

Abstract
Rationale:
Dozens of preclinical studies have reported cannabinoid agonist potentiation of the analgesic effects of µ-opioid agonists.
Objectives:
The aim of this study was to determine if a cannabinoid agonist could potentiate opioid analgesia in humans using several laboratory pain models.
Methods:
Healthy participants (n=10) without current drug use/pain conditions completed this within-subject, double blind, placebo-controlled, randomized outpatient study. Nine 8-hr sessions were completed during which dronabinol (0, 2.5, 5 mg, p.o.) was administered 1 hr before oxycodone (0, 5, 10 mg, p.o.) for a total of 9 test conditions. Outcomes included sensory threshold and tolerance from four experimental pain models (cold pressor, pressure algometer, hot thermode, cold hyperalgesia), along with participant- and observer-rated, performance and physiological effects.
Results:
Oxycodone produced miosis (p<.05) and analgesic responses (e.g., pressure algometer, [p<.05]), while dronabinol did not (p>.05). Depending on the dose combination, dronabinol attenuated or did not alter oxycodone analgesia; for example, dronabinol (2.5 mg) decreased the analgesic effects of oxycodone (10 mg) on pressure tolerance. Conversely, dronabinol increased oxycodone subjective effects (e.g., drug liking) (p<.05); oxycodone (5 mg) ratings of “high” were potentiated by 5 mg dronabinol (p<.05; placebo=1.1 [±0.7]; 5mg oxycodone =4.7 [±2.2]; 5mg dronabinol=9.9 [±8.4]; 5mg oxycodone + 5mg dronabinol= 37.4 [±11.3]).
Conclusions:
This study indicates that dronabinol did not enhance the analgesic effects of oxycodone and increased abuse- and impairment-related subjective effects. These data suggest that dronabinol may not be an effective or appropriate opioid adjuvant; it could potentially increase opioid dose requirements, while increasing psychoactive opioid effects.
Keywords: cannabinoid, dronabinol, human, opioid, opioid sparing, pain
INTRODUCTION
Opioids are generally recognized to be efficacious for acute pain; despite their efficacy, there are important public health harms associated with widespread use of opioids (e.g., sedation/impairment, misuse and dependence, unintentional poisoning, non-fatal or fatal overdose). There is a great public need to develop opioid alternatives (Califf et al, 2016; U.S. FDA, 2018; Dowell et al., 2016) and find medications that boost the analgesic effects of opioids without increasing the opioid dose (known as opioid sparing), both of which are current initiatives of the U.S. Food and Drug Administration (US FDA, 2018; Congressional Research Service, 2018). The latter strategy relies on an adjuvant drug, that when combined with ongoing opioid therapy, reduces the opioid dose required while maintaining full analgesia and mitigating opioid-related side effects, abuse potential, impairment or respiratory depression (Kahn et al., 2011). Many opioid adjuvant drugs are used in clinical practice (e.g., topical analgesics, acetaminophen/ NSAIDs, corticosteroids, anxiolytics, antidepressants, anticonvulsants, benzodiazepines, gabapentinoids, sodium channel blockers). However, more effective, and safer/non-toxic compounds are needed, particularly agents that exhibit a high therapeutic index, do not produce untoward pharmacokinetic/pharmacodynamic interactions with opioids, and are suitable for acute or chronic co-administration with opioids (Khan et al., 2011; Moulin et al., 2007).
Dozens of studies conducted in rodents and monkeys have demonstrated that cannabinoid agonist pretreatment, often with doses that are otherwise inactive alone, produce clear, greater-than-additive, often synergistic interactions with opioid agonists, increasing opioid analgesic potency in the range of 0.5 – 25 fold (e.g., Welch & Eads, 1999; Welch, 2009; Cichewicz et al., 1999; 2003; 2004; Maguire et al., 2013). For example, in a comprehensive drug-drug interaction study in mice, pretreatment with an inactive, placebo-like dose (when given alone) of the CB1/CB2 agonist, delta-9-tetrahydrocannabinol (Δ9 THC; 20 mg/kg, p.o.), increased the analgesic potency of a wide range of doses of several µ-opioid drugs (e.g., hydromorphone, codeine) on the tail flick assay of nociception (Cichewicz et al., 1999). Compared to placebo pre-treatment, THC administration produced a 2.2 to 25.8-fold increase in analgesic potency of nine distinct µ-opioid agonists, decreasing the opioid dose necessary for analgesia (ED50) between 55–98%. In a similar study, monkeys were administered a non-active dose of THC (1.0 mg/kg, s.c.) prior to opioid agonist injection (e.g., fentanyl, morphine); THC produced a 0.55 to 20.6-fold leftward shift in the µ-opioid dose response curves, decreasing the opioid ED50 by 69–94% (Maguire et al., 2013). Interestingly, the synergistic effects of opioid/cannabinoid combinations appear to be rather selective for nociception in animals, as other direct effects of cannabinoids (hypothermia, catalepsy, hypoactivity) and opioids (respiratory depression) are not potentiated by their co-administration (Welch, 2009). Only a few non-human studies have directly assessed outcomes related to the reinforcing effects of these dose combinations, and the findings have been mixed. Several rodent studies indicate that cannabinoids enhance the reinforcing effects of opioids (e.g., self-administration, drug reinstatement) (e.g., Fattore et al., 2011; Haghparast et al., 2014; Solinas et al., 2005). However, a series of studies conducted in non-human primates indicate that acute administration of cannabinoid agonists (e.g., THC) do not alter and, in some cases, decrease heroin self-administration (Li et al., 2012; Maguire et al., 2013; 2016); similar effects were reported with daily THC administration (Maguire and France, 2016). When available together for self-administration, opioid-cannabinoid combinations (e.g., THC + heroin [Li et al., 2012]; THC + remifentanil [Maguire and France, 2018]) are self-administered less than or equal to the opioid alone. Cannabinoid agonists also attenuate the discriminative stimulus effects of morphine (e.g., Maguire et al., 2013); the authors indicate that this profile of effects suggests that cannabinoid agonists do not change or decrease the reinforcing efficacy of µ-opioid agonists in non-human primates (e.g, Maguire and France, 2016).
