unlabeled competitor, RPAGΔSLII, that lacks the SLII region did not block binding as effectively (Fig. 5E). This shows an affinity by AtxA for the region containing SLII in the pagA promoter. These results provide evidence that the hypothesized SLII is important for AtxA binding, and the removal of even two nucleotides from the beginning of SLII results in decreased binding of AtxA to the DNA.
FIG 5.

EMSA analysis indicates that the predicted SLII is important for DNA binding. (A) Nucleotide sequences of the pRSP pagA promoter region. The sequence shown is located immediately upstream of the translation start codon ATG (uppercase bold). The −35 and −10 regions of a σA-like promoter are denoted in underlined bold, while the putative Shine-Dalgarno sequence is underlined (37). The hypothesized SLI is presented in bold and SLII is in bold italic. Transcription start site A is indicated in uppercase bold italic. (B) Map of PAG4-1 and PAG4-2 fragments within PAG4 in relation to the hypothesized stem-loops. Numbers indicate the distance from the transcriptional start site. The figure is drawn to scale. (C) EMSA on a 2% agarose EABE gel with AtxA WT incubated with PAG4 fragment and unlabeled competitor of either PAG4-1 or PAG4-2 generated via PCR. An excess of 10×, 30×, 60×, and 100× was added in increasing amounts to the EMSAs. (D) Map of RPAGΔSLI and RPAGΔSLII fragments in relation to the hypothesized stem-loops on the labeled RPAG700 fragment. Numbers indicate the distance from the transcriptional start site. The figure is drawn to scale. (E) EMSA on a 2% agarose EABE gel with AtxA WT incubated with the RPAG700 fragment and unlabeled competitor of either RPAGΔSLI or RPAGΔSLII generated via PCR. Excesses of 10×, 30×, and 60× were added in increasing amounts to the EMSA mixtures.
FIG 6.

Genetic and phenotypic consequences of various deletions within the pRSP pagA promoter region. (A) Map of pRSP pagA promoter region deletions in comparison to unaltered pRSP pagA promoter region and the hypothesized stem-loops in this promoter region. The right column shows relative GFP fluorescence, the fluorescence of which is activated by the transcription of wild-type AtxA on different forms of the pagA promoter region demonstrated on the left. Numbers indicate the distance from the transcriptional start site. (B) Graphical representation and statistical analysis by Welch's t test of the GFP fluorescence levels due to transcriptional activation of the pagA promoter in the various altered plasmids.
The hypothesized SLII in the pagA promoter region is important for AtxA-induced expression.
Once the region containing the two potential stem-loops within the pagA promoter region was identified as potentially important for AtxA binding, further analysis was done by deleting various regions in this area (Fig. 6A). Deletion analyses were done on pRSP, which has a shortened pagA promoter region but is otherwise the same as pMRC4 (Fig. S1). Interestingly, deletion of the hypothesized SLI alone from the promoter region did not result in a significant loss of expression compared to the unaltered pRSP pagA promoter or the pMRC4 promoter (Fig. 6B). However, increasing deletions of the full-potential SLI and regions extending into the hypothesized SLII resulted in a gradual decrease in expression. The deletion of half of SLII resulted in an almost complete loss of expression. Deletion of the region containing the suspected SLII alone resulted in a complete loss of expression (Fig. 6B). These results indicate that while the hypothesized SLI is not important for AtxA-induced expression of the gene, the predicted SLII is essential for the full activity of the protein on the pagA promoter.
DISCUSSION
AtxA plays a vital role in B. anthracis biology as the master virulence regulator. Its regulation of the key virulence factors, anthrax toxin and the polyglutamic acid capsule, makes the study of AtxA essential to better understand the regulation of B. anthracis gene expression. Here, we demonstrate that AtxA binds directly to the 371-bp region upstream of the pagA gene, which encodes the key protective antigen component of the anthrax toxin. EMSAs reveal AtxA’s affinity for a specific area of the pagA promoter region, consistent with expectations that this somehow regulates gene activation. It is still unclear whether AtxA binds to a specific DNA sequence motif or to a complex folded or bent DNA structure that has little dependence on a specific DNA sequence.
EMSAs with overlapping fragments clearly demonstrated the selectivity of AtxA binding for a specific part of the pagA promoter region. Although DNase I footprinting did not reveal one specific area of protection, there still appears to be consistent binding of the protein to selected regions of the DNA. Previously, another group used promoter deletions to identify areas of importance in the pagA promoter (9). Another previous study identified a region of the pagA promoter necessary for AtxA-induced pagA transcription (15). Although their proposed TSS differs from the TSS designated in our study, that study also found a region of importance for pagA transcription that encompasses the two hypothesized stem-loops identified in our study. Both earlier studies of the pagA promoter found similar regions of importance but did not identify the stem-loops presented in this study. EMSA analysis done in this study of four overlapping segments of a 1,260-bp segment of the region upstream of our predicted TSS showed that AtxA binds specifically to the 432-bp region containing the predicted RNA polymerase binding site and the area directly upstream of it. These data, along with those from the previous studies, prompted our analysis of the secondary structure of this region directly upstream of the predicted RNA polymerase binding site using the mfold Web server DNA folding tool (19). We determined that the most likely secondary structure for the DNA in this region consists of two stem-loops. Homodimer HTH transcription factors like AtxA are known to bind specifically to palindromes (20). Thus, this area likely plays a role in AtxA’s regulation of pagA activation, perhaps because AtxA binds to the hypothesized stem-loops.
