

Abstract
OBJECTIVES
Genetic disorders are a leading contributor to mortality in the neonatal and pediatric intensive care unit (ICU) in the United States. Although individually rare, there are over 6200 single gene diseases, which may preclude a genetic diagnosis prior to ICU admission. Rapid whole genome sequencing (rWGS) is an emerging method of diagnosing genetic conditions in time to affect ICU management of neonates; however its clinical utility has yet to be adequately demonstrated in critically ill children. This study evaluates next-generation sequencing in pediatric critical care.
DESIGN
Retrospective cohort study
SETTING
Single-center PICU in a tertiary children’s hospital
PATIENTS
Children 4 months to 18 years admitted to the PICU who were nominated between July 2016 and May 2018.
INTERVENTIONS
rWGS with targeted phenotype-driven analysis was performed on patients and their parents, when parental samples were available.
MEASUREMENTS AND MAIN RESULTS
A molecular diagnosis was made by rWGS in 17 of 38 children (45%). In four of the 17 patients (24%), the genetic diagnoses led to a change in management while in the PICU, including genome-informed changes in pharmacotherapy and transition to palliative care. Nine of the 17 diagnosed children (53%) had no dysmorphic features or developmental delay.
Eighty-two percent of diagnoses affected the clinical management of the patient and/or family after PICU discharge, including avoidance of biopsy, administration of factor replacement, and surveillance for disorder-related sequelae.
CONCLUSIONS
This study demonstrates a retrospective evaluation for undiagnosed genetic disease in the PICU and clinical utility of rWGS in a portion of critically ill children. Further studies are needed to identify PICU patients who will benefit from rWGS early in PICU admission when the underlying etiology is unclear.
Keywords: whole genome sequencing, genomics, pediatric critical care, diagnostic utility, clinical utility, precision medicine
INTRODUCTION
The rapid pace of advancement in understanding the molecular basis of genetic diseases has led to a growing appreciation for the prevalence of these disorders and the impact of genetic diagnosis on provision of medical care. Though the incidence of individual monogenic diseases is low, more than 6250 disorders have been described and over 250 new monogenic diseases are discovered each year (1, 2). Genetic disorders are a leading contributor to mortality in the NICU and PICU in the United States (3–11).
Provision of optimal clinical care for affected children depends on timely ascertainment of the underlying genetic cause, facilitating a shift from empiric treatment to definitive management of an identified disorder when feasible (12–14). Previously, the slow turnaround time for genetic tests precluded their real-time application to critical care medicine. Recently, technological advances have yielded rapid whole genome sequencing (rWGS) coupled with a focused phenotype-driven analysis of WGS data, which is capable of making a provisional molecular diagnosis in one day (15–19). Thus, applicability of WGS to critical care medicine may no longer be hindered by time constraints.
The use of WGS as a comprehensive genetic testing approach has shown superiority for detecting pathogenic variation when compared to more targeted standard genetic approaches and can detect copy number variation equivalent in sensitivity to microarray (20–23). When used appropriately in combination with clinical information, WGS is able to interrogate the genome for disorders that may not have been included in the treating clinician’s differential diagnosis (13, 17). This includes analyzing recently discovered genes, genes not found in standard gene panels, and identification of atypical and previously unrecognized presentations of classic disorders (13, 20, 24). Although its technical reliability has been established in multiple studies (20–23), one reservation surrounding WGS is the possibility that a pathogenic variant will be identified that is not the cause of the particular disease process in question, potentially resulting in incorrect management decisions, psychological distress, and concerns surrounding insurability. Focused analysis of WGS data on a specific clinical phenotype (as done in rWGS) helps to mitigate the risk of “false positives”. Thus, an accurate bioinformatic interface that synthesizes the clinical presentation and the genetic data provides the best opportunity for the proper interpretation and appropriate use of pertinent rWGS findings. Overall, the benefits make rWGS an attractive testing approach for certain high-acuity diagnostic challenges in the intensive care unit.
Recent studies have demonstrated the clinical utility of WGS in the NICU (13–19). Thirty-six to 73% of infants tested received a molecular diagnosis, and changes in management occurred in up to 72% of diagnosed patients (19). Thirty-eight to 45% of diagnoses were disorders that had not been considered by clinicians at the time of enrollment, due to atypical or early presentation of disease (16). However, only one of these studies included patients admitted to the PICU, and all were less than four months old. Applicability of rWGS to critically ill children outside of the NICU is just beginning to be explored. Mestek-Boukhibar et al recently evaluated utility of rWGS in a cohort of critically ill children in the PICU in the United Kingdom (25). Of 24 patients tested, 42% received a molecular diagnosis and 30% of those were reported to have clinical utility (25). We hypothesized that rWGS would have similar clinical utility in PICU patients from four months to 18 years of age.
METHODS
Study Design
We retrospectively analyzed an rWGS cohort of PICU patients in order to evaluate patient characteristics and clinical utility at a tertiary PICU in the United States. The outcomes studied were changes in PICU management, palliative care decisions, non-ICU clinical management, and family screening. This study was approved by the institutional review board at the University of California San Diego and a non-significant risk determination lead to a waiver of need for an investigational device exemption by the Food and Drug Administration (ClinicalTrials.gov ). Inpatient children in the PICU at Rady Children’s Hospital that did not have an etiologic diagnosis for their illness were identified by a PICU physician or subspecialty consultant between July 2016 and May 2018. The majority (74%) of patients who received rWGS were nominated by pediatric intensivists (Figure 1). Inclusion criteria were: age 4 months to 18 years, suspicion for an underlying monogenic disease, and ability to obtain parental consent in English or Spanish. Exclusion criteria were death prior to study enrollment or a nongenetic alternative etiology was diagnosed. Nominations were screened by the Senior Medical Director at Rady Children’s Institute of Genomic Medicine. Written informed consent was obtained from the parent or guardian. During the time period in which this study was conducted, rWGS testing was available only as part of a research protocol. This cohort represents all patients in the PICU over four months of age that underwent rWGS during this time period.
Figure 1.