To date, there have only been three human laboratory studies examining opioid-cannabinoid analgesic interactions on experimental pain outcomes, also with mixed results (Naef et al., 2003; Roberts et al., 2006, Cooper et al., 2018). One study examined the effects of morphine (30 mg, p.o.), dronabinol (20 mg, p.o.) and their combination on pain threshold and tolerance on heat, cold, pressure and electrical stimulation assays. Dronabinol alone did not decrease pain and induced hyperalgesia on several outcomes, morphine alone produced mild analgesic effects; however, there were no interactions suggesting that dronabinol potentiated morphine analgesia (Naef et al., 2003). A similar study administered dronabinol (5 mg, p.o.), morphine (0.02 mg/kg, i.v.) and their combination to healthy participants and assessed subjective pain response from a thermal pain assay. There was one modest decrease in ratings of “bad” pain effects after the active dose combination (relative to either drug alone), but no other indications of analgesic interactions. However, a recent experimental pain study enrolling heavy cannabis smokers administered oxycodone (2.5, 5 mg, p.o.) alone and in combination with smoked cannabis (5.6% THC) and measured cold pressor response. The combination of 2.5 mg oxycodone and active cannabis increased cold pressor threshold and tolerance outcomes (i.e., increased analgesic effect), relative to either drug alone; however, this dose combination also increased the abuse liability of oxycodone (i.e., increases in ratings of drug liking, wanting to take the drug again; Cooper et al., 2018).
The objective of the current study was to enroll healthy participants with no current pain conditions or current illicit drug use to determine if low/therapeutic doses of oral dronabinol (0, 2.5, 5 mg), a CB1/CB2 receptor agonist, could potentiate the analgesic effects of oral oxycodone (0, 5, 10 mg), a µ-opioid agonist, using laboratory models of acute pain. Secondary outcomes assessed the safety/tolerability and subjective effect profile of these dose combinations using an array of physiological, subject- and observer-rated and psychomotor performance outcome measures.
METHODS
Participants
Participants were healthy adults, ages 18–50, without acute or chronic pain conditions (e.g., recent wounds/surgery, migraine, chronic lower back pain) and no current (e.g., past-60 day) opioid or cannabinoid drug use/misuse. All participants completed in-person screening evaluations that included substance use and psychiatric assessments, medical history and physical exam, blood chemistry, urinalysis, and ECG. Participants provided observed urine samples during each screening visit. Those individuals testing positive for cannabinoids or opioids, or participants repeatedly testing positive for other drugs (e.g., cocaine, methamphetamine) were disqualified. Other exclusion criteria included current physiological drug dependence requiring medical intervention, pregnancy, significant medical (e.g., seizure disorder, chronic pain) or psychiatric problems (e.g., bipolar disorder) and insensitivity/inability to tolerate the experimental pain procedures. All participants provided sober, written informed consent prior to participation and were paid for their participation. The study was approved by the University of Kentucky Institutional Review Board and was conducted in accordance with the Helsinki guidelines for ethical research. A Certificate of Confidentiality was obtained from the U.S. Food and Drug Administration (FDA).
Drugs
This study was conducted under an investigator-initiated Investigational New Drug Application from the FDA (#69,214). All drug doses were prepared by the University of Kentucky (UK) Investigational Pharmacy. Commercially available doses of immediate-release oxycodone hydrochloride tablets (5 mg; Mallinckrodt Inc., Hazelwood, MO) and dronabinol capsules (2.5 mg capsules: Par Pharmaceutical, Woodcliff Lake, NJ; 5 mg capsules: Pharmaceutics International Inc., Hunt Valley, MD) were obtained. These were over-encapsulated with two uniform size 00 gelatin capsules (Health Care Logistics, Circleville, OH) to blind the doses. Lactose monohydrate powder (Medisca Pharmaceuticals, Plattsburgh, NY) was used for the placebo condition and as filler in the active dose capsules.
Dose Selection Rationale
The recommended starting doses of each drug (5, 10 mg oxycodone; 2.5 mg, 5 mg dronabinol) (American Pain Society Guidelines, 2016; Marinol® package insert) were selected to evaluate doses that are available to clinicians and are safe for those who are naïve/non-tolerant to both drug classes. These doses were also selected to model those used in animal studies that have demonstrated low/non-sedating active cannabinoid and opioid doses produce enhanced analgesia when given in combination.
The study was initiated using dronabinol doses of 5, 10 mg; however, administration of 10 mg dronabinol produced anxiety/panic and tachycardia on one occasion (n=1) and was, therefore, discontinued. We modified the dronabinol doses (2.5, 5 mg) and administered these doses alone and in combination with oxycodone (5, 10 mg) to all participants (n=10).
