

Abstract
Small-molecule cationic amphiphiles (CAms) were designed to combat the rapid rise in drug-resistant bacteria. CAms were designed to target and compromise the structural integrity of bacteria membranes, leading to cell rupture and death. Discrete structural features of CAms were varied, and structure-activity relationship studies were performed to guide the rational design of potent antimicrobials with desirable selectivity and cytocompatibility profiles. In particular, the effects of cationic conformational flexibility, hydrophobic domain flexibility, and hydrophobic domain architecture were evaluated. Their influence on antimicrobial efficacy in Gram-positive and Gram-negative bacteria was determined, and their safety profiles were established by assessing their impact on mammalian cells. All CAms have a potent activity against bacteria, and hydrophobic domain rigidity and branched architecture contribute to specificity. The insights gained from this project will aid in the optimization of CAm structures.
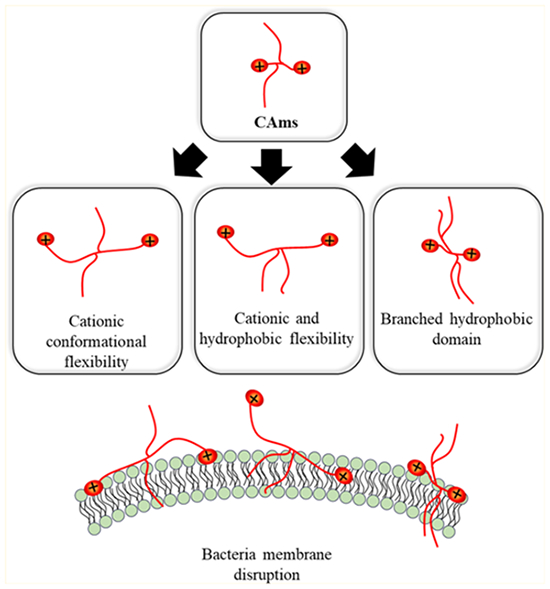
Graphical Abstract
1. INTRODUCTION
The overuse of antibiotics has resulted in a rapid rise in antibiotic resistance and emergence of multidrug-resistant bacteria, sparking significant global health concerns.1,2 While ongoing efforts are being taken to promote antimicrobial stewardship and slow the progression of resistant bacterial strains, the scientific community has also initiated a movement to understand innate immune responses to bacterial infections and to develop alternatives to traditional antibiotics.1–4
Antimicrobial peptides (AMPs) are naturally existing membrane-active molecules that have gained significant attention as alternatives to traditional antibiotics.4,5 While the primary sequences and lengths of AMPs vary greatly, the vast majority contains a net cationic charge and adopts amphipathic secondary structures segregating hydrophobic and charged residues in the presence of lipid bilayers.5,6 Although additional molecular targets exist, most AMPs initially interact with the negatively charged components of bacterial cell membranes (e.g., phosphatidylglycerol, lipopolysaccharides, and teichoic acids) and hydrophobic residue insertion leads to membrane integrity disruption and cell death.5,7–9 Their unique membrane-targeting mechanism results in lower resistance rates as compared to traditional antibiotics that target specific biological pathways (e.g., DNA or cell wall synthesis) or inhibit enzymatic or metabolic activities.1,9–12 However, several shortcomings limit the applicability of AMPs as viable antimicrobial therapeutics. AMPs are associated with high production costs because of multiple low-yielding peptide coupling reactions or expression systems; the resulting peptide bonds are susceptible to native proteases, and they commonly elicit undesirable toxicity to mammalian cells.11,13,14 To address these limitations, several groups have evaluated the antibacterial potential of nonpeptide-derived AMP mimics. For example, peptidomimics developed with nonnatural amino acid structures aid in protease susceptibility; arylamide oligomers designed de novo have been synthesized from inexpensive monomers; Jennings et al. designed simple quaternary ammonium amphiphiles via cost-effective syntheses that additionally overcame obstacles associated with polymer and oligomer folding.6,15–18
This work builds upon these efforts by designing small-molecule cationic amphiphiles (CAms) with high potency against Gram-positive and Gram-negative bacteria.19,20 CAms were designed from technologically cost-effective starting materials and possess physicochemical features similar to AMPs, namely, cationic charge and amphiphilicity, while additionally being resistant to protease activity. CAm structures are based on a linear sugar backbone that serves as both a spacer to two cationic moieties as well as the branch point for two hydrophobic domains derived from fatty acids or analogous aliphatic alcohols (Figure 1). Comparable to the key features that impart AMPs with antimicrobial efficacy, CAms have net cationic charges and structural flexibility required to fold into facially amphipathic structures that may lend to selective activity against bacterial membranes.
Figure 1.

Chemical structures of three CAm series indicating nomenclature (bold underlined text) and structural variations. All CAm structures are based on parent CAm (top) with strategic chemical changes. Charge flexibility is varied by increasing the linker length between cationic charges and the sugar backbone (left). Charge and hydrophobic arm flexibility are altered though increasing charged end-group linker lengths in addition to ether-linkages for hydrophobic arms (center). The branched hydrophobic architecture was generated by using dendritic branch points for hydrophobic domains.
Herein, the design, synthesis, and subsequent antimicrobial characterization of CAms with strategic structural variations (Figure 1) is reported to better understand the chemical features that contribute to antimicrobial efficacy and specificity. Molecular flexibility has been shown to strongly influence the antimicrobial activity and global amphiphilicity in a variety of molecular architectures, including AMPs, small-molecule amphiphiles, and polymers.21–24 In particular, Palermo et al. designed methacrylate copolymers with varying cationic linker lengths to the polymer backbone. In doing so, they found that shorter linker lengths had desirable selectivity profiles for bacterial cells over mammalian cells, whereas longer cationic linker lengths resulted in polymers with nonselective membrane activity.21 As such, two series of CAms were designed to evaluate the influence of flexibility in our molecular platform. The conformational flexibility of cationic moieties, namely, charge flexibility, was introduced in CAm structures through step-wise increases in the length of cationic linkers (i.e., two, four, or six methylene units) extending from the sugar backbone. Hydrophobic domains are incorporated onto the backbone as side chains. Also, to explore the possible variations of chemical structures with bio activities, one of the two series of CAms with charge flexibility was designed to have ester-linked hydrophobic domains, while additional chain flexibility was introduced into the other series by replacing ester with ether linkages.
A third series of CAms was designed to probe the effect of hydrophobic domain architecture on antimicrobial efficacy and selectivity. Several studies, including those performed in the Uhrich group, have identified hydrocarbon chains that elicit desirable antimicrobial selectivity profiles, frequently to be of intermediate lengths.19,20,25,26 Lind et al. demonstrated that increased branching density and charge flexibility resulted in improved bacterial potency and specificity in antimicrobial dendrimers.27 Similarly, Chen et al. found that higher generation dendrimers exhibited more potent antibacterial efficacy, suggesting that a branched architecture may influence the membrane activity.26 As such, additional Cams were designed with branched hydrophobic domain architectures and varying hydrophobic chain lengths to assess the relative contributions of hydrophobic domain branching, hydrophilic-lipophilic balance (HLB), and alkyl chain length.
Upon successful synthesis, all compounds were screened for antimicrobial efficacy in representative Gram-positive and Gram-negative bacteria, as well as for hemolytic activity in human-derived red blood cells. In vitro membrane specificity experiments were then carried out for selected compounds, demonstrating enhanced activity against bacterial membrane mimics compared to mammalian cell membrane mimics. Together, these experiments identify several key features of CAms that contribute to antimicrobial potency and specificity.
2. MATERIAL AND METHODS
2.1. Materials.
All reagents were purchased from Sigma-Aldrich (St. Louis, MO) and used as received unless stated otherwise. Di-tert-butyl tartrate, N-Boc-ethylenediamine, N-Boc-l,4-diaminobutane, and N-Boc-1,6-diaminohexane were purchased from TCI (Portland, OR). Whole blood was purchased from New Jersey Blood Center (East Orange, Nj). Lipids and polycarbonate membranes were purchased from Avanti Polar Lipids (Birmingham, AL).