Our work suggests that the second of the two potential stem-loops, SLII, is important for AtxA to bind to the pagA promoter region and that this interaction may be structurally specific rather than sequence specific. The deletion of even 2 nucleotides from the hypothesized stem region of SLII results in a notable decrease in binding, potentially due to the destabilization of the structure of the stem-loop. Increasing destruction of the suspected SLII by deleting increasing numbers of nucleotides from the upstream region results in steadily decreased AtxA-induced expression, and complete deletion of the SLII region results in a lack of AtxA binding to the DNA and a complete loss of activity. Additionally, deletion of exactly half of the region containing the potential SLII results in an almost complete loss of activity, supporting the idea that the structure rather than the sequence of SLII is important. Interestingly, the region containing the hypothesized SLII necessary for pagA promoter activation contains 94% AT content. Both stem-loops are generally well conserved and unique for all B. anthracis pXO1 plasmids independent of orientation of the pathogenicity island in the plasmid (Ames ancestor and Sterne strains). However, the pXO1-like plasmids of certain Bacillus cereus strains (pBCX01 from G9241 and pCI-XO1 from B. cereus bv. anthracis CI) contain one additional T in the SLI region (Fig. S4).
A recent report by Toyomane et al. (16) showed that (i) the upstream region of pagA suppressed transcription of the downstream gene and (ii) AtxA recovered suppressed transcription. The upstream region contains 1,093 bp and overlaps only 13 bp of the most upstream part of the pRSP pagA promoter region that we identified as being the most important for AtxA binding and activity. Our pMRC4 pagA promoter region contains 1,257 bp that completely include the region described by Toyomane et al. (16). However, we did not find a significant difference in the activation of the gfp gene between the pagA promoter regions of pMRC4 and pRSP, which contradicts the result from Toyomane et al. that pagA transcription is suppressed by the sequence studied.
Previous work suggested that AtxA binds to the curvature of the DNA (9). Subsequently, it was suggested that recognition of DNA curvature is a characteristic of other proteins in the Mga/AtxA family (10). Although there is predicted to be intrinsic curvature in the AtxA promoter region (9), footprinting data in this study did not appear to have a correlation to the curvature of the promoter. It is possible that AtxA relies on a combination of curvature and sequence to identify its targets. This phenomenon has been demonstrated in other systems (21–23). Analysis of AtxA binding to the promoters of other AtxA-regulated genes may provide further insight into the mechanism of AtxA regulation.
Phosphorylation in AtxA has been previously shown to alter its activity (5, 6). Our work confirms previous reports that phosphorylation at H199 is important for efficient binding of AtxA to its targets. This is evidenced by the reduced binding affinity of the phosphoablative substitution at this site. However, we did not see a difference between the binding of AtxA H199D and AtxA WT in footprinting assays, and thus, it is not clear how phosphorylation increases binding activity. The complete lack of PA expression and binding to pagA by the AtxA H379D phosphomimetic substitution, which was previously shown to be defective in dimerization, affirms the importance of oligomerization to AtxA function and adds support to previous work published on this mutant (5, 6). Furthermore, restoration of activity by replacement of the aspartic acid at the 379 site in the AtxA H379D with glycine provides new data on the importance of this residue and supports the view that the negative charge is responsible for the lack of activity rather than the absence of the His. Our sedimentation velocity work provides direct evidence of AtxA dimerization under physiological conditions and is consistent with previous work done using chemical cross-linking in bacteria.
There is still much we do not understand about AtxA, particularly in the context of its interaction with CO2. Unlike reports on other CO2-activated regulators (24, 25), we did not see a difference in the binding affinity of AtxA in the presence of sodium bicarbonate. This suggests that, at least in the limits of our in vitro experiment, AtxA and CO2 do not interact directly. Future work may employ in vivo techniques to assess the interaction of AtxA and CO2 in more detail.
In this study, we present evidence that AtxA binds to its targets and that this interaction is regulated by phosphorylation. Future work can clarify the manner in which AtxA binds to its targets and further the understanding of the relationship between AtxA and CO2.
MATERIALS AND METHODS
Plasmids for production of AtxA and His substitution mutants.
AtxA wild-type (WT) and mutant proteins were expressed with an N-terminal, TEV protease-cleavable His6 tag in Escherichia coli from the plasmid pProEX HTc-AtxA, a gift of Neha Dhasmana (see Fig. S1 in the supplemental material for plasmid details). Histidines located at residues 199 and 379 within AtxA were substituted with either aspartic acid or alanine to create phosphomimetic and phosphoablative substitutions, respectively. The Q5 site-directed mutagenesis kit (New England BioLabs, Ipswich, MA) was used, following the manufacturer’s protocol. Mutagenic primers were designed using NEBaseChanger v1.2.7 (https://nebasechanger.neb.com/) (Table 1) and were synthesized by Integrated DNA Technologies (IDT, Inc., Coralville, IA). When appropriate, medium was supplemented with ampicillin to give a final concentration of 100 μg/ml. Plasmids were isolated using the QIAprep Spin miniprep kit (Qiagen, Hilden, Germany) and transformed into E. coli BL21(DE3)RIPL competent cells (Agilent Technologies, Santa Clara, CA), following the manufacturer’s protocol. All plasmid DNA sequences were confirmed.
TABLE 1.