Dronabinol was administered 1 hr before oxycodone in order to align the peak effects of each drug (dronabinol Tmax = 2–3 hrs, oxycodone Tmax = 1–1.5 hrs; e.g., Lile et al., 2013; Babalonis et al., 2015).
Study Design
This study utilized a within-subject crossover, randomized, double-blind placebo controlled design and examined oral dronabinol (0, 2.5, 5 mg), oral oxycodone (0, 5, 10 mg) and their combination for a total of 9 test conditions: 1) 0 mg dronabinol + 0 mg oxycodone; 2) 2.5 mg dronabinol + 0 mg oxycodone; 3) 5 mg dronabinol + 0 mg oxycodone; 4) 0 mg dronabinol + 5 mg oxycodone; 5) 0 mg dronabinol + 10 mg oxycodone; 6) 2.5 mg dronabinol + 5 mg oxycodone; 7) 2.5 mg dronabinol + 10 mg oxycodone; 8) 5 mg dronabinol + 5 mg oxycodone; 9) 5 mg dronabinol + 10 mg oxycodone. Each participant completed a total of 9 outpatient experimental sessions (8.5 hrs/session) with a minimum of 48 hrs separating each session.
General Methods
Upon arrival at the laboratory, participants were provided a light, standardized breakfast (including one standardized cup of coffee for heavy caffeine users only) 1.25 hr prior to drug administration. Urine samples were collected each morning; they were tested for drugs of abuse (Discover™ Drug Test Card; American Screening LLC, Shreveport, LA) and for pregnancy with females (hCG Test Card; Teco Diagnostics, Anaheim, CA). Breath samples were obtained prior to each session (AlcoMate Premium AL7000, Advance Safety Devices LLC, Chatswort, CA) and were tested for the presence of alcohol. Sessions were canceled if urine samples tested positive for illicit drugs or pregnancy or if breath samples were positive for alcohol.
Physiological Measures
Heart rate, blood pressure, oxygen saturation (Dinamap Non-Invasive Patient Monitor, GE Medical Systems, Tampa, FL), expired end-tidal carbon dioxide (EtCO2), respiration rate (N-85 Capnograph, Nellcor, Boulder, CO) and pupil diameter measurements (PLR-200, NeurOptics, Irvine, CA) were collected at baseline and in 30-min intervals after dronabinol administration for 6 hrs.
Pain Assessments
Each pain assay was conducted once during study screening to assess participants’ sensitivity and tolerability of the procedures; the tests were also conducted several times during training sessions (no study drug was administered) to familiarize participants to the procedures.
During test sessions, the full battery of pain assays was administered before and 1, 2, 3, 4 and 6 hrs after dronabinol administration. Primary outcome measures included 1) pain threshold (point at which pain was initially detected), and 2) pain tolerance (the point at which pain was no longer tolerable), the latter signaled termination of the trial.
Pressure Algometer Test
A computerized algometer device (Medoc AlgoMed, U.S.A, Durham, N.C.) was used to apply pressure to the palmar surface of the thenar eminence of the dominant hand at the rate of 40 kilopascals (kPa)/second. The maximum pressure was 1500 kPa (predetermined safety cut-off). During each administration, two trials were conducted, with a minimum of 5 minutes separating each pressure application. Participants indicated pain threshold and tolerance via a two-button response apparatus connected to the AlgoMed system; these outcome measures are reported in kPa.
Cold Pressor Test
Participants immersed their non-dominant forearm in a room-temperature circulating bath for a total of 2 minutes; immediately afterward, they placed their forearm in a circulating bath of cold water (1.0°C ± 0.5°C). They were instructed not to touch the sides or bottom of the water bath container. Maximum cold water immersion time was 5 minutes (pre-determined cut-off to avoid tissue damage). Participants verbally indicated pain threshold and removed their arm from the cold water to indicate pain tolerance. These outcomes were recorded in seconds (e.g., time elapsed since cold water immersion; adapted from Compton et al., 2010).
Hot Thermode Test
The TSA-II Neurosensory Analyzer (Medoc Inc.) was used to deliver heat stimulation through a 3×3 cm thermode that was placed on the skin over the lateral head of the gastrocnemius muscle of the dominant leg (i.e., leg lateral to the dominant arm). The thermode temperature began at 25°C and increased at a linear rate of 1°C/second. The highest possible temperature was 52°C (safety cut-off). Participants used a hand-held two-button response box to indicate threshold and tolerance, measured in degrees Celsius. During each administration, two trials were conducted, with a minimum of 5 minutes separating each application of heat.
Topical Menthol/Cold Hyperalgesia Test
At baseline, the cold thermode (TSA-II, Medoc Inc.) was placed on the volar forearm of the dominant arm. The thermode temperature began at 30°C and decreased at a linear rate of 1°C/second (lowest temp = 0°C). Participants responded on a hand-held two-button response box to indicate threshold and tolerance (measured in °C). Sensory probes were applied to assess mechanical hyperalgesia (5.46 von Frey; 26 g of force) and allodynia (standardized soft brush). The total area of mechanical hyperalgesia (von Frey) and allodynia (brush) was measured (total cm2) using GSP® software.
At 280 minutes post-dronabinol dose, a patch of gauze soaked with high potency menthol solution (1 mL, 40% l-menthol solution) was applied and secured (Tegaderm®) on the skin for 20 min. The cold thermode was applied immediately after, 45 and 90 min after patch removal; cold threshold and tolerance and sensory outcomes were assessed (adapted and modified from Binder et al., 2011).
