

This work focuses on the characterization of cis- and trans-acting elements essential for the induction of the cidABC operon in S. aureus. The results of this study are the first to demonstrate the synergistic control of cidABC expression by transcriptional regulators CidR and CcpA during carbohydrate metabolism. We established that the full induction of cidABC expression depends on the metabolic state of bacteria and requires both CidR and CcpA. Together, these findings delineate regulatory control of cidABC expression under different metabolic conditions and provide important new insights into our understanding of cell death mechanisms during biofilm development in S. aureus.
KEYWORDS: regulation of gene expression, glycolysis, carbon catabolite repression, Staphylococcus aureus
ABSTRACT
The death and lysis of a subpopulation of Staphylococcus aureus cells during biofilm development benefit the whole bacterial population through the release of an important component of the biofilm matrix, extracellular DNA. Previously, we have demonstrated that these processes are affected by the gene products of the cidABC operon, the expression of which is controlled by the LysR-type transcriptional regulator, CidR. In this study, we characterized cis- and trans-acting elements essential for the induction of the cidABC operon. In addition to a CidR-binding site located within the cidABC promoter region, sequence analysis revealed the presence of a putative catabolite responsive element (cre box), suggestive of the involvement of the catabolite control protein A (CcpA) in the regulation of cidABC expression. This was confirmed using electrophoretic mobility shift assays and real-time reverse transcriptase PCR analysis demonstrating the direct positive control of cidABC transcription by the master regulator of carbon metabolism. Furthermore, the importance of CcpA and the identified cre site for the induction of the cidABC operon was demonstrated by examining the expression of PcidABC-lacZ reporter fusions in various mutant strains in which the genes involved in carbon metabolism and carbon catabolite repression were disrupted. Together the results of this study demonstrate the necessity of both transcriptional regulators, CidR and CcpA, for the induction of the cidABC operon and reveal the complexity of molecular interactions controlling its expression.
IMPORTANCE This work focuses on the characterization of cis- and trans-acting elements essential for the induction of the cidABC operon in S. aureus. The results of this study are the first to demonstrate the synergistic control of cidABC expression by transcriptional regulators CidR and CcpA during carbohydrate metabolism. We established that the full induction of cidABC expression depends on the metabolic state of bacteria and requires both CidR and CcpA. Together, these findings delineate regulatory control of cidABC expression under different metabolic conditions and provide important new insights into our understanding of cell death mechanisms during biofilm development in S. aureus.
INTRODUCTION
Staphylococcus aureus infections often involve the formation of a biofilm, a structured community of bacteria held together by a self-produced matrix that consists of extracellular polymeric substances (EPS) (1). The EPS matrix plays an important role in the adherence between cells and surface materials, retains water within the biofilm, and serves as a barrier protecting bacteria from antimicrobial agents and the host immune response (2). The composition of the EPS matrix is diverse in nature and varies between bacterial species and strains. It is typically comprised of different molecules, including polysaccharides, lipids, proteins, and extracellular DNA (eDNA) (3, 4). In S. aureus, eDNA is an important structural component of the biofilm matrix that is released by the lysis of a subpopulation of cells via a process termed programmed cell death (PCD) (4–7).
Although the understanding of bacterial PCD in S. aureus envisions a holin-antiholin action of the membrane-associated cidA and lrgA gene products (8–10), several studies have revealed that activities of metabolic enzymes of the CidR regulon, consisting of the cidABC and alsSD operons, also have a direct impact on bacterial cell death during the stationary phase of growth (5, 11, 12). It has been demonstrated that the cidC-encoded pyruvate:menaquinone oxidoreductase (13, 14) contributes to the cell death through acetate-mediated acidification of the growth medium leading to protonation of bacterial cytoplasm (12). In contrast, the gene products of the alsSD operon, involved in acetoin biosynthesis, promote cell survival under these conditions by consuming protons and, thus, increasing cytoplasmic pH (11, 12). These findings are consistent with a previously proposed hypothesis that the balance between the prolife and prodeath functions of these enzymes determines cell fate during growth (8). In accordance with this, our recent work demonstrated alternative functions of the CidA and CidB proteins in modulating CidC activity and revealed the involvement of the CidR regulon in limiting acetate-mediated cell death in staphylococcal populations (15). Specifically, it has been shown that under conditions of excess glucose, both CidB and CidC promote the generation of acetate and cell death, whereas the activation of cidA and alsSD expression by CidR opposes the effects of CidB and CidC by directing carbon flux toward the production of acetoin (15).
The cidABC and alsSD operons represent the only genes directly controlled by the transcriptional regulator CidR (11, 12, 16). CidR is a member of the LysR-type transcriptional regulator (LTTR) family of proteins, which are typically activated through the binding of a specific small molecule to a conserved ligand-binding domain (16, 17). It has been suggested that pyruvate might serve as an inducer molecule for activation of CidR, as its intracellular concentrations under acidic stress conditions, in the presence of excess glucose, will modulate the metabolic status of the cells (10). While CidR is a major transcriptional regulator of the cidABC and alsSD operons, the regulatory network controlling expression of these operons is more complex. Recently, we have shown that expression of cidABC genes is also controlled by the SrrAB two-component system and affected by cellular respiration (18). This regulation appears to be superimposed on the regulation mediated by CidR, providing an exquisite level of transcriptional control of the cidABC operon. Furthermore, in agreement with the necessity of glucose for the induction of the cidABC operon (16), earlier work has suggested involvement of the master regulator of carbon metabolism, CcpA, in cidABC expression (19). In particular, using Northern blot analysis, Seidl et al. (19) demonstrated that inactivation of the ccpA gene in S. aureus leads to a significant decrease in the accumulation of cidABC transcripts during the exponential phase of growth. Interestingly, in addition to the transcriptional control of cidABC expression, recent work by Hussein et al. (20) revealed the presence of a functional thermosensor within the 5′ untranslated region (UTR) of the cidA gene, which posttranscriptionally facilitates increased cidABC expression at lower temperatures.
In the current study, we investigated in greater detail the cis- and trans-acting regulatory elements critical for the control of cidABC expression. We identified the nucleotide sequences of the CidR-binding site and catabolite responsive element (cre box) within the cidABC promoter region and demonstrated the importance of these cis-acting elements in the control of cidABC expression. These findings were supported by the demonstration of specific interactions of CidR and CcpA with these cis-acting elements. In agreement with this, a transposon mutagenesis approach to identify additional factors involved in the induction of cidABC expression revealed genes involved in carbon metabolism and carbon catabolite repression (CCR). Overall, the results of this study suggest a model in which CcpA and CidR work synergistically to induce cidABC expression under conditions of carbon overflow.
RESULTS
Identification of a cis-acting element required for CidR-mediated induction of cidABC expression.