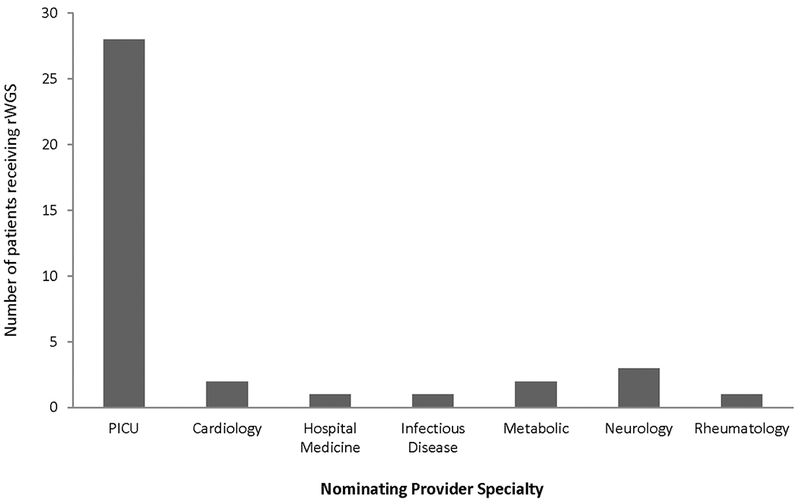
Number of probands nominated for rWGS by specialty
Analysis Pipeline
Complete methods are described in the supplementary digital content, including sequencing and bioinformatics, phenotyping information, variant filtering, variant interpretation, and statistical analysis. Briefly, rWGS with targeted phenotype-driven analysis was performed. If a potentially causative variant was identified, literature curation was performed with regard to evidence of pathogenicity. All identified causative variants were confirmed by orthogonal methods. No variants failed confirmation and no false positives were detected. In all cases, rWGS analysis also found variants of uncertain significance that did not explain the etiology of the patient’s illness and/or lacked sufficient evidence of pathogenicity, and these were not reported. Per IRB protocol, only pathogenic and likely pathogenic variants were reported (variants of uncertain significance could not be reported). The reports provided to treating clinicians were limited to confirmed variants that explained the presenting phenotype of the patient’s acute illness. Additionally, for well-characterized Mendelian disorders, diagnostic findings were only reported if identified pathogenic variants or likely pathogenic variants were consistent with classical inheritance pattern. The analysis pipeline utilized was specifically designed to identify variants causal for the patients’ presenting symptoms and not to investigate other incidental aspects of the genome. During the consent process, families were offered the choice to opt out of receiving medically actionable incidental findings that were inadvertently discovered. All families received genetic counseling.
Clinical Utility
Acute clinical utility of diagnoses (i.e. implementation of precision medicine interventions) was assessed according to ACMG recommendations (26), and impact on outcomes was evaluated by interviews with treating physicians and/or consensus determinations of at least two physicians, of whom one was a relevant pediatric subspecialist and one a medical geneticist. A modified Delphi method was used to determine consensus for counterfactual trajectories (Supplemental Methods). Outcomes were assessed until September 11 2018.
RESULTS
Forty-nine children were nominated between July 2016 and May 2018. Forty children were eligible and 38 were consented and received rWGS (Figure 2). Of those 38 children, rWGS was performed on 24 trios, 4 parent-child duos, and 10 singletons. The average age of the probands was 5.73 years (range 4 months to 17 years; Figure 3A) and the median was 2.96 years. Forty-two percent were of Hispanic/Latino descent (Table 1), in keeping with population characteristics of the greater San Diego area. The most common primary reasons for PICU admission were respiratory failure (18%), shock (16%), altered mental status (13%), and cardiac arrest (13%) (Table 1). Seventy-one percent of patients (27 of 38) needed positive pressure ventilation, with 85% (23 of 27) requiring endotracheal intubation (Table 2). Fifty percent received ionotropic support and overall mortality during PICU admission was 13% (Table 2).
Figure 2:
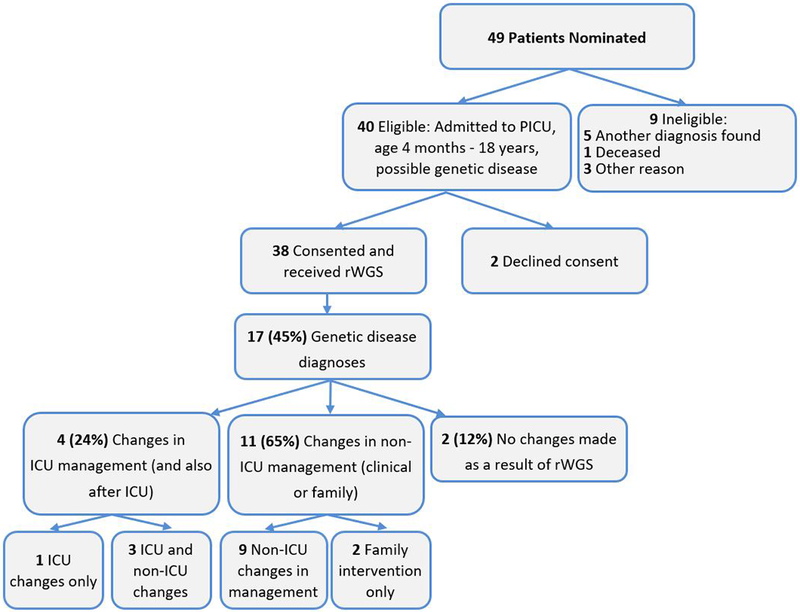
Flow diagram of the proportion of PICU patients who were enrolled, received genetic disease diagnoses by rWGS, and had consequent changes in management (precision medicine)
Figure 3.
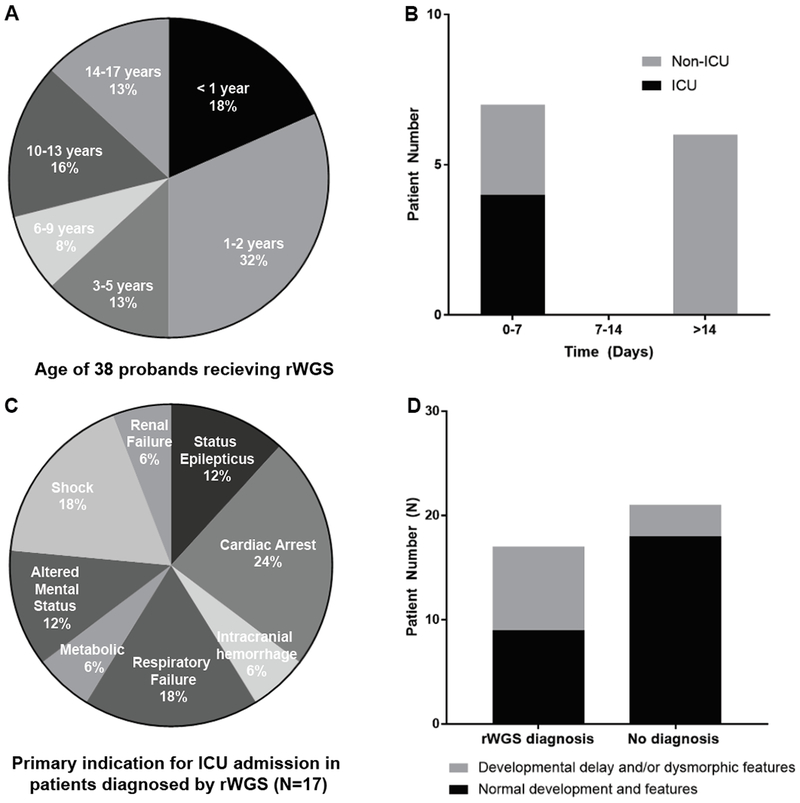
Characteristics of PICU cohort. A, Age of 38 probands receiving rWGS. B, Time from diagnosis to change in clinical management. C, Primary indication for PICU admission in 18 patients in whom a diagnosis was made. D, Presence or absence of developmental delay and/or dysmorphic features at the time of PICU admission.