This model was included as a measure of cold hyperalgesia (i.e., menthol sensitizes the skin and produces pain at temperatures that [at baseline] were not painful). Preclinical data suggests that cannabinoids can alleviate cold hyperalgesia (e.g., Kinsey et al., 2010); this assay was included to determine if acute THC administration (or acute opioid administration) could alter cold hyperalgesia in a human experimental pain model.
Subjective Ratings of Pain
Immediately upon completion of each pain trial, participants were asked four visual analog questions (VAS) on a 100-mm line that was anchored with the terms “not at all” and “extremely” and rated the degree to which the sensation was 1) painful, 2) intense, 3) unpleasant and 4) bothersome. During the menthol patch administration, participants answered three additional VAS questions, rating the degree to which the menthol sensation was: 1) hot, 2) cold, and 3) tingly.
Subjective and Observer-Rated Measures
Subjective effects measures included a five-item Visual Analog Scale (VAS; Walsh et al., 2008) that presented the following questions: “Do you feel any drug effect?”, “How high are you?”, “Does the drug have any good effects?”, “Does the drug have any bad effects?” and “How much do you like the drug effects?”. These items were presented at baseline and at 15-min intervals for the first 2.5 hr, then every 30 min for the remaining 3.5 hr. A 47-item VAS questionnaire was collected at baseline and every 1 hr after dronabinol administration and included items from the Participant-Rated Opioid Adjective Scale (Fraser et. al., 1961) and the Marijuana Drug Effect Questionnaire (Haney et al., 2016). Trained research assistants rated signs of opioid (e.g., nodding, itching) and cannabinoid agonist effects (e.g., red/bloodshot eyes) on an Observer Rated Checklist at baseline and at 30-min intervals after dronabinol administration.
Psychomotor Performance Measure
A 90-second computerized version of the DSST (adopted from McLeod et al., 1982) was collected at baseline and at 30-min intervals for 6 hrs after dronabinol administration.
Statistical Analyses
All measures were initially analyzed as raw time course data using a two-factor repeated measures model (drug condition, time) with an AR(1) covariance structure. Physiological measures, collected minute-by-minute, were initially averaged across 15–30 min intervals corresponding to the subjective reporting intervals. Peak/trough scores and peak change from baseline scores were calculated for individual participants within each dose condition and analyzed in a one-factor model (drug condition). In this model, a main effect of dose indicates that dose-related variance was detected. Tukey’s post-hoc tests were completed to determine if an individual dose was significantly different from placebo and if a given dose combination was significantly different from the comparator dose (e.g., determining if an effect from dronabinol + oxycodone was different from the comparator dose of oxycodone alone). All models were conducted with Proc Mixed in SAS 9.3 (Cary, NC) with significance at p < 0.05. All means are reported with the standard error of the mean (±SEM).
Two trials per measurement were conducted for the pressure algometer and the hot thermode test to ensure that at least one trial measurement was valid due to occasional participant or researcher errors (e.g., probe/thermode slipping off surface of skin); however, in the current study, there were minimal issues and the first exposure of each measurement (i.e., trial one) was retained for data analysis.
RESULTS
A total of 43 participants were screened for the study; a total of 11 participants were enrolled. One participant did not complete due to schedule conflicts. A total of 10 participants completed the protocol (6 female [4 Caucasian, 1 African American, 1 Asian] and 4 male [3 Caucasian, 1 Asian]); mean age of 26.3 (± 0.3) years old and 15.8 (± 0.2) years of education. All participants were non-smokers. None of the participants reported opioid or cannabinoid misuse in the 60 days prior to enrollment. All participants tested negative for these drugs during screening and study enrollment.
Pressure Algometer
There were no dose-related effects detected on pressure threshold, relative to placebo (F (8, 72) = 0.9, p>0.05). A main effect of dose was detected on pressure tolerance (F (8, 72) = 3.1; p>0.05). Mean baseline tolerance = 716 (± 56) kPa. As displayed in Figure 1 (left panel) oxycodone alone produced dose-related increases in pressure tolerance (displayed as peak change from baseline), with 10 mg oxycodone producing a significant effect relative to placebo (p<0.05). Both doses of dronabinol (2.5, 5 mg) were placebo-like when administered alone (p>0.05). Dronabinol did not increase the effects of oxycodone. Depending on the dose combination, dronabinol pre-treatment did not alter or decreased the effects of oxycodone. For example, 5 mg dronabinol did not alter the effects of 10 mg oxycodone – this dose combination produced an effect similar to 10 mg oxycodone alone (p<0.05; Fig. 1). However, 2.5 mg dronabinol decreased the effects of 10 mg oxycodone (peak change from baseline values, 10 mg oxycodone alone: 186 [± 28] kPa; 2.5 mg dronabinol + 10 mg oxycodone = 66 [±55] kPa; as displayed in Figure 1).
Figure 1.

Pain tolerance outcomes for Panel A: pressure algometer (measured in kPa, expressed as mean peak change from baseline), Panel B: cold pressor (measured in seconds, expressed as mean peak effects), and Panel C: hot thermode (measured in degrees Celsius, expressed as mean peak change from baseline) models, displayed as a function of dose condition (n=10; ±1 SEM). Oxycodone doses (mg) are displayed on the x-axis and the doses of dronabinol pre-treatment are displayed as the three line functions: dronabinol 0 mg = circle, 2.5 mg = triangle, 5 mg = square symbols. The filled symbols indicate a significant difference from the placebo condition (Tukey post-hoc, p<0.05). A time course analyses detected a significant effect of dose x time interaction on algometer tolerance (F(40, 357) = 1.5, p<0.05).