To characterize the molecular interactions associated with CidR-mediated regulatory control, we first sought to identify potential cis-acting DNA elements important for the binding of CidR to the promoter region of the cidABC operon. Although LTTR binding sites typically encompass the consensus sequence, T-N11-A motif (17), the identification of the putative CidR-binding site within the cidABC promoter region is difficult because of the AT-rich nature of the S. aureus DNA. We, therefore, constructed reporter plasmids with a nested set of truncations of the sequences spanning the cidABC promoter region (Fig. 1A) that were fused to the lacZ reporter gene and then introduced these constructs into the S. aureus wild-type (wt) strain, UAMS-1. Because the CidR-mediated induction of cidABC transcription exhibits glucose-inducible expression under conditions of acidic stress (11, 21), we grew each reporter strain in the presence or absence of 35 mM glucose and measured the β-galactosidase activity associated with the cells after 6 h of growth. Consistent with previous results (16), cidABC promoter activity was induced by the full-length construct (designated −177) after 6 h of growth in the presence of glucose (Fig. 1B) in a CidR-dependent manner, suggesting that the cis-acting element important for the CidR-mediated control was intact in this construct. As can be seen in Fig. 1B, removal of the regions 5′ to the nucleotides at positions −177, −138, and −93 upstream of the transcription start site had a minimal effect on cidABC expression, indicating the absence of cis-acting regulatory elements involved in cidABC induction upstream of the −93 position. In contrast, the −57 construct does not demonstrate induction of cidABC expression, suggesting that a required cis-acting element is present in the region between nucleotides −93 and −57. Analysis of the sequence within this region revealed the presence of a 12-bp element (TAGTAATACAAA) (Fig. 1A) that is one base pair shorter than typical LTTR binding sites. Interestingly, this sequence is located in a region that contains what was previously predicted to be the cidABC promoter (22).
FIG 1.

Identification of cis-acting elements in the cidA promoter region. (A) Nucleotide sequence of the cidA promoter. The −35 and −10 elements, Shine-Dalgarno (SD) sequence and translational start codon are underlined. The transcriptional start point is marked by an asterisk and presented in bold. The positions of truncated DNA fragments used for the deletion analysis of cidA promoter activity, the putative CidR binding site, and identified catabolite responsive elements (cre) are underlined and presented in bold. A 60-bp biotin-labeled DNA fragment used in EMSAs (IW110/113) is marked by a dotted line. (B) Deletion and mutational analysis of cidA promoter activity. Wild-type S. aureus cells (strain UAMS-1) containing lacZ reporter fusions with truncated cidA promoter fragments or the full-length cidA promoter with mutations in the putative CidR-binding site were grown under inducing conditions for 6 h and subsequently assayed for β-galactosidase activity. The length of the cidA promoter fragments used for the deletion analysis and the wild-type sequence of the CidR-binding site with generated mutations are shown in bold and underlined on the left. The results are representative of at least three independent experiments. Statistical significance in β-galactosidase activity between the wild‐type strain containing cidA promoter fragments in the presence or absence of glucose (*) was determined by Student's t test (P ≤ 0.01).
To test the importance of the identified cis-acting element for cidABC expression, we altered the nucleotide sequence of this element by introducing different mutations into the −177 PcidABC-lacZ reporter construct and then examined the impact of these mutations on induction of cidABC promoter activity by measuring β-galactosidase activity in the wild-type cells as performed above. As shown in Fig. 1B, all generated mutations in the CidR-binding site abolished expression of the reporter gene, confirming the critical role of the identified cis-acting element for the induction of cidABC expression.
CidR binds to the identified specific cis-acting element within the cidABC promoter region.
Although the above mutagenesis experiments demonstrated that the TAGTAATACAAA element is important for the induction of cidABC promoter activity, these results alone are not sufficient to conclude that this sequence represents the CidR-binding site. To address this, we performed electrophoretic mobility shift assays (EMSAs) using a C-terminal His tag-labeled CidR protein and a 60-bp biotin-labeled DNA fragment spanning the region containing the putative CidR-binding site (Fig. 1A). As shown in Fig. 2A, incubation of purified CidR protein with this DNA fragment resulted in a dose-dependent shift in the migration of the target DNA. Furthermore, addition of 200-fold excess of unlabeled specific competitor DNA effectively blocked formation of the CidR-DNA complexes, suggesting that CidR binding to the target DNA was specific (Fig. 2A). In agreement with this, CidR binding was not observed in the EMSAs with DNA fragments originating from sequences located upstream and downstream of the 60-bp target region and not containing the putative CidR-binding site (see Fig. S1 in the supplemental material). To further demonstrate the specificity of the protein-DNA interactions between CidR and its cis-acting element, we performed EMSAs with a cidABC promoter fragment containing a mutated CidR-binding site. As seen in Fig. 2B, competitor DNA in which the entire CidR-binding element in the cidABC promoter region was replaced with a random nucleotide sequence failed to block binding of CidR to the labeled target DNA. Combined, these data demonstrate that CidR directly regulates cidABC expression by specifically binding to the identified 12-bp TAGTAATACAAA nucleotide sequence.
FIG 2.

CidR binds to the 12-bp cis-acting element within the cidA promoter. (A) Electrophoretic mobility-shift assay (EMSA) was performed using increasing amounts of purified CidR protein and biotin-labeled cidA promoter DNA as a target. Lanes 1 and 10 contain a no-protein control and a 200-fold excess of unlabeled competitor DNA control, respectively. (B) EMSA performed with cidA promoter using increasing concentrations of mutated competitor DNA and a constant amount of purified CidR protein. Lane 1 contains wild-type biotin-labeled cidA promoter fragment only. Reaction mixtures were incubated for 30 min at room temperature and separated in a 6% TBE polyacrylamide gel.
CcpA is a positive regulator of cidABC transcription.
Previously, the inactivation of the ccpA gene in S. aureus was shown to cause a drastic decrease in cidABC transcript accumulation under conditions of excess glucose, suggesting the involvement of the master regulator of carbon metabolism, CcpA, in the control of cidABC expression (19). Consistent with this, the cidABC promoter region was found to contain a putative cre box upstream of the CidR-binding site (Fig. 1A), indicative of the direct positive regulation of this operon by CcpA. To demonstrate that CcpA is involved in the direct transcriptional control of cidABC expression, we performed a quantitative real‐time reverse transcriptase PCR (RT‐PCR) analysis using primers specific to the cidA and cidR genes. Using this approach, we found that the lack of CcpA during growth under conditions of acetate-mediated acidic stress in the presence of glucose (inducing conditions) abolishes cidABC expression, even though it leads to a slight increase in the cidR transcript accumulation (Fig. 3A).
FIG 3.

CcpA is required for induction of cidABC expression. (A) Relative transcript levels of the cidA and cidR genes determined by quantitative RT‐PCR after 6 h of growth. Transcript levels in the wild-type strain, cidR, and ccpA mutants grown in TSB containing 35 mM glucose (inducing conditions) are presented as a fold difference compared to those for the wild-type strain grown in TSB without glucose. Statistical significance between the wild‐type strain grown with or without glucose (*) as well as between the cidR mutant grown under inducing conditions and wild‐type strain grown in the absence of glucose (**) was determined by Student's t test (P ≤ 0.01). (B) Relative cidA transcript levels determined by quantitative RT‐PCR after 3 h of growth in TSB containing 14 mM glucose. Transcript levels in the cidR, ackA, ccpA, ackA cidR, and ackA ccpA mutants are presented as fold differences compared to those in the wild‐type strain. Statistical significance between the wild‐type strain and ackA mutant (*) and between the wild‐type strain and ccpA mutant (**) was determined by Student's t test (P ≤ 0.005). (C) Effect of alternative carbon sources on cidABC expression. Wild‐type strain cultures containing the PcidABC-lacZ reporter plasmid were grown either in plain NZY medium or NZY medium supplemented with 35 mM glucose, 42 mM fructose, or 70 mM glycerol. After 6 h of growth, samples were collected and assayed for β-galactosidase activity. Statistical significance between the wild‐type strain grown in plain NZY and NZY supplemented with glucose (*) or fructose (**) was determined by Student's t test (P ≤ 0.01).