2.2. Chemical Characterization.
Proton (1H) and carbon (13C) nuclear magnetic resonance (NMR) spectra were obtained using a Varian 500 MHz spectrometer. Samples were dissolved in deuterated chloroform (CDCl3) with trimethylsilane as an internal reference or deuterated methanol (CD3OD). Fourier transform infrared spectra were obtained by solvent-casting small molecules onto sodium chloride (NaCl) plates from dichloromethane (DCM) solutions, and 32 scans were averaged and processed using OMNIC software on a Thermo Scientific Nicolet iS10 spectrophotometer. Molecular weights of CAms and intermediates were determined using a ThermoQuest Finnigan (LCQ-DUO system equipped with a syringe pump, optional divert/inject valve, atmospheric pressure ionization source, and mass spectrometer (MS) detector). Samples were dissolved in methanol (10 μg/mL) and spectra were processed using Xcalibur.
2.3. Synthesis.
2.3.1. CAm-Ethers with Extended Cationic Linkers (3).
Di-Boc-amino ether tartramide (2)—T10 ether (1) was prepared as previously described.28 Mono-Boc-protected diamines with various linker lengths (i.e., N-Boc-ethylenediamine, N-Boc-1,4-diaminobutane, N-Boc-1,6-diaminohexane) were conjugated to 1 via carbodiimide coupling following modified published procedures.19,20 Synthesis of 2a is previously reported.20 Synthesis of 2c is presented as an example. T10 ether (0.27 mmol, 0.12 g) and 4-(dimethylamino)pyridine (DMAP, 0.70 mmol, 0.09 g) were dissolved in anhydrous DCM (5 mL) under nitrogen and stirred for 10 min. N-Boc-1,6-diaminohexane (0.70 mmol, 0.15 mL) and N-(3-dimethyla-minopropyl)-N′-ethylcarbodiimide hydrochloride (EDCI, 0.70 mmol, 0.13 g) were sequentially added to the reaction flask and stirred overnight at room temperature. The reaction mixture was washed with 10% potassium bisulfate (KHSO4, 2×, 10 mL) and brine (1×, 10 mL). The organic layer was dried over magnesium sulfate (MgSO4), filtered, and concentrated in vacuo to yield pure 2c.
2b: Yield: 93% (white solid) 1H NMR (500 MHz, CDCl3): δ 0.87 (t, 6H), 1.24 (m, 28H), 1.42 (s, 18H), 1.52 (m, 8H), 1.67 (m, 4H), 3.12 (m, 4H), 3.31 (m, 4H), 3.47 (m, 4H), 4.24 (s, 2H), 4.55 (br, 2H), 6.69 (br, 2H). 13C NMR (500 MHz, CDCl3): δ 14.09, 22.65, 26.01, 26.39, 26.57, 28.40, 29.29, 29.37, 29.54, 29.59, 29.69, 31.86, 39.03, 73.37, 81.08, 165.96, 169.85. ESI-MS m/z: [M + 23]+.
2c: Yield: 86% (white solid) 1H NMR (500 MHz, CDCl3): δ 0.87 (t, 6H), 1.24 (m, 28H), 1.33 (m, 4H), 1.43 (s, 18H), 1.52 (m, 8H), 1.71 (m, 4H), 3.09 (m, 4H), 3.27 (m, 4H), 3.47 (m, 4H), 4.25 (s, 2H), 4.50 (br, 2H), 6.71 (br, 2H). 13C NMR (500 MHz, CDCl3): δ 14.06, 23.12, 25.98, 26.13, 26.32, 26.53, 27.99, 29.22, 29.41, 29.51, 29.54, 29.63, 32.00, 39.03, 73.33, 81.09, 166.87, 169.91. ESI-MS m/z: 793.5 [M + 23]+.
CAm ether (3)—Boc protecting groups were removed under acidic conditions following literature precedence.19,29 Briefly, 2 was dissolved in hydrochloric acid in dioxane (4 M HCl, 10 mL) and cooled to 0 °C on ice. The reaction was stirred for 1 h on ice and then allowed to warm to room temperature and proceed overnight. Volatiles were removed in vacuo, and 3 was reconstituted in a minimum amount of methanol (<4 mL), precipitated in hexanes (15 mL), and isolated by centrifugation (3500 rpm, 5 min) using a Hettich EBA 12 Centrifuge (Beverly, MA).
3a: Yield: quantitative (tan waxy solid) 1H NMR (500 MHz, CD3OD): δ 0.89 (t, 6H), 1.29 (m, 28H), 1.56 (m, 4H), 1.63 (m, 4H), 1.69 (m, 4H), 2.96 (m, 4H), 3.19 (m, 4H), 3.30 (m, 8H), 3.59 (m, 2H), 4.08 (s, 2H). 13C NMR (500 MHz, CD3OD): δ 13.03, 22.31, 24.41, 25.71, 26.09, 29.38, 29.34, 29.25, 29.20, 29.06, 31.65, 37.97, 38.89, 72.41, 81.23, 171.18. ESI-MS m/z: 286.3 [(M + 2)/2]+.
3b: Yield: quantitative (off-white waxy solid) 1H NMR (500 MHz, CD3OD): δ 0.89 (t, 6H), 1.29 (m, 28H), 1.41 (m, 8H), 1.55 (m, 8H), 1.65 (m, 4H), 2.91 (m, 4H), 3.17 (m, 4H), 3.30 (m, 8H), 3.65 (m, 2H), 4.08 (s, 2H). 13C NMR (500 MHz, CD3OD): δ 13.03, 22.32, 25.98, 25.74, 25.60, 27.08, 29.37, 29.32, 29.24, 29.18, 29.07, 28.96, 31.64, 38.60, 39.23, 72.45, 81.05, 170.97. ESI-MS m/z: 314.4 [(M + 2)/2]+.
2.3.2. CAm Esters with Extended Cationic Linkers (6).
Di-Boc-amino tartramide (4)—Tartaric acid was subjected to an aminolysis reaction with mono-Boc-protected diamines with various linker lengths (i.e., N-Boc-ethylenediamine, N-Boc-1,4-diaminobutane, and N-Boc-1,6-diaminohexane) following methods described in previously reported procedures.19,20 Synthesis of 4a–6a has previously been reported.20 Reactions will be described using N-Boc-1,6-diaminohexane as an example. Briefly, dimethyl tartrate (0.56 mmol, 0.1 g) was dissolved in 3.0 mL of tetrahydrofuran and heated to 40 °C. N-Boc-1,6-diaminohexane (1.57 mmol, 0.35 mL) was added, and the reaction stirred overnight at 40 °C. Volatiles were removed in vacuo, and 4c was precipitated in diethyl ether (15 mL) and isolated via filtration.
4b: Yield: 91% (white solid) 1H NMR (500 MHz, DMSO): δ 1.41 (s, 18), 1.51 (m, 8H), 3.10 (m, 4H), 3.28 (m, 4H), 4.31 (s, 2H), 4.75 (br, 2H), 7.15 (br, 2H). 13C NMR (500 MHz, DMSO): δ 28.93, 39.17, 39.20, 40.03, 73.71, 79.14, 156.12, 172.21. ESI-MS m/z: 513.4 [M + 23]+.
4c: Yield: 79% (white solid) 1H NMR (500 MHz, DMSO): δ 1.32 (s, 8H), 1.44 (m, 30H), 1.61 (m, 4H), 3.09 (m, 4H), 3.25 (m, 4H), 4.26 (s, 2H), 4.60 (br, 2H), 5.37 (br, 2H), 7.10 (br, 2H). 13C NMR (500 MHz, DMSO): δ 26.42, 26.54, 28.93, 39.15, 39.24, 40.00, 73.72, 79.14, 156.07, 172.28. ESI-MS m/z: 569.4 [M + 23]+.
Di-Boc-amino ester tartramide (5)—4c was acylated by dissolving decanoic acid (0.46 mmol, 0.08 g) and DMAP (0.46 mmol, 0.06 g) in anhydrous DCM, followed by addition of the 4c (0.12 mmol, 0.08 g) and EDC (0.46 mmol, 0.09 g). The reaction was stirred overnight under nitrogen and washed as described earlier for synthesis of 2.