Cold Pressor Test
A main effect of dose was detected on cold water threshold; however, post-hoc testing did not reveal any significant differences between the doses (p>0.05). Mean baseline tolerance = 14 (± 0.8) seconds. As displayed in Figure 1 (middle panel), peak effects on cold water tolerance increased as a function of oxycodone dose; for example, 5 mg oxycodone increased mean cold water tolerance (relative to placebo) by 16 seconds; 10 mg oxycodone increased this time by 25.5 seconds (these increases were not statistically significant, p>0.05). In contrast, dronabinol produced mean decreases in latency by approximately 3 seconds when administered alone (2.5, 5 mg) (p>0.05). When dose combinations were administered, dronabinol did not enhance the effects oxycodone (p>0.05). For example, 2.5 mg dronabinol + 5 mg oxycodone decreased tolerance (17.9 [± 2.4] sec), compared the 5 mg oxycodone dose alone (34.3 [±17.7] sec; not statistically significant; p>0.05).
Hot Thermode
There were no significant dose related effects detected on hot thermode threshold (p>0.05) or tolerance (p>0.05). Baseline tolerance = 48.8 (± 0.15) °C. Figure 1 (right panel) displays hot thermode tolerance as peak change from baseline (in degrees Celsius). There was a slight non-significant increase in heat tolerance as a function of oxycodone dose 5 and 10 mg doses. Dronabinol alone (2.5 mg) produced placebo-like effects, while the higher dose (5 mg) produced some small, non-significant increases in tolerance (p>0.05). Neither dronabinol dose of dronabinol increased the effects of oxycodone (p>0.05).
Cold Thermode/Topical Menthol Test
Menthol reliably increased cold sensitivity, such that participants detected pain at higher temperatures after menthol application (mean = 10.9 [± 3.4] °C) relative to baseline (mean = 3.7 [± 2.4] °C) (mean threshold data from placebo condition); however, cold detection was not modified by any of the active dose conditions (p>0.05; data not displayed). Similarly, there were no changes relative to placebo on the sensory (mechanical pain, allodynia) outcomes (p>0.05).
Subjective Ratings of Pain
All of the pain assays increased ratings on the VAS pain outcomes (painful, intense, bothersome, unpleasant), but these ratings did not change as a function of dose (p>0.05). Overall, there was little variability in the ratings across the dose conditions (with no clear differences from placebo). For example, mean ratings of pressure pain (on a scale from 0–100) ranged from 37.8 (±3.0) to 43.5 (±3.1) (despite pressure tolerance outcomes being sensitive to dose). Similarly, acute menthol administration increased ratings of cold, hot and intense, but these ratings did not change as a function of dose (p>0.05).
Physiological Effects
Oxycodone produced dose-related miosis (5, 10 mg) (p<0.05; Table 1). Dronabinol alone (2.5 mg, 5 mg) did not produce changes in pupil diameter, and did not alter the effects of oxycodone (p>0.05; Table 1).
Table 1.
Physiological and Subject-Rated Measures
| Outcome Measure | 0 DRO | 2.5 DRO | 5 DRO | 0 DRO | 0 DRO | 2.5 DRO | 2.5 DRO | 5 DRO | 5 DRO | |
|---|---|---|---|---|---|---|---|---|---|---|
| F (8,72) | 0 OXY | 0 OXY | 0 OXY | 5 OXY | 10 OXY | 5 OXY | 10 OXY | 5 OXY | 10 OXY | |
| Physiologic Measures | ||||||||||
| Heart Rate | 1.5 | 76.2 (2.3) | 76.9 (3.7) | 82.2 (5.0) | 74.4 (3.1) | 76.2 (2.9) | 76.3 (3.5) | 78.1 (2.9) | 77.0 (3.9) | 78.4 (4.1) |
| Systolic Blood Pressure | 1.4 | 132.7 (2.8) | 126.4 (2.9) | 129.1 (3.2) | 130.2 (2.8) | 128.7 (4.0) | 130.2 (3.3) | 132.9 (3.2) | 129.4 (3.3) | 127.9 (3.4) |