Table 1:
Demographic and clinical characteristics of the 38 probands
| Demographics | Rapid WGS (n=38) | Diagnostica Rapid WGS (n=17) | Negative Rapid WGS (n=21) | |
|---|---|---|---|---|
| Sex | Female | 19 (50%) | 8 (47%) | 11 (52%) |
| Male | 19 (50%) | 9 (53%) | 10 (48%) | |
| Race and Ethnicity | Caucasian | 10 (24%) | 4 (24%) | 6 (29%) |
| Hispanic/Latino | 16 (42%) | 9 (53%) | 7 (33%) | |
| African/ African American | 2 (5%) | 1 (6%) | 1 (5%) | |
| Asian/Native American/Pacific Islander | 2 (5%) | 0 (0%) | 2 (10%) | |
| Other (including multi-racial) | 8 (21%) | 3 (18%) | 5 (24%) | |
| Indication for ICU admission | Status epilepticus | 3 (8%) | 2 (12%) | 1 (5%) |
| Respiratory failure | 7 (18%) | 3 (18%) | 4 (19%) | |
| Cardiac arrest | 5 (13%) | 4 (24%) | 1 (5%) | |
| Altered mental status | 5 (13%) | 2 (12%) | 3 (14%) | |
| Intracranial hemorrhage | 3 (8%) | 1 (6%) | 2 (10%) | |
| Metabolic derangement | 3 (8%) | 1 (6%) | 2 (10%) | |
| Acute liver failure | 1 (3%) | 0 (0%) | 1 (5%) | |
| Pulmonary hemorrhage | 1 (3%) | 0 (0%) | 1 (5%) | |
| Cavernous sinus thrombosis | 1 (3%) | 0 (0%) | 1 (5%) | |
| Atrial tumor | 1 (3%) | 0 (0%) | 1 (5%) | |
| Shock | 6 (16%) | 3 (18%) | 3 (14%) | |
| Acute heart failure | 1 (3%) | 0 (0%) | 1 (5%) | |
| Renal failure | 1 (3%) | 1 (6%) | 0 (0%) | |
Values shown are number (percentage) of subjects, except as indicated;
Includes partial diagnoses
Table 2:
Comparison of diagnosis, clinical utility, and characteristics between NICU and PICU patients who received rWGS
| Comparative Factor | All patients | Diagnosed patients | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| NICU (n=24) | PICU (n=38) | p | NICU (n=10) | PICU (n=17) | p | |||||
| Diagnostic Utility | 10 | 41.7% | 18 | 47.4% | 0.66 | |||||
| Clinical Utility | 7 | 29.2% | 13 | 34.2% | 0.68 | 7 | 70.0% | 12 | 70.6% | 1 |
| Multiple congenital anomalies | 9 | 37.5% | 5 | 13.2% | 0.03 | 3 | 30.0% | 3 | 17.6% | 0.64 |
| Primary system involved | ||||||||||
| Neurologic | 6 | 25.0% | 8 | 21.1% | 0.72 | 3 | 30.0% | 5 | 29.4% | 1 |
| Hepatic | 2 | 8.3% | 1 | 2.6% | 0.55 | 1 | 10.0% | 0 | 0.0% | 0.37 |
| Cardiac | 2 | 8.3% | 6 | 15.8% | 0.47 | 0 | 0.0% | 5 | 29.4% | 0.12 |
| Hematologic | 1 | 4.2% | 1 | 2.6% | 1 | 0 | 0.0% | 0 | 0.0% | 1 |
| Gastrointestinal | 1 | 4.2% | 0 | 0.0% | 0.39 | 1 | 10.0% | 0 | 0.0% | 0.37 |
| Endocrine/Biochemical | 1 | 4.2% | 3 | 7.9% | 0.65 | 1 | 10.0% | 1 | 5.9% | 1 |
| Musculoskeletal | 1 | 4.2% | 0 | 0.0% | 0.39 | 1 | 10.0% | 0 | 0.0% | 0.37 |
| Pulmonary | 1 | 4.2% | 2 | 5.3% | 1 | 0 | 0.0% | 0 | 0.0% | 1 |
| Support required | ||||||||||
| Ionotropic Support | 12 | 50.0% | 19 | 50.0% | 1 | 4 | 40.0% | 6 | 35.3% | 1 |
| Respiratory Support | 23 | 95.8% | 27 | 71.1% | 0.02 | 10 | 100.0% | 11 | 64.7% | 0.06 |
| Intubated | 19 | 79.2% | 23 | 60.5% | 0.12 | 7 | 70.0% | 9 | 52.9% | 0.45 |
| ECMO | 2 | 8.3% | 4 | 10.5% | 1 | 2 | 20.0% | 2 | 11.1% | 0.61 |
| Antimicrobial Treatment | 21 | 87.5% | 33 | 86.8% | 1 | 10 | 100.0% | 14 | 82.4% | 0.27 |
| >5 Subspecialty consults | 10 | 41.7% | 11 | 28.9% | 0.41 | 5 | 50.0% | 2 | 11.8% | 0.06 |
| Mortality | 6 | 25.0% | 5 | 13.2% | 0.31 | 1 | 10.0% | 0 | 0.0% | 0.37 |
Diagnostic Sensitivity of rWGS
rWGS diagnosed a genetic disease in 17 of 38 critically ill children (45%; Tables 3, S1). Diagnostic rates were highest in patients whose primary reason for PICU admission was cardiac arrest, shock, status epilepticus, and renal failure (Figure 3C). Fifty-three percent of diagnosed patients had no known developmental delay or dysmorphic features (Figure 3D). Eighty-two percent of diagnosed patients did not have multiple congenital anomalies (Table 2). Of the seventeen diagnosed patients, the genetic result was felt to completely explain the phenotype in fifteen and provide a partial explanation of disease for two (Table 3). A partial diagnosis designation was assigned if the identified mutation did not provide a satisfactory explanation for the patient’s complete phenotype (19, 25). For example, the PUS3 variant for patient 6205 explained the child’s intellectual disability and developmental delay, but thus far has not been associated with Wilms tumor.