5b: (white solid) 1H NMR (500 MHz, CDCl3):δ 0.86 (t, 6H), 1.25 (m, 24H), 1.42 (s, 18H), 1.49 (m, 4H), 1.61 (m, 4H), 2.38 (t, 4H), 3.10 (m, 4H), 3.25 (m, 4H), 4.68 (br, 2H), 5.56 (s, 2H), 6.40 (br, 2H) 13C NMR (500 MHz, CDCl3): δ 14.03, 22.63, 24.73, 25.79, 26.44, 27.33, 28.36, 29.01, 29.24, 29.41, 31.80, 33.87, 36.85, 39.14, 40.01, 72.12, 79.14, 156.06, 166.31, 172.27. ESI-MS m/z: 822.2 [M + 23]+.
5c: (off-white solid) 1H NMR (500 MHz, CDCl3): δ 0.86 (t, 6H), 1.30 (m, 24H), 1.25 (m, 8H), 1.43 (s, 18H), 1.62 (m, 8H), 2.38 (t, 4H), 3.09 (m, 4H), 3.22 (m, 4H), 4.55 (br, 2H), 5.57 (s, 2H), 6.22 (br, 2H). 13C NMR (500 MHz, CDCl3): δ 14.07, 22.63, 24.78, 26.04, 26.11, 28.43, 29.06, 29.19, 29.26, 29.40, 29.93, 31.80, 33.89, 39.19, 40.23, 72.13, 79.81, 157.21, 166.24, 172.30. ESI-MS m/z: 855.1 [M + 1]+.
CAm ester (6)—Boc groups were removed as previously described using 4 M HCl in dioxane. Products were isolated via precipitation in cold diethyl ether (15 mL) followed by centrifugation as previously described to isolate the products.
6b: Yield: quantitative (off-white waxy solid) 1H NMR (500 MHz, CD3OD): δ 0.89 (t, 6H), 1.29 (m, 24H), 1.59 (m, 12H), 2.46 (4H), 2.94 (t, 4H), 3.23 (m, 4H), 5.54 (s, 2H). 13C NMR (500 MHz, CD3OD): δ 13.03, 22.31, 24.33, 24.40, 25.82, 28.75, 29.01, 29.05, 29.18, 31.63, 33.20, 38.17, 38.89, 72.28, 167.50, 172.63. ESI-MS m/z: 300.2 [(M + 2)/2]+.
6c: Yield: quantitative (yellowish waxy solid) 1H NMR (500 MHz, CD3OD): δ 0.89 (t, 6H), 1.30 (m, 32H), 1.51 (t, 4H), 1.65 (m, 8H), 2.46 (m, 4H), 2.91 (t, 4H), 3.19 (m, 4H), 5.54 (s, 2H). 13C NMR (500 MHz, CD3OD): δ 13.03, 22.31, 24.41, 25.50, 25.74, 27.01, 28.66, 28.76, 29.01, 29.05, 29.18, 31.62, 33.19, 38.75, 39.20, 72.27, 167.35, 172.51. ESI-MS m/z: 328.4 [(M + 2)/2]+.
2.3.3. Branched CAms (11).
Branched arm (7)—Branched arms were synthesized by acylating 2,2-bis(hydroxymethyl)propionic acid (bis-MPA) following modified literature procedures.30 Synthesis of 11b will be provided as an example. Bis-MPA (7.4 mmol, 1.0 g) was dissolved in a solution of dimethylformamide (2.0 mL) and pyridine (0.2 mL) under nitrogen. Valeroyl chloride (18.6 mmol, 2.24 mL) was added, and the reaction was stirred overnight at room temperature. Chloroform (5.0 mL) and HCl (1 N, 5.0 mL) were added to the resulting slurry and stirred for 10 min. The aqueous layer was extracted with chloroform (3×, 10 mL), and the combined organic layers were washed with a 1:1 mixture of saturated sodium bicarbonate (NaHCO3) and brine (1×, 10 mL). The organic layer was dried over MgSO4, filtered, and concentrated in vacuo. The crude product was recrystallized from hexanes to purify 7b.
7a: Yield: 82% (clear oil) 1H NMR (500 MHz, CDCl3): δ 0.87 (t, 6H), 1.24 (s, 3H), 1.29 (m, 4H), 1.55 (q, 4H), 2.28 (t, 4H), 4.23 (d, 4H). 13C NMR (500 MHz, CDCl3): δ 13.62, 17.70, 22.14, 26.82, 33.78, 46.14, 64.99, 173.32, 178.85. ESI-MS m/z: 301.0 [M - 1]−.
7b: Yield: 71% (white solid) 1H NMR (500 MHz, CDCl3): δ 0.88 (t, 6H), 1.26 (m, 24H), 1.30 (s, 3H), 1.59 (qn, 4H), 2.31 (t, 4H), 4.23 (s, 4H). 13C NMR (500 MHz, CDCl3): δ 14.05, 17.23, 22.65, 24.79, 29.10, 29.23, 29.25, 29.40, 31.84, 33.97, 47.87, 64.54, 167.50, 173.05. ESI-MS m/z: 441.2 [M - 1]−.
Di-branched dibenzyl tartrate (DBT) (8)—7 was conjugated to DBT via carbodiimide coupling. 7b (0.66 mmol, 0.19 g) was dissolved in DCM (5.0 mL) followed by addition of DMAP (0.66 mmol, 0.08 g) and allowed to stir for 10 min under nitrogen. DBT (0.3 mmol, 0.10 g) was added to the reaction followed by EDC (0.66 mmol, 0.13 g) and stirred overnight. The resulting solution was washed with 10% KHSO4 (2×, 10 mL) and a 1:1 mixture of saturated NaHCO3 and brine (1×, 10 mL). The organic layer was dried over MgSO4, filtered, and concentrated in vacuo. 8a required no further purification. 8b was subjected to flash column chromatography using 12% ethyl acetate in hexanes as the mobile phase to yield pure product.
8a: Yield: 75% (white solid) 1H NMR (500 MHz, CDCl3): δ 0.88 (t, 12H), 1.15 (s, 6H), 1.30 (m, 8H), 1.55 (m, 8H), 2.26 (m, 8H), 4.19 (m, 8H), 5.13 (s, 4H), 5.73 (s, 2H), 7.31 (m, 10H). 13C NMR (500 MHz, CDCL3): δ 13.68, 17.45, 22.20, 26.82, 33.69, 46.33, 64.63, 68.00, 70.84, 128.32, 128.67, 128.75, 134.43, 164.93, 171.40, 173.15. ESI-MS m/z: 921.4 [M + 23]+.
8b: Yield: 65% (white solid) 1H NMR (500 MHz, CDCl3): δ 0.86 (t, 12H), 1.16 (s, 6H), 1.25 (m, H), 1.57 (m, 8H), 2.24 (m, 8H), 4.17 (m, 8H), 5.13 (s, 4H), 5.73 (s, 2H), 7.31 (m, 10H). 13C NMR (500 MHz, CDCl3): δ 14.08, 17.46, 22.65, 24.78, 29.12, 29.26, 29.43, 31.84, 33.95, 33.97, 46.34, 64.60, 67.97, 70.86, 128.34, 128.70, 128.74, 134.44, 164.90, 171.40, 173.13. ESI-MS m/z: 1201.9 [M + 23]−.
Di-branched tartaric acid (9): Benzyl groups were removed via hydrogenolysis with palladium on carbon (Pd/C, 10% w/w) as the catalyst for 24 h in DCM (10 mL). The reaction mixture was then filtered through celite to remove Pd/C and 9 concentrated in vacuo.
9a: Yield: quantitative (white solid) 1H NMR (500 MHz, CDCl3): δ 0.87 (t, 12H), 1.31 (m, 14H), 1.57 (m, 8H), 2.32 (m, 8H), 4.20 (m, 8H), 5.62 (s, 2H). 13C NMR (500 MHz, CDCl3): δ 13.64, 17.36, 22.16, 26.78, 26.82, 71.04, 167.30, 171.11, 173.77, 174.00. ESI-MS m/z: 717.1 [M - 1]−.