Primers used in this study
| Primer | Sequence (5′–3′)a | Dye | Relevant property |
|---|---|---|---|
| H199A_F | CTATTCAAAAgcaAAATTGTGTGTGTTGTTCGC | Used in Q5 mutagenesis for H199A substitution | |
| H199D_F | CTATTCAAAAgatAAATTGTGTGTGTTG | Used in Q5 mutagenesis for H199D substitution | |
| H199AD_R | GTGTACATTTGTACATTTAAAATTTTTTC | Used in Q5 mutagenesis for H199A and H199D substitutions | |
| H379A_F | ATTAACAATGgcaTTTGAAACTCAACGTATGC | Used in Q5 mutagenesis for H379A substitution | |
| H379D_F | ATTAACAATGgatTTTGAAACTCAAC | Used in Q5 mutagenesis for H379D substitution | |
| H379AD_R | AATGAAATTTCATAGCAGTC | Used in Q5 mutagenesis for H379A and H379D substitutions | |
| BamHI F | AAGCGACGGGTGATTCACTAGTTCTAG | Primer pair used in Q5 deletion of BamHI site from pPAPK-AtxA | |
| BamHI R | CTCCTCAATAAACTCAAAACTAATTG | ||
| C4R5 F | ACTGCAGCTGAAATCCTTACATACGGCGATATCTTC | Primer pair used in creation of pMRC43 plasmid | |
| C4N1 R | GGGGGGCATATGCGTTCTCCTTTTTGTATAAAATTA | ||
| 700PAG1_F | 700-TCTAGATGAAGATGTAAATCAAGCACTATCTGGA | IR700 | Primer pair used in PCR to create PAG1 fragment |
| 700PAG1_R | 700-GTCCAAGCTAACCAACGTTCAATAAAATACACTA | ||
| 700PAG2_F | 700-TAGTGTATTTTATTGAACGTTGGTTAGCTTGGAC | IR700 | Primer pair used in PCR to create PAG2 fragment |
| 700PAG2_R | 700-GTAACCCAAAAACAACCTGAAAGAAAATAGG | ||
| 700PAG3_F | 700-CTGCCCACCAAGCTAAACCTAAATAATCTT | IR700 | Primer pair used in PCR to create PAG3 fragment |
| 700PAG3_R | 700-GCTAAAATACGTGTATCAGTTCGGATAAAAGAAC | ||
| 700PAG4_F | 700-ATGGTTCTTTAGCTTTCTGTAAAACAGCCTT | IR700 | Primer pair used in PCR to create PAG4 fragment |
| 700PAG4_R | 700-TGCTTGAAACTAATATCGTAGACAATGCCA | ||
| PAG4_F | ATGGTTCTTTAGCTTTCTGTAAAACAGCCTT | Primer pair used in PCR to create PAG4 fragment used as unlabeled competitor in EMSA | |
| PAG4_R | TGCTTGAAACTAATATCGTAGACAATGCCA | ||
| FAMPAG4_F | 6-FAM-ATGGTTCTTTAGCTTTCTGTAAAACAGCCTT | FAM | Primer pair used in PCR to create PAG4 fragment for footprinting |
| VICPAG4_R | VIC-TGCTTGAAACTAATATCGTAGACAATGCCA | VIC | |
| PAG4-1_F | TTTATAAAAGTTCTGTTTAAAAAGCC | Used with PAG4R to create EMSA competitor | |
| PAG4-2_F | CTATTAAACATATAAATTCTTTTTTATGTT | Used with PAG4R to create EMSA competitor | |
| SBPF | CGGATTCATCCTATTAAACATATAAATTC | Used in Q5 deletion of upstream portion of pagA promoter to make pRSP from pMRC4 plasmid | |
| SBPR | TTCCGGATCCGGTGATTG | ||
| RSPdel_R | TAATACAATGGCCAGTAC | Used in Q5 deletion of SLI and SLI with downstream portions in pRSP plasmid | |
| RSPdel_F | ATATATTTATAAAAGTTCTGTTTAAAAAG | Used in Q5 deletion of SLI in pRSP plasmid | |
| 4RSPdel_F | ATTTATAAAAGTTCTGTTTAAAAAGC | Used in Q5 deletion of SLI and downstream 4 bp in pRSP plasmid | |
| 8RSPdel_F | ATAAAAGTTCTGTTTAAAAAGCC | Used in Q5 deletion of SLI and downstream 8 bp in pRSP plasmid | |
| 12RSPdel_F | AAGTTCTGTTTAAAAAGCC | Used in Q5 deletion of SLI and downstream 12 bp in pRSP plasmid | |
| 17RSPdel_F | CTGTTTAAAAAGCCAAAAATAAATAATTATC | Used in Q5 deletion of SLI and downstream 17 bp in pRSP | |
| 23RSPdel_F | AAAAAGCCAAAAATAAATAATTATCTC | Used in Q5 deletion of SLI and downstream 23 bp in pRSP plasmid | |
| SL2del_F | ATTATCTCTTTTTATTTATATTATATTGAAAC | Used in Q5 deletion of SLII in pRSP plasmid | |
| SL2del_R | TATAACATAAAAAAGAATTTATATGTTTAATAG | ||
| SwaI F | TTGAAATATAATAGCATTTGTCAGGTCATC | Used with GeneMorph II kit to create random mutations in gene for AtxA H379D | |
| BspMI R | TACGTTTTTAACACAAACCCTTTTATACAC | ||
| 700_pagSLF | GGTCCCTCGAAGAGGTTCACTAGT | IR700 | Primer pair used in PCR to create RPAG700 fragment |
| 700_pagSLR | AATTTCATTCCTTCCACGTCACCCT | IR700 | |
| pagSL_F | GGTCCCTCGAAGAGGTTCACTAGT | Primer pair used in PCR to create unlabeled competitor RPAG fragments | |
| pagSL_R | AATTTCATTCCTTCCACGTCACCCT |
Lowercase letters denote inserted mutation. Restriction sites are underlined.
Production and purification of AtxA proteins.