Table 3:
Phenotype and inheritance pattern in children diagnosed (Dx) with genetic disorders by rWGS (N=17)
| Subject ID | Age | Sex | Dx Type | Phenotype | Diagnosis Name(s) | Gene | Inheritance | de novo or inherited | Variant Chromosomal (Chr)/Gene (c.) Coordinate(s) | HGVS nomenclature (c. and p.) | Zygosity | Minor Allele Frequency (MAF) in ExAC | HPO terms |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 6007 | 14 mo | F | C | Refractory complex partial epilepsy | Early infantile epileptic encephalopathy | PCDH19 | X-linked | de novo | chrX:88361395- 99758442 11.4 Mb deletion (Xq21.3q22.1) |
NA | heterozygous | NA | HP:0002373 |
| 6031 | 16 yr | F | C | Ventricular fibrillation, cardiac arrest | Catecholaminergic polymorphic ventricular tachycardia | RYR2 | AD | de novo | chr1:237632425 | c.1646C>T (p.Ala549Val) |
heterozygous | NF (not found) | HP:0001695 |
| 6052 | 3 yr | F | C | Complex partial epilepsy and developmental delay, new onset lactic acidosis | Metabolic encephalomyopathic crises, recurrent, with rhabdomyolysis, cardiac arrhythmias, and neurodegeneration (MECRCN) | TANGO2 | AR | Inherited | chr22:20049207; chr22:20029260- 20062700×1 22q11.21 |
c.605+1G>A; del exons 3-9 |
compound heterozygous | 0.00032, NA |
HP:0011342 HP:0006846 HP:0030235 HP:0003128 |
| 6118a | 16 mo | M | C | Idiopathic pulmonary hypertension | Hereditary hemorrhagic telangiectasia, type 2 | ACVRL1 | AD | de novo | chr12:52314615 | c.1450C>T (p.Arg484Trp) |
heterozygous | NF | HP:0002092 |
| 6147 | 4 mo | M | C | Bilateral sensorineural hearing loss, transaminitis, anemia, new onset lethargy | Sideroblastic anemia with B-cell immunodeficiency, periodic fevers, and developmental delay (SIFD) | TRNT1 | AR | Inherited | chr3:3182294 | c.443C>T (p.Ala148Val) |
homozygous | 0.00002 | HP:0002910 HP:0003128 HP:0008619 HP:0001935 |
| 6153a | 5 yr | F | C | Hypoparathyroidism, hypocalcemic status epilepticus | Autoimmune polyendocrinopathy syndrome, type I | AIRE | AR | Inherited | chr21:45706575; chr21:45713043 |
c.268T>C (p.Tyr90His); c.1265delC (p.Pro422LeufsTer58) |
compound heterozygous | NF, 0.00003 |
HP:0000829 HP:0002901 |
| 6159a | 5 yr | F | P | Pan serositis (ascites, peritonitis, pleural effusion, pericardial effusion), glomerulonephritis | Thin basement membrane nephropathy/ Alport syndrome | COL4A4 | AD | Inherited | chr2:227872828 |
c.4715C>T (p.Pro1572Leu) |
heterozygous | 0.00014 | HP:0000123 HP:0001903 HP:0003256 HP:0012115 HP:0031123 |
| 6180a | 19 mo | M | C | Ecthyma gangrenosum, sepsis | Agammaglobulinemia, X-linked | BTK | X-linked | de novo | chrX: 100615605 | c.726dupT (p.Ile243TyrfsTer15) |
heterozygous | NF | HP:0002721 HP:0001875 HP:0003256 HP:0010703 HP:0100806 |
| 6183 | 5 mo | M | C | Hypotonia, hypopnea, failure to thrive, and abnormal eye movements | Rett syndrome | MECP2 | X-linked | de novo | chrX:153296525 | c.789dupC (p.Gly264ArgfsTer7) |
heterozygous | NF | HP:0001319 HP:0040213 |
| 6193 | 22 mo | F | C | Distal arthrogryposis, developmental delay, apnea | Congenital contractures of the limbs and face, hypotonia, developmental delay (CLIFAHDD) | NALCN | AD | de novo | chr13:101828691 | c.1799A>G (p.Asp600Gly) |
heterozygous | NF | HP:0001263 HP:0005684 |
| 6205 | 11 yr | M | P | Wilms tumor, mental retardation, seizures | Mental retardation, autosomal recessive | PUS3 | AR | Inherited | chr11:125766179 | c.1A>G (p.Met1?) |
homozygous | 0.00002 | HP:0001249 HP:0000252 HP:0001003 HP:0002667 HP:0004322 |
| 6207a | 9 mo | M | C | Spontaneous intracranial hemorrhage | Factor XIIIA deficiency | F13A1 | AR | Inherited | chr6:6182326 | c.1352_1353delAT (p.His451ArgfsTer29) |
homozygous | NF | HP:0001048 HP:0001269 |
| 7002 | 10 yr | M | C | Ventricular fibrillation, cardiac arrest | Long QT syndrome, type 1 | KCNQ1 | AD | Inherited | chr11:2591949 | c.573_577delGCGCT (p.Arg192CysfsTer91) |
heterozygous | 0.00003 | HP:0001657 |
| 7004 | 14 yr | M | C | Ventricular fibrillation, cardiac arrest, hypertrophic cardiomyopathy | Cardiomyopathy, hypertrophic | MYH7 | AD | Inherited | chr14:23900677 | c.746G>A (p Arg249Gln) |
heterozygous | NF | HP:0001639 |
| 7031 | 5 yr | F | C | Refractory epilepsy, intermittent R hemiplegia | Alternating hemiplegia of childhood | ATP1A3 | AD | unknown | chr19:42490329 | c.449C>T (p.Ser150Phe) |
heterozygous | NF | HP:0001250 |
| 7039 | 11 mo | F | C | Apnea, hypotonia, global developmental delay, decreased cerebral white matter | Progressive encephalopathy with brain atrophy and thin corpus callosum | TBCD | AR | unknown | chr17:80763767; chr17:80828121 |
c.967C>T (p.Arg323Ter); c.1340C>T (p.Ala447Val) |
compound heterozygous | 0.00002, NF | HP:0002194 HP:0001601 HP:0002500 HP:0010314 HP:0012411 |
| 7043 | 17 yr | M | C | Ventricular fibrillation, cardiac arrest | Cardiomyopathy | ACTC1 | AD | Unknown | chr15:35086982 | c.28C>A (p.Leu10Met) |
heterozygous | 0.00003 | HP:0001638 HP:0001663 HP:0001695 HP:0004756 HP:0030149 |
C: Complete diagnosis; P: Partial diagnosis; AD: autosomal dominant; AR: autosomal recessive; XLD: X-linked dominant; NF: not found in ExAC.
Previously published cases (44, rS23-rS26)
The inheritance patterns for probands with identified genetic disorders are reported in Table 3. Six mutations were de novo, eight were inherited, and in three patients inheritance was unable to be determined. Additionally, the number of each variant type identified is provided in Table S2 (Supplemental Digital Content).
Clinical Utility
Clinical utility of rWGS results was determined based on the guidelines outlined in the ACMG 2015 policy statement on clinical utility (26). Specific changes in ICU management occurred as a result of molecular diagnosis in four of the 17 diagnosed children (24%; 11% of tested children; Table 4, Figure 3B). Three patients had medication changes made during their PICU admission directly due to the genetic result. For 14 of the 17 children (82%; 37% of tested children), the genetic diagnosis resulted in a subacute (non-ICU) change in the clinical management of the patient or had important implications for family screening (Table 4, Supplemental Digital Content). These subacute changes were judged by consensus of an expert panel using a modified Delphi method (Supplemental Methods).