9b: Yield: quantitative (white solid) 1H NMR (500 MHz, CDCl3): δ 0.87 (t, 12H), 1.26 (m, H), 1.32 (s, 6H), 1.58 (m, 8H), 2.31 (m, 8H), 4.20 (m, 8H), 5.63 (s, 2H). 13C NMR (500 MHz, CDCl3): δ 14.08, 17.37, 22.69, 24.75, 24.80, 29.11, 29.26, 29.43, 31.85, 34.07, 46.43, 64.91, 65.18, 71.01, 166.73, 171.05, 173.73, 174.00. ESI-MS m/z: 998.2 [M - 1]−.
Di-branched, di-Boc-amino tartramide (10)—Mono-Boc-protected diamines were conjugated following a modified literature procedure.31 9b (0.34 mmol, 0.34 g) was dissolved in DCM (5.0 mL) and cooled to 0 °C on ice. HOBt (0.68 mmol, 0.09 g) and N-Boc-ethylenediamine were added sequentially. DCC (1 M, 0.68 mmol, 0.68 mL) was then slowly added, and the reaction was stirred for 15 min on ice. The resulting mixture was allowed to warm to room temperature and stirred overnight. The reaction was cooled to −20 °C to facilitate the urea byproduct precipitation, which was removed via filtration. The filtrate was concentrated in vacuo and subjected to flash column chromatography with 5% methanol in DCM as the eluent. If further purification was required, the crude 10b was recrystallized in cold hexanes.
10a: Yield: 76% (white solid) 1H NMR (500 MHz, CDCl3): δ 0.90 (t, 12H), 1.30 (m, H), 1.42 (m, 18H), 1.58 (m, 8H), 1.66 (s), 2.32 (m, 8H), 3.26–3.40 (m, 8H), 4.13–4.43 (m, 8H), 5.27 (s, 2H), 5.56 (s, 2H), 6.96 (s, 2H). 13C NMR (500 MHz, CDCl3): δ 13.72, 17.69, 22.19, 28.41, 26.83, 33.77, 40.49, 39.82, 46.67, 64.92, 64.80, 72.83, 79.45, 165.88, 171.23, 173.52. ESI-MS m/z: 1025.6 [M + 23]+.
10b: Yield: 66% (white solid) 1H NMR (500 MHz, CDCl3): δ 0.86 (t, 12H), 1.25 (m, H), 1.28 (s, 6H), 1.59 (m, 18H), 1.64 (m, 8H), 2.29 (8H), 3.25–3.38 (m, 8H), 4.11–4.41 (m, 8H), 5.25 (s, 2H), 5.55 (s, 2H), 6.93 (s, 2H). 13C NMR (500 MHz, CDCl3): 14.08, 17.65, 22.64, 24.80, 28.38, 29.09, 29.24, 29.41, 31.85, 34.05, 46.68, 64.79, 64.92, 72.85, 79.44, 165.92, 171.27, 173.47, 173.59. ESI-MS m/z: 1305.6 [M + 23]+.
Branched CAm (11)—Boc groups were removed to generate the final products using 4 M HCl in dioxane following the aforementioned procedure. Products were isolated in vacuo.
11a: Yield: quantitative (off-white paste) 1H NMR (500 MHz, CDCl3): δ 0.91 (t, 12H), 1.33 (m, 14H), 1.57 (m, 8H), 2.35 (m, 8H), 3.10 (m, 4H), 3.43–3.59 (d of m, 4H), 4.18–4.43 (m, 8H), 5.63 (s, 2H). 13C NMR (500 MHz, CDCl3): 13.18, 16.39, 22.01, 24.85, 33.70, 39.69, 40.82, 45.15, 65.17, 75.73, 167.30, 171.73, 173.95, 174.01. ESI-MS m/z: 402.4 [(M + 2)/2]+.
11b: Yield: quantitative (off-white sticky solid) 1H NMR (500 MHz, CDCl3): δ 0.89 (t, 12H), 1.33 (m, 48 H), 1.29 (s, 6H), 1.58 (m, 8H), 2.34 (m, 8H), 3.10 (m, 4H), 3.38–3.73 (d of m, 4H), 4.12–4.45 (m, 8H), 5.57 (s, 2H). 13C NMR (500 MHz, CDCl3):δ13.09, 16.65, 22.32, 24.59, 28.84, 29.06, 29.08, 29.22, 31.70, 33.50, 36.82, 39.07, 46.43, 64.56, 72.52, 167.31, 171.72, 173.43, 173.51. ESI-MS m/z: 542.8 [(M + 2)/2]+.
2.3.4. Cytotoxicity.
In vitro cytotoxicity was evaluated in 3T3 fibroblasts. Cells were cultured in the Dulbecco’s modified Eagle medium supplemented with 10% fetal bovine serum (complete media) at 37 °C, 95% humidity, and 5% CO2. Samples were solubilized in sterile 10 mM HEPES buffer, pH 7.4 and diluted in complete media to the desired concentrations. Cells were seeded onto a 96-well tissue culture plate at 5000 cells/well in 100 μL of complete media and allowed to attach for 24 h. Media was aspirated and replaced with CAm solutions and controls (HEPES buffer diluted with complete media) and cells were incubated under standard conditions for additional 24 h. Cell viability was then determined using a CellTiter 96 Aqueous One Solution Proliferation Assay following the manufacturer’s suggested protocol and absorbance was recorded using an Infinite M200 PRO plate reader (Tecan Group Ltd., Männedorf, Switzerland).
2.3.5. Hemolysis.
Hemolytic activity of CAms was determined in human red blood cells. Red blood cells were isolated from whole blood by centrifugation at 400×g min (Allegra 21 centrifuge, Beckman Coulter, Brea, CA) for 15 min. The supernatant was carefully removed via pipette aspiration, and the red blood cells were washed with isotonic HEPES buffer (5×, 10 mM, 0.8% NaCl, pH 7.4). The red blood cells were resuspended in isotonic HEPES buffer to 5% hematocrit. CAm stock solutions were prepared in isotonic HEPES buffer and serial diluted to predetermined concentrations for the hemolysis assays. CAm solutions (400 μL) and controls (ddH2O and isotonic HEPES buffer) were mixed with red blood cell suspensions (100 μL) and incubated for 1 h at 37 °C. Red blood cells were pelleted via centrifugation (Labenet Spectrafuge 16 M microcentrifuge, Labnet International, Inc., Edison, NJ, 400×g, 10 min), and the supernatant absorbance was read at 410 nm using an Infinite M200 PRO plate reader (Tecan Group Ltd., Männedorf, Switzerland). Percent hemolysis was calculated with the following equation
The concentration of CAms was plotted against the calculated percent hemolysis, and the point on the curve corresponding to the concentration required for 50% hemolysis was taken as the HC50.
2.4. Bacteria Cell Culture Preparation.
Bacteria were inoculated onto brain-heart infusion (BHI) agar (Becton Dickinson, Franklin Lakes, NJ) and propagated under aerobic conditions at 37 °C for 24 h. A single colony of each bacterial strain was transferred to BHI broth (Becton Dickinson, Franklin Lakes, NJ) and incubated under aerobic conditions at 37 °C for 18–24 h. Bacterial growth suspensions were diluted in a fresh BHI medium to a concentration of 106 CFU/mL for microbroth dilution assays.
2.5. Minimum Inhibitory Concentration Determination.
Minimum inhibitory concentrations (MICs) were determined using a broth microdilution assay according to CLSI standards32 and following a procedure adopted in our previous study.33 The microbial strains used in this study were representative of Gram-positive (Listeria monocytogenes Scott A) and Gram-negative (Escherichia coli O157:H7) human pathogens, both from our laboratory culture collection. CAm stock solutions were prepared in ddH2O and sterilized under UV light for 25 min. Stock solutions were serial diluted into a 96-well microplate in triplicate (Becton Dickinson, Franklin Lakes, NJ) with BHI broth to 100 μL. Aliquots (100 μL) of bacterial suspensions were added to wells, and incubated at 37 °C for 24 h under aerobic conditions. To prevent evaporation of the culture medium, 75 μL of mineral oil (Sigma-Aldrich, St. Louis, MO) was added to each well before incubation. The OD595 for each well was tracked using a microplate reader (model 550, Bio-Rad Laboratories, Hercules, CA). The MIC was determined as the lowest CAm concentration that produced no visible growth after 24 h incubation.