The BL21(DE3)RIPL strains containing the pProEX HTc-AtxA plasmids were grown in 50 ml LB medium with 100 μg/ml ampicillin and 30 μg/ml chloramphenicol for 8 h. This starter culture was diluted 1:100 into each of 12 5,000-ml portions of autoinduction medium (26) containing 100 μg/ml ampicillin and 30 μg/ml chloramphenicol in 2.5-liter shake flasks. The cultures were grown at 250 rpm and 37°C approximately for 3 h and then shifted to 30°C overnight. The cultures were chilled on ice, and the cells were recovered by centrifugation at 5,000 × g for 15 min and stored at −80°C. The cells were thawed and lysed in an EmulsiFlex C3 homogenizer device (Avestin, Inc., Ottawa, Canada) at 15,000 lb/in2. To the lysate, 10 μl of Benzonase was added to degrade the DNA, and the cell debris was removed by centrifugation at 18,000 × g for 30 min. All procedures afterward were performed at 4 to 8°C. The His6-TEV-AtxA cleared lysate was loaded on a nickel-nitrilotriacetic acid (Ni-NTA) resin which was then washed with 20 mM Tris-HCl (pH 8), 500 mM NaCl (pH 8), and 0.50% IGEPAL CA-630 detergent to remove endotoxin. After further washing with 20 mM Tris-HCl (pH 8), 500 mM NaCl, and 40 mM imidazole (pH 8), the desired protein was eluted with 20 mM Tris-HCl, 500 mM NaCl, and 500 mM imidazole (pH 8). The desired fractions containing the protein were pooled, EDTA was added to a final concentration of 0.25 mM, and β-mercaptoethanol was added to a final concentration of 5 mM. The protein was digested with 0.15 mg TEV protease (27) per 20 mg purified protein for 16 h at 4°C. The His6-TEV(S219V)-Arg5 variant TEV protease was produced in this lab from plasmid pRK793, a gift of David Waugh, obtained from Addgene (Cambridge, MA) as plasmid no. 8827. The digested AtxA protein was loaded directly on an Akta system equipped with a Macro-Prep ceramic hydroxyapatite column (2.5-cm diameter by 10 cm, type 1, 40 μm; Bio-Rad Laboratories, Hercules, CA) preequilibrated with 0.01 M potassium phosphate, 0.5 M NaCl buffer (pH 7.0), and 5 mM β-mercaptoethanol. The protein was then eluted at 1.0 ml/min by a 0.01 to 0.50 M potassium phosphate gradient with 0.5 M NaCl buffer (pH 7.0) and 5 mM β-mercaptoethanol. The fractions were analyzed by an 8 to 25% SDS-PAGE gel. The pooled protein was dialyzed overnight against 2 liters of 10 mM HEPES, 0.50 M NaCl, and 5 mM β-mercaptoethanol. The dialyzed protein was finally passed through a Ni-NTA column to remove the TEV protease and the cleaved N-terminal tag. The pure fractions were again dialyzed in the above-described buffer system. The proteins were quantified using a DS-11 fluorometer/spectrophotometer (DeNovix, Inc., Wilmington, NC) at an optical density at 280 nm (OD280). The yield of the AtxA WT and mutant proteins was in the range of 3.0 to 11.5 mg/liter of culture. For verification purposes, proteins were run on SDS-PAGE gels, as previously described (17). The molecular masses of the purified proteins were estimated by time of flight-liquid chromatogram mass spectrometry (TOF-LC/MS) using an HP/Agilent G6224A mass selective detector (MSD) instrument (Hewlett-Packard, Palo Alto, CA) at the NHLBI core facility in Bethesda, MD. Also, N-terminal sequencing data were generated at the Research Technologies Branch (RTB) core facility of the NIAID in Rockville, MD.
Sedimentation velocity.
Sedimentation velocity experiments were carried out at 20°C on a Beckman Optima XL-A or Beckman Coulter ProteomeLab XI-I analytical ultracentrifuge, following standard protocols (28). Samples of AtxA and of AtxA having the H199D, H199A, and H379A substitutions were prepared in 500 mM NaCl, 10 mM HEPES (pH 7.2), and 5 mM 2-mercaptoethanol and were studied at different concentrations ranging from approximately 2 to 20 μM. Samples were loaded in 2-channel centerpiece cells, and sedimentation data were collected using the absorbance (280 nm) and interference (655 nm, when available) optical detection systems. Time-corrected data (29) were analyzed in SEDFIT 15.01c (30) in terms of a continuous c(s) distribution of sedimenting species with a maximum entropy regularization confidence level of 0.68. Excellent fits were observed, with a root mean standard deviation (RMSD) of 0.0026 to 0.0084 absorbance units and 0.0043 to 0.015 fringes. The solution density and viscosity were measured experimentally at 20°C on an Anton Paar DMA 5000 density meter and Anton Paar AMVn automated rolling ball viscometer, and the protein partial specific volumes were determined based on the composition in SEDNTERP (31). Sedimentation coefficients “s” were corrected to s20,w values under standard conditions.
Plasmid construction for complementation of pXO1 atxA deletion in Ames 35 strain.
The atxA deletion strain (11) was complemented with the pKA2 plasmid (Fig. S1) and its derivatives. To create the pKA2 plasmid, we used restriction digests to cut and modify pPAGK (17) in order to insert the atxA gene from pSC (17). We then used the Q5 site-directed mutagenesis kit (New England BioLabs, Inc.) and further digestions to remove pagA and its promoter. The final version of the plasmid pKA2 contains the pUB110 replicon, a kanamycin resistance (Kmr) gene for selection both in E. coli and B. anthracis, and a functionally active atxA gene (Fig. S1). pKA2 plasmid variants encoding AtxA having the H199 and H379 substitutions were created by replacing a fragment of pKA2-AtxA with the corresponding fragments from the pProEX HTc-AtxA plasmids described above that have the H199 and H379 substitutions.
Preparation of DNA for electrophoretic mobility shift assays.
IRDye 700-labeled DNA fragments (Fig. 2A and 5B and D) were prepared using 5′-end-labeled IRDye 700 primers (IDT, Inc.) listed in Table 1 at a final concentration of 40 μM. Unlabeled excess competitor fragments were prepared by using primers with the same sequence at a final concentration of 4 mM. PCR using OneTaq 2× mastermix with standard buffer (New England BioLabs) and pPAGK DNA (17) and pRSP (this study) as the template was conducted under the following conditions: 30 s at 94°C, 30 cycles of 30 s at 94°C, 30 s at 53°C, and 1 min at 68°C, followed by 5 min at 68°C. Infrared-labeled PCR products were purified using the PureLink PCR purification kit (Invitrogen, Carlsbad, CA). Unlabeled PCR products were purified using the DNA Clean & Concentrator kit (Zymo Research, Irvine, CA), following the manufacturer’s protocol.
Electrophoretic mobility shift assays.
Infrared dye end-labeled probe was incubated with various concentrations of AtxA and reagents from the Odyssey EMSA buffer kit (Li-Cor Biosciences, Lincoln, NE) in 20-μl reaction mixtures. Final reactions contained 2.5 nM or 1.67 nM IRDye 700-labeled DNA probe, 50 μg/ml poly(dI-dC), 500 μg/ml bovine serum albumin (New England BioLabs), 10 mM Tris-HCl, 50 mM KCl, 3.5 mM dithiothreitol (DTT), 10 mM EDTA, 0.25% Tween 20 (pH 7.5), and various concentrations of AtxA proteins, typically 50 to 500 nM. The reaction mixtures were incubated at room temperature in the dark for 30 min. 1× orange dye was added, and the samples were run on 2% agarose gels in 1× EABE buffer (90 mM ethanolamine, 90 mM boric acid, 1 mM EDTA [pH 8.0]). The gels and buffer were previously chilled in an ice-water bath and then run in the dark at 100 V for 15 min, followed by 80 V for 45 min, and imaged on an Odyssey infrared imaging system (Li-Cor Biosciences).