Table 4:
Patient and family interventions secondary to genomic diagnosis
| Subject ID | Causal Gene | ICU Intervention | Non-ICU Intervention | Family Intervention |
|---|---|---|---|---|
| 6007 | PCDH19 | Pulse methylprednisolone | ||
| 6031 | RYR2 | Flecainide initiated (CPVT-RYR2 indication) | Exercise testing/ECG for immediate family | |
| 6052 | TANGO2 | Riboflavin initiated, ubiquinol and B50 increased; Carries letter describing diagnosis/treatment recommendations. subsequent acute encephalopathic episode improved secondary to recommendations | ||
| 6118 | ACVRL1 | Brain MRI every 3-5 years as surveillance to monitor for intracranial AVMs | ||
| 6147 | TRNT1 | Palliative care, code status modified to DNR | ||
| 6153 | AIRE | Periodic screening for adrenal insufficiency | ||
| 6159 | COL4A4 | Avoided renal biopsy. Ongoing screening for vision problems (keratoconus), neurosensory hearing loss, and hypertension/kidney failure | ||
| 6180 | BTK | IVIG to maintain IgG > 800 mg/dL | Avoidance of live vaccines, risk of infection | 3 yr old male sibling tested for XLA(negative) |
| 6183 | MECP2 | Palliative care, discharged on 1L/min nasal cannula oxygen for apnea | ||
| 6193 | NALCN | Palliative care initiated, discharged on BiPAP | ||
| 6207 | F13A1 | Factor XIII A-subunit replacement administered monthly | ||
| 7002 | KCNQ1 | Nadolol initiated | Mother had ECG with upper limit of normal QTc, referred for genetic testing | |
| 7004 | MYH7 | Maternal variant, mother referred for echocardiogram | ||
| 7039 | TBCD | Palliative care, home supportive services initiated | ||
| 7043 | ACTC1 | Echocardiogram and ECG ordered for female sibling |
Cases with changes in PICU management
Patient 6031 was a previously healthy 16 year-old who had sudden cardiac arrest at home. The presenting rhythm was ventricular fibrillation, which was successfully defibrillated. During the post-arrest recovery period, electrocardiogram and echocardiogram were unrevealing of potential etiologies. She was treated empirically with lidocaine and esmolol. rWGS identified a de novo, likely pathogenic missense variant in RYR2 (c.1646C>T;p.Ala549Val), which allowed prompt tailoring of care for catecholaminergic polymorphic ventricular tachycardia (CPVT), specifically, to the transition to longer-acting beta-blockade with nadolol and addition of flecainide for the stabilizing effect on sarcoplasmic calcium release (27). High probability of the diagnosis of CPVT also allowed formulation of a disease-specific algorithm to treat potential breakthrough arrhythmias, including sedation and avoidance of calcium or vasopressors. Though this RYR2 mutation would have been discovered through standard genetic testing, the turnaround time (three to eight weeks) would have precluded tailoring initial treatment to this patient’s specific arrhythmia.
Patient 6180 was a previously healthy 19 month-old male admitted with pseudomonal septic shock. An immunodeficiency was suspected and lymphocyte subset analyses suggested a lack of B cells as an etiology, but sequencing the BTK gene to confirm X-linked agammaglobulinemia (XLA) typically takes four to six weeks. rWGS identified a de novo, pathogenic frameshift variant in BTK (c.726dupT;p.Ile243TyrfsTer15) in four days. Prior to molecular diagnosis, the consulting immunologist recommended obtaining IgG levels every seven days and replacing for a level less than 400 mg/dL. With knowledge of the BTK mutation, the recommendation was to obtain levels every 24 to 48 hours during the period of severe sepsis and administer IVIG if under 800 mg/dL. This resulted in ten administrations of IVIG over the first four weeks of PICU admission.
Patient 7002 was a previously healthy 10 year-old found unresponsive in a pool. The presenting rhythm was ventricular fibrillation, which was successfully defibrillated. ECGs demonstrated prolonged QTc (510-560ms by Bazett’s formula), though the specificity of this finding was diminished in setting of post-arrest acidosis and therapeutic hypothermia (28). rWGS results revealed a maternally-inherited, pathogenic frameshift variant in KCNQ1 (c.573_577delGCGCT;p.Arg192CysfsTer91). The molecular diagnosis permitted prompt transition to first-line medical therapy with nadolol (29). Data is emerging that even within the spectrum of long QT syndrome, knowledge of the specific causal gene (KCNQ1, KCNH2, or SCN5A in 80-90% of cases) affects the decision of whether or not to initiate a beta blocker, and if used, which specific beta blocker will be most effective (30). For patient 7002, nadolol was specifically chosen as the most appropriate therapy because the mutation was in KCNQ1 (30). A standard gene panel would generally be performed if the QTc on ECG remained prolonged during the post-arrest recovery period and would have identified this KCNQ1 variant, but the turnaround time for is generally three to eight weeks, and thus would not have been available at the time of critical clinical decision-making. Additionally, a substantial number of patients with Long QT syndrome have a normal QTc on resting ECG, including this patient’s mother (31).
For Patient 6193, molecular diagnosis facilitated the transition to palliative care during the concurrent ICU admission. This was a 22 month-old female with a history of distal arthrogryposis and developmental delay who was admitted after a cyanotic episode at home. Her parents reported prior apneic spells during sleep, such that they slept next to her so they could periodically stimulate her breathing. In the hospital, evaluation revealed both central and obstructive apnea. Bi-level positive pressure ventilation (BiPAP) was initiated during sleep with improvement. However, without an explanation for the disordered breathing, her parents were reluctant to make decisions about goals of care. rWGS revealed a de novo, likely pathogenic missense variant in NALCN (c.1799A>G;p.Asp600Gly), which causes a syndrome of Congenital Contractures of the Limbs, Hypotonia, and Developmental Delay (CLIFAHDD) as well as respiratory insufficiency in some patients (32). This syndrome has only been recently described and could not have been identified without WGS as no commercial test is available. The molecular diagnosis facilitated engagement of palliative care and the transition to planning for home BiPAP prior to the patient leaving the ICU. The palliative care physician noted that prior to molecular diagnosis, the parents stated that the hardest thing was not knowing exactly what was wrong with their daughter, the father stated, “it’s like they are guessing” (referring to the physicians). After the molecular diagnosis of CLIFAHDD was conveyed to her parents, their priorities shifted from searching for answers to taking her home, keeping her comfortable, and treating her apnea with Bipap.
Time to Molecular Diagnosis
The turnaround time from receipt of specimen to delivery of a result averaged 13.6 days (Supplemental Digital Content, Table S3; range 1 to 56 days). The turnaround time for the cases in 2016 and early 2017 was more variable while the workflow and pipeline were becoming established.
Comparison to rWGS in NICU patients
Our diagnostic rate was similar to that of a recently published cohort of NICU patients undergoing rWGS as part of a randomized controlled trial (Table 2; 19). There was not sufficient evidence to detect differences between NICU and PICU cohorts in terms of inotropic support, endotracheal intubation, extracorporeal membrane oxygenation, or antimicrobial treatment (Table 2). While there was not sufficient power to detect a difference in the level of patient complexity (measured by number of subspecialty consults), we see a trend that PICU patients that had six or more subspecialty services involved were less likely to be diagnosed by rWGS than NICU patients (6% vs 50%; p=0.06).
DISCUSSION
The pace of clinical decision-making for critically ill children requires expeditious changes in management and therefore any new diagnostic test must likewise be available in a timely fashion to be practically useful in the practice of critical care medicine. Only recently, with the advent of rapid whole genome sequencing has technology for genetic testing reached this threshold (16, 17, 20). Recent studies have established that rWGS is rapid enough to facilitate interventions leading to decreased morbidity and mortality in critically ill infants under four months of age in the NICU (13, 16, 17, 19, 33). Utility for acutely ill children outside of the neonatal ICU is just beginning to be explored (25). This study endeavored to determine the rate of molecular diagnosis and the clinical utility of rWGS for a retrospective cohort of 38 patients admitted to the PICU.