| Diastolic Blood Pressure | 0.8 | 79.7 (2.2) | 79.6 (2.7) | 80.2 (2.7) | 79.6 (2.4) | 81.3 (3.1) | 83.5 (2.3) | 81.4 (2.9) | 80.8 (2.3) | 78.7 (2.0) |
| Oxygen Saturation | 1.1 | 97.8 (0.2) | 98.0 (0.3) | 98.0 (0.2) | 97.7 (0.4) | 97.5 (0.3) | 97.9 (0.3) | 97.5 (0.3) | 97.8 (0.4) | 97.3 (0.5) |
| End Tidal Carbon Dioxide | 1.8 | 37.0 (1.3) | 38.1 (0.9) | 36.1 (1.0) | 37.9 (0.9) | 38.0 (1.2) | 37.5 (1.1) | 38.5 (1.1) | 38.1 (0.9) | 37.2 (0.7) |
| Pupils | 25.6 | 4.2 (0.2) | 4.2 (0.3) | 4.2 (0.2) | 3.5 (0.2) | 3.1 (0.1) | 3.5 (0.2) | 2.9 (0.2) | 3.4 (0.2) | 2.8 (0.1) |
| Subject-Rated Measures | ||||||||||
| Visual Analog Scales | ||||||||||
| Drug Effect | 8.9 | 5.5 (2.5) | 6.3 (2.2) | 12.7 (8.9) | 10.4 (3.7) | 34.0 (8.5) | 15.9 (6.9) | 47.3 (9.1) | 45.8 (11.2) | 48.5 (10.2) |
| High | 4.3 | 1.1 (0.7) | 2.5 (1.7) | 9.9 (8.4) | 4.7 (2.2) | 21.1 (9.6) | 12.5 (5.1) | 26.4 (9.9) | 37.4 (11.3) | 34.9 (10.6) |
| Good Drug Effect | 2.1 | 5.4 (5.0) | 6.5 (4.9) | 7.1 (4.7) | 6.6 (3.5) | 15.9 (4.8) | 8.2 (4.9) | 22.3 (8.5) | 16.1 (5.5) | 24.4 (7.8) |
| Bad Drug Effect | 3.8 | 3.2 (2.2) | 1.9 (1.3) | 9.4 (7.9) | 6.9 (3.7) | 21.5 (10.7) | 6.1 (3.0) | 33.7 (12.6) | 34.3 (13.3) | 23.7 (11.1) |
| Like Drug Effect | 2.4 | 5.8 (4.8) | 7.6 (5.8) | 7.8 (5.5) | 7.4 (3.9) | 14.0 (4.2) | 8.6 (4.6) | 17.9 (6.3) | 12.8 (4.4) | 28.8 (8.0) |
| Drug Effect VAS | ||||||||||
| Nauseous | 4.0 | 0.9 (0.9) | 1.9 (1.3) | 0.0 (0.0) | 5.0 (3.4) | 19.1 (10.7) | 1.8 (1.2) | 29.6 (13.1) | 23.6 (10.9) | 24.7 (11.2) |
| Nodding | 3.6 | 0.7 (0.7) | 8.0 (6.6) | 3.0 (3.0) | 0.0 (0.0) | 10.3 (7.2) | 0.0 (0.0) | 19.1 (10.3) | 35.6 (12.1) | 11.8 (7.7) |
| Coasting or Spaced Out | 3.4 | 10.3 (7.5) | 5.2 (5.2) | 5.7(3.8) | 1.0 (1.0) | 14.3 (9.8) | 5.1 (3.4) | 29.7 (10.9) | 36.9 (11.9) | 31.1 (11.6) |
| Heavy or Sluggish | 3.0 | 4.3 (3.8) | 3.4 (2.4) | 9.5 (6.7) | 6.2 (4.2) | 21.9 (10.9) | 11.5 (6.5) | 36.8 (10.8) | 34.3 (12.5) | 27.2 (11.2) |
| Tired/Sleepy | 3.2 | 23.1 (8.3) | 29.5 (9.1) | 27.9 (10.6) | 29.5 (11.8) | 48.5 (12.8) | 26.6 (8.4) | 57.8 (11.6) | 61.6 (10.4) | 40.5 (11.0) |
| Sedated | 2.8 | 2.9 (2.5) | 2.0 (1.5) | 7.3 (6.8) | 2.6 (2.6) | 14.6 (8.2) | 3.9 (3.1) | 26.3 (10.1) | 28.6 (13.3) | 12.1 (4.8) |
| Dizzy/Light-Headed | 2.8 | 0.0 (0.0) | 0.0 (0.0) | 5.4 (5.4) | 6.8 (4.5) | 22.6 (13.0) | 5.6 (4.4) | 27.5 (12.3) | 25.3 (10.8) | 13.6 (10.2) |
| Difficulty Concentrating | 2.5 | 5.0 (3.8) | 1.0 (1.0) | 5.2 (3.8) | 4.4 (4.4) | 12.2 (8.2) | 1.4 (1.4) | 21.0 (10.3) | 29.8 (11.2) | 13.8 (8.5) |
| Unmotivated/Lazy | 2.5 | 12.2 (6.6) | 11.6 (7.8) | 16.6 (8.9) | 17.6 (10.4) | 25.8 (13.0) | 13.5 (8.4) | 32.4 (11.8) | 43.6 (14.2) | 25.7 (12.5) |
| Clumsy | 2.2 | 0.0 (0.0) | 0.0 (0.0) | 4.1 (4.1) | 0.0 (0.0) | 17.9 (10.9) | 0.0 (0.0) | 10.2 (8.6) | 20.4 (8.4) | 17.8 (12.0) |
| Impaired | 3.1 | 0.0 (0.0) | 2.0 (1.4) | 3.0 (2.0) | 0.0 (0.0) | 4.6 (3.0) | 0.8 (0.8) | 14.7 (6.2) | 13.8 (6.7) | 8.1 (3.5) |
All measures were analyzed as peak maximum score, with the exception of pupil diameter and oxygen saturation (which are trough or minimum scores). The values displayed each of the dose conditions (oxycodone (OXY), dronabinol (DRO); numbers next to drug abbreviations are doses expressed in mg). Bolded F values indicate a significant main effect of dose (p < 0.05); bolded mean values indicate the mean is significantly different from the corresponding placebo value (p < 0.05, Tukey post-hoc); boxed values indicate the dronabinol/oxycodone condition was significantly different from the comparator condition (the same dose of oxycodone administered alone) (p < 0.05, Tukey post-hoc). The subject-rated outcomes that are displayed are those for which a significant drug effect was detected.