Due to the fact that inactivation of the ccpA gene in staphylococci diminishes production of acetate by directing carbon flux into the tricarboxylic acid (TCA) cycle (23, 24), we asked whether the observed decrease in cidABC transcription in the ccpA mutant was simply a result of insufficient acidification of the medium or a direct effect of this transcription factor on the cidABC promoter. Circumstantial evidence against the former model was previously generated by demonstrating that disruption of the major acetate-generating pathway, the Pta-AckA pathway, in S. aureus causes a significant increase in cidABC transcription while simultaneously reducing the ability of the bacteria to acidify the culture medium in the presence of excess glucose (25, 26). To more directly examine the role of culture acidification in the CcpA-mediated control of cidABC expression, we combined the ackA and ccpA mutations in a single strain and performed a quantitative RT‐PCR analysis of cidABC expression during exponential growth. Of note, similar to the single mutants, the ackA ccpA double mutant exhibited a severe impairment of acetate generation, resulting in a reduced acidification of the culture medium (Fig. 4). As shown in Fig. 3B, the previously observed effect of the ackA mutation on cidABC expression was completely eliminated by the presence of the ccpA mutation, demonstrating that CcpA-mediated induction of cidABC expression does not require acidification of the culture medium. Furthermore, in agreement with the results of Northern blot analysis (19), the absence of CcpA during exponential growth significantly decreases the levels of cidABC transcription compared to those of the wild-type strain, underlying the necessity for glucose in the induction of cidABC expression (Fig. 3B).
FIG 4.

Inactivation of ccpA and Pta-AckA pathway decreases production of acetate and consequent acidification of the culture medium. (A) Acetate production rates of the UAMS-1 wild-type strain and ccpA, ackA, pta, ackA ccpA, and pta ccpA mutants determined after 6 h of growth on TSB supplemented with 35 mM glucose. (B) pH values of the culture medium of the UAMS-1 wild-type strain and ccpA, ackA, pta, ackA ccpA, and pta ccpA mutants determined after 6 h of growth on TSB supplemented with 35 mM glucose. Statistical significance between the wild‐type strain and the mutants (*) was determined by Student's t test (P ≤ 0.01).
In Gram-positive bacteria, the DNA-binding activity of CcpA is triggered or enhanced by interactions with its coregulator, histidine-containing protein (HPr), which in the presence of the glycolytic intermediate, fructose-1,6-bisphosphate, is phosphorylated on residue Ser46 by its cognate HPr kinase (27). To demonstrate the essentiality of the glycolytic flux (i.e., fructose-1,6-bisphosphate) for the CcpA-mediated regulation of cidABC expression, we compared the impact of alternative carbon sources on cidABC induction using a PcidABC-lacZ reporter plasmid. In this approach, we grew wild-type cells in medium containing either glucose (35 mM), fructose (42 mM), or glycerol (70 mM) as a carbon source and then measured β-galactosidase activity after 6 h of growth to assess the activity of the cidABC promoter. As anticipated, the addition of fructose to the culture medium induced cidABC promoter activity in a similar manner to that of glucose since catabolism of both of these sugars yields fructose-1,6-bisphosphate (Fig. 3C). In contrast, glycerol, which catabolism bypasses the formation of fructose-1,6-bisphosphate, did not induce cidABC promoter activity (Fig. 3C). Combined, the results of these experiments demonstrate that CcpA-mediated regulation of cidABC expression requires carbon flux through the glycolytic machinery and occurs independently of acidification of the culture medium during growth.
CcpA directly regulates cidABC expression by binding to the cidABC promoter.
The above experiments and the presence of a putative cre site within the cidABC promoter region strongly suggest direct positive regulation of cidABC expression by CcpA. To provide experimental evidence that CcpA directly binds within the cidABC promoter region, we performed EMSA experiments similar to those described above for CidR. In this approach, a C-terminal His tag-labeled CcpA protein was affinity purified and incubated with the same 60-bp biotin-labeled cidABC promoter fragment (Fig. 1A) that was previously used in the experiments to assess DNA binding by CidR. The results presented in Fig. 5A demonstrate that CcpA binds to the target DNA in a dose-dependent manner. Furthermore, the addition of a 200-fold excess of unlabeled specific competitor DNA effectively blocked the formation of higher-order CcpA-DNA complexes, demonstrating the specificity of the CcpA interaction with the target DNA.
FIG 5.

CcpA directly regulates cidABC expression by binding to the cre site within the cidA promoter. (A) EMSA was performed using increasing concentrations of purified CcpA protein (100 to 7,000 nM) and biotin-labeled wild-type cidA promoter DNA as a target. Reaction mixtures were incubated for 30 min at room temperature and separated on a 6% TBE polyacrylamide gel. Lanes 1 and 9 contain a no-protein control and a 200-fold excess of unlabeled competitor DNA control, respectively. (B) Effect of cre site presence on cidABC expression. S. aureus cultures with lacZ reporter plasmids containing cidA promoter DNA fragment with native or mutated (cre*) cre site were grown in plain TSB or TSB medium supplemented with 35 mM glucose. After 6 h of growth, samples were collected and assayed for β-galactosidase activity. Statistical significance between the wild‐type strain grown in plain TSB and wild‐type strain (*) or pta mutant (**) grown under inducing conditions was determined by Student's t test (P ≤ 0.001).
Analysis of the nucleotide sequence spanning the cidABC promoter region revealed the presence of a potential cre box sequence, TAGAGAGCGTTTCCA, upstream of the CidR-binding site (Fig. 1A), which has only one mismatched nucleotide (underlined) (see Fig. S2 in the supplemental material) from the cre site consensus in Bacillus subtilis, WTGNAANCGNWNNCW (28). To demonstrate the ability of CcpA to recognize and bind this putative cre site, we constructed a PcidABC-lacZ transcriptional reporter fusion in which this element was substituted with random sequence GCAGTGTATCCAGGC and then analyzed its ability to generate β-galactosidase activity in S. aureus. As shown in Fig. 5B, the elimination of the putative cre site from the cidABC promoter region drastically decreased β-galactosidase production in wild-type cells under inducing conditions. Furthermore, the absence of this site eliminates the positive impact of the pta mutation on cidABC promoter activity similarly to the lack of CcpA (Fig. 5B). Combined, the results of these experiments demonstrate that the CcpA-mediated control of cidABC expression occurs through the direct binding of CcpA to the cidABC promoter region, which requires the presence of a functional cre site.
