

Abstract
Photodynamic therapy (PDT) is an effective, noninvasive therapeutic modality against local tumors that are accessible to the source of light. However, it remains challenging to apply PDT for the treatment of disseminated, metastatic cancer. On the other hand, cancer immunotherapy offers a promising approach for generating systemic antitumor immune responses against disseminated cancer. Here we report a multifunctional nanomaterial system for the combination of PDT and personalized cancer immunotherapy and demonstrate their potency against local as well as disseminated tumors. Specifically, we have synthesized uniform and biodegradable mesoporous silica nanoparticles (bMSN) with an average size of ~80 nm and large pore size of 5–10 nm for theranostic positron emission tomography (PET)-guided PDT and neoantigen-based cancer vaccination. Multiple neoantigen peptides, CpG oligodeoxynucleotide adjuvant, and photosensitizer chlorin e6 were coloaded into a bMSN nanoplatform, and PET imaging revealed effective accumulation of bMSN in tumors (up to 9.0% ID/g) after intravenous administration. Subsequent PDT with laser irradiation recruited dendritic cells to PDT-treated tumor sites and elicited neoantigen-specific, tumor-infiltrating cytotoxic T-cell lymphocytes. Using multiple murine models of bilateral tumors, we demonstrate strong antitumor efficacy of PDT-immunotherapy against locally treated tumors as well as distant, untreated tumors. Our findings suggest that the bMSN is a promising platform for combining imaging and PDT-enhanced personalized immunotherapy for the treatment of advanced cancer.
Keywords: mesoporous silica nanoparticles, cancer immunotherapy, positron emission tomography, photodynamic therapy, neoantigen, vaccine
Graphical Abstract
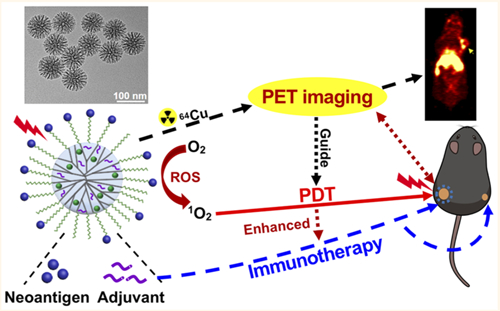
Photodynamic therapy (PDT) is a noninvasive tumor ablation approach that has been investigated extensively for cancer treatment.1–3 PDT employs activation of a light-sensitive molecule (photosensitizer) by a specific wavelength of laser to generate cytotoxic reactive oxygen species (ROS), which induces tumor cell death and vascular shut down.1–3 In addition, image-guided therapy has recently shown great promise for cancer treatment.4,5 Image-guided therapy could provide important information on the size and location of tumors, optimal time window for treatment, and response to cancer treatment. In particular, positron emission tomography (PET) is a highly sensitive and noninvasive imaging method that is ideally suited for image-guided therapy.6 With radiolabeled theranostic nanoplatforms, PET imaging-guided PDT could facilitate a quantitative diagnosis of cancer while allowing for personalized therapeutic treatments utilizing the same nanoplatforms.7 However, while PDT is highly effective against superficial tumors that are accessible to the source of laser irradiation, it remains ineffective against disseminated, metastatic cancers that are beyond the reach of light-induced activation, thus precluding its wide application. On the other hand, cancer immunotherapy, which aims to harnesses the body’s own immune system to combat cancer, elicits and sustains systemic immune responses against disseminated tumor cells.8 Importantly, as PDT can induce the release of tumor-associated antigens and other immunogenic factors from dying tumor cells,9 cancer immunotherapy combined with PDT may offer a complementary strategy for treating advanced cancer. In fact, recent studies have demonstrated robust antitumor efficacy of PDT combined with cancer immunotherapy.9–12 Yet, these previous reports focused on combination PDT–immunotherapy with coadministration of immune checkpoint blockers, which may trigger immune-related adverse events, especially in the context of combination immunotherapy.13
In contrast, cancer vaccination can elicit cytotoxic T-cell lymphocytes (CTLs) that are specifically targeted against tumor cells with minimal off-target toxicity.14 In particular, recent developments in the next-generation DNA/RNA sequencing allow for identification of patient-specific, tumor-specific mutations, termed neoantigens.14–16 As neoantigens are not expressed among healthy tissues, they provide excellent targets for personalized cancer vaccination, and early stage clinical trials have shown their potential.17,18 However, conventional vaccines composed of free soluble neoantigens and adjuvants are not ideal, as they exhibit rapid in vivo clearance with limited immunogenicity.19,20 To overcome these challenges, various multifunctional nanoparticles (NPs), based on nanodiscs,21,22 liposomes,23–25 polymers,9 and inorganic NPs,11,26 have been developed with varying success for delivery of cancer antigens and adjuvants.27,28
In this work, we report the development of mesoporous silica nanoparticles (MSNs) for personalized cancer immunotherapy. Notably, MSNs, composed of amorphous silicon dioxide with well-define mesopores, are an emerging platform for various drug delivery applications due to their tunable size, biocompatibility, and controlled release of cargo materials.29–31 However, conventional MSNs are not ideally suited for neoantigen vaccination, as they have limited loading capacity of peptide antigens due to their relatively small mesopores (0.5–3.0 nm) and have a typical particle size of 200–300 nm, which can compromise their ability to target lymphoid tissues.31–33 In addition, conventional MSNs could cause chronic tissue damage due to slow biodegradation and long-term retention in major organs.34,35
To address these issues, we have developed small MSNs (~80 nm in diameter) with large 5–10 nm pore size and fast biodegradation rate as a multifunctional nanoplatform for combination immunotherapy (Figure 1). We coloaded these biodegradable MSNs (bMSNs) with CpG oligodeoxynucleotide (CpG ODN, a potent Toll-like receptor-9 agonist) and photosensitizer chlorin e6 (Ce6) for combination PDT–immunotherapy. Neoantigen peptides were conjugated on the surface of bMSNs via disulfide bonds, which can be rapidly cleaved in the highly reductive tumor intracellular environment. Using PET imaging with radioisotope 64Cu, we have shown that bMSN nanoplatforms loaded with neoantigens and adjuvants accumulated effectively in tumors after intravenous (i.v.) administration. Subsequent application of PDT induced recruitment of dendritic cells (DCs) to PDT-treated tumor sites and elicited strong neoantigen-specific CD8α+ CTL responses. Compared with single PDT or vaccination, the combination of PDT and personalized cancer vaccination achieved strong synergy and exerted potent antitumor efficacy against local as well as distant tumors in multiple murine tumor models, demonstrating their potential for the treatment of advanced cancer.
Figure 1.

Schematic illustration of fabrication of bMSN(CpG/Ce6)-neoantigen and mechanism of bMSN(CpG/Ce6)-neoantigen nanovaccines for PDT-enhanced cancer immunotherapy. We synthesized bMSN using the heterogeneous oil–water biphasic reaction system. CpG and Ce6 were loaded into the mesopores of bMSNs through electrostatic and hydrophobic interactions, respectively. After surface PEGylation of bMSNs with PDP-PEG5k-NHS, neoantigen peptides were conjugated to bMSNs via formation of disulfide bonds. Laser irradiation (660 nm) was applied to generate cytotoxic ROS and eliminate tumor cells, while triggering local immune activation for antitumor immunity.
RESULTS AND DISCUSSION
Synthesis and Characterization of the bMSN Nanoplatform.