Other physiological outcomes, including heart rate, respiration rate, oxygen saturation, and blood pressure did not vary as a function of dose condition (p>0.05; Table 1).
Psychomotor Task Performance
A main effect of dose was detected on the number of trials that were attempted (i.e., the speed of the emitted responses) (F(8,72) =2.5; p<0.05), with most of the active drug conditions decreasing the number of trials attempted compared to placebo; however, post-hoc testing did not detect a difference between the dose conditions (p>0.05). There were no dose-related effects on accuracy outcomes (e.g., number of correct trials) (p>0.05).
Subjective Ratings of Drug Effects
As displayed in Figure 2, oxycodone alone produced dose-related increases in ratings on the VAS question, “Do you feel any drug effect?” (p<0.05; Table 1), with 10 mg oxycodone producing a significant effect relative to placebo (p<0.05; Table 1). Dronabinol alone produced placebo-like effects (2.5 mg) and small non-significant increases (5 mg) on these ratings (p>0.05; Table 1). Oxycodone effects were potentiated when combined with dronabinol (p<0.05; Table 1); specifically, 5 mg dronabinol greatly increased the effects of 5 mg oxycodone, producing effects that were significantly greater than 5 mg of oxycodone alone (p<0.05; Table 1). Both doses of dronabinol (2.5, 5 mg) increased the effects of 10 mg oxycodone; these dose combinations were significantly different from placebo (p<0.05; Table 1).
Figure 2.
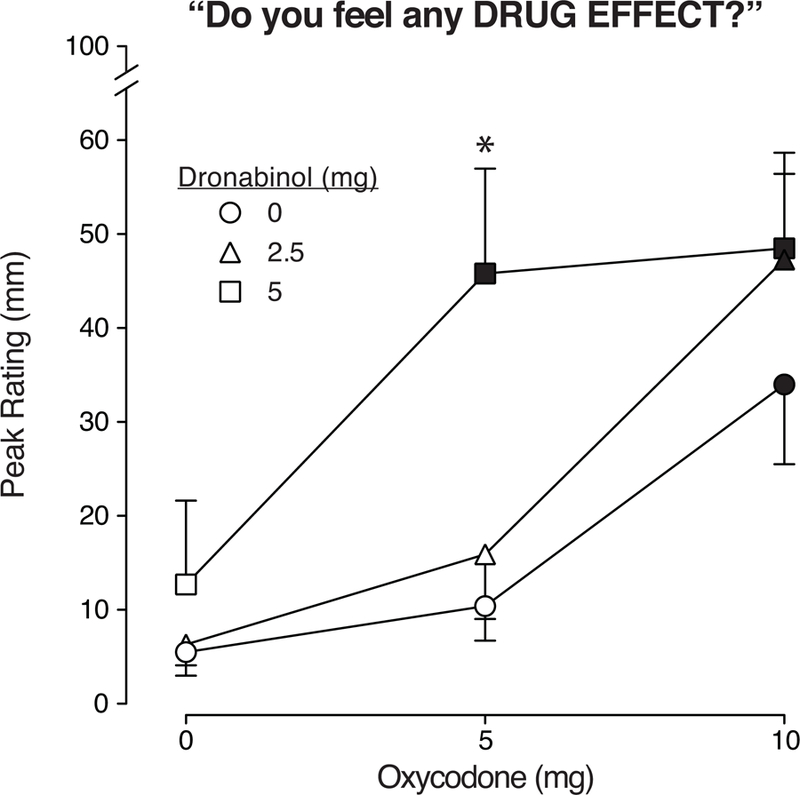
Mean peak VAS ratings for the subjective measure “Do you feel any drug effect?” are displayed as a function of dose condition (n=10; ±1 SEM). Oxycodone doses (mg) are displayed on the x-axis and the doses of dronabinol pre-treatment are displayed as the three line functions: dronabinol 0 mg = circle, 2.5 mg = triangle, 5 mg = square symbols. The filled symbols indicate a significant difference from the placebo condition (Tukey post-hoc, p<0.05). The asterisk indicates the dronabinol/oxycodone combination condition was significantly different from the comparator condition (the same dose of oxycodone administered alone) (p < 0.05, Tukey post-hoc). Time course analyses detected a significant main effect of dose (F(8,72) = 3.5, p<0.01) and dose x time interaction (F(96, 864) = 1.7, p<.0001).
Figure 3 displays ratings of VAS questions, “How much do you like the drug effect?” (left panel), “How high are you?” (middle panel) and “Does the drug have any bad effects?” (right panel). Dronabinol alone (2.5 mg) was placebo-like, while the 5 mg dose produced small non-significant increases in ratings of high and bad drug effects (p>0.05; Table 1). Oxycodone alone also produced small, non-significant increases in ratings (p>0.05; Table 1). However, when combined, dronabinol increased oxycodone effects on all measures displayed (p<0.05; see Table 1). The low dose of dronabinol (2.5 mg) increased the effects of 10 mg oxycodone on ratings of bad drug effects (p<0.05). The high dose of dronabinol (5 mg): 1) increased the effects of 10 mg oxycodone on ratings of drug liking (p<0.05), 2) increased the effects of both 5 and 10 mg oxycodone on ratings of high (p<0.05), with the 5 mg oxycodone/5 mg dronabinol combination producing greater effects than 5 mg oxycodone alone (p<0.05), and 3) increased the effects of 5 mg oxycodone on ratings of bad drug effect (p<0.05). The active dose combinations also increased ratings of nauseous, nodding, coasting/spaced out, tired/sleepy, unmotivated/lazy, and impaired, relative to placebo (p<0.05; Table 1). Ratings of nodding and coasting/spaced out were significantly greater after the 5 mg dronabinol/5 mg oxycodone condition, compared to 5 mg oxycodone alone (p<0.05; Table 1).