2.6. Hydrophilic Dye Release from Large Unilamellar Vesicles.
CAm’s ability to target and lyse membranes was evaluated in large unilamellar vesicles (LUVs) mimicking mammalian and bacterial cell membranes following modified literature procedures.34 LUVs were prepared via the thin film hydration and extrusion method. 1,2-Dioleoyl-sn-glycero-3-phosphocholine (DOPC, 25 m/mL) or a 1:1 mixture of DOPC/1,2-dioleoyl-sn-glycero-3-phospho-(1′-rac-glycerol) (DOPG) was mixed in 20 mL scintillation vials, concentrated in vacuo, and further dried overnight in a vacuum desiccator. Lipid films were hydrated with 70 mM calcein in HEPES release buffer (10 mM, 150 mM NaCl, 1 mM ethylenediaminetetraacetic acid, pH 7.4) for 1 h at 40 °C with gentle agitation. Hydrated films were subjected to five freeze/thaw cycles by heating vesicles at 40 °C in a water bath and freezing in dry ice at −78 °C for 15 min each. Multilamellar vesicles were extruded 11 times through a 100 nm polycarbonate membrane at 40 °C with an Avanti Mini-Extruder. Unencapsulated calcein was removed via size exclusion chromatography using Sephadex G50 Fine resin as the stationary phase and HEPES release buffer as the eluent. Phospholipid concentration was determined via a Phospholipid Assay Kit (Sigma-Aldrich) as per the manufacturer’s suggested protocol. LUVs were diluted to 10 μM in HEPES release buffer for experiments.
CAm solutions were prepared in HEPES release buffer. Background fluorescence of LUV suspensions was read on a RF-5301PC spectrofluorimeter (Shimadzu Scientific Instruments, Columbia, MD) at an excitation wavelength of 495 nm and an emission wavelength of 515 nm. CAm solutions were added to 1 mL of LUV suspensions, and fluorescence was immediately read and monitored for 2 min. At end of experiments, 20 μL of 5% Triton X was added to samples to fully release dye from the LUV suspensions. Percent lysis was calculated using the following equation
where Fsample represents the stabilized fluorescence reading after CAm addition, F0 represents the background fluorescence, and Ftriton represents the fluorescence recorded after full LUV lysis with Triton X.
3. RESULTS
3.1. Synthesis.
CAm ethers (3) with charged end-group flexibility were synthesized in a total of four reactions (Figure 2) from readily available and inexpensive starting materials.20 Ether linkages were generated by alkylation of di-terf-butyl tartrate and quantitative deprotection of t-butyl groups to yield T10 ether in high yield as previously described.28 End group conformational flexibility was achieved via carbodiimide coupling reactions of mono-Boc-protected diamines with T10 ether to synthesize 2. The linker between amines and the tartramide backbone was either two, four, or six methylene units in length, as correlations with antimicrobial efficacy and specificity have been previously identified in this range.21,23 Boc groups were then removed with HCl in dioxane to generate the final products (3) as chloride salts in quantitative yields.
Figure 2.

Synthetic approach used to generate CAm-ethers in two reaction steps from T10-ether.
CAm esters (6) with charged end-group flexibility were synthesized via an analogous approach (Figure 3).19 Di-Boc-amino tartramides (4) were first synthesized by conjugating mono-Boc-protected diamines with varying linker lengths to dimethyl tartrate via an aminolysis reaction. The intermediates were then acylated via carbodiimide coupling to generate 5, and the final products (6) were generated via deprotection under acidic conditions as described earlier. This synthetic approach generates the final products in a three-step reaction sequence, each step generating high yields of products with facile isolation procedures.
Figure 3.

Synthetic approach used to generate CAm-esters in three reaction steps from readily available starting materials.
Branched CAms resulted from a novel synthetic approach (Figure 4). To generate these amphiphiles, bis-MPA was utilized as a dendritic branch point for hydrophobic domains to generate 7. Alkyl chlorides with 5 or 10 total carbons in length (5C or 10C, respectively) were used to maintain analogous HLB with CAm esters or equivalent lengths of hydrophobic chains, respectively. Branched arm 7 was conjugated to DBT via carbodiimide coupling, and the benzyl groups were removed via hydrogenolysis in quantitative yields. N-Boc-1,2-ethylenediamine was successfully conjugated to the backbone following modified literature precedence to yield 10 in high yield using HOBt as the catalyst and DCC as the coupling reagent.30 Last, the deprotection of Boc groups was performed by using HCl solution (4.0 M in dioxane) to generate the branched CAms (11) in quantitative yields.
Figure 4.

Synthetic approach used to generate branched CAms.
3.2. Antimicrobial Activity.
CAms were evaluated for antimicrobial efficacy against representative Gram-positive (i.e., L. monocytogenes Scott A) and Gram-negative (i.e., E. coli O157:H7) bacteria. Assays were carried out using the microbroth dilution method, and the lowest concentration of a given CAm that resulted in no bacterial growth after 24 h was taken as the MIC. The vast majority of tested CAms had MICs in relevant therapeutic ranges (<50 μg/mL) against the tested Gram-positive pathogen. In the tested Gram-negative bacteria, MICs were generally higher, but still less than 50 μg/mL with the exceptions of 2C ester and 4C ether (Table 1). As Gram-negative bacteria have an additional outer membrane structure (Figure 5), they are more difficult to permeabilize and eradicate.35 This trend is reflected in our experiment results, as MIC values are lower against L. monocytogenes compared to E. coli for both linear CAms. Most CAms have MIC values against Gram-negative bacteria between 15 and 50 μg/mL, which is comparable to or better than what is commonly reported in the literature.21,25,27,36,37 These values are generally below the typical critical micelle concentrations of CAms with similar chemical structures, suggesting that the bioactivity comes from mainly monomeric amphiphile molecules.20 One the other hand, the branched CAms exhibited opposite behaviors. The 5C branched CAm (11a) exhibited a 2× lower MIC against E. coli (15.6 μg/mL) as compared to L. monocytogenes (31.3 μg/mL) and was the same as the lowest MIC value against E. coli of all CAms that were evaluated (6C ether and ester). The corresponding 10C branched CAm (11b) exhibited no effect on either species at the highest concentrations tested (250 μg/mL).
Table 1.
Selectivity Properties of CAms for Gram-Positive (G+) and Gram-Negative (G−) Bacteria as Compared to Hemolytic Potential in Human Red Blood Cellsa
| MIC (μg/mL) | |||||
|---|---|---|---|---|---|
| CAm | L. monocytogenes—(G+) | E. coli—(G−) | HC50 (μg/mL) | SI (G+) (μg/mL) | SI (G−) (μg/mL) |
| 2C-ether (3a) | 4.7 ± 0 | 50 ± 3.4 | 18 | 3.8 | 0.36 |
| 4C-ether (3b) | 3.1 ± 0 | 28.1 ± 4.1 | 23 | 7.4 | 0.81 |
| 6C-ether (3c) | 4.2 ± 1.8 | 15.6 ± 0 | 13 | 3.1 | 0.83 |
| 2C-ester (6a) | 2.6 ± 0.9 | 43.8 ± 3.4 | 50 | 19.2 | 1.14 |
| 4C-ester (6b) | 2.6 ± 0.9 | 56.3 ± 2.8 | 53 | 20.3 | 0.94 |
| 6C-ester (6c) | 3.1 ± 0 | 15.6 ± 0 | 28 | 9.0 | 1.8 |
| 5C-branched (11a) | 31.3 ± 0 | 15.6 ± 0 | 98 | 3.1 | 6.2 |
| 10C-branched (11b) | >250 | >250 | 56 | <0.2 | <0.2 |
SIs represent the ratio of the concentration required to lyse 50% of red blood cells (HC50) to the MIC of bacteria.