The schematic for PET-guided PDT–immunotherapy combination using a bMSN platform is shown in Figure 1. Briefly, bMSNs were synthesized using a heterogeneous oil–water biphase reaction system,34 and the resulting bMSNs were surface-modified with (3-aminopropyl)triethoxysilane (APS) to introduce amine groups. CpG and Ce6 were then loaded into mesopores of bMSNs via electrostatic and hydrophobic interactions, respectively. To improve their colloidal stability in vitro and in vivo, bMSNs were PEGylated by reacting surface-displayed amines with PDP-PEG5k-NHS. Finally, bMSNs surface-decorated with pyridyl functional groups were incubated with neoantigen peptides modified with cysteine-serine-serine at the N-terminus.36 We evaluated the therapeutic potential of the bMSN platform in tumor-bearing animals after systemic administration, followed by 660 nm laser irradiation to tumors. We also examined the fate of bMSNs after systemic administration in tumor-bearing mice using PET imaging.
As visualized by transmission electron microscopy (TEM), bMSNs exhibited uniform, highly porous dendritic nanostructures, with an average diameter of 78 ± 23 nm (Figure 2a,b). As measured by dynamic light scattering (DLS), PEGylation increased the size of bMSNs to 87 ± 31 nm (Figure 2c). The zeta potential of bMSNs increased from −31 ± 3 mV to 21 ± 5 mV after surface amino modification (Figure 2d), which would allow efficient complexation with anionic CpG ODN. The N2 adsorption–desorption isotherm of bMSNs indicated a Brunauer–Emmett–Teller (BET) surface area of 608 m2/g and a pore volume of 2.29 cm3/g (Figure 2e). The pore size of bMSNs ranged from 5 to 10 nm with a minor peak at 2 nm (Figure 2f). Compared with the average pore size of 2.0–4.0 nm for conventional MSNs in the literature29,30,37 and those from commercial sources, bMSNs with the unique 3D dendritic and hierarchical nanostructures have a higher average pore size, pore volume, and surface area (Figure S1a,b), thus potentially allowing a high loading capacity of macromolecular drugs, such as proteins and peptides.34
Figure 2.

Morphology, structure, and characterization of bMSNs. (a and b) TEM images of bMSNs; (c) hydrodynamic size analysis of bMSNs (black line) and bMSN-PEG (green line) by DLS; (d) surface zeta potential of bMSNs, bMSN-NH2, and bMSN-PEG; nitrogen adsorption and desorption isotherms (e) and pore size distributions (f) of bMSNs; (g) in vitro biodegradation profile of bMSNs in simulated body fluid (Krebs–Henseleit solution) at 37 °C for 9 days. At indicated time points, TEM images of (h) were obtained.
Conventional MSNs are reported to undergo slow degradation over several weeks.35 As our bMSNs were highly porous compared with conventional MSNs, we sought to investigate the degradation kinetics of bMSNs. When incubated in simulated body fluid (Krebs–Henseleit solution) at 37 °C, bMSNs underwent rapid degradation as measured by inductively coupled plasma mass spectrometry (ICP-MS), releasing >81% of its Si content within 9 days (Figure 2g). TEM images indicated that the degradation process proceeded from the outer silica matrix to the inner core (Figure 2h). In contrast, conventional MSNs obtained from commercial sources appeared intact even after 12 days of incubation in simulated body fluid and released only 4–13% Si content (Figure S1c,d). We speculate that the highly porous core of bMSNs with a less dense and low cross-linking Si–O–Si matrix allows for this rapid degradation.34
Drug Loading, Release, and in Vitro PDT by bMSN Nanoplatforms.
Ce6 in DMSO showed the characteristic absorption peak at ~660 nm as measured by the UV–vis spectrum (Figure 3a); however, this characteristic peak was absent for Ce6 diluted in phosphate-buffered saline (PBS) due to its poor aqueous solubility. Notably, Ce6-loaded bMSNs (bMSN(Ce6)) in PBS exhibited the characteristic peak of Ce6 at ~660 nm, and bMSN(Ce6) in PBS appeared in dark green color (Figure 3a, inset), thus showing that bMSNs significantly improved the dispersibility of Ce6. Indeed, HPLC analyses indicated very efficient Ce6 loading in bMSNs, with the loading efficiency of 50–90% for the range of bMSN:Ce6 tested and the loading capacity of 50 ± 6 μg Ce6 per 100 μg of bMSNs at a bMSN:Ce6 weight ratio of 1:1, which are significantly higher than the previously reported MSN-based nanocarrier systems.29,30 Furthermore, bMSN(Ce6) was also efficiently loaded with CpG and neoantigen peptide of MC-38 tumor, Adpgk. The simultaneous loading capacity for Ce6, CpG, and Adpgk in 100 μg of bMSN was 28 ± 5, 16 ± 2, and 15 ± 3 μg, respectively (Figure 3b, c). Consistent with the increased pore size and volume, bMSNs exhibited significantly increased loadings of Ce6, CpG, and Adpgk, compared with conventional MSNs (Figure S1e). Ce6, CpG, and Adpgk coloaded in bMSNs (termed bMSN vaccine) were gradually released over time, resulting in a retention of 43%, 70%, and 82% of initial cargo, respectively, in bMSNs after 48 h in PBS (Figure 3d). Ce6-loaded bMSNs showed efficient singlet oxygen generation by 660 nm laser irradiation as the free form of Ce6 (Figure 3e). Notably, laser irradiation and generation of singlet oxygen species did not influence the release of antigen from bMSNs (Figure S2). Next, we examined the biocompatibility of various formulations and cytotoxicity of PDT using in vitro culture of MC-38 tumor cells. Notably, Ce6 mixed in CpG and Adpgk peptide soluble vaccine caused cytotoxicity even without laser irradiation (Figure 3f); however, no cytotoxicity was observed with the bMSN nanoplatform itself or bMSN vaccine even at a high concentration of 0.5 mg/mL. After laser irradiation, the bMSN vaccine efficiently killed MC-38 tumor cells, demonstrating selective, laser-responsive PDT of tumor cells.
Figure 3.

(a) UV–vis absorption spectra of Ce6 (in PBS), Ce6 (in DMSO), bMSN(Ce6) (in PBS), and bMSN (in PBS). Inset: Photographs of Ce6 (in PBS) and bMSN(Ce6) (in PBS) with a Ce6 concentration of 0.2 mg/mL. (b) Loading efficiency of CpG, Ce6, and Adpgk peptide by bMSNs in various weight ratios. (c) Simultaneous loading capacity of CpG, Ce6, and Adpgk peptide by 100 μg of bMSNs in PBS. (d) Release profile of bMSN(CpG/Ce6)-Adpgk in PBS at 37 °C. (e) Singlet oxygen production by bMSNs, free Ce6, and bMSN(Ce6) after 660 nm laser irradiation (25 mW/cm2) as measured by the changes in the fluorescence intensity of SOSG (Singlet Oxygen Sensor Green). (f) In vitro PDT assay after incubating MC-38 tumor cells with bMSNs (0.5 mg/mL), Ce6 (0.5 μg/mL), CpG (1.0 μg/mL), Adpgk (10 μg/mL), soluble vaccine (CpG (1.0 μg/mL), Ce6 (1.0 μg/mL), Adpgk (10 μg/mL)) with or without laser irradiation (660 nm, 10 mW/cm2, 2 min), and bMSN vaccine (bMSN(CpG/Ce6)-Adpgk, same Ce6, CpG, Adpgk concentration with soluble vaccine) with or without laser irradiation (660 nm, 10 mW/cm2, 2 min).