Identification of potential effectors of cidABC expression.
The results of RT-PCR analysis (Fig. 3B) and β-galactosidase assays (Fig. 5B) revealed that induction of cidABC promoter activity by CcpA and CidR requires both acidification of the culture medium and the presence of glucose, suggesting that expression of the cidABC operon under conditions of carbon overflow is modulated by the metabolic status of the cells. Therefore, to demonstrate the direct linkage of cidABC expression to carbohydrate metabolism and to identify metabolic genes that affect expression of this operon, we used an unbiased transposon mutagenesis approach. For this purpose, we generated a reporter strain, a derivative of S. aureus JE2 (29), that contains a PcidABC-lacZ fusion construct inserted into the geh gene within the S. aureus chromosome (strain KB6001). To confirm the suitability of this reporter strain for screening, we measured β-galactosidase activity after 6 h of growth in the presence of glucose (inducing conditions) and demonstrated that the reporter strain reflects the anticipated cidR-dependent control of the cidABC promoter activity (Fig. 6A). Next, we performed transposon mutagenesis of the KB6001 strain as previously described (29) and screened mutants on X-Gal (5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside) indicator plates for the colonies that displayed a decrease in the β-galactosidase activity. To verify screening results, we measured β-galactosidase activity in mutant cultures grown for 6 h in tryptic soy broth (TSB) supplemented with 35 mM glucose as described above. Using this approach, we identified 23 genes where transposon insertions led to decreased cidABC promoter activity (see Table S3 in the supplemental material). Of particular interest, the results of this screen together with subsequent β-galactosidase activity assay (Fig. 6A) revealed three genes involved in carbohydrate metabolism that affect cidABC promoter activity. Two of these genes, ptsI and ptsH, encode the components of a phosphoenolpyruvate-dependent sugar phosphotransferase system (PTS) (i.e., phosphoenolpyruvate protein phosphotransferase and phosphocarrier protein HPr [30]), where seryl-phosphorylated HPr under conditions of excess glucose also acts as a corepressor in CcpA-mediated carbon catabolite repression (CCR) (31). The third gene, glkA, encodes glucokinase, which together with glucose permease (encoded by glkU) comprises a PTS-independent system that transports glucose and catalyzes the formation of the very first glycolytic intermediate, glucose-6-phosphate (32). Interestingly, the β-galactosidase activity assays and RT-PCR analysis of two different S. aureus strains revealed a range of effects of these genes on cidABC expression. As can be seen in Fig. 6A and B, inactivation of either ptsI or ptsH genes completely abolished induction of cidABC promoter activity, while the lack of glucokinase during growth decreases its activity approximately 2.5 fold, suggestive of the different contributions of products of these genes to glucose transport and subsequent carbon flux through the glycolytic machinery (33). Together, the results of these experiments reveal a strong correlation of cidABC expression with glucose uptake and glycolytic activity, which are required for the induction of cidABC expression by CcpA, demonstrating the critical role of carbohydrate metabolism in the regulation of this operon.
FIG 6.

Relative induction of cidABC expression in isolated transposon mutants. (A) Mutants from the transposon mutagenesis screen were tested for cidABC expression under inducing conditions (NZY medium supplemented with 35 mM glucose). After 6 h of growth, samples were collected and assayed for β-galactosidase activity. Statistical significance between the wild‐type strain grown under inducing conditions and wild-type strain grown in plain NZY (*), as well as between wild‐type strain and ptsI, ptsH, and glkA mutants grown under inducing conditions (**), was determined by Student's t test (P ≤ 0.01). (B) Relative cidA transcript levels determined by quantitative RT‐PCR after 6 h of growth in TSB containing 35 mM glucose. Statistical significance between the UAMS-1 wild‐type strain grown with or without glucose (*), as well as between the UAMS-1 wild‐type strain and ptsI, ptsH, and glkA mutants grown under inducing conditions (**), was determined by Student's t test (P ≤ 0.01).
DISCUSSION
In S. aureus, cell death and lysis during biofilm development are known to be affected by the gene products of the cidABC operon (5, 22). Based on growth conditions, this operon can be expressed in a form of two overlapping transcripts designated cidABC and cidBC (34). It has been shown that expression of the smaller cidBC transcript depends on sigma factor B (21), while induction of the full-length cidABC transcript is controlled by the LTTR family transcriptional regulator, CidR, and requires both acidification of the culture medium and excess glucose (16, 34). Microarray analysis revealed two operons, alsSD and cidABC, that are directly controlled by the transcriptional regulator, CidR (11), and whose gene products are involved in catabolism of pyruvate formed under excess glucose conditions. The products of the alsSD operon are acetolactate synthase and acetolactate decarboxylase of the 2,3-butanediol pathway, converting pyruvate to the neutral by-product, acetoin, whereas the cidC-encoded pyruvate:menaquinone oxidoreductase decarboxylates pyruvate to acetate, leading to acidification of the culture medium that causes acidic stress and subsequent cell death (11, 12, 14). It has been hypothesized that an intricate balance in the metabolic activities of the alsSD and cidC gene products, directing carbon flux at the pyruvate node, determines cell fate during aerobic growth with an excess of glucose (8). In support of this, the results of our recent study provided a new mechanistic insight into the role of the CidR regulon during overflow metabolism and have extended the understanding of the contribution of individual Cid proteins to carbon flux at the pyruvate node (15). Specifically, we have shown that the cidB gene product enhances the generation of acetate by CidC, while, in contrast, CidR-mediated induction of the cidA and alsSD expression has a negative effect on CidC activity and directs carbon away from acetate and toward the production of acetoin (15). Therefore, by revealing opposing functions of the CidA and CidB proteins in modulating CidC activity, we established that the major function of the CidR regulon is to limit acetate-mediated cell death in the bacterial population during growth under conditions of carbon overflow (15).
In the present study, we further characterized the regulatory network controlling cidABC expression in S. aureus and clarified the metabolic requirements essential for the induction of this operon. Using computational analyses, mutagenesis approaches, and subsequent β-galactosidase activity assays, we identified cis-acting elements within the cidABC promoter region (Fig. 1A) and then confirmed their necessity for the binding of the transcriptional regulators, CidR and CcpA (Fig. 2A and Fig. 5A). Importantly, β-galactosidase activity assays and RT-PCR analyses revealed that CcpA and CidR are involved in the direct positive control of cidABC expression (Fig. 3A and B), where the complete induction of the cidABC promoter depends on acidification of the culture medium in the presence of glucose and requires the synergistic contribution of both transcriptional regulators (Fig. 3A and Fig. 5B). Interestingly, in contrast to the CidR-mediated induction of cidABC expression during acidic stress, the positive regulation of the cidABC promoter by CcpA is independent of acidification of the culture medium (Fig. 3A and B; Fig. 5B) but requires the presence of glycolytic intermediate fructose-1,6-bisphosphate (Fig. 3C). In agreement with this, the transposon mutagenesis approach revealed genes involved in glucose transport and CCR, the inactivation of which has a negative impact on cidABC expression as it affects glycolytic flux and the regulatory function of CcpA (Fig. 6).