Figure 5.

Cartoon depicting the basic membrane structure of Gram-negative and Gram-positive bacteria. Gram-negative bacteria have a double membrane structure and additional lipopolysaccharide layer compared with Gram-positive bacteria. This figure was adapted from a previous Rutgers thesis.45
3.3. Hemolytic Activity in Human Red Blood Cells.
The hemolytic activity of the CAms was evaluated in human red blood cells at different concentrations, and the results are shown in Figure 6. For all three series of CAms, the hemolytic activity increased with the concentration. Ether 3c showed highest hemolytic activity, with lowest HC50 value. More than 90% of the hemolytic activity was observed for all esters and ethers with linear side chains (compound 3 and 6) at the concentration of 75 μg/mL. Overall, a global trend was evident demonstrating comparably lower hemolysis for CAm esters compared with CAm ethers. CAms with a branched architecture showed a general lower hemolytic activity compared with the ones with linear side chains.
Figure 6.

Hemolytic activity of CAms. CAm ethers and CAm esters hemolytic potential (left). CAm ethers have filled markers and CAm esters have open markers. Hemolytic activity of branched CAms is shown on the right in addition to hemolytic activity of analogous CAm ether for comparison.
3.4. Cytocompatibility in Model Mammalian Cell Cultures.
The cytocompatibility of CAms were also evaluated in model mammalian cell cultures, and the results are shown in Figure 7. The relative viability of three series of CAms was measured at different concentrations. All CAms with ester-linkages, including CAms with extended cationic linkers and those with branched hydrophobic domains, displayed high levels of cytocompatibility at different concentrations. The ethers, on the other hand, showed a significant decrease in cytocompatibility when reaching 30 μg/mL. As all tested concentrations resulted in 90% or greater viability, there was no observable trends correlating cationic conformational flexibility or branched architecture with cytocompatibility.
Figure 7.

Cytocompatibility of CAms evaluated against mammalian fibroblasts.
3.5. Selectivity Index Determination.
Structural features leading to the selectivity of CAms were evaluated by screening the selectivity (activity against bacteria vs effects on red blood cells) within the library of CAms. The selectivity index (SI) of CAms was used to evaluate their propensity to interact with bacterial membranes over human cells (Table 1). The ratio of the concentration required to lyse 50% of red blood cells (HC50) relative to the MIC (HC50/MIC) was calculated to represent the SI of a CAm. Sis higher than 1.0 indicate specificity toward bacterial cell membranes, with higher specificity correlating with higher SI magnitudes. The CAms had SI values greater than 1.0 for the representative Gram-positive organism, with 6a and 6b exhibiting the highest selectivity. SI values for the tested Gram-negative pathogen were generally below 1.0, with the exceptions of 6a and 6c, which were close to 1.0, and 11a having the highest activity against Gram-negative bacteria.
3.6. Calcein Leakage Experiments.
In vitro experiments were performed with LUVs mimicking the lipid contents of bacterial cells and mammalian cells. To quickly visualize the effect of the CAms on both cell membrane types and to understand the mechanisms of interaction the cell types, we designed experiments using a simplified model, where DOPC served to mimic mammalian cell membranes and DOPC/DOPG (1:1 mol ratio) served to mimic bacterial membranes. The LUVs were loaded with a self-quenching dye, calcein, and exposed to increasing CAm concentrations. Upon loss of membrane integrity, a dye was released, and an increase in fluorescence was taken as an indication of membrane rupture. Calcein leakage experiments were performed for CAm ethers and esters with cationic groups two methylene units extended from the backbone (i.e., 3a and 6a). These two CAms were chosen as they have high potency against bacteria and sufficient selectivity, and the N-Boc-ethylenediamine is the least expensive linker and is thus the least costly CAms to synthesize for translational purposes. Results for 3a are shown in Figure 8, which confirm membrane activity and specificity. Concentrations ≥100 μg/mL resulted in membrane lysis for bacterial membrane mimics (i.e., DOPC/DOPG) (Figure 8B), but the highest concentration evaluated, 500 μg/mL, showed <5% lysis of membranes mimicking mammalian cells (i.e., DOPC) only, demonstrating specificity (Figure 8C). The total amount of membrane lysis induced by CAms at the end of experiments clearly indicates that 3a has a greater affinity for negatively charged membranes. Similar results were obtained for 6a and are shown in Figure 9. CAm concentrations as low at 50 μg/mL resulted in 15% lysis of negatively charged membranes, and a positive relationship was observed between CAm concentrations and total lysis. However, same as 3a, no mammalian mimic LUV disruption was observed for 6a below 500 μg/mL, with the highest concentration of CAm tested resulted in only 7% lysis of zwitterionic membranes (Figures 8 and 9C).
Figure 8.

Calcein leakage experiments of CAm ethers. Leakage of dye from experiments performed with 3a (A) and negatively charged membranes (B) mimicking bacterial cells and neutral membranes (C) mimicking mammalian host cell membranes indicate membrane specificity. A comparison of total dye leakage at the end of the experiment for each membrane type (D) shows influence of CAm concentration.
Figure 9.

Calcein leakage experiments for CAm-esters. Results from experiments performed with 6a (A) and negatively charged membranes (B) mimicking bacterial cells and neutral membranes (C) mimicking mammalian host cell membranes indicate membrane specificity. A comparison of total dye leakage at the end of the experiment for each membrane type (D) shows influence of CAm concentration.
4. DISCUSSION
The syntheses used in the generation of each CAm series demonstrate improvement upon AMP synthetic protocols. In general, AMPs require a minimum of seven residues to fold into an amphipathic secondary structure.14 To generate synthetic AMPs of this length using traditional peptide synthesis, addition of each Fmoc-protected amino acid and its subsequent deprotection is required, resulting in at least double the number of reaction steps as are amino acid residues.38–40 In contrast, both series of CAms can be generated in five or less reactions with high product yields, significantly reducing production costs. Additionally, tartrates, fatty acids, and alkyl alcohols are inexpensive and readily available and can be found on the list of Generally Recognized as Safe (GRAS) ingredients or are approved by the Food and Drug Administration for direct addition to food products for human consumption.33,41 As such, low or negligible toxicity is anticipated from degradation products.
In the MIC determination experiment, some CAms exhibited comparable or better antimicrobial activities than values commonly reported in the literature.21,25,27,36,37 The 5C branched CAm (11a) exhibited lower MIC against E. coli as compared to L. monocytogenes and was one of the lowest MICs against E. coli of all of the CAms. However, the corresponding 10C branched CAm (11b) exhibited no effect on either species at the highest concentrations tested (250 μg/mL). This result reveals a critical finding regarding the CAm chemical structure and activity: branched architecture better permeabilizes Gram-negative bacteria membranes relative to linear alkyl chains.
Conformational flexibility of the cationic end-group, however, had a less pronounced influence on antimicrobial efficacy in the tested Gram-positive pathogen, whereas it does appear to impact activity against Gram-negative pathogens. It is possible that the increased flexibility of 3c and 6c results in electrostatic repulsion between charges, preventing simultaneous interaction with the negatively charged components of bacterial membranes. This intriguing observation may indicate that CAm’s membrane disrupting mechanism is primarily governed by electrostatic interactions at shorter charged end-group linker lengths. At longer linker lengths, hydrophobic interactions may dominate and are enhanced by hydrophobic domain flexibility. CAms with different cationic linker lengths (i.e., 3a–3c and 6a–6c) all had MICs <7 μg/mL against Gram-positive bacteria, while moderate linker length (i.e., 3b and 6b with four methylene units) showed slightly better efficacy (Table 1). Conversely, when the effect of cationic spacer length was evaluated in Gram-negative bacteria, MICs were similar for two (i.e., 3a, 6a) or four carbon spacers (i.e., 6a, 6b), but were much lower for CAms with six carbon spacers (i.e., 3c, 6c). This data suggests that increased charge flexibility may have a different impact in Gram-positive and Gram-negative bacteria, which is not wholly surprising given the differing membrane structures between Gram-negative and Gram-positive organisms. To further explore these observed differences, it may be useful to look at the activity of these substances against a Gram-negative microorganism with a removed outer membrane.