Dendritic Cell Activation and Cytokine Secretion.
Successful activation of T-cells by DCs requires upregulation of costimulatory markers, including CD40, CD80, and CD86, and secretion of cytokines, such as IL-12.38 Therefore, we next investigated whether CpG-loaded bMSNs could induce DC activation and promote cytokine secretion. Mouse bone marrow-derived dendritic cells (BMDCs) incubated with either plain bMSNs, Ce6, or Adpgk peptide alone exhibited no obvious signs of DC maturation (Figure S3a–c). In contrast, incubation of BMDCs with CpG either in a free soluble or bMSN form significantly increased the expression levels of CD40, CD80, and CD86 on DCs (Figure S3a–c). There was no significant effect of laser irradiation on the expression levels of costimulatory markers. BMDCs incubated with either plain bMSN, Ce6, or Adpgk peptide alone did not secrete any detectable levels of IL-12p70 or TNF-α (Figure S3d,e). In contrast, incubation of DCs with CpG either in a free soluble or bMSN form led to strong production of IL-12p70 and TNF-α. Notably, bMSN loaded with CpG and Ce6 (bMSN(CpG/Ce6)) induced more robust secretion of IL-12p70 and TNF-α than free CpG, probably due to increased cellular uptake of bMSN(CpG/Ce6). Overall, CpG-loaded bMSNs promoted DC maturation, activation, and secretion of inflammatory cytokines.
In Vivo PET Imaging and Biodistribution.
We next examined the biodistribution of bMSN(CpG/Ce6)-Adpgk nanocomplexes after intravenous administration in tumor-bearing mice. We employed radioisotope 64Cu (t1/2 = 12.7 h)6 and NOTA chelator to visualize and quantify the biodistribution of peptide either in a free form or incorporated in bMSNs. C57BL/6 mice were inoculated with 3 × 105 of MC-38 colon carcinoma cells on day 0. On day 19, when tumors were ~260 mm3, mice were administered intravenously with 10 μg of Adpgk peptide in either 64Cu-NOTA-Adpgk or 64Cu-NOTA-bMSN(CpG/Ce6)-Adpgk form, both labeled with ~200 μCi of 64Cu. We then performed serial PET scans and quantitated 64Cu signal in the major organs over 25 h (Figure 4a–c). In mice administered with 64Cu-NOTA-Adpgk, 64Cu signal was mainly detected in the bladder and intestines at the first two time points (0.5 and 2.5 h). This indicated rapid clearance of soluble 64Cu-NOTA-Adpgk with an estimated blood circulation half-life of less than 30 min. At 25 h post injection (p.i.), 64Cu signal was very weak (<0.3% ID/g) in MC-38 tumors or major organs except for the kidneys, with residual 64Cu signal (3.2% ID/g) (Figure 4a,b). In stark contrast, mice administered with 64Cu-NOTA-bMSN(CpG/Ce6)-Adpgk exhibited high 64Cu signal in the heart at 0.5 and 2.5 h p.i. with 64Cu signal at 23.2% ID/g at and 19.3% ID/g, respectively. Even at 17 h p.i., we detected 8.8% ID/g 64Cu signal in the heart, giving an estimated blood circulation half-life of ~14.2 h (Figure S4). Since the passive tumor targeting by the EPR (enhanced permeability and retention) effect is achieved via extravasation from systemic circulation, long-circulating nanoparticles could exhibit higher passive tumor accumulation efficiency.5 bMSN vaccine efficiently accumulated in MC-38 tumors over time, with 7.4% ID/g and 9.0% ID/g signal detected in tumor tissues at 17 and 25 h p.i., respectively (Figure 4a,c). As anticipated for any intravenously administered nanomaterials larger than the cutoff of renal filtration (~5.5 nm),39 bMSN(CpG/Ce6)-Adpgk accumulated in the spleen and liver; however, the toxicity profile of bMSNs is expected to be minimal due to its rapid biodegradation kinetics (Figure 2g,h). To validate the in vivo PET results, we directly measured the radioactivity of 64Cu ex vivo from the major organs harvested at 25 h p.i. using a gamma counter (Figure 4d). In line with the quantitative ROI analysis of PET imaging (Figure 4b,c), we detected ~20-fold higher signal for 64Cu-NOTA-bMSN(CpG/Ce6)-Adpgk in MC-38 tumors, compared with that of 64Cu-NOTA-Adpgk (P < 0.001, Figure 4d). We also detected ~11-fold higher 64Cu signal at 25 h p.i. in the peripheral blood of animals administered with nanocomplexes, compared with free peptide (P < 0.001).
Figure 4.

(a) Serial PET images of MC-38 tumor-bearing mice at various time points postinjection of 64Cu-NOTA-Adpgk or 64Cu-NOTA-bMSN(CpG/Ce6)-Adpgk. Tumors are indicated by yellow arrowheads. Time–radioactivity curves of MC-38 tumor, blood, liver, spleen, kidney, and muscle after i.v. injection of 64Cu-NOTA-Adpgk (b) and 64Cu-NOTA-bMSN(CpG/Ce6)-Adpgk (c). (d) Biodistribution studies in MC-38 tumor-bearing mice at 25 h postinjection of 64Cu-NOTA-Adpgk and 64Cu-NOTA-bMSN(CpG/Ce6)-Adpgk.
Therapeutic Efficacy of bMSN Vaccination Combined with PDT.
Having confirmed accumulation and retention of bMSNs in tumors, we next investigated their therapeutic potential for combined cancer vaccination and PDT. We first employed a bilateral two-tumor model with MC-38 colon carcinoma and examined the abscopal effect of the combination immunotherapy (Figure 5a). C57BL/6 mice were inoculated with 3 × 105 MC-38 cells in the right flank on day 0, followed by inoculation of 2 × 105 MC-38 cells in the contralateral flank on day 8. On day 9 when the average primary tumor size was ~50 mm3, the animals were randomly divided into the following six treatment groups: (1) PBS control; (2) soluble vaccine (CpG, Ce6, and Adpgk peptide); (3) soluble vaccine with laser irradiation; (4) bMSN(Ce6) with laser irradiation; (5) bMSN vaccine (bMSN(CpG/Ce6)-Adpgk); and (6) bMSN vaccine with laser irradiation. Prime vaccination was administered intravenously via tail vein on day 9, and after 24 h, only the right flank tumors were treated with laser irradiation (660 nm, 50 mW/cm2 for 15 min), whereas the left flank tumors were left untreated. The animals were treated with booster vaccination and a second laser irradiation on days 16 and 17, respectively.
Figure 5.

Antitumor therapy study in MC-38 tumor-bearing mice. (a) C57BL/6 mice were randomly divided into the following six treatment groups: (1) PBS control; (2) soluble vaccine (CpG, Ce6, and Adpgk peptide); (3) soluble vaccine with laser irradiation; (4) bMSN(Ce6) with laser irradiation; (5) bMSN vaccine (bMSN(CpG/Ce6)-Adpgk); and (6) bMSN vaccine with laser irradiation. Laser irradiation (660 nm, 50 mW/cm2 for 15 min) was conducted over the tumors at 24 h after each injection. The frequency of Adpgk-specific CD8α+ T-cells in peripheral blood was measured 7 days after the prime and booster vaccination. The representative scatter plots (c) and percentage of Adpgk-specific CD8α+ T-cells (b) on day 16 (prime) and day 23 (booster) are shown. (d) Average primary and contralateral MC-38 tumor growth curves of each group. (e) Overall survival curves of each group.