As indicated above, the CidR protein belongs to the LTTR family of transcriptional regulators. The canonical LTTR binding region contains two distinct binding sites separated by a few nucleotides, termed the recognition binding site (RBS) and the activation binding site (ABS) (17). It has been suggested that the LTTR initially binds each site as a dimer, then dimers interact to form a tetramer, and finally the tetramer acts to bend the DNA, where the high angle of the bent DNA impedes the formation of the transcription bubble (17). In the presence of a specific ligand, the LTTR undergoes a conformational change, causing the tetramer to shift on the DNA and relax the angle. The new angle then allows for the formation of the transcription bubble, thus facilitating the initiation of transcription. This model of LTTR regulation has been termed the “sliding dimer” model (35).
Although the proposed LTTR-mediated regulation of gene expression requires two separate binding sites, to our surprise, the results of the current work revealed the presence of only one cis-acting element within the cidABC promoter region that is critical for the binding of CidR, suggesting that regulation of cidABC expression by this LTTR might vary from the classical model. Interestingly, in a close proximity (7 bp upstream) to the cidR binding site, we identified another cis-acting element critical for the induction of the cidABC promoter (Fig. 1A and Fig. 5B). This element has only a single mismatch from the canonical cre box sequence (28), and it is required for the binding of the master regulator of carbon metabolism, CcpA (27). Therefore, considering the mechanisms of LTTR-mediated regulation of gene expression (17) and based upon the results of current work, we propose a model that predicts the induction of cidABC expression through a synergistic interplay between CidR and CcpA and links its expression to the metabolic status during glucose catabolism (Fig. 7). In this model, we speculate that the complete induction of cidABC expression will occur in two steps. First, during growth in medium with excess glucose, carbon flux through the glycolytic machinery, yielding fructose-1,6-bisphosphate, would lead to the formation of the CcpA-HPr (Ser-P) complex. Then, the CcpA-HPr (Ser-P) dimer will bind to the cre site within the cidABC promoter region, which will act as a functional analog of RBS (Fig. 7A). Simultaneously, the CidR dimer will bind to the ABS sequence. Subsequent protein-protein interactions between CidR and CcpA-HPr (Ser-P) dimers will bend the DNA leading to a basal level of expression of the cidABC operon (Fig. 7A). At the second stage, over time, the excretion of acetate into the culture medium during glucose catabolism will decrease external pH values close to the pKa of acetate (4.74), leading to formation of acetic acid (Fig. 7B). The generated weak acid will translocate across the membrane and dissociate within the bacterial cytoplasm resulting in its protonation. Then, the decrease in intracellular pH (pHi) will stimulate binding of a yet to be identified inducer molecule (acetate or pyruvate) to the conserved ligand-binding domains within the CidR dimer. The conformational changes caused by interaction of CidR with its inducer will affect protein-protein interactions with the CcpA-HPr (Ser-P) dimer, leading to relaxation of the DNA-bending angle and facilitating transcription (Fig. 7B).
FIG 7.
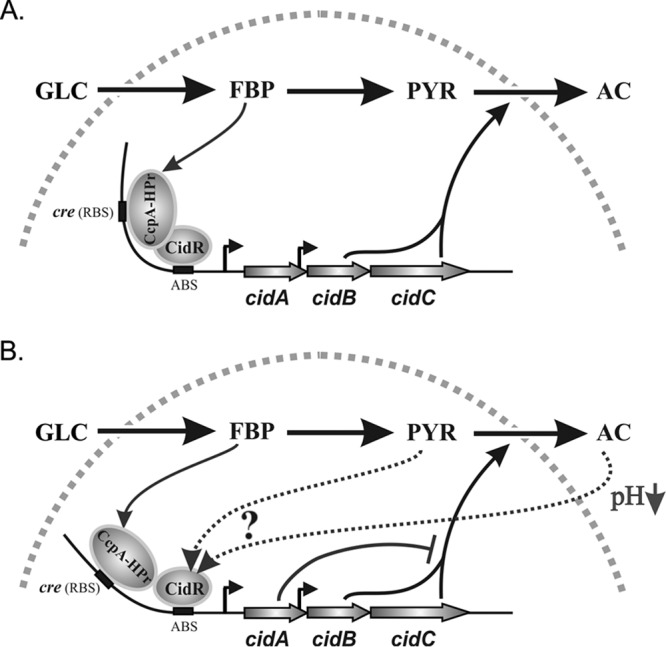
A conceptual model demonstrating synergistic regulation of cidABC expression by CidR and CcpA-HPr (Ser-P) complex under conditions of carbon overflow without (A) and with (B) acidic stress. Metabolites presented in the scheme include the following: GLC, glucose; FBP, fructose 1,6-bisphosphate; PYR, pyruvate; AC, acetate.
Overall, the results of this study provide important insight into the signals required for the induction of cidABC expression during carbohydrate metabolism. Previously, we have considered the presence of glucose during growth mainly as a prerequisite for the acetate-mediated acidification of the culture medium that causes induction of the cidABC promoter activity by CidR (16, 34). The results of this work, however, reveal that glucose, per se, is an important component of this regulation, as its catabolism through the glycolytic machinery leads to the accumulation of fructose-1,6-bisphosphate that is required for CcpA-mediated control of cidABC expression. Furthermore, we established that the full induction of cidABC expression requires the synergistic contribution of CidR and CcpA, and proposed a conceptually novel model where the interaction of a LTTR with CcpA-HPr (Ser-P) complex under different metabolic conditions modulates the activity of the cidABC promoter. To support this model, we are currently planning in vitro experiments that will reveal protein-protein interactions between the CcpA-HPr (Ser-P) complex and CidR within the cidABC promoter region under conditions mimicking those observed in the current study.
MATERIALS AND METHODS
Bacterial strains, plasmids, and growth conditions.
Bacterial strains and plasmids used in this study are listed in Table S1 in the supplemental material. The UAMS-1-ccpA mutant as well as the UAMS-1-pta ccpA and UAMS-1-ackA ccpA double mutants were generated by bacteriophage Φ11-mediated transduction (36) of the ccpA::tetL allele from the JE2 ccpA::tetL strain (37) into UAMS-1, UAMS-1-pta, and UAMS-1-ackA, correspondingly. The replacement of the ccpA gene by the ccpA::tetL allele was verified by PCR using primers SAV1737-f and acuC-f. The UAMS-1-pta cidR and UAMS-1-ackA cidR double mutants were generated by bacteriophage Φ11-mediated transduction (36) of the cidR::tet allele from the KB1090 strain into UAMS-1-pta and UAMS-1-ackA, correspondingly. The UAMS-1 background in all mutants was confirmed by PCR using primers cna-f and cna-r (25).
S. aureus strains were grown in tryptic soy broth (TSB) medium (BD Biosciences) at 37°C with aeration at 250 rpm and a flask-to-medium ratio of 10:1. Escherichia coli DH5α was grown in Luria-Bertani medium (Fisher Scientific). Antibiotics were purchased from Sigma-Aldrich and Fisher Scientific and were used at the following concentrations: kanamycin, 50 μg/ml; chloramphenicol, 10 μg/ml; ampicillin, 100 μg/ml; and erythromycin, 5 μg/ml.