While it is crucial that antimicrobials exhibit efficacy at low concentrations, it is also important that they demonstrate specificity for bacteria over mammalian cell membranes. This specificity is largely accomplished through the initial electrostatic interaction between cationic moieties and the negatively charged components of the bacterial cell membranes.5,7 However, many AMPs suffer from toxicity: in fact many AMPs of eukaryotic origin are known to have an increased toxicity correlating with increased antimicrobial activity, which has also been correlated with high levels of hydrophobicity.42,43 The CAm esters exhibited lower hemolysis compared with CAm ethers evaluated in human red blood cells. These results indicate that hydrophobic arm flexibility reduces membrane specificity, with more rigid hydrophobe conformations (i.e., ester linkage) favoring interactions with bacterial membranes. In both series, CAms with four methylene units between the backbone and charged end group (3b, 6b) were slightly less hemolytic than CAms with longer or shorter cationic linker lengths. These results are consistent with those obtained by Palermo et al. demonstrating an increase in hemolytic activity in methacrylate based-copolymers with six carbon linkers between cationic moieties and the polymer backbone.21 Interestingly, this result is just the opposite of antimicrobial efficacy, making 3b and 6b the most potent compounds in their own series. On the other hand, branched CAms had the lowest hemolytic activity evaluated in this study, indicating that they have lower potential to interact with mammalian cell membranes. Consistent with trends in the literature, the 10C branched CAm (11b) had comparatively higher hemolytic activity than the 5C branched CAm (11a) because of increased hydrophobicity.42
As red blood cells do not have many of the cellular components and metabolic activity of normal mammalian cells, we also evaluated the CAms cytocompatibility in model mammalian cell cultures. A similar trend was observed where CAm ethers were more toxic to regular mammalian cells compared with esters, especially at higher concentrations. These results are consistent with the results from red blood cell experiments, suggesting additional hydrophobic flexibility increases toxicity and reduce bacterial cell selectivity. There were no observable trends correlating cationic conformational flexibility or branching architecture with cytocompatibility.
The simplified model membrane used in this study required much higher concentrations of antimicrobials to achieve membrane damages, as compared to live cells and/or membrane vesicles.44 This simplified model provides a preliminarily evaluation of the potential effectiveness of these CAms. However, further experiments using different membrane mimicking liposome-based models (including membrane vesicles) will be utilized in future studies aimed at better understanding of the mechanisms of interaction between the membrane and CAms.
5. CONCLUSIONS
Three series of CAms were synthesized, and their membrane activity was evaluated to assess the impact of select chemical features, namely, flexibility and hydrophobic architecture, on antimicrobial efficacy and selectivity. The CAms were synthesized via high yielding synthetic approaches that require fewer reactions than traditional peptide syntheses, resulting in larger quantities of product for lower financial investments. Several key features of CAms were identified that have significant influences on antimicrobial activity. First, CAm esters had the highest activity against bacteria with the most desirable compatibility profiles. Second, CAm esters with moderate charge flexibility (i.e., linker length of four methylene) were most potent against both Gram-positive and Gram-negative bacteria, and third, the branched hydrophobic architecture had the most detrimental effect against Gram-negative bacteria. Together, these data highlight the potential for CAms as membrane-active agents with specificity against bacteria to combat the surge of antibiotic resistance.
ACKNOWLEDGMENTS
Funding from the National Institutes of Health (R21 AI126053) is gratefully acknowledged. We also thank Dr. Yue Cao and Dr. Shuang Song for their assistance with this manuscript.
Footnotes
The authors declare no competing financial interest.
REFERENCES
- (1).Llor C; Bjerrum L Antimicrobial resistance: risk associated with antibiotic overuse and initiatives to reduce the problem. Ther. Adv. Drug Saf 2014, 5, 229–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Dellit TH; Owens RC; McGowan JE; Gerding DN; Weinstein RA; Burke JP; Huskins WC; Paterson DL; Fishman NO; Carpenter CF; Brennan M; Hooton TM Infectious Diseases Society of America and the Society for Healthcare Epidemiology of America guidelines for developing an institutional program to enhance antimicrobial stewardship. Clin. Infect. Dis 2007, 44, 159–177. [DOI] [PubMed] [Google Scholar]
- (3).Boucher HW; Talbot GH; Bradley JS; Edwards JE; Gilbert D; Rice LB; Scheld M; Spellberg B; Bartlett J Bad bugs, no drugs: no ESKAPE! An update from the Infectious Diseases Society of America. Clin. Infect. Dis 2009, 48, 1–12. [DOI] [PubMed] [Google Scholar]
- (4).Zasloff M Antimicrobial peptides of multicellular organisms. Nature 2002, 415, 389–395. [DOI] [PubMed] [Google Scholar]
- (5).Brogden KA Antimicrobial peptides: pore formers or metabolic inhibitors in bacteria? Nat. Rev. Microbiol 2005, 3, 238. [DOI] [PubMed] [Google Scholar]
- (6).Chongsiriwatana NP; Patch JA; Czyzewski AM; Dohm MT; Ivankin A; Gidalevitz D; Zuckermann RN; Barron AE Peptoids that mimic the structure, function, and mechanism of helical antimicrobial peptides. Proc. Natl. Acad. Sci. U.S.A 2008, 105, 2794–2799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Shai Y Mode of action of membrane active antimicrobial peptides. Pept. Sci 2002, 66, 236–248. [DOI] [PubMed] [Google Scholar]
- (8).Shai Y Mechanism of the binding, insertion and destabilization of phospholipid bilayer membranes by α-helical antimicrobial and cell non-selective membrane-lytic peptides. Biochim. Biophys. Acta, Biomembr 1999, 1462, 55–70. [DOI] [PubMed] [Google Scholar]
- (9).Guilhelmelli F; Vilela N; Albuquerque P; Derengowski L. d. S.; Silva-Pereira I; Kyaw CM Antibiotic development challenges: the various mechanisms of action of antimicrobial peptides and of bacterial resistance. Front. Microbiol 2013, 4, 353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Hancock REW; Lehrer R Cationic peptides: a new source of antibiotics. Trends Biotechnol. 1998, 16, 82–88. [DOI] [PubMed] [Google Scholar]
- (11).Marr A; Gooderham W; Hancock R Antibacterial peptides for therapeutic use: obstacles and realistic outlook. Curr. Opin. Pharmacol 2006, 6, 468–472. [DOI] [PubMed] [Google Scholar]
- (12).Alanis AJ Resistance to Antibiotics: Are We in the Post-Antibiotic Era? Arch. Med. Res 2005, 36, 697–705. [DOI] [PubMed] [Google Scholar]
- (13).Sieprawska-Lupa M; Mydel P; Krawczyk K; Wojcik K; Puklo M; Lupa B; Suder P; Silberring J; Reed M; Pohl J; Shafer F; Foster T; Travis J; Potempa J Degradation of human antimicrobial peptide LL-37 by Staphylococcus aureus-derived proteinases. Antimicrob. Agents Chemother 2004, 48, 4673–4679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Bahar A; Ren D Antimicrobial peptides. Pharmaceuticals 2013, 6, 1543–1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Jennings MC; Ator LE; Paniak TJ; Minbiole KPC; Wuest WM Biofilm-Eradicating Properties of Quaternary Ammonium Amphiphiles: Simple Mimics of Antimicrobial Peptides. ChemBioChem 2014, 15, 2211–2215. [DOI] [PubMed] [Google Scholar]