We examined the animals for the induction of neoantigen-specific CD8α+ T-cells by examining peripheral blood mononuclear cells (PBMCs) on 7 days after each prime and booster vaccination. Mice administered with soluble vaccine composed of CpG, Ce6, and Adpgk peptide induced an average of 1.0% Adpgk-specific CD8α+ T-cells among PBMCs on day 7 after the boost vaccination (day 23, Figure 5b,c). Mice that received soluble vaccine plus laser irradiation had an average of 2.1% Adpgk-specific CD8α+ T-cells among PBMCs on day 23. In stark contrast, mice vaccinated with bMSN-(CpG/Ce6)-Adpgk elicited 8.9% antigen-specific CD8α+ T-cell response by day 23 (9.1-fold and 4.1-fold greater than soluble vaccine and soluble vaccine + laser, respectively, P < 0.05, Figure 5b,c). The combination of bMSN vaccine with PDT further amplified CD8α+ T-cell responses, achieving 22.9% antigen-specific CD8α+ T-cell response among PBMCs (23-fold greater than soluble vaccine, P < 0.0001; and 10.5-fold greater than soluble vaccine + laser, P < 0.0001, Figure 5b,c). This represented 2.6-fold greater frequency of Adpgk-specific CD8α+ T-cells, compared with bMSN vaccine without PDT combination (P < 0.01). On the other hand, the control group with PDT treatment alone (bMSN(Ce6) + laser) generated baseline antigen-specific CD8α+ T-cell response. Taken together, PDT significantly enhanced antitumor T-cell immune responses elicited by bMSN vaccination.
In parallel with T-cell assays, we measured the sizes of primary and contralateral tumors every 2 days. Soluble vaccine groups with or without laser treatment as well as PDT treatment alone (bMSN(Ce6) + laser group) showed rapid growth of primary and contralateral tumors, with the median survival of 24–26 days (Figure 5d, e), exhibiting only marginal differences from the PBS-treated control group. Vaccination with bMSNs resulted in moderate suppression of tumor growth and extended animal survival, with a median survival of 40 days (Figure 5d,e). In stark contrast, administration of bMSN(CpG/Ce6)-Adpgk vaccine combined with laser irradiation exhibited significantly improved antitumor efficacy, compared with either bMSN(Ce6)-based PDT alone (P < 0.001), soluble vaccine (P < 0.001), or bMSN vaccine without laser irradiation (P < 0.05) (Figure 5d). Moreover, mice treated with bMSN vaccine + laser treatment lived significantly longer than the other groups (P < 0.05, compared with bMSN vaccine group; P < 0.0001, compared with the other four groups, Figure 5e). Inclusion of antigen in the bMSN vaccine + laser combination therapy was crucial, as the bMSN(CpG/Ce6) + laser control group induced limited Adpgk-specific T-cell response with minimal antitumor efficacy (Figure 6). In addition, compared with MSN(Ce6/CpG)-Adpgk vaccine + laser, the bMSN(Ce6/CpG)-Adpgk vaccine + laser treatment group elicited significantly stronger T-cell responses (P < 0.01) and extended animal survival (P < 0.05) (Figure 6), suggesting the potency of bMSNs for combined immunotherapy.
Figure 6.

Antitumor therapy study in MC-38 tumor-bearing mice. C57BL/6 mice were randomly divided into the following four treatment groups: (1) PBS control; (2) bMSN(CpG/Ce6) with laser irradiation; (3) bMSN vaccine (bMSN(CpG/Ce6)-Adpgk) with laser irradiation; and (4) MSN1 vaccine (MSN(CpG/Ce6)-Adpgk) with laser irradiation. Laser irradiation (660 nm, 50 mW/cm2 for 15 min) was conducted over the tumors at 24 h after each injection. (a, b) Average primary and contralateral MC-38 tumor growth curves of each group. (c) Average bodyweight of mice. (d) Frequency of Adpgk-specific CD8α+ T-cells in peripheral blood was measured 7 days after the prime and booster vaccination. Percentages of Adpgk-specific CD8α+ T-cells (e) and CD11c+CD86+ dendritic cells (f) in the MC-38 tumor microenvironment 7 days after booster vaccination. (g) Overall survival curves of each group.
Analysis of Systemic and Local Immune Responses.
Having observed strong systemic antitumor efficacy of bMSN vaccine combined with PDT, we repeated the treatment regimen as outlined in Figure 5a and euthanized animals on day 22 for immunological analysis of systemic and local compartments. Analysis of splenocytes by IFN-γ ELISPOT (enzyme-linked immunospot) assay showed that the bMSN vaccine + laser group generated significantly enhanced antigen-specific CD8α+ T-cell responses, compared with the soluble vaccine groups (with or without laser, 5.5-fold increase) and the bMSN(Ce6) + laser group (>100-fold increase) (P < 0.001, Figure 7a). We next examined the impact of various treatments on the local tumor microenvironment. Mice treated with bMSN(CpG/Ce6)-Adpgk + laser had a high frequency of tumor-infiltrating CD8α+ T-cells (2.9-fold and 3.9-fold greater than soluble vaccine + laser and bMSN-based PDT alone, respectively, P < 0.01, Figure 7b). Among tumor-infiltrating CD8α+ T-cells, bMSN vaccine + laser generated robust Adpgk-specific CD8α+ T-cell responses (11.7-fold and 23.7-fold greater than soluble vaccine + laser and bMSN-based PDT alone, respectively, P < 0.01, Figure 7c), suggesting strong synergy between bMSN vaccination and PDT. Interestingly, compared with other control groups, bMSN vaccine + laser treatment significantly increased the frequency of activated intratumoral CD11c+CD86+ DCs (P < 0.01, Figure 7d), whereas their impact on natural killer (NK) cells was minimal (Figure 7e). Notably, throughout our studies, we did not observe any overt sign of systemic toxicity, bodyweight loss, abnormal liver function, or tissue damage after bMSN vaccine + laser treatment (Figure 7f, Figures S5 and S6).
Figure 7.

Tumor microenvironment analysis, ELISPOT (enzyme-linked immunospot) assay, and H&E staining of major organs. C57BL/6 mice were randomly divided into the following six treatment groups: (1) PBS control; (2) soluble vaccine (CpG, Ce6, and Adpgk peptide); (3) soluble vaccine with laser irradiation; (4) bMSN(Ce6) with laser irradiation; (5) bMSN vaccine (bMSN(CpG/Ce6)-Adpgk); and (6) bMSN vaccine with laser irradiation. The laser irradiation (660 nm, 50 mW/cm2 for 15 min) was conducted to the tumor area 24 h after each injection in the laser irradiation group. (a) Seven days postimmunization, the IFN-γ ELISPOT assay was performed by ex vivo restimulation of splenocytes with Adpgk peptides (10 μg/mL). In parallel, tumor tissues were analyzed for the frequencies of CD8α+ T-cells (b), Adpgk-specific CD8α+ T-cells (c), activated CD11c+CD86+ DCs (d), and NK cells (e) using flow cytometry. (f) Hematoxylin–eosin (H&E) staining images of major mice organs in the PBS group and the bMSN(CpG/Ce6)-Adpgk group on day 30 after immunization.