DNA manipulations.
Oligonucleotides used in this study are listed in Table S2 in the supplemental material. Genomic DNA was isolated from S. aureus using Wizard Plus kits from Promega, Inc. Restriction endonucleases, DNA polymerases, and T4 DNA ligase used in this study were purchased from New England Biolabs and Invitrogen Life Technologies. Recombinant plasmids were isolated using the Wizard Plus SV Minipreps DNA purification system (Promega, Inc.). PCR was performed using the KOD polymerase kit (Novagen) in the Applied Biosystems GeneAmp PCR system 9700 (Life Technologies Corporation) with primers purchased from Integrated DNA Technologies and Eurofins Operon. DNA fragments were purified using the DNA Clean and Concentrator-5 kit (Zymo Research), and recombinant DNA plasmid products were sequenced at the University of Nebraska Medical Center DNA Sequencing Core facility and analyzed using Vector NTI (Invitrogen).
The cidABC promoter was amplified using the primers IW11 and IW15 and UAMS-1 genomic DNA as a template following digestion by BamHI and NheI and ligation into pIHW7 (18) to yield the cidABC reporter plasmid, pIHW10-lac.
A deletion analysis was performed by creating nested sets of DNA fragments spanning the cidABC promoter region, fusing them to the lacZ reporter gene, and then measuring β-galactosidase activity (see below) under inducing and noninducing conditions. To create 5′ end shortened DNA fragments of the cidABC promoter region, the reverse primer IW11 was paired with the forward primers, IW10, IW16, IW31, and IW22. Each generated PCR promoter fragment was digested with BamHI and NheI and then ligated into pIHW7 (18), yielding the plasmids, pIHW9, pIHW11-lac, pIHW22-lac, and pIHW17-lac. Specific point mutations in the cidABC promoter were made as previously described (38) using pIHW25 plasmid as a template.
For the substitution of the cre site in the pIHW10-lac plasmid with the sequence GCAGTGTATCCAGGC, we used outward primers cre*-cidABC-f and cre*-cidABC-r followed by self-ligation and transformation of the PCR fragment into E. coli DH5α to generate the plasmid pSW1. The elimination of the cre site in the plasmid pSW1 was confirmed by DNA sequencing using primers pCN51-S2-f and B-gal-S-r.
Construction of the lacZ reporter strain for transposon mutagenesis.
For screening and identification of transposon mutants with altered cidABC expression, we created S. aureus strain KB6001 containing the PcidABC-lacZ reporter fusion integrated into the geh gene within the bacterial chromosome. The 5′ region upstream of geh from LAC-13c was amplified (using the primers IW27 and IW28) and ligated into the EcoRI and SacI sites of pCL52.2 (39) to generate the plasmid, pIHW20. The 3′ region downstream of geh was amplified using the primers IW29 and IW30 and ligated into the PstI and NheI sites of pIHW20 to generate the plasmid pIHW21. The PcidABC-lacZ reporter fusion construct was generated by PCR using the primers IW10 and IW44 and the pIHW9 plasmid as a template. Then, the PCR product was ligated into the PstI and BamHI sites of pIHW21 to produce the temperature-sensitive integration plasmid pIHW25. The JE2 cidABC reporter strain with the pIHW25 plasmid integrated into the chromosome was generated using allelic replacement methodology as described previously (40), selecting for blue colonies in the presence of 35 mM glucose and 50 μg/ml X-Gal. The reporter strain was confirmed by PCR and used as a host for further transposon mutagenesis as described previously (29).
β-Galactosidase assays.
β-Galactosidase assays were performed as previously described (41). Briefly, 1.0 ml of cell culture collected by centrifugation was suspended in 1 ml of Z-buffer (60 mM Na2HPO4, 40 mM NaH2PO4, 10 mM KCl, 1 mM MgSO4, 50 mM β-mercaptoethanol, pH 7.0) and lysed using a FastPrep FP120 instrument (MP Biomedicals). The lysates were then centrifuged, and 700 μl of the supernatants was transferred to a 1.5-ml microcentrifuge tube. After the addition of 140 μl of ONPG (o-nitrophenyl-β-d-galactopyranoside; 4 mg ml−1), the samples were incubated at 37°C until they turned slightly yellow (under an optical density at 420 nm [OD420] of 1.0). To stop the reactions, 500 μl of 1 M sodium carbonate was added and the OD420 was measured. Protein concentrations were determined by performing Bradford assays using the protein assay dye solution from Bio-Rad. Miller units were calculated using protein concentration instead of OD600 (42).
Expression and purification of the CidR and CcpA proteins.
To generate a plasmid for expression and purification of the CidR protein, the S. aureus cidR gene was amplified using the primers IW128cc and IW129cc and UAMS-1 genomic DNA as a template. Then, the PCR product was cloned into the NheI and XhoI sites of pET24b, and its integrity was confirmed by DNA sequencing. The resulting plasmid (pET24b-cidR) was transformed into the rare-codon enhanced E. coli strain BL21 derivative Rosetta 2 (Invitrogen) for protein expression. Similarly, to construct a plasmid for expression and purification of the CcpA protein, the ccpA gene was amplified from the UAMS-1 genomic DNA via PCR using the primers IW116 and IW117 and cloned into pET24b as described above. The resulting plasmid (pET24b-ccpA) was transformed into the E. coli strain BL21 (Invitrogen) for protein expression.
To produce large quantities of the CidR protein, the Rosetta 2 (pET24b-cidR) strain was grown in 1.0 liter of LB medium containing kanamycin and chloramphenicol at 37°C until the culture reached mid-exponential phase. IPTG (isopropyl-β-d-thiogalactopyranoside) was added to a final concentration of 0.4 mM to induce expression of CidR. The culture was transferred to a 20°C incubator and expressed overnight. To produce the CcpA protein, a similar approach was utilized, with the exception that the overnight cultures were grown at 30°C. Purification of the CidR and CcpA proteins was achieved by collecting the expressed cells by centrifugation and resuspending them in 50 ml of lysis buffer (100 mM phosphate and 300 mM NaCl). Phenylmethylsulfonyl fluoride (PMSF) was added to a final concentration of 1.0 mM to inhibit protease activity. The cells were lysed by serial passage through an EmulsiFlex C3 system. The released recombinant proteins were purified using a HisPur Cobalt purification kit (Pierce Biotechnology) and desalted by ultrafiltration using an exchange buffer (100 mM Tris-HCl [pH 8.0], 150 mM KCl, 1 mM EDTA, and 0.1 mM dithiothreitol [DTT]). The purified proteins were stored at –20°C in an exchange buffer containing 40% glycerol.
Protein-DNA interactions.