- (16).Black JW; Jennings MC; Azarewicz J; Paniak TJ; Grenier MC; Wuest WM; Minbiole KPC TMEDA-derived biscationic amphiphiles: An economical preparation of potent antibacterial agents. Bioorg. Med. Chem. Lett 2014, 24, 99–102. [DOI] [PubMed] [Google Scholar]
- (17).Som A; Vemparala S; Ivanov I; Tew GN Synthetic mimics of antimicrobial peptides. Pept. Sci 2008, 90, 83–93. [DOI] [PubMed] [Google Scholar]
- (18).Ong ZY; Wiradharma N; Yang YY Strategies employed in the design and optimization of synthetic antimicrobial peptide amphiphiles with enhanced therapeutic potentials. Adv. Drug Delivery Rev 2014, 78, 28–45. [DOI] [PubMed] [Google Scholar]
- (19).Faig A; Arthur TD; Fitzgerald PO; Chikindas M; Mintzer E; Uhrich KE Biscationic tartaric acid-based amphiphiles: charge location impacts antimicrobial activity. Langmuir 2015, 31, 11875–11885. [DOI] [PubMed] [Google Scholar]
- (20).Zhang Y; Algburi A; Wang N; Kholodovych V; Oh DO; Chikindas M; Uhrich KE Self-assembled cationic amphiphiles as antimicrobial peptides mimics: Role of hydrophobicity, linkage type, and assembly state. Nanomedicine 2017, 13, 343–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Palermo EF; Vemparala S; Kuroda K Cationic Spacer Arm Design Strategy for Control of Antimicrobial Activity and Conformation of Amphiphilic Methacrylate Random Copolymers. Biomacromolecules 2012, 13, 1632–1641. [DOI] [PubMed] [Google Scholar]
- (22).Vermeer LS; Lan Y; Abbate V; Ruh E; Bui TT; Wilkinson LJ; Kanno T; Jumagulova E; Kozlowska J; Patel J; McIntyre CA; Yam WC; Siu G; Atkinson RA; Lam JKW; Bansal SS; Drake AF; Mitchell GH; Mason AJ Conformational Flexibility Determines Selectivity and Antibacterial, Antiplasmodial and Anticancer Potency of Cationic α-Helical Peptides. J. Biol. Chem 2012, 287, 34120–34133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Kolodkin-Gal I; Cao S; Chai L; Böttcher T; Kolter R; Clardy J; Losick R A self-produced trigger for biofilm disassembly that targets exopolysaccharide. Cell 2012, 149, 684–692. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- (24).Mowery BP; Lee SE; Kissounko DA; Epand RF; Epand RM; Weisblum B; Stahl SS; Gellman SH Mimicry of antimicrobial host-defense peptides by random copolymers. J. Am. Chem. Soc 2007, 129, 15474–15476. [DOI] [PubMed] [Google Scholar]
- (25).Grenier MC; Davis RW; Wilson-Henjum KL; LaDow JE; Black JW; Caran KL; Seifert K; Minbiole KPC The antibacterial activity of 4, 4′-bipyridinium amphiphiles with conventional, bicephalic and gemini architectures. Bioorg. Med. Chem. Lett 2012, 22, 4055–4058. [DOI] [PubMed] [Google Scholar]
- (26).Chen CZ; Beck-Tan NC; Dhurjati P; van Dyk TK; LaRossa RA; Cooper SL Quaternary Ammonium Functionalized Poly(propylene imine) Dendrimers as Effective Antimicrobials: Structure-Activity Studies. Biomacromolecules 2000, 1, 473–480. [DOI] [PubMed] [Google Scholar]
- (27).Lind T; Polcyn P; Zielinska P; Cardenas M; Urbanczyk-Lipkowska Z On the Antimicrobial Activity of Various Peptide-Based Dendrimers of Similar Architecture. Molecules 2015, 20, 738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Zhang Y; Li Q; Welsh WJ; Moghe PV; Uhrich KE Micellar and structural stability of nanoscale amphiphilic polymers: implications for anti-atherosclerotic bioactivity. Biomaterials 2016, 84, 230–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Han G; Tamaki M; Hruby VJ Fast, efficient and selective deprotection of the tert-butoxycarbonyl (Boc) group using HCl/dioxane (4 m). Chem. Biol. Drug Des 2001, 58, 338–341. [DOI] [PubMed] [Google Scholar]
- (30).Liu Y; Fan Y; Liu X-Y; Jiang S-Z; Yuan Y; Chen Y; Cheng F; Jiang S-C Amphiphilic hyperbranched copolymers bearing a hyperbranched core and dendritic shell: synthesis, characterization and guest encapsulation performance. Soft Matter 2012, 8, 8361–8369. [Google Scholar]
- (31).Buijnsters PJJA; Garcia Rodriguez CL; Willighagen EL; Sommerdijk NAJM; Kremer A; Camilleri P; Feiters MC; Nolte RJM; Zwanenburg B Cationic gemini surfactants based on tartaric acid: synthesis, aggregation, monolayer behaviour, and interaction with DNA. Eur. J. Org. Chem 2002, 2002, 1397–1406. [Google Scholar]
- (32).CLSI. Performance standards for antimicrobial susceptibility testing, 27th ed CLSI supplement M100; Clinical and Laboratory Standards Institute: Wayne, PA, 2017. [Google Scholar]
- (33).Code of Federal Regulations. Title 21, Chapter 1, Subchapter B--Food for human consumption, Volume 3; USF Administration, 2017. [Google Scholar]
- (34).Arias M; Vogel HJ Fluorescence and Absorbance Spectroscopy Methods to Study Membrane Perturbations by Antimicrobial Host Defense Peptides. Methods Mol. Biol 2017, 1548, 141–157. [DOI] [PubMed] [Google Scholar]
- (35).Delcour AH Outer Membrane Permeability and Antibiotic Resistance. Biochim. Biophys. Acta 2009, 1794, 808–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).LaDow JE; Warnock DC; Hamill KM; Simmons KL; Davis RW; Schwantes CR; Flaherty DC; Willcox JAL; Wilson-Henjum K; Caran KL; Minbiole KPC; Seifert K Bicephalic amphiphile architecture affects antibacterial activity. Eur. J. Med. Chem 2011, 46, 4219–4226. [DOI] [PubMed] [Google Scholar]
- (37).Rotem S; Mor A Antimicrobial peptide mimics for improved therapeutic properties. Biochim. Biophys. Acta, Biomembr 2009, 1788, 1582–1592. [DOI] [PubMed] [Google Scholar]
- (38).Suwandecha T; Srichana T; Balekar N; Nakpheng T; Pangsomboon K Novel antimicrobial peptide specifically active against Porphyromonas gingivalis. Arch. Microbiol 2015, 197, 899–909. [DOI] [PubMed] [Google Scholar]
- (39).Duval E; Zatylny C; Laurencin M; Baudy-Floc’h M; Henry J KKKKPLFGLFFGLF: A cationic peptide designed to exert antibacterial activity. Peptides 2009, 30, 1608–1612. [DOI] [PubMed] [Google Scholar]
- (40).Cochrane SA; Lohans CT; Brandelli JR; Mulvey G; Armstrong GD; Vederas JC Synthesis and structure-activity relationship studies of N-terminal analogues of the antimicrobial peptide tridecaptin A1. J. Med. Chem 2014, 57, 1127–1131. [DOI] [PubMed] [Google Scholar]
- (41).Code of Federal Regulations. Title 21, Chapter I Subchapter B, Part 184-Direct Food Substances Affirmed as Generally Recognized as Safe; USF Administration, 2017. [Google Scholar]
- (42).Wieprecht T; Dathe M; Beyermann M; Krause E; Maloy WL; MacDonald DL; Bienert M Peptide hydrophobicity controls the activity and selectivity of magainin 2 amide in interaction with membranes. Biochemistry 1997, 36, 6124–6132. [DOI] [PubMed] [Google Scholar]
- (43).Dagan A; Efron L; Gaidukov L; Mor A; Ginsburg H In vitro antiplasmodium effects of dermaseptin S4 derivatives. Antimicrob. Agents Chemother 2002, 46, 1059–1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Chikindas ML; García-Garcerá MJ; Driessen AJ; Ledeboer AM; Nissen-Meyer J; Nés IF; Abee T; Konings WN; Venema G Pediocin PA-1, a bacteriocin from Pediococcus acidilactici PAC1.0, forms hydrophilic pores in the cytoplasmic membrane of target cells. Appl Environ Microbiol 1993, 59, 3577–3584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Faig AM Design, synthesis, and characterization of bioactive amphiphiles for therapeutic applications. Ph.D. thesis, Rutgers University-Graduate School-New Brunswick, 2015. [Google Scholar]