Therapeutic Efficacy in a Highly Aggressive B16F10 Model.
Lastly, to further evaluate the potency of our strategy, we examined the efficacy of the combination immunotherapy using a highly aggressive and immune-suppressive B16F10 melanoma model. M27 and M30 neoantigens identified from B16F10 tumor cells40 were loaded in bMSN nanoplatforms using the same approach as shown in Figure 1. C57BL/6 mice were inoculated with 3 × 105 B16F10 cells in the right flank (day 0) and 2 × 105 B16F10 cells in the contralateral flank (day 6) (Figure 8a). Prime and booster vaccines were administered intravenously via tail vein on days 7 and 14. At 24 h after each injection, only the right flank tumors were treated with laser irradiation (660 nm, 100 mW/cm2 for 15 min), whereas the left flank tumors were left untreated. The soluble vaccine groups (with or without laser), the bMSN-(Ce6) + laser group, and the bMSN(CpG/Ce6) + laser group (without antigen) all exhibited rapid growth of primary and contralateral tumors, with negligible antitumor effects as in the PBS-treated control group (Figure 8c, Figure S7a–c). In contrast, vaccination with bMSN significantly delayed the primary tumor growth, compared with the soluble vaccine groups (with or without laser) (P < 0.01, Figure 8c). Importantly, the combination of bMSN vaccine and PDT further slowed the growth of primary and contralateral tumors (P < 0.05, Figure 8c). Overall, mice treated with bMSN vaccine + laser exhibited a significantly higher survival rate, compared with mice treated with either bMSN vaccine alone (P < 0.01) or all other control treatments (P < 0.0001, Figure 8b).
Figure 8.

Antitumor therapy study in B16F10 tumor-bearing mice. (a) C57BL/6 mice were randomly divided into the following six treatment groups: (1) PBS control; (2) soluble vaccine (CpG, Ce6, M27, and M30 peptides); (3) soluble vaccine with laser irradiation; (4) bMSN(Ce6) with laser irradiation; (5) bMSN vaccine (bMSN(CpG/Ce6)-M27/M30); and (6) bMSN vaccine with laser irradiation. Laser irradiation (660 nm, 100 mW/cm2 for 15 min) was conducted over the tumors at 24 h after each injection. (b) Overall survival curves of each group. (c) Average primary and contralateral B16F10 tumor growth curves of each group. (d) On day 21, IFN-γ ELISPOT assay was performed by ex vivo restimulation of splenocytes with M27 and M30 peptides (10 μg/mL). In parallel, tumor tissues were analyzed for the frequencies of CD3+CD8α+ T-cells (e) and CD11c+CD86+ DCs (f) using flow cytometry.
We also analyzed immunological responses in the systemic and local compartments in the B16F10 tumor model on day 21 (7 day after the second immunization) (Figure 8d–f). The bMSN vaccine + laser group elicited potent neoantigen-specific, IFN-γ + T-cell responses in spleen (Figure 8d) and induced the highest frequency of tumor-infiltrating CD8α+ T cells (Figure 8e) and CD11c+CD86+ DCs (Figure 8f), compared with other groups. We have also shown that inclusion of neoantigens in the bMSN vaccine + laser treatment was crucial for strong induction of CD8α+ T-cells and antitumor efficacy, as bMSN(CpG/Ce6) + laser failed to expand Adpgk-specific CD8α+ T-cell response or extend animal survival, compared with the PBS group (Figure S7d,e). Lastly, we have observed that MSN vaccine + laser group exhibited significantly reduced B16F10 neoantigen-specific T-cell responses (P < 0.001) with minimal survival benefit (P < 0.05), compared with the bMSN vaccine + laser treatment group (Figure S7d,e), confirming the results observed in the MC-38 tumor model (Figure 6).
In summary, these results demonstrate that PDT combined with personalized bMSN vaccination elicits robust neoantigen-specific T-cell immune responses within the systemic compartment and local tumor microenvironment, thereby exerting strong therapeutic efficacy in multiple murine tumor models. While there are recent reports of large-pore mesoporous-silica-coated upconversion nanoparticles for vaccination + PDT41 as well as other biomaterials for PDT combined with systemic immune checkpoint therapy,9–12 here we have demonstrated a therapeutic strategy of personalized cancer immunotherapy based on bMSNs combined with PDT. Without the use of immune checkpoint blockers, bMSN vaccine + PDT generated robust neoantigen-specific T-cell responses with strong abscopal effect against untreated, distal tumors, thus potentially broadening the applicability of PDT–immunotherapy for local as well as disseminated, advanced cancer.
CONCLUSION
We have developed a facile and effective approach for delivering personalized neoantigen peptides and adjuvants. Using PET imaging, we have shown that bMSNs effectively accumulated in tumors after intravenous administration. The combination of bMSN vaccination and PDT synergistically promoted antitumor T-cell immunity and achieved robust antitumor efficacy. These results indicate that nanomaterials may offer a powerful therapeutic platform for personalized cancer immunotherapy.
MATERIALS AND METHODS
Reagents and Materials.
All reagents used in this work were analytical or higher grade. Tetraethyl orthosilicate (TEOS), triethylamine (TEA), cetyltrimethylammonium chloride (CTAC), (3-aminopropyl)triethoxysilane (APS), and Chelex 100 resin were purchased from Sigma-Aldrich (St. Louis, MO, USA). Orthopyridyl disulfide PDP-PEG-succinimidyl ester (OPSS-PEG-NHS; molecular weight: 5 kDa) was purchased from Creative PEGworks (Winston Salem, NC, USA). S-2-(4-Isothiocyanatobenzyl)-1,4,7-triazacyclononane-1,4,7-triacetic acid (p-SCN-Bn-NOTA) was purchased from Macrocyclics, Inc. (Dallas, TX, USA). Chlorin e6 was purchased from Thermo Fisher Scientific. Antigen peptides, including Adpgk mutant peptide CSSASMTNMELM, M27 neoantigen peptide LCPGNKYEM, and M30 neoantigen peptide CSSVDWENVSPELNSTDQ, were synthesized by RS Synthesis (Louisville, KY, USA). Murine class B CpG ODN 1826 was purchased from Integrated DNA Technologies (Coralville, IA, USA). “MSN1” (748161) was purchased from Sigma-Aldrich. “MSN2” (SHAD-100) was purchased from Nanocomposix (San Diego, CA, USA). All other chemicals and reagents were purchased from Thermo Fisher Scientific (Fair Lawn, NJ, USA).
Characterization.
TEM images were obtained by Jeol 1400-PLUS, 120 kV field emission. Nitrogen (N2) adsorption–desorption isotherms were measured by a NOVA 4200e system. Pore size and surface areas were determined by the BET method.42 Nanoparticles’ zeta potential and size analysis were performed on a Nano-ZS90 Zetasizer (Malvern Instruments Ltd.). UV–vis spectra were recorded on a Biotek Synergy microplate reader.