Rapid WGS identified a genetic disease in seventeen of 38 critically ill children (45%), which is consistent with previous outcome reports for WGS and whole exome sequencing (WES). In published studies, 25-45% of patients received a molecular diagnosis (13, 34–37), as did 36 to 73% of NICU cohorts (13, 15, 19). In 2018, Mestek-Boukhibar et al. in the United Kingdom (U.K.) published the first true cohort of rWGS in 24 PICU patients and established a molecular diagnosis in 42% (25). The patients’ average age in our study was 5.73 years, with a median of 2.96 years and a range of 4 months to 17 years (Figure 3A). This substantially separates our cohort from previously described critically ill infants analyzed by rWGS (13, 16, 19, 24) and is also older on average than the mean age of 15.86 months (median of 2.5 months) in the U.K. study (25). Direct comparison of our patients was made to a cohort of NICU patients at the same institution who recently received rWGS as part of a separate clinical trial (19) and confirmed that acuity of critical illness, as indicated by requirement for ionotropic support, significant respiratory support, extracorporeal membrane oxygenation, and overall mortality did not differ significantly between critically ill neonates (NICU) and critically ill infants and children (PICU). Larger cohorts of both NICU and PICU patients are needed to determine if these trends persevere in an adequately powered prospective study.
WGS has shown superiority in detecting pathogenic variation when compared to more targeted standard genetic approaches (21, 22) and can detect copy number variation equivalent in sensitivity to microarray (23), making it an attractive testing approach in the intensive care unit, as the turnaround time is significantly faster and may eliminate the need for multiple tests. Historically, WES was more commonly used, but recent ICU studies have preferentially used WGS (16, 19, 24, 25) for diagnosis. WES primarily examines exons, while WGS analyzes exons and approximately 90% of the genome (20). Recent meta-analysis did not find a significant difference between the two technologies, though WGS has the inherent ability to detect intronic variants, single nucleotide variants in noncoding RNA, small copy number variants (CNVs), and mitochondrial DNA variants that will not be captured by WES (20, 22). These diagnostic advantages along with the faster turnaround time (despite larger amount of data to analyze) are predicted to shift testing preferentially in favor of WGS despite its higher cost (20, 22). WGS diagnosis with automated phenotyping and interpretation using natural language processing software is further decreasing time to diagnosis (38).
The impact of rWGS on patient care was evaluated for clinical utility (19, 26). In this cohort, 24% of diagnosed patients had a resultant change in the clinical care provided during ICU admission directly as a result of rWGS diagnosis, including three genome-informed changes in pharmacotherapy. Also reported here are the documented alterations to medical management that occurred in 82% of patients after transfer out of the ICU, as judged by a Delphi panel (Supplemental Digital Content). Previous studies of critically ill infants have found clinical utility rates of 49 to 72% (13, 16, 19, 24). In the U.K. PICU cohort, diagnostic utility was calculated at 30%, though no specific distinction was made between ICU and non-ICU changes in management (25). Which subsets of PICU patients will benefit from rWGS and lead to active changes in management is an area which will become increasingly pertinent as testing continues to become less expensive and more rapid.
An interesting distinction between the U.K. cohort and this study are the notable differences in nominating clinicians. While the U.K. patients were mostly nominated for rWGS by a variety of subspecialty consultants (2½4 or 88%; 25), the majority of patients in this study were nominated for rWGS by a treating intensivist (28/38 or 74%, Figure 1). Though only an observation presently, it would be interesting to parse out in future studies whether nomination by an intensivist portends any relationship, positive or negative, to diagnostic or clinical utility in critically ill children.
As exemplified by the NALCN case, for many newly described genetic disorders there are no commercial tests available, essentially making diagnosis impossible without WGS/WES (15). Supplemental Digital Content Table S4 illustrates the exhaustive and inconclusive testing that some probands received prior to undergoing rWGS. rWGS is especially powerful for critically ill patients who are in need of a timely diagnosis and particularly useful for those disorders lacking a physically visual phenotype. In this cohort in particular, it is worth noting that only 18% of patients diagnosed by rWGS had multiple congenital anomalies. In multiple previous WGS/WES studies, 49-64% of patients had multiple congenital anomalies and/or neurodevelopmental delay (22, 34, 35). For some of the significant molecular diagnoses in this study, including RYR2, KCNQ1, F13A1, and ACVRL1, neither the patient’s physical appearance nor family history would have prompted the treating intensivist to search for a genetic disorder. Rather, the presenting symptoms suggested a potential genetic etiology. Though some of these molecular diagnoses would have potentially been identified in two or three months on gene panels ordered based on the patient’s presenting symptoms, we found that time-to-diagnosis is clinically relevant in certain cases. The discovery of approximately 20 new genetic diseases every month (1) makes it increasingly difficult to exclude the possibility of monognenic disease based on negative family and developmental histories.
Whole genome and exome sequencing identify atypical and heretofore unknown presentations of genetic disorders. For example, the patient in this cohort who had a MECP2 variant was male and presented when he was 5 months old, which is younger than the average age at diagnosis of 3.8 years for atypical Rett syndrome.
The data presented here quantify the molecular diagnostic rate and clinical utility of rWGS in a cohort of 38 children admitted to the pediatric ICU. Additional studies are needed to confirm these data. Our study has some notable limitations, in addition to those already touched upon. The sample size of 38 patients, and although similar in size to recent ICU trials (16, 19, 25), renders the determination of which ICU patients would benefit from rages impossible. Larger, preferably prospective, trials in the PICU will be required to further investigate the findings from this study. WGS itself has inherent limitations, including challenges in identifying trinucleotide repeat disorders and disease-causing deep intronic mutations (39). Additionally, though one of the strengths of WGS is hypothesis-free interrogation of the genome, an accurate phenotype is critical to refining the HPO terms used in genomic data analysis (25). Though not a specific limitation of this paper per se, there are certain instances in which standard genetic or biochemical testing may be the more suitable step in evaluation of a disorder. For example, if there is a high suspicion for an amino acid disorder in an acutely ill patient (perhaps due to an abnormal newborn screen or a known affected sibling), obtaining basic plasma amino acid and urine organic acid levels would likely be a more appropriate initial choice. This is potentially also the case for other situations in which one specific monogenic disease is highly likely to be causing the acute illness in question.
Another recurring concern regarding WGS/WES is the challenge that variants of uncertain significance and incidental findings present. The ACMG has identified 59 genes for adult-onset conditions recommended for inclusion on all clinical WES/WGS reports, with the goal of facilitating preventative treatment, despite lack of relationship between these genes and a given patient’s clinical phenotype (birthing the term “incidental finding”; 40, 41). Controversy surrounding this recommendation persists, especially as it applies to children, due to a pretest probability for the 59 ACMG genes of less than one in 1000 and thus an estimated twenty false positives for every true positive (41). This may result in undue stress placed on families who engaged in WGS as a means of diagnosing their child’s acute illness, not necessarily what that child may experience in the decades to come, in addition to raising ethical concerns regarding consent in children (21, 41). In this study we did not specifically evaluate for any “incidental finding genes”, and during the consent process families were offered the choice to opt out of receiving medically actionable incidental findings that may be inadvertently discovered.