The binding of purified proteins to target DNA was demonstrated by electrophoretic mobility shift assays (EMSAs) using a LightShift chemiluminescent kit (Pierce Biotechnology) according to the manufacturer’s instructions. The target DNA used for the CidR and CcpA EMSAs was made by annealing biotin-labeled 60-bp primers IW110 and IW113 (spanning the CidR and CcpA binding sites in the cidABC promoter region). Specific competitor DNA (annealed oligonucleotides IW110 and IW113) was added in 200-fold excess as per the manufacturer’s instructions. Mutant-specific competitor DNA was made by annealing the primers IW176 and IW177 for the complete substitution of the CidR-binding site. The binding reaction mixture for each sample contained 10 mM Tris (pH 7.5), 50 mM KCl, 1 mM DTT, 2.5% glycerol, 1 μg of salmon sperm DNA, 5 mM MgCl2, 0.05% NP-40, 10 mM EDTA, and 5.0 fmol of labeled DNA in a total volume of 20 μl. After 30 min of incubation at room temperature, the samples were separated in a 6% polyacrylamide gel in 0.5× Tris-borate-EDTA (TBE) buffer at 85 V. The DNA was then transferred and cross-linked to the nylon membrane using a UV Stratalinker 1800 (Stratagene) cross-linker instrument. The labeled DNA fragments were visualized using an SRX-101a Imager (Konica Minolta).
Quantification of the mRNA transcripts.
RNA isolation was carried out as described previously (43). Quantitative real-time PCR was performed using the cidA-, cidR-, and sigA-specific primers listed in Table S2. Total RNA (500 ng) was converted to cDNA using the QuantiTect reverse transcription kit (Qiagen). The samples were then diluted 1:50, and the cDNA products were amplified using the LightCycler DNA master SYBR green I kit (Roche Applied Science) following the manufacturer’s instructions. The relative transcript levels were calculated using the comparative threshold cycle (CT) method (44) with normalization to the amount of sigA transcripts present in the RNA samples. The results were recorded in duplicate and are representative of three independent experiments.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by research grants from the National Institute of Allergy and Infectious Diseases P01-AI83211 and R01-AI125589 (to K.W.B.).
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/JB.00371-19.
REFERENCES
- 1.Davey ME, O'toole GA. 2000. Microbial biofilms: from ecology to molecular genetics. Microbiol Mol Biol Rev 64:847–867. doi: 10.1128/mmbr.64.4.847-867.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Costerton JW, Lewandowski Z, Caldwell DE, Korber DR, Lappin-Scott HM. 1995. Microbial biofilms. Annu Rev Microbiol 49:711–745. doi: 10.1146/annurev.mi.49.100195.003431. [DOI] [PubMed] [Google Scholar]
- 3.Whitchurch CB, Tolker-Nielsen T, Ragas PC, Mattick JS. 2002. Extracellular DNA required for bacterial biofilm formation. Science 295:1487. doi: 10.1126/science.295.5559.1487. [DOI] [PubMed] [Google Scholar]
- 4.Flemming HC, Wingender J. 2010. The biofilm matrix. Nat Rev Microbiol 8:623–633. doi: 10.1038/nrmicro2415. [DOI] [PubMed] [Google Scholar]
- 5.Bayles KW. 2007. The biological role of death and lysis in biofilm development. Nat Rev Microbiol 5:721–726. doi: 10.1038/nrmicro1743. [DOI] [PubMed] [Google Scholar]
- 6.Otto M. 2013. Staphylococcal infections: mechanisms of biofilm maturation and detachment as critical determinants of pathogenicity. Annu Rev Med 64:175–188. doi: 10.1146/annurev-med-042711-140023. [DOI] [PubMed] [Google Scholar]
- 7.Moormeier DE, Bayles KW. 2017. Staphylococcus aureus biofilm: a complex developmental organism. Mol Microbiol 104:365–376. doi: 10.1111/mmi.13634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rice KC, Bayles KW. 2003. Death’s toolbox: examining the molecular components of bacterial programmed cell death. Mol Microbiol 50:729–738. doi: 10.1046/j.1365-2958.2003.t01-1-03720.x. [DOI] [PubMed] [Google Scholar]
- 9.Rice KC, Bayles KW. 2008. Molecular control of bacterial death and lysis. Microbiol Mol Biol Rev 72:85–109. doi: 10.1128/MMBR.00030-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sadykov MR, Bayles KW. 2012. The control of death and lysis in staphylococcal biofilms: a coordination of physiological signals. Curr Opin Microbiol 15:211–215. doi: 10.1016/j.mib.2011.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yang SJ, Dunman PM, Projan SJ, Bayles KW. 2006. Characterization of the Staphylococcus aureus CidR regulon: elucidation of a novel role for acetoin metabolism in cell death and lysis. Mol Microbiol 60:458–468. doi: 10.1111/j.1365-2958.2006.05105.x. [DOI] [PubMed] [Google Scholar]
- 12.Thomas VC, Sadykov MR, Chaudhari SS, Jones J, Endres JL, Widhelm TJ, Ahn JS, Jawa RS, Zimmerman MC, Bayles KW. 2014. A central role for carbon-overflow pathways in the modulation of bacterial cell death. PLoS Pathog 10:e1004205. doi: 10.1371/journal.ppat.1004205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Patton TG, Rice KC, Foster MK, Bayles KW. 2005. The Staphylococcus aureus cidC gene encodes a pyruvate oxidase that affects acetate metabolism and cell death in stationary phase. Mol Microbiol 56:1664–1674. doi: 10.1111/j.1365-2958.2005.04653.x. [DOI] [PubMed] [Google Scholar]
- 14.Zhang X, Bayles KW, Luca S. 2017. Staphylococcus aureus CidC is a pyruvate:menaquinone oxidoreductase. Biochemistry 56:4819–4829. doi: 10.1021/acs.biochem.7b00570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chaudhari SS, Thomas VC, Sadykov MR, Bose JL, Ahn DJ, Zimmerman MC, Bayles KW. 2016. The LysR-type transcriptional regulator, CidR, regulates stationary phase cell death in Staphylococcus aureus. Mol Microbiol 101:942–953. doi: 10.1111/mmi.13433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yang SJ, Rice KC, Brown RJ, Patton TG, Liou LE, Park YH, Bayles KW. 2005. A LysR-type regulator, CidR, is required for induction of the Staphylococcus aureus cidABC operon. J Bacteriol 187:5893–5900. doi: 10.1128/JB.187.17.5893-5900.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Maddocks SE, Oyston PC. 2008. Structure and function of the LysR-type transcriptional regulator (LTTR) family proteins. Microbiology 154:3609–3623. doi: 10.1099/mic.0.2008/022772-0. [DOI] [PubMed] [Google Scholar]