Synthesis of bMSN(CpG/Ce6)-Adpgk Nanocomposite.
bMSNs were synthesized by an oil–water biphase reaction approach.34 In a typical process, 20 mL of CTAC solution (25 wt %) and 0.01 g of TEA were added to 30 mL of water and gently stirred at 50 °C for 1 h in a 150 mL round-bottom flask. Then 15 mL of (5% v/v) TEOS in cyclohexane was slowly added to the CTAC–TEA solution and kept at 50 °C for 18 h. Afterward, bMSNs were collected by centrifugation at 14000g for 15 min. The precipitates were washed four times (24 h/time) with 1% (wt %) NaCl/methanol solution to remove CTAC. The surface amino group modification was achieved by adding 1 mL of APS to 10 mL of bGNR@MSN in absolute ethanol solution (1 mg/mL) and stirring in 45 °C water bath for 72 h, followed by three centrifugation steps (14000g for 15 min) and an ethanol wash to remove any unreacted APS.
Next, CpG and Ce6 were added to a bMSN suspension in water and stirred for 0.5 h at room temperature (RT). bMSN(CpG/Ce6) were collected by centrifugation (14000g for 15 min) and washed twice with PBS. A 5 mg amount of OPSS-PEG-NHS was added into a 10 mL bMSN(CpG/Ce6) suspension in water (1.2 mg/mL) and stirred for 2 h at RT for the PEGylation process followed by centrifugation (14000g for 15 min) and washed twice with PBS. Finally, neoantigen peptides (and the chelator NOTA for the PET imaging study) were added to the bMSN(CpG/Ce6)-PEG suspension, stirred for 1 h at RT, and washed twice with PBS to remove free peptide.
BMDC Activation, Cytokine Secretion, and in Vitro PDT.
BMDCs were prepared as described in a previous report.43 Immature BMDCs were plated at 2 × 106 cells per well in six-well plates and incubated overnight. CpG, Ce6, or bMSN(CpG/Ce6) were added to the wells and incubated for 6 h. The medium was replaced with fresh cell culture medium and incubated for another 18 h. Finally, the medium was collected and measured by an IL-12p70 and TNF-α ELISA (enzyme-linked immunosorbent assay) kit (R&D System), and BMDCs were harvested and stained with fluorophore-labeled antibodies against CD40, CD80, and CD86. For in vitro PDT, MC-38 cells were plated at 5 × 104 cells per well in 96-well plates and incubated overnight. CpG, Ce6, Adpgk, or bMSN(CpG/Ce6)-Adpgk were added to the wells and incubated for 6 h. The medium was replaced with fresh cell culture medium and incubated for another 18 h. In the laser treatment group, a 660 nm laser with a power density of 10 mW/cm2 was directly applied to cells for 2 min. Finally, the cell viability was measured by the WST-1 assay (Sigma-Aldrich). The method for singlet oxygen detection was reported previously.44 In brief, 5 mg of SOSG (Molecular Probes, USA) was dissolved in 16.5 mL of methanol, and 5 μL of SOSG was added to 1 mL of bMSN(Ce6) or free Ce6 PBS/DMSO solution (DMSO:PBS = 1:9) containing 0.2 μM Ce6. Next, cells were irradiated with a 660 nm laser at a power density of 10 mW/cm2. The fluorescence intensity of SOSG was measured with an excitation and emission wavelength of 495 and 525 nm, respectively.
In Vivo PET Imaging and Biodistribution Study.
Radioisotope 64Cu was purchased from University of Wisconsin, Madison.6 64CuCl2 (74 MBq) was diluted in 0.2 mL of 0.1 M sodium acetate buffer (pH 5.0) and mixed with 0.4 mg of NOTA-bMSN(CpG/Ce6)-Adpgk. The reaction was conducted at 37 °C for 1 h with constant shaking. Then 10 μL of 0.1 M EDTA (ethylenediaminetetraacetic acid) was added and incubated for 15 min to remove free 64Cu. The resulting 64Cu-NOTA-bMSN(CpG/Ce6)-Adpgk were purified by centrifugation (14000g for 15 min) and washed three times with PBS. Free Adpgk peptide was labeled with 64Cu using the same method but purified by centrifugation filtration (2k Da) to remove free 64Cu.
PET scans of MC-38 tumor-bearing mice were performed using a microPET Inveon rodent model scanner (Siemens Medical Solutions USA, Inc.) at various time points after intravenous tail vein injection of 4–6 MBq of 64Cu-NOTA-bMSN(CpG/Ce6)-Adpgk or 64Cu-NOTA-Adpgk. Detailed procedures for data acquisition and region-of-interest (ROI) analysis of PET imaging data have been reported previously.6 Quantitative PET data for tumors and major organs were presented as percentage injected dose per gram of tissue (% ID/g). To correlate the ROI values of PET imaging and distribution of radioactivity in mice, we collected tumors and major organs/tissues at 25 h postinjection, weighed them, and measured the radioactivity of each sample with a gamma counter (PerkinElmer).
In Vivo Immunization and Cancer PDT-Enhanced Immunotherapy Study.
All animal experiments were in accordance with and approved by the University Committee on Use and Care of Animals (UCUCA) at University of Michigan, Ann Arbor. Female C57BL/6 mice of age 6–8 weeks (Jackson Laboratories) were inoculated subcutaneously with MC-38 or B16F10 cells on both sides of the flank. When the primary tumor volume reached ~40–50 mm3, mice were immunized intravenously with the indicated vaccine formulations, including PBS, soluble vaccine (free CpG, Ce6 and Adpgk), bMSN(Ce6), bMSN(CpG/Ce6), and bMSN vaccine (bMSN(CpG/Ce6)-Adpgk). The doses for CpG, Ce6, Adpgk peptide, and bMSN were 20, 30, 19, and 120 μg of bMSNs per mouse, respectively. MSN vaccine was also used as control nanoparticles. After 24 h, a subset of animals received a laser treatment at the tumor area for 15 min with a laser power density of 50 and 100 mW/cm2 for MC-38 and B16F10 tumors, respectively. Tumor volume was measured every other day with the following equation: tumor volume = length × width2 × 0.5. Animals were euthanized when tumors reached 1.5 cm in any dimension.
Immunological Assays.
For the analysis of neoantigen-specific CD8α+ T-cells among peripheral blood, submandibular bleeding was performed 7 day after vaccination, and PBMCs were collected by removing red blood cells with ACK lysis buffer. A tetramer staining assay was performed by peptide-MHC tetramer tagged with PE (H-2Db-restricted ASMTNMELM, from NIH Tetramer Core Facility) as described previously.21 For the analysis of T-cells, DCs, and NK cells in the tumor microenvironment, tumor tissues were harvested 7 days after immunization, cut into small pieces, and digested by incubation with 1 mg/mL type IV collagenase and 0.1 mg/mL DNase I for 20 min at 37 °C. The cell suspensions were filtered using 70 μm strainers and washed with buffer (1% BSA (bovine serum albumin) in PBS). Cells were then stained with the following reagents: CD8α-APC and Adpgk-tetramer-PE for T-cells; CD45-APC, CD11c-FITC, and CD86-PE for DCs; CD45-APC, NK1.1-Percp-Cy5.5, and CD3-FITC for NK cells. Cells were also stained with DAPI and analyzed by a CytoFLEX flow cytometer (Beckman Coulter, Indianapolis, IN, USA). In all flow cytometry analyses, only live and intact cells were analyzed. The ELISOPOT (enzyme-linked immunospot) assay was performed with splenocytes obtained on day 7 after immunization as described previously.45 For histopathological analysis, the major organs were harvested on day 30 after immunization and processed for hematoxylin–eosin staining.
Statistical Analysis.