Analysis of PA production by Western blotting.
Ames 35, Ames 35 ΔAtxA, and Ames 35 ΔAtxA(pKA2) variants were inoculated into nutrient broth yeast extract (NBY) (11) with 0.8% (wt/vol) sodium bicarbonate containing 20 μg/ml kanamycin for strains containing pKA2 plasmid and grown overnight at 37°C in 15% CO2. The overnight cultures were diluted to an A600 of 0.3 and grown under the same conditions until reaching an A600 of 2.0. The cultures were then centrifuged at 10,000 × g for 10 min at 4°C. Supernatants were isolated, mixed with 500 μM EDTA (pH 8.0), frozen on dry ice, and stored at –80°C until further analysis. Supernatant samples were mixed with 4× Laemmli sample buffer (Bio-Rad Laboratories) and boiled for 5 min at 95°C. Samples were then analyzed by Novex WedgeWell 4 to 20% Tris-glycine gels (Invitrogen) with 1× Tris-glycine SDS-PAGE buffer (National Diagnostics, Atlanta, GA) and run at 140 V for 90 min in the XCell SureLock mini-cell electrophoresis system (Thermo Fisher Scientific, Waltham, MA). Proteins were either stained with Coomassie blue R250 or were transferred to nitrocellulose paper using the iBlot dry blotting system (Thermo Fisher Scientific). After transferring, the blot was blocked for 45 min at room temperature in Odyssey blocking buffer (phosphate-buffered saline [PBS]; Li-Cor Biosciences) and incubated with anti-PA rabbit primary antibody at a 1:2,500 dilution (32) in the blocking buffer. After overnight incubation at room temperature, membranes were rinsed 3 times in 1× PBS-T (PBS plus 0.05% Tween 20) and washed another 3 times with 1× PBST for 5 min per wash. An anti-rabbit Rockland 700Dx IgG F secondary antibody was diluted 1:5,000 in the blocking buffer as before and added to membrane for 45 min at room temperature. Membrane was then rinsed as described previously and imaged using the Odyssey imaging system.
Analysis of pagA promoter activation by different forms of AtxA.
The plasmid pSW4-DasherC4 was used as a source of green fluorescent protein (GFP) gene. In this plasmid, expression of the gfp gene (Dasher C4; Atum, Newark, CA) is controlled by the pSW4-pagA promoter (33). A fragment of this plasmid containing the gfp gene and promoter was inserted into a large fragment of the pMR plasmid (34). The resulting pMRC41 plasmid was cut and ligated to produce pMRC42 containing the gfp gene without a promoter. A short fragment upstream of the gfp gene in the pMRC42 plasmid was replaced with a PCR-created fragment of the pPAGK plasmid that contains the pagA promoter region. The resulting pMRC43 plasmid was combined with a fragment of pMRC41 to produce the final pMRC4 plasmid, which contains a gfp gene under the control of the 1,257-bp pPAGK pagA promoter region, and an ampicillin resistance gene, a spectinomycin resistance gene, and the appropriate replicons for replication in E. coli and B. anthracis (Table 2 and Fig. S1). All restriction enzymes were obtained from New England BioLabs.
TABLE 2.
Plasmids used in this studya
| Plasmid | Relevant characteristic(s) | Source or reference |
|---|---|---|
| pSC-A | pSC plasmid containing atxA | 17 |
| pPAGK | Plasmid containing pagA under control of full-length pagA promoter region | 17 |
| pKA2 | Plasmid containing pUB110 replicon, Kmr for selection both in E. coli and B. anthracis, and functionally active atxA | This study |
| pKA2-AtxA H199A | pKA2 with H199A substitution in AtxA | This study |
| pKA2-AtxA H199D | pKA2 with H199D substitution in AtxA | This study |
| pKA2-AtxA H379A | pKA2 with H379A substitution in AtxA | This study |
| pKA2-AtxA H379D | pKA2 with H379D substitution in AtxA | This study |
| pPROEx HTc-AtxA | Plasmid pPROEx HTc (Addgene, Cambridge, MA) containing his6-tev-atxA under control of Trc promoter | N. Dhasmana and Y. Singh |
| pPROEx HTc-AtxA H199A | Plasmid pPROEx HTc-AtxA with substitution H199A | This study |
| pPROEx HTc-AtxA H199D | Plasmid pPROEx HTc-AtxA with substitution H199D | This study |
| pPROEx HTc-AtxA H379A | Plasmid pPROEx HTc-AtxA with substitution H199A | This study |
| pPROEx HTc-AtxA H379D | Plasmid pPROEx HTc-AtxA with substitution H199A | This study |
| pSW4 | B. anthracis/E. coli shuttle vector containing pagA promoter region; Kmr in B. anthracis, Apr in E. coli | 33 |
| pSW1 | pSW4 converted into an Electra Vector System (Atum, Menlo Park, CA) | Nate Hepler |
| pSW4-DasherC4 | pSW1 encoding optimized Dasher variant of GFP (Atum) under control of pSW4 pagA promoter region | Catherine Vrentas |
| pMR | pXO1 minireplicon, Spr in B. anthracis, Apr in E. coli | 34 |
| pMRC4 | pMR encoding DasherC4 under control of full-length pagA promoter region | This study |
| pRSP | pMRC4 plasmid with upstream portion of full-length pagA promoter region deleted; the residual 162-bp pagA promoter sequence is the same as pYS5 pagA promoter region (38) | This study |
| pRSPΔSLI | pRSP plasmid with stem-loop I deleted | This study |
| pRSPΔSLI4 | pRSP plasmid with stem-loop I and downstream 4 bp deleted | This study |
| pRSPΔSLI8 | pRSP plasmid with stem-loop I and downstream 8 bp deleted | This study |
| pRSPΔSLI12 | pRSP plasmid with stem-loop I and downstream 12 bp deleted | This study |
| pRSPΔSLI17 | pRSP plasmid with stem-loop I and downstream 17 bp deleted | This study |
| pRSPΔSLI23 | pRSP plasmid with stem-loop I and downstream 23 bp deleted | This study |
| pRSPΔSLII | pRSP plasmid with stem-loop II deleted | This study |
Maps of relevant plasmids are in Fig. S1. Apr, apramycin resistance.