- 18.Windham IH, Chaudhari SS, Bose JL, Thomas VC, Bayles KW. 2016. SrrAB modulates Staphylococcus aureus cell death through regulation of cidABC transcription. J Bacteriol 198:1114–1122. doi: 10.1128/JB.00954-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Seidl K, Goerke C, Wolz C, Mack D, Berger-Bachi B, Bischoff M. 2008. Staphylococcus aureus CcpA affects biofilm formation. Infect Immun 76:2044–2050. doi: 10.1128/IAI.00035-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hussein H, Fris ME, Salem AH, Wiemels RE, Bastock RA, Righetti F, Burke CA, Narberhaus F, Carroll RK, Hassan NS, Mohamed SA, Fahmy AS, Murphy ER. 2019. An unconventional RNA-based thermosensor within the 5′ UTR of Staphylococcus aureus cidA. PLoS One 14:e0214521. doi: 10.1371/journal.pone.0214521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rice KC, Patton T, Yang SJ, Dumoulin A, Bischoff M, Bayles KW. 2004. Transcription of the Staphylococcus aureus cid and lrg murein hydrolase regulators is affected by sigma factor B. J Bacteriol 186:3029–3037. doi: 10.1128/JB.186.10.3029-3037.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rice KC, Firek BA, Nelson JB, Yang SJ, Patton TG, Bayles KW. 2003. The Staphylococcus aureus cidAB operon: evaluation of its role in regulation of murein hydrolase activity and penicillin tolerance. J Bacteriol 185:2635–2643. doi: 10.1128/JB.185.8.2635-2643.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Seidl K, Stucki M, Ruegg M, Goerke C, Wolz C, Harris L, Berger-Bachi B, Bischoff M. 2006. Staphylococcus aureus CcpA affects virulence determinant production and antibiotic resistance. Antimicrob Agents Chemother 50:1183–1194. doi: 10.1128/AAC.50.4.1183-1194.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sadykov MR, Hartmann T, Mattes TA, Hiatt M, Jann NJ, Zhu Y, Ledala N, Landmann R, Herrmann M, Rohde H, Bischoff M, Somerville GA. 2011. CcpA coordinates central metabolism and biofilm formation in Staphylococcus epidermidis. Microbiology 157:3458–3468. doi: 10.1099/mic.0.051243-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sadykov MR, Thomas VC, Marshall DD, Wenstrom CJ, Moormeier DE, Widhelm TJ, Nuxoll AS, Powers R, Bayles KW. 2013. Inactivation of the Pta-AckA pathway causes cell death in Staphylococcus aureus. J Bacteriol 195:3035–3044. doi: 10.1128/JB.00042-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Marshall DD, Sadykov MR, Thomas VC, Bayles KW, Powers R. 2016. Redox imbalance underlies the fitness defect associated with inactivation of the Pta-AckA pathway in Staphylococcus aureus. J Proteome Res 15:1205–1212. doi: 10.1021/acs.jproteome.5b01089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Warner JB, Lolkema JS. 2003. CcpA-dependent carbon catabolite repression in bacteria. Microbiol Mol Biol Rev 67:475–490. doi: 10.1128/MMBR.67.4.475-490.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Miwa Y, Nakata A, Ogiwara A, Yamamoto M, Fujita Y. 2000. Evaluation and characterization of catabolite-responsive elements (cre) of Bacillus subtilis. Nucleic Acids Res 28:1206–1210. doi: 10.1093/nar/28.5.1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fey PD, Endres JL, Yajjala VK, Widhelm TJ, Boissy RJ, Bose JL, Bayles KW. 2013. A genetic resource for rapid and comprehensive phenotype screening of nonessential Staphylococcus aureus genes. mBio 4:e00537-12. doi: 10.1128/mBio.00537-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Deutscher J, Francke C, Postma PW. 2006. How phosphotransferase system-related protein phosphorylation regulates carbohydrate metabolism in bacteria. Microbiol Mol Biol Rev 70:939–1031. doi: 10.1128/MMBR.00024-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gorke B, Stulke J. 2008. Carbon catabolite repression in bacteria: many ways to make the most out of nutrients. Nat Rev Microbiol 6:613–624. doi: 10.1038/nrmicro1932. [DOI] [PubMed] [Google Scholar]
- 32.Lakshmi HP, Yeswanth S, Prasad UV, Vasu D, Swarupa V, Kumar PS, Narasu ML, Sarma P. 2013. Cloning, expression and characterization of glucokinase gene involved in the glucose-6- phosphate formation in Staphylococcus aureus. Bioinformation 9:169–173. doi: 10.6026/97320630009169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jahreis K, Pimentel-Schmitt EF, Brückner R, Titgemeyer F. 2008. Ins and outs of glucose transport systems in eubacteria. FEMS Microbiol Rev 32:891–907. doi: 10.1111/j.1574-6976.2008.00125.x. [DOI] [PubMed] [Google Scholar]
- 34.Rice KC, Nelson JB, Patton TG, Yang SJ, Bayles KW. 2005. Acetic acid induces expression of the Staphylococcus aureus cidABC and lrgAB murein hydrolase regulator operons. J Bacteriol 187:813–821. doi: 10.1128/JB.187.3.813-821.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Porrua O, Garcia-Jaramillo M, Santero E, Govantes F. 2007. The LysR-type regulator AtzR binding site: DNA sequences involved in activation, repression and cyanuric acid-dependent repositioning. Mol Microbiol 66:410–427. doi: 10.1111/j.1365-2958.2007.05927.x. [DOI] [PubMed] [Google Scholar]
- 36.Novick RP. 1991. Genetic systems in staphylococci. Methods Enzymol 204:587–636. doi: 10.1016/0076-6879(91)04029-n. [DOI] [PubMed] [Google Scholar]
- 37.Halsey CR, Lei S, Wax JK, Lehman MK, Nuxoll AS, Steinke L, Sadykov M, Powers R, Fey PD. 2017. Amino acid catabolism in Staphylococcus aureus and the function of carbon catabolite repression. mBio 8:e01434-16. doi: 10.1128/mBio.01434-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bose JL, Lehman MK, Fey PD, Bayles KW. 2012. Contribution of the Staphylococcus aureus Atl AM and GL murein hydrolase activities in cell division, autolysis, and biofilm formation. PLoS One 7:e42244. doi: 10.1371/journal.pone.0042244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sau S, Sun J, Lee CY. 1997. Molecular characterization and transcriptional analysis of type 8 capsule genes in Staphylococcus aureus. J Bacteriol 179:1614–1621. doi: 10.1128/jb.179.5.1614-1621.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lehman MK, Bose JL, Bayles KW. 2016. Allelic exchange. Methods Mol Biol 1373:89–96. doi: 10.1007/7651_2014_187. [DOI] [PubMed] [Google Scholar]
- 41.Lehman MK, Bose JL, Sharma-Kuinkel BK, Moormeier DE, Endres JL, Sadykov MR, Biswas I, Bayles KW. 2015. Identification of the amino acids essential for LytSR-mediated signal transduction in Staphylococcus aureus and their roles in biofilm-specific gene expression. Mol Microbiol 95:723–737. doi: 10.1111/mmi.12902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Haskell RE, Hughes SM, Chiorini JA, Alisky JM, Davidson BL. 2003. Viral-mediated delivery of the late-infantile neuronal ceroid lipofuscinosis gene, TPP-I to the mouse central nervous system. Gene Ther 10:34–42. doi: 10.1038/sj.gt.3301843. [DOI] [PubMed] [Google Scholar]
- 43.Sadykov MR, Olson ME, Halouska S, Zhu Y, Fey PD, Powers R, Somerville GA. 2008. Tricarboxylic acid cycle-dependent regulation of Staphylococcus epidermidis polysaccharide intercellular adhesin synthesis. J Bacteriol 190:7621–7632. doi: 10.1128/JB.00806-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schmittgen TD, Livak KJ. 2008. Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc 3:1101–1108. doi: 10.1038/nprot.2008.73. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.