Sample sizes were chosen based on preliminary data from pilot experiments. For animal studies, the mice were randomized to match similar primary tumor volume, and all procedures were performed in a nonblinded fashion. Statistical analysis was performed with one-way or two-way ANOVA (analysis of variance) with Bonferroni multiple comparisons post-test with Prism 8.0 software (GraphPad Software). Statistical significance is indicated as *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported in part by the NIH (R01AI127070, R01EB022563, R01CA210273, R01CA223804, U01CA210152), MTRAC for Life Sciences Hub, UM Forbes Institute for Cancer Discovery Pilot Grant, and Emerald Foundation. J.J.M. is a Young Investigator supported by the Melanoma Research Alliance (348774), DoD/CDMRP Peer Reviewed Cancer Research Program (W81XWH-16-1-0369), and NSF CAREER Award (1553831). We acknowledge the NIH Tetramer Core Facility (contract HHSN272201300006C) for provision of MHC-I tetramers. Opinions, interpretations, conclusions, and recommendations are those of the author and are not necessarily endorsed by the Department of Defense.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsnano.9b06691.
Data set on in vitro degradation of bMSNs, MSN1, and MSN2; their drug loading and release profiles; in vitro activation and cytokine secretion by BMDCs; in vivo blood circulation half-life curve of 64Cu-NOTA-bMSN-(CpG/Ce6)-Adpgk; safety profiles of bMSN treatment; and therapeutic study in B16F10 tumor-bearing mice (PDF)
The authors declare no competing financial interest.
REFERENCES
- (1).Lucky SS; Soo KC; Zhang Y Nanoparticles in Photodynamic Therapy. Chem. Rev 2015, 115, 1990–2042. [DOI] [PubMed] [Google Scholar]
- (2).Abrahamse H; Hamblin MR New Photosensitizers for Photodynamic Therapy. Biochem. J 2016, 473, 347–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Wang C; Tao H; Cheng L; Liu Z Near-Infrared Light Induced In Vivo Photodynamic Therapy of Cancer Based on Upconversion Nanoparticles. Biomaterials 2011, 32, 6145–6154. [DOI] [PubMed] [Google Scholar]
- (4).Yuan Y; Zhang CJ; Gao M; Zhang R; Tang BZ; Liu B Specific Light-Up Bioprobe with Aggregation-Induced Emission and Activatable Photoactivity for the Targeted and Image-Guided Photodynamic Ablation of Cancer Cells. Angew. Chem., Int. Ed 2015, 54, 1780–1786. [DOI] [PubMed] [Google Scholar]
- (5).Xu C; Shi S; Feng L; Chen F; Graves SA; Ehlerding EB; Goel S; Sun H; England CG; Nickles RJ Long Circulating Reduced Graphene Oxide–Iron Oxide Nanoparticles for Efficient Tumor Targeting and Multimodality Imaging. Nanoscale 2016, 8, 12683–12692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Hong H; Yang K; Zhang Y; Engle JW; Feng L; Yang Y; Nayak TR; Goel S; Bean J; Theuer CP In Vivo Targeting and Imaging of Tumor Vasculature with Radiolabeled, Antibody-Conjugated Nanographene. ACS Nano 2012, 6, 2361–2370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Xu C; Chen F; Valdovinos HF; Jiang D; Goel S; Yu B; Sun H; Barnhart TE; Moon JJ; Cai W Bacteria-Like Mesoporous Silica-Coated Gold Nanorods for Positron Emission Tomography and Photoacoustic Imaging-Guided Chemo-Photo-thermal Combined Therapy. Biomaterials 2018, 165, 56–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Ngwa W; Irabor OC; Schoenfeld JD; Hesser J; Demaria S; Formenti SC Using Immunotherapy to Boost the Abscopal Effect. Nat. Rev. Cancer 2018, 18, 313–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Chen Q; Xu L; Liang C; Wang C; Peng R; Liu Z Photothermal Therapy with Immune-Adjuvant Nanoparticles Together with Checkpoint Blockade for Effective Cancer Immunotherapy. Nat. Commun 2016, 7, 13193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).He C; Duan X; Guo N; Chan C; Poon C; Weichselbaum RR; Lin W Core-Shell Nanoscale Coordination Polymers Combine Chemotherapy and Photodynamic Therapy to Potentiate Checkpoint Blockade Cancer Immunotherapy. Nat. Commun 2016, 7, 12499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Yang G; Xu L; Chao Y; Xu J; Sun X; Wu Y; Peng R; Liu Z Hollow MnO2 as a Tumor-Microenvironment-Responsive Biodegradable Nano-Platform for Combination Therapy Favoring Antitumor Immune Responses. Nat. Commun 2017, 8, 902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Xu J; Xu L; Wang C; Yang R; Zhuang Q; Han X; Dong Z; Zhu W; Peng R; Liu Z Near-Infrared-Triggered Photodynamic Therapy with Multitasking Upconversion Nanoparticles in Combination with Checkpoint Blockade for Immunotherapy of Colorectal Cancer. ACS Nano 2017, 11, 4463–4474. [DOI] [PubMed] [Google Scholar]
- (13).Michot J; Bigenwald C; Champiat S; Collins M; Carbonnel F; Postel-Vinay S; Berdelou A; Varga A; Bahleda R; Hollebecque A Immune-Related Adverse Events with Immune Checkpoint Blockade: a Comprehensive Review. Eur. J. Cancer 2016, 54, 139–148. [DOI] [PubMed] [Google Scholar]
- (14).Yarchoan M; Johnson BA III; Lutz ER; Laheru DA; Jaffee EM Targeting Neoantigens to Augment Antitumour Immunity. Nat. Rev. Cancer 2017, 17, 209–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Yadav M; Jhunjhunwala S; Phung QT; Lupardus P; Tanguay J; Bumbaca S; Franci C; Cheung TK; Fritsche J; Weinschenk T Predicting Immunogenic Tumour Mutations by Combining Mass Spectrometry and Exome Sequencing. Nature 2014, 515, 572–576. [DOI] [PubMed] [Google Scholar]
- (16).Sahin U; Tureci O Personalized Vaccines for Cancer Immunotherapy. Science 2018, 359, 1355–1360. [DOI] [PubMed] [Google Scholar]
- (17).Ott PA; Hu Z; Keskin DB; Shukla SA; Sun J; Bozym DJ; Zhang W; Luoma A; Giobbie-Hurder A; Peter L An Immunogenic Personal Neoantigen Vaccine for Patients with Melanoma. Nature 2017, 547, 217–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Sahin U; Derhovanessian E; Miller M; Kloke B-P; Simon P; Löwer M; Bukur V; Tadmor AD; Luxemburger U; Schrörs B Personalized RNA Mutanome Vaccines Mobilize Poly-Specific Therapeutic Immunity Against Cancer. Nature 2017, 547, 222–226. [DOI] [PubMed] [Google Scholar]