To analyze the activity of the pMRC4-pagA promoter depending on different forms of AtxA, pMRC4 was transformed into the B. anthracis BH500 strain (17) containing the pKA2, pKA2-atxA H199A, pKA2-atxA H199D, pKA2-atxA H379A, or pKA2-atxA H379D plasmid. BH500 was transformed as previously described (34) and plated onto selective LB agar containing 15 μg/ml kanamycin to maintain pKA2 and its derivatives and 150 μg/ml spectinomycin to maintain pMRC4. All kanamycin-resistant (Kmr) and spectinomycin-resistant (Spr) isolated strains were verified for the presence of intact plasmids by sequencing.
To assess the role of AtxA in the pagA promoter-dependent expression of GFP, overnight cultures of plasmid-containing B. anthracis were diluted to an A600 of 0.1 in fresh NBY supplemented with 0.8% sodium bicarbonate and corresponding antibiotics and grown at 37°C in 14% CO2. Fluorometric measurements of bacteria grown to different optical densities were performed in a Victor3 reader (PerkinElmer) using a GFP filter set (excitation at 485 nm and emission at 535 nm), as previously described (35). Fluorescence intensities were compared with the fluorescence of the BH500 strain containing only the pMRC4 plasmid. This control strain was grown under the same conditions in medium containing only 150 μg/ml spectinomycin.
Construction and analysis of plasmids with shortened and modified pagA promoter region.
The Q5 site-directed mutagenesis kit (New England BioLabs) was used, following the manufacturer’s protocol, to produce specific deletions in the pMRC4 plasmid to create nine additional plasmids, as follows: pRSP, pRSPΔSLI, pRSPΔSLI4, pRSPΔSLI8, pRSPΔSLI12, pRSPΔSLI17, pRSPΔSLI23, pRSPΔSLII, and pRSPΔhalfSLII (Table 2 and Fig. 6A and S1). The primers used to create these modified plasmids were created using NEBaseChanger v1.2.7 (https://nebasechanger.neb.com/) (Table 1) and were synthesized by Integrated DNA Technologies (IDT, Inc.). The region upstream of the pagA promoter was deleted using the SBPF and SBPR primers to create a shortened promoter region in the pRSP plasmids for a comparison of AtxA activation to the pMRC4 pagA promoter (Fig. S1). Various segments of two suspected stem-loops in the region upstream of the RNAP binding site in the pagA promoter region in pRSP were deleted using the Q5 site-directed mutagenesis kit and various specific primers for each deletion (Table 1 and Fig. 6A). When appropriate, media were supplemented with spectinomycin to 150 μg/ml and ampicillin to 100 μg/ml. Plasmids were isolated using the QIAprep Spin miniprep kit (Qiagen), and all plasmids were transformed into E. coli One Shot TOP10 competent cells and then into E. coli SCS110 competent cells, following the manufacturer’s protocol for each.
pMRC4 and these various pRSP plasmids were then transformed into a B. anthracis BH500 (17) strain containing pKA2, as previously described (34), and plated onto selective NBY agar containing both 15 μg/ml kanamycin to maintain pKA2 and 150 μg/ml spectinomycin to select for pMRC4/pRSP and its derivatives. These strains were then analyzed by fluorometric assays as described above.
DNase I footprinting.
DNA was labeled at both ends via PCR with 6-FAM dye on the forward primer (IDT, Inc.) and VIC dye on the reverse primer (Applied Biosystems, Foster City, CA) under the same conditions as for the infrared probe (see “Preparation of DNA for electrophoretic mobility shift assays,” above), except that primers were used at a final concentration of 400 μM. Labeled fragments were purified using the previously mentioned Wizard SV gel and PCR cleanup system (Promega, Madison, WI). DNA was then incubated with 675 nM AtxA protein for 30 min at room temperature under the same conditions as for EMSAs. After 30 min, 0.07 U of DNase I (New England BioLabs) and 1× DNase buffer were added to the mixture for a 10-min incubation at room temperature. DNase was inactivated by incubation for 10 min at 75°C. The sample was then purified using the Qiagen MinElute kit (Qiagen). Samples were sent to the Plant and Microbe Genetics Facility at The Ohio State University for analysis on the 3730 DNA analyzer (Applied Biosystems), as previously reported (36).
Random mutagenesis of AtxA H379D gene in pKA2 plasmid.
The GeneMorph II random mutagenesis kit (Agilent Technologies, Inc.) was used following the manufacturer’s protocol to generate random mutations in the portion of the gene encoding amino acids (aa) 135 to 475 of AtxA H379D. Error-prone PCR used 1 μl each of 100 μM SwaI F and 100 μM BspMI R primers and 100 ng of pKA2-AtxA H379D linearized by digestion with EcoRI-HF (New England BioLabs) in a 100-μl reaction mixture. The modified gene was then cut out with SwaI and BspMI enzymes (New England BioLabs) and ligated into appropriately digested pKA2 plasmid. The mutated pKA2 plasmid pool was transformed into E. coli One Shot TOP10 competent cells (Invitrogen Corporation), following the manufacturer’s protocol, and then into E. coli SCS110 competent cells (Agilent Technologies). Where appropriate, media were supplemented with kanamycin to 15 μg/ml. The plasmid pool isolated from SCS110 was transformed into B. anthracis BH500 (17) containing the pMRC4 plasmid, as previously described (34), and transformants were plated on selective NBY medium with 15 μg/ml kanamycin and 150 μg/ml spectinomycin. Isolated colonies were inspected for fluorescence using an UV illuminator (PrepOne Sapphire PI-1000; Embi Tec, San Diego, CA). Colonies having increased fluorescence were isolated and expanded, and the plasmid was isolated and sequenced.