Another controversy related to WGS concerns the handling of variants of uncertain significance (VUS). In accordance with the 2015 ACMG guidelines, an analyzed variant is categorized as “pathogenic”, “likely pathogenic”, “VUS”, “likely benign”, or “benign” based on a 28-point scoring system (42). In general, a pathogenic or likely pathogenic variant has met sufficient criteria such that a clinician can apply the information in clinical decision making when combined with clinical phenotype (42). Applicability to asymptomatic patients, such as in prenatal screening, is beyond the scope of this paper. A VUS should not be used in clinical decision making because the current evidence is insufficient, though in years to come the variant may potentially be re-classified as new evidence is available (42). In this study the IRB protocol did not allow for VUS reporting. Reporting of only pathogenic and likely pathogenic variants was intended to ensure that rWGS results would meet the recommendations of clinical significance, and thus medical actionability to could be taken per the physician’s discretion, lessening the burden on the clinician. However, some clinical laboratories do report VUS results, and managing those results poses a challenge worth considering for clinicians with limited exposure to clinical genetics.
Finally, not every institution has access to rWGS, though the expectation is that availability will increase exponentially in the near future (43). This study was not able to quantify the cost savings resulting from rWGS diagnosis, but emerging prospective data suggest that early use of WGS/WES can result in cost savings compared with traditional genetic and non-genetic testing (22), and cost-effectiveness of rapid WGS has been demonstrated in acutely ill neonates specifically by two groups (19, 44).
CONCLUSION
As with any new medical technology, the period of its emergence is replete with challenges in deploying the test responsibly and to the appropriate population. For rWGS in critically ill children, these questions certainly require further exploration, as many remain unanswered. While we anticipate that in time such questions will be clarified with further study, it would behoove us in the interim to encourage thoughtful utilization over indiscriminate application, thinking of rWGS as a useful tool, and not as a diagnostic panacea. Implementation of precision medicine in the ICU must be guided by both beneficence and minimization of risk to the patient and family. Even when ordered as a clinical test outside of the research setting, respect for families as manifested by education regarding WGS/WES technology and the implication of WGS/WES findings is paramount to complying with medical ethics principles. Overall, this study was able to demonstrate undiagnosed genetic disease in PICU patients and the potential impact of rWGS for influencing care of critically ill children both during and after ICU admission.
The continuing inevitable transformation of medical practice towards personalization of treatment and therapy based on individual genetic data will increasingly become applicable to critical care medicine, and rWGS can be expected to be at the forefront of this movement. We retrospectively assessed the rate of molecular diagnosis and clinical utility of rWGS for patients aged four months to 18 years admitted to the pediatric ICU, and found rates similar to those seen in neonatal ICU populations. Our results suggest that rWGS should be studied further as a diagnostic test for critically ill children, including children without preexisting concern for a genetic disorder. As technological advances continue and are implemented clinically, expansion of the phenotypic spectrum of disorders may include less severely affected individuals with atypical presentations. In the interim period prior to the elucidation of a complete phenotypic spectrum for a given disease, when our collective diagnostic ability exceeds our collective prognostic ability, it may be increasingly challenging to predict the disease course for disorders with phenotypic heterogeneity, and to counsel the families who are weighing decisions regarding goals of care and potentially palliation.
Supplementary Material
Acknowledgements
John Bradley, Susan Duthie, Brock Fisher, Michael Gottschalk, Richard Haas, Helen Harvey, Charlotte Hobbs, Sandeep Khanna, Bradley Peterson, Raveen Raviendran, Michael Worthen
Funding: Grant U19HD077693 from NICHD and NHGRI
Copyright form disclosure: Drs. Sanford Kobayashi, Briggs, Watkins, and Dimmock’s institution received funding from Grant U19HD077693 from National Institute of Child Health and Human Development and National Human Genome Research Institute (NHGRI). Drs. Sanford Kobayashi, Clark, Farnaes, Bainbridge, Chowdhury, Watkins, Dimmock, and Kingsmore received support for article research from the National Institutes of Health (NIH). Dr. Sanford Kobayashi also received support for article research from NHGRI. Dr. Briggs disclosed that he is employed by the US Navy. Drs. Bainbridge and Kingsmore’s institutions received funding from the NIH. Dr. Bainbridge he disclosed that he is a founder of Codified Genomics LLC. Dr. Dimmock received funding from Biomarin (consultant for Pegvaliase trials), Audentes Therapeutics (Scientific Advisory Board), and Ichorion Therapeutics (consultant for mitochondrial disease drugs). The remaining authors have disclosed that they do not have any potential conflicts of interest.
Footnotes
The views expressed in this article are those of the author(s) and do not necessarily reflect the official policy or position of the Department of the Navy, Department of Defense, or the United States Government.
References
- 1.Online Mendelian Inheritance in Man. McKusick-Nathans Institute of Genetic Medicine, Johns Hopkins University; Baltimore, MD: available at www.omim.org/statistics [accessed 21 June 2018] [Google Scholar]
- 2.Chong JX, Buckingham KJ, Jhangiani SN, et al. : Centers for Mendelian Genomics. The genetic basis of Mendelian phenotypes: discoveries, challenges, and opportunities. Am J Hum Genet 2015; 97:199–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.March of Dimes Foundation Data Book for Policy Makers. Maternal, Infant, and Child Health in the United States 2016. http://www.marchofdimes.org/March-of-Dimes-2016-Databook.pdf. Accessed 27 May 2016. [Google Scholar]
- 4.Berry MA, Shah PS, Brouillette RT, Hellmann J. Predictors of mortality and length of stay for neonates admitted to children’s hospital neonatal intensive care units. J. Perinatol. 2008;28:297–302. doi: 10.1038/sj.jp.7211904. [DOI] [PubMed] [Google Scholar]
- 5.Kochanek KD, Murphy SL, Xu J, Arias E. Mortality in the United States, 2016. NCHS Data Brief. 2017;293:1–8Khokha MK, Mitchell LE, Wallingford JB. White paper on the study of birth defects. Birth Defects Res 2017; 109:180–185 [Google Scholar]
- 6.Weiner J, Sharma J, Lantos J, et al. : How infants die in the neonatal intensive care unit: trends from 1999 through 2008. Arch Pediatr Adolesc Med 2011; 165:630–634 [DOI] [PubMed] [Google Scholar]
- 7.Wilkinson DJ, Fitzsimons JJ, Dargaville PA, et al. : Death in the neonatal intensive care unit: changing patterns of end of life care over two decades. Arch Dis Child Fetal Neonatal Ed 2006; 91:F268–271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hagen CM, Hansen TW: Deaths in a neonatal intensive care unit: a 10-year perspective. Pediatr Crit Care Med 2004; 6:463–468 [DOI] [PubMed] [Google Scholar]
- 9.Berger TM, Hofer A: Causes and circumstances of neonatal deaths in 108 consecutive cases over a 10-year period at the Children’s Hospital of Lucerne, Switzerland. Neonatology 2009; 95:157–163 [DOI] [PubMed] [Google Scholar]
- 10.O’Malley M, Hutcheon RG: Genetic disorders and congenital malformations in pediatric long-term care. J Am Med Dir Assoc 2007; 8:332–334 [DOI] [PubMed] [Google Scholar]