- (19).Li AW; Sobral MC; Badrinath S; Choi Y; Graveline A; Stafford AG; Weaver JC; Dellacherie MO; Shih T-Y; Ali OA A Facile Approach to Enhance Antigen Response for Personalized Cancer Vaccination. Nat. Mater 2018, 17, 528–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Zhu G; Lynn GM; Jacobson O; Chen K; Liu Y; Zhang H; Ma Y; Zhang F; Tian R; Ni Q Albumin/Vaccine Nanocomplexes that Assemble In Vivo for Combination Cancer Immunotherapy. Nat. Commun 2017, 8, 1954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Kuai R; Ochyl LJ; Bahjat KS; Schwendeman A; Moon JJ Designer Vaccine Nanodiscs for Personalized Cancer Immunotherapy. Nat. Mater 2017, 16, 489–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Kuai R; Sun X; Yuan W; Xu Y; Schwendeman A; Moon JJ Subcutaneous Nanodisc Vaccination with Neoantigens for Combination Cancer Immunotherapy. Bioconjugate Chem 2018, 29, 771–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Moon JJ; Suh H; Bershteyn A; Stephan MT; Liu H; Huang B; Sohail M; Luo S; Um SH; Khant H Interbilayer-Crosslinked Multilamellar Vesicles as Synthetic Vaccines for Potent Humoral and Cellular Immune Responses. Nat. Mater 2011, 10, 243–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Nakamura T; Miyabe H; Hyodo M; Sato Y; Hayakawa Y; Harashima H Liposomes Loaded with a STING Pathway Ligand, Cyclic Di-GMP, Enhance Cancer Immunotherapy Against Metastatic Melanoma. J. Controlled Release 2015, 216, 149–157. [DOI] [PubMed] [Google Scholar]
- (25).Koshy ST; Cheung AS; Gu L; Graveline AR; Mooney DJ Liposomal Delivery Enhances Immune Activation by STING Agonists for Cancer Immunotherapy. Adv. Biosyst 2017, 1, 1600013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Xu C; Yang D; Mei L; Li Q; Zhu H; Wang T Targeting Chemophotothermal Therapy of Hepatoma by Gold Nanorods/Graphene Oxide Core/Shell Nanocomposites. ACS Appl. Mater. Interfaces 2013, 5, 12911–12920. [DOI] [PubMed] [Google Scholar]
- (27).Nam J; Son S; Park KS; Zou W; Shea LD; Moon JJ Cancer Nanomedicine for Combination Cancer Immunotherapy. Nat. Rev. Mater 2019, 4, 389–414. [Google Scholar]
- (28).Scheetz L; Park KS; Li Q; Lowenstein PR; Castro MG; Schwendeman A; Moon JJ Engineering Patient-Specific Cancer Immunotherapies. Nat. Biomed. Eng 2019, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Su J; Sun H; Meng Q; Zhang P; Yin Q; Li Y Enhanced Blood Suspensibility and Laser-Activated Tumor-Specific Drug Release of Theranostic Mesoporous Silica Nanoparticles by Functionalizing with Erythrocyte Membranes. Theranostics 2017, 7, 523–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Yang X; Wen Y; Wu A; Xu M; Amano T; Zheng L; Zhao L Polyglycerol Mediated Covalent Construction of Magnetic Mesoporous Silica Nanohybrid with Aqueous Dispersibility for Drug Delivery. Mater. Sci. Eng., C 2017, 80, 517–525. [DOI] [PubMed] [Google Scholar]
- (31).Li Z; Barnes JC; Bosoy A; Stoddart JF; Zink JI Mesoporous Silica Nanoparticles in Biomedical Applications. Chem. Soc. Rev 2012, 41, 2590–2605. [DOI] [PubMed] [Google Scholar]
- (32).Benne N; van Duijn J; Kuiper J; Jiskoot W; Slütter B Orchestrating Immune Responses: How Size, Shape and Rigidity Affect the Immunogenicity of Particulate Vaccines. J. Controlled Release 2016, 234, 124–134. [DOI] [PubMed] [Google Scholar]
- (33).Tang F; Li L; Chen D Mesoporous Silica Nanoparticles: Synthesis, Biocompatibility and Drug Delivery. Adv. Mater 2012, 24, 1504–1534. [DOI] [PubMed] [Google Scholar]
- (34).Shen D; Yang J; Li X; Zhou L; Zhang R; Li W; Chen L; Wang R; Zhang F; Zhao D Biphase Stratification Approach to Three-Dimensional Dendritic Biodegradable Mesoporous Silica Nanospheres. Nano Lett. 2014, 14, 923–932. [DOI] [PubMed] [Google Scholar]
- (35).Cauda V; Schlossbauer A; Bein T Bio-Degradation Study of Colloidal Mesoporous Silica Nanoparticles: Effect of Surface Functionalization with Organo-Silanes and Poly (Ethylene Glycol). Microporous Mesoporous Mater. 2010, 132, 60–71. [Google Scholar]
- (36).Hirosue S; Kourtis IC; van der Vlies AJ; Hubbell JA; Swartz MA Antigen Delivery to Dendritic Cells by Poly (Propylene Sulfide) Nanoparticles with Disulfide Conjugated Peptides: Cross-Presentation and T Cell Activation. Vaccine 2010, 28, 7897–7906. [DOI] [PubMed] [Google Scholar]
- (37).Kwon D; Cha BG; Cho Y; Min J; Park E-B; Kang S-J; Kim J Extra-Large Pore Mesoporous Silica Nanoparticles for Directing In Vivo M2Macrophage Polarization by Delivering IL-4. Nano Lett. 2017, 17, 2747–2756. [DOI] [PubMed] [Google Scholar]
- (38).Fujii S.-i.; Liu K; Smith C; Bonito AJ; Steinman RM The Linkage of Innate to Adaptive Immunity via Maturing Dendritic Cells In Vivo Requires CD40 Ligation in Addition to Antigen Presentation and CD80/86 Costimulation. J. Exp. Med 2004, 199, 1607–1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Ehlerding EB; Chen F; Cai W Biodegradable and Renal Clearable Inorganic Nanoparticles. Adv. Sci 2016, 3, 1500223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Kreiter S; Vormehr M; Van de Roemer N; Diken M; Löwer M; Diekmann J; Boegel S; Schrörs B; Vascotto F; Castle JC Mutant MHC Class II Epitopes Drive Therapeutic Immune Responses to Cancer. Nature 2015, 520, 692–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Ding B; Shao S; Yu C; Teng B; Wang M; Cheng Z; Wong KL; Ma P. a.; Lin J Large-Pore Mesoporous-Silica-Coated Upconversion Nanoparticles as Multifunctional Immunoadjuvants with Ultrahigh Photosensitizer and Antigen Loading Efficiency for Improved Cancer Photodynamic Immunotherapy. Adv. Mater 2018, 30, 1802479. [DOI] [PubMed] [Google Scholar]
- (42).Hu Y; Cai K; Luo Z; Jandt KD Layer-by-Layer Assembly of β-Estradiol Loaded Mesoporous Silica Nanoparticles on Ttanium Substrates and Its Implication for Bone Homeostasis. Adv. Mater 2010, 22, 4146–4150. [DOI] [PubMed] [Google Scholar]
- (43).Lutz MB; Kukutsch N; Ogilvie AL; Rößner S; Koch F; Romani N; Schuler G An Advanced Culture Method for Generating Large Quantities of Highly Pure Dendritic Cells from Mouse Bone Marrow. J. Immunol. Methods 1999, 223, 77–92. [DOI] [PubMed] [Google Scholar]
- (44).Cheng L; Kamkaew A; Sun H; Jiang D; Valdovinos HF; Gong H; England CG; Goel S; Barnhart TE; Cai W Dual-Modality Positron Emission Tomography/Optical Image-Guided Photodynamic Cancer Therapy with Chlorin e6-Containing Nano-micelles. ACS Nano 2016, 10, 7721–7730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Anthony DD; Lehmann PV T-Cell Epitope Mapping Using the ELISPOT Approach. Methods 2003, 29, 260–269. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.