Structural analysis of pagA promoter region.
The pagA promoter region DNA sequence was examined for structural motifs using the mfold Web server (http://unafold.rna.albany.edu/?q=mfold/DNA-Folding-Form) (19). The NCBI BLAST program (https://blast.ncbi.nlm.nih.gov/Blast.cgi?PROGRAM=blastn&PAGE_TYPE=BlastSearch&LINK_LOC=blasthome) was used for DNA homology searches in the GenBank database.
Supplementary Material
ACKNOWLEDGMENTS
This research was supported by the intramural research programs of the National Institute of Allergy and Infectious Diseases and the National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health.
We thank Anthony McCoy at The Ohio State University Plant-Microbe Genomics Facility for assistance with footprinting assays. We thank Brian Martin at the NIAID Research Technologies Branch for N-terminal sequencing and Duck-Yeon Lee from the NHLBI for molecular mass determination of the purified proteins. Catherine Vrentas provided the pSW4-DasherC4 plasmid, Nathan Hepler provided the pSW1 plasmid, and Neha Dhasmana provided pPro-Ex HTC-AtxA.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/JB.00569-19.
REFERENCES
- 1.Fouet A, Mock M. 2006. Regulatory networks for virulence and persistence of Bacillus anthracis. Curr Opin Microbiol 9:160–166. doi: 10.1016/j.mib.2006.02.009. [DOI] [PubMed] [Google Scholar]
- 2.Leppla SH. 2013. Chapter 281–anthrax lethal factor, p 1257–1261. In Rawlings ND, Salvesen GS (ed), Handbook of proteolytic enzymes, 3rd ed Academic Press, London, United Kingdom. [Google Scholar]
- 3.Uchida I, Hornung JM, Thorne CB, Klimpel KR, Leppla SH. 1993. Cloning and characterization of a gene whose product is a trans-activator of anthrax toxin synthesis. J Bacteriol 175:5329–5338. doi: 10.1128/jb.175.17.5329-5338.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fouet A. 2010. AtxA, a Bacillus anthracis global virulence regulator. Res Microbiol 161:735–742. doi: 10.1016/j.resmic.2010.09.006. [DOI] [PubMed] [Google Scholar]
- 5.Hammerstrom TG, Horton LB, Swick MC, Joachimiak A, Osipiuk J, Koehler TM. 2015. Crystal structure of Bacillus anthracis virulence regulator AtxA and effects of phosphorylated histidines on multimerization and activity. Mol Microbiol 95:426–441. doi: 10.1111/mmi.12867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tsvetanova B, Wilson AC, Bongiorni C, Chiang C, Hoch JA, Perego M. 2007. Opposing effects of histidine phosphorylation regulate the AtxA virulence transcription factor in Bacillus anthracis. Mol Microbiol 63:644–655. doi: 10.1111/j.1365-2958.2006.05543.x. [DOI] [PubMed] [Google Scholar]
- 7.Hondorp ER, Hou SC, Hause LL, Gera K, Lee CE, McIver KS. 2013. PTS phosphorylation of Mga modulates regulon expression and virulence in the group A streptococcus. Mol Microbiol 88:1176–1193. doi: 10.1111/mmi.12250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Solano-Collado V, Lurz R, Espinosa M, Bravo A. 2013. The pneumococcal MgaSpn virulence transcriptional regulator generates multimeric complexes on linear double-stranded DNA. Nucleic Acids Res 41:6975–6991. doi: 10.1093/nar/gkt445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hadjifrangiskou M, Koehler TM. 2008. Intrinsic curvature associated with the coordinately regulated anthrax toxin gene promoters. Microbiology 154:2501–2512. doi: 10.1099/mic.0.2007/016162-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ruiz-Cruz S, Moreno-Blanco A, Espinosa M, Bravo A. 2018. DNA-binding properties of MafR, a global regulator of Enterococcus faecalis. FEBS Lett 592:1412–1425. doi: 10.1002/1873-3468.13032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McKenzie AT, Pomerantsev AP, Sastalla I, Martens C, Ricklefs SM, Virtaneva K, Anzick S, Porcella SF, Leppla SH. 2014. Transcriptome analysis identifies Bacillus anthracis genes that respond to CO2 through an AtxA-dependent mechanism. BMC Genomics 15:229. doi: 10.1186/1471-2164-15-229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dale JL, Raynor MJ, Dwivedi P, Koehler TM. 2012. cis-acting elements controlling expression of the master virulence regulatory gene atxA in Bacillus anthracis. J Bacteriol 194:4069–4079. doi: 10.1128/JB.00776-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bongiorni C, Fukushima T, Wilson AC, Chiang C, Mansilla MC, Hoch JA, Perego M. 2008. Dual promoters control the expression of the Bacillus anthracis virulence factor AtxA. J Bacteriol 190:6483–6492. doi: 10.1128/JB.00766-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dai Z, Koehler TM. 1997. Regulation of anthrax toxin activator gene (atxA) expression in Bacillus anthracis: temperature, not CO2/bicarbonate, affects AtxA synthesis. Infect Immun 65:2576–2582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dai Z, Sirard JC, Mock M, Koehler TM. 1995. The atxA gene product activates transcription of the anthrax toxin genes and is essential for virulence. Mol Microbiol 16:1171–1181. doi: 10.1111/j.1365-2958.1995.tb02340.x. [DOI] [PubMed] [Google Scholar]