- 11.Stevenson DA, Carey JC: Contribution of malformations and genetic disorders to mortality in a children’s hospital. Am J Med Genet A 2004; 126A:393–397 [DOI] [PubMed] [Google Scholar]
- 12.Bainbridge MN, Wiszniewski W, Murdock DR, et al. : Whole-genome sequencing for optimized patient management. Sci Transl Med 2011; 3:87re3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Soden SE, Saunders CJ, Willig LK, et al. : Effectiveness of exome and genome sequencing guided by acuity of illness for diagnosis of neurodevelopmental disorders. Sci Transl Med 2014; 6:265ra168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stavropoulos DJ, Merico D, Jobling R, et al. : Whole-genome sequencing expands diagnostic utility and improves clinical management in paediatric medicine. NPJ Genom Med 2016; 1:15012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Saunders CJ, Miller NA, Soden SE, et al. : Rapid whole-genome sequencing for genetic disease diagnosis in neonatal intensive care units. Sci Transl Med 2012; 4:154ra35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Willig LK, Petrikin J, Smith LD, et al. : Whole-genome sequencing for identification of Mendelian disorders in critically ill infants: a retrospective analysis of diagnostic and clinical findings. Lancet Respir Med 2015; 3:377–387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Miller NA, Farrow EG, Gibson M, et al. : A 26-hour system of highly sensitive whole genome sequencing for emergency management of genetic diseases. Genome Med 2015; 7:100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Petrikin JE, Cakici JA, Clark MM, et al. The NSIGHT1-randomized controlled trial: rapid whole-genome sequencing for accelerated etiologic diagnosis in critically ill infants. NPJ Genom Med 2018; 3:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Farnaes L, Hildreth A, Sweeney NM, et al. Rapid whole-genome sequencing decreases infant morbidity and cost of hospitalization. NPJ Genomic Medicine 2018; 3:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Clark MM, Stark Z, Farnaes L, et al. Meta-analysis of the diagnostic and clinical utility of genome and exome sequencing and chromosomal microarray in children with suspected genetic diseases. Npj Genomic Medicine 2018; 3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bagnall RD, Ingles J, Dinger ME, et al. : Whole genome sequencing improves outcomes of genetic testing in patients with hypertrophic cardiomyopathy. J Am Coll Cardiol 2018; 72(4):419–429 [DOI] [PubMed] [Google Scholar]
- 22.Lionel AC, Costain G, Monfared N, et al. : Improved diagnostic yield compared with targeted gene sequencing panels suggests a role for whole-genome sequencing as a first-tier genetic test. Genet Med 2018; 20(4):435–443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gross MM, Ajay SS, Rajan V, et al. : Copy number variants in clinical genome sequencing: deployment and interpretation for rare and undiagnosed disease. Genet Med 2018; doi: 10.1038/s41436-018-0295-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Petrikin JE, Willig LK, Smith LD, et al. : Rapid whole genome sequencing and precision neonatology. Semin Perinatol . 2015; 39:623–631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mestek-Boukhibar L, Clement E, Jones WD , et al. : Rapid Paediatric Sequencing (RaPS): comprehensive real-life workflow for rapid diagnosis of critically ill children. Journal of Medical Genetics 2018; 55:721–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Directors ABo. Clinical utility of genetic and genomic services: a position statement of the American College of Medical Genetics and Genomics. Genet Med 2015; 17:505–507 [DOI] [PubMed] [Google Scholar]
- 27.Kannankeril PJ, Moore JP, Cerrone M, et al. : Efficacy of flecainide in the treatment of catecholaminergic polymorphic ventricular tachycardia: a randomized clinical trial. JAMA Cardiol . 2017;2(7):759–766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Khan JN, Prasad N, Glancy JM: QTc prolongation during therapeutic hypothermia: are we giving it the attention it deserves? EP Europace, 2010; 12(2):266–270 [DOI] [PubMed] [Google Scholar]
- 29.Ackerman MJ, Priori SG, Dubin AM, et al. : Beta-blocker therapy for Long QT Syndrome and Catecholaminergic Polymorphic Ventricular Tachycardia: are all beta-blockers equivalent? Heart Rhythm, 2017;14:e41–e44. [DOI] [PubMed] [Google Scholar]
- 30.Ahn J, Kim HJ, Choi JI, et al. : Effectiveness of beta-blockers depending on the genotype of congenital long-QT syndrome: A meta-analysis. PLoS One 2017; 12(10):e0185680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Taggar NW, Haglund CM, Ackerman MJ: Diagnostic miscues in congenital Long QT Syndrome. Circulation, 2007;115:2613–2620. [DOI] [PubMed] [Google Scholar]
- 32.Chong JX, McMillin MJ, Shively KM, et al. : De novo mutations in NALCN cause a syndrome characterized by congenital contractures of the limbs and face, hypotonia, and developmental delay. Am J Hum Genet 2015; 96:462–473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Phillips KA, Deverka PA, Sox HC, et al. : Making genomic medicine evidence-based and patient-centered: a structured review and landscape analysis of comparative effectiveness research. Genet Med 2017; 19:1081–1091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Farewell KD, Shahmirzadi L, El-Khechen D, et al. : Enhanced utility of family-centered diagnostic exome sequencing with inheritance model-based analysis: results from 500 unselected families with undiagnosed genetic conditions. Genetics in Med 2015; 17:578–586 [DOI] [PubMed] [Google Scholar]
- 35.Lee H, Deignan JL, Dorrani N, et al. : Clinical exome sequencing for genetic identification of rare Mendelian disorders. JAMA 2014; 312:1880–1887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yang Y, Muzny DM, Reid JG, et al. : Clinical whole-exome sequencing for the diagnosis of mendelian disorders. N Eng J Med 2013; 369:1502–1511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yang Y, Muzny DM, Xia F, et al. : Molecular findings among patients referred for clinical whole-exome sequencing. JAMA 2014; 312:1870–1879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Clark M, Hildreth A, Batalov S, et al. : Diagnosis of genetic diseases in seriously ill children by rapid whole-genome sequencing and automated phenotyping and interpretation. Sci Transl Med 2019; 11(489) doi: 10.1126/scitranslmed.aat6177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Smith LD, Willig LK, Kingsmore SF: Whole-exome sequencing and whole-genome sequencing in critically ill neonates suspected to have single-gene disorders. Cold Spring Harb Perspect Med 2016; 6:1–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kalia SS, Adelman K, Bale SJ, et al. : Recommendations for reporting of secondary findings in clinical exome and genome sequencing, 2016 update (ACMG SF v2.0): a policy statement of the American College of Medical Genetics and Genomics. Genet Med 2017; 19(2):249–255 [DOI] [PubMed] [Google Scholar]
- 41.Kingsmore SF: Incidental swimming with millstones. Sci Transl Med 2013; 5(194):194ed10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Richards S, Aziz N, Bale S, et al. : Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in Medicine 2015; 17(5):405–424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Grosse SD, Farnaes L: Genomic sequencing in acutely ill infants: what will it take to demonstrate clinical value? Genet Med 2018; doi: 10.1038/s41436-018-0124-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Stark Z, Lunke S, Brett GR, et al. : Meeting the challenges of implementing rapid genomic testing in acute pediatric care. Genet Med 2018; doi: 10.1038/gim.2018.37 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.