- 16.Toyomane K, Furuta Y, Fujikura D, Higashi H. 2019. Upstream sequence-dependent suppression and AtxA-dependent activation of protective antigens in Bacillus anthracis. PeerJ 7:e6718. doi: 10.7717/peerj.6718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pomerantsev AP, McCall RM, Chahoud M, Hepler NK, Fattah R, Leppla SH. 2017. Genome engineering in Bacillus anthracis using tyrosine site-specific recombinases. PLoS One 12:e0183346. doi: 10.1371/journal.pone.0183346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chitlaru T, Gat O, Gozlan Y, Ariel N, Shafferman A. 2006. Differential proteomic analysis of the Bacillus anthracis secretome: distinct plasmid and chromosome CO2-dependent cross talk mechanisms modulate extracellular proteolytic activities. J Bacteriol 188:3551–3571. doi: 10.1128/JB.188.10.3551-3571.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zuker M. 2003. mfold Web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res 31:3406–3415. doi: 10.1093/nar/gkg595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Luscombe NM, Austin SE, Berman HM, Thornton JM. 2000. An overview of the structures of protein-DNA complexes. Genome Biol 1:REVIEWS001. doi: 10.1186/gb-2000-1-1-reviews001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Deng Z, Wang Q, Liu Z, Zhang M, Machado AC, Chiu TP, Feng C, Zhang Q, Yu L, Qi L, Zheng J, Wang X, Huo X, Qi X, Li X, Wu W, Rohs R, Li Y, Chen Z. 2015. Mechanistic insights into metal ion activation and operator recognition by the ferric uptake regulator. Nat Commun 6:7642. doi: 10.1038/ncomms8642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Al-Zyoud WA, Hynson RM, Ganuelas LA, Coster AC, Duff AP, Baker MA, Stewart AG, Giannoulatou E, Ho JW, Gaus K, Liu D, Lee LK, Böcking T. 2016. Binding of transcription factor GabR to DNA requires recognition of DNA shape at a location distinct from its cognate binding site. Nucleic Acids Res 44:1411–1420. doi: 10.1093/nar/gkv1466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ding P, McFarland KA, Jin S, Tong G, Duan B, Yang A, Hughes TR, Liu J, Dove SL, Navarre WW, Xia B. 2015. A novel AT-Rich DNA recognition mechanism for bacterial xenogeneic silencer MvaT. PLoS Pathog 11:e1004967. doi: 10.1371/journal.ppat.1004967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Thomson JJ, Withey JH. 2014. Bicarbonate increases binding affinity of Vibrio cholerae ToxT to virulence gene promoters. J Bacteriol 196:3872–3880. doi: 10.1128/JB.01824-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yang J, Hart E, Tauschek M, Price GD, Hartland EL, Strugnell RA, Robins-Browne RM. 2008. Bicarbonate-mediated transcriptional activation of divergent operons by the virulence regulatory protein, RegA, from Citrobacter rodentium. Mol Microbiol 68:314–327. doi: 10.1111/j.1365-2958.2008.06171.x. [DOI] [PubMed] [Google Scholar]
- 26.Studier FW. 2005. Protein production by auto-induction in high density shaking cultures. Protein Expr Purif 41:207–234. doi: 10.1016/j.pep.2005.01.016. [DOI] [PubMed] [Google Scholar]
- 27.Tropea JE, Cherry S, Waugh DS. 2009. Expression and purification of soluble His6-tagged TEV protease. Methods Mol Biol 498:297–307. doi: 10.1007/978-1-59745-196-3_19. [DOI] [PubMed] [Google Scholar]
- 28.Zhao H, Brautigam CA, Ghirlando R, Schuck P. 2013. Overview of current methods in sedimentation velocity and sedimentation equilibrium analytical ultracentrifugation. Curr Protoc Protein Sci Chapter 20:Unit20.12. doi: 10.1002/0471140864.ps2012s71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ghirlando R, Balbo A, Piszczek G, Brown PH, Lewis MS, Brautigam CA, Schuck P, Zhao H. 2013. Improving the thermal, radial, and temporal accuracy of the analytical ultracentrifuge through external references. Anal Biochem 440:81–95. doi: 10.1016/j.ab.2013.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schuck P. 2000. Size-distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and Lamm equation modeling. Biophys J 78:1606–1619. doi: 10.1016/S0006-3495(00)76713-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cole JL, Lary JW, P Moody T, Laue TM. 2008. Analytical ultracentrifugation: sedimentation velocity and sedimentation equilibrium. Methods Cell Biol 84:143–179. doi: 10.1016/S0091-679X(07)84006-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Moayeri M, Wiggins JF, Leppla SH. 2007. Anthrax protective antigen cleavage and clearance from the blood of mice and rats. Infect Immun 75:5175–5184. doi: 10.1128/IAI.00719-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pomerantsev AP, Kalnin KV, Osorio M, Leppla SH. 2003. Phosphatidylcholine-specific phospholipase C and sphingomyelinase activities in bacteria of the Bacillus cereus group. Infect Immun 71:6591–6606. doi: 10.1128/iai.71.11.6591-6606.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pomerantsev AP, Camp A, Leppla SH. 2009. A new minimal replicon of Bacillus anthracis plasmid pXO1. J Bacteriol 191:5134–5146. doi: 10.1128/JB.00422-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sastalla I, Chim K, Cheung GY, Pomerantsev AP, Leppla SH. 2009. Codon-optimized fluorescent proteins designed for expression in low GC Gram-positive bacteria. Appl Environ Microbiol 75:2099–2110. doi: 10.1128/AEM.02066-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zianni M, Tessanne K, Merighi M, Laguna R, Tabita FR. 2006. Identification of the DNA bases of a DNase I footprint by the use of dye primer sequencing on an automated capillary DNA analysis instrument. J Biomol Tech 17:103–113. [PMC free article] [PubMed] [Google Scholar]
- 37.Guiziou S, Sauveplane V, Chang HJ, Clerte C, Declerck N, Jules M, Bonnet J. 2016. A part toolbox to tune genetic expression in Bacillus subtilis. Nucleic Acids Res 44:7495–7508. doi: 10.1093/nar/gkw624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sharma AK, Leppla SH, Pomerantsev AP, Shiloach J. 2018. Effect of over expressing protective antigen on global gene transcription in Bacillus anthracis BH500. Sci Rep 8:16108. doi: 10.1038/s41598-018-34196-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.