Figure 3.

Mean peak VAS ratings for the subjective measures “How much do you like the drug effect?” (left panel), “How high are you?” (middle panel), and “Does the drug have any bad effects?” (right panel). Values are displayed as a function of dose condition (n=10; ±1 SEM); oxycodone doses (mg) are displayed on the x-axis and the doses of dronabinol pre-treatment are displayed as the three line functions: dronabinol 0 mg = circle, 2.5 mg = triangle, 5 mg = square symbols. The filled symbols indicate a significant difference from the placebo condition (Tukey post-hoc, p<0.05). The asterisk indicates the dronabinol/oxycodone combination condition was significantly different from the comparator condition (the same dose of oxycodone administered alone) (p < 0.05, Tukey post-hoc). Time course analyses detected a significant main effect of dose (F(8,72) = 2.3, p<0.05) and dose x time interaction (F(96, 864) = 1.3, p<.05) for drug liking ratings; a dose x time interaction (F(96, 864) = 1.7, p<0.0001) for ratings of high; and a main effect of dose (F(8,72) = 2.4, p<0.05) for bad drug effect ratings.
Observer-Rated Drug Effects
Several observer-rated outcomes increased as a function of dose. For example, the active dose combinations produced effects that were significantly different from placebo on ratings of relaxed, heavy/sluggish, sleepy and nauseous (p<0.05). Ratings of heavy/sluggish and sleepy were significantly greater after the 5 mg dronabinol/5 mg oxycodone condition, compared to 5 mg oxycodone alone (p<0.05).
DISCUSSION
This study sought to replicate findings from non-human studies showing that cannabinoid (CB1/CB2) agonists can potentiate the analgesic effects of mu opioid agonists in humans (e.g., Cichewicz et al., 1999; Maguire et al., 2013). This randomized, placebo-controlled study enrolled healthy volunteers and examined the effects of oxycodone, dronabinol and their combination on experimental pain outcomes, physiologic response, and self-reported drug effects. The results revealed that oxycodone dose-dependently decreased pain response (e.g., pressure algometer [p<0.05]), while dronabinol alone (2.5, 5 mg) did not produce analgesic effects (e.g., no changes in threshold, tolerance outcomes) in any of the models (p>0.05). Critically, this controlled study provided no evidence that dronabinol enhanced the analgesic effects of oxycodone. Moreover, dronabinol increased the subjective effects of oxycodone, including ratings of feeling high and drug liking. These findings are inconsistent with several preclinical reports, but have important implications for the potential utility of cannabinoid agonists as adjunct or “opioid-sparing” therapy in the treatment of pain.
With respect to the pain assays employed here, oxycodone increased pain tolerance on the pressure algometer (p<0.05). Oxycodone-related increases in pain tolerance were also observed on the cold pressor and hot thermode tests; however, these effects were not statistically significant. Overall, these results replicate prior work (Staahl et al., 2009; Olesen et al., 2012; Babalonis, et al., 2016), suggesting that the present approach was sensitive to pain production and its suppression. The menthol/cold thermode assay produced cold hyperalgesia, but this was unaltered by oxycodone. The menthol assay is less commonly employed, although at least one prior study reported pain reduction with this method after 100 mg tramadol (Altis et al., 2009). Here, dronabinol alone (2.5, 5 mg) was placebo-like on all pain outcomes. Prior studies have reported mixed results of cannabinoid agonists on experimental pain outcomes (Redmond et al., 2008; Wallace et al., 2007; Naef et al., 2003; Greenwald & Stitzer, 1999; Hill et al., 1974; Walter et al., 2015). Several studies have reported no cannabinoid agonist-induced analgesia with acute heat (Naef et al., 2003, Redmond et al., 2008), cold pressor (Naef et al., 2003, Redmond et al., 2008), or pressure (Naef et al., 2003) assays (acute oral doses: 0.5 mg, 1 mg nabilone [Naef et al., 2003]; 20 mg THC [Redmond et al., 2008]). Others have reported analgesia from radiant heat pain after smoked cannabis (3.55% THC) (Greenwald & Stitzer, 1999) and a small reduction in heat hyperalgesia in women only after 1 mg nabilone administration (Redmond et al., 2008). Importantly, others have reported cannabinoid agonist-induced hyperalgesia (i.e., increased pain) with heat/capsaicin (8% THC smoked cannabis [Wallace et al., 2007]) and electrical stimulation assays (15 mg oral dronabinol [Kraft et al., 2008], 12 mg smoked THC [Hill et al., 1974]; 20 mg oral THC [Walter et al., 2011]; see brief review by Walter et al., 2014).
Dronabinol in combination with oxycodone reliably failed to enhance oxycodone analgesia (e.g., no changes in time to peak effect, duration of peak effect [data not shown]) in contrast to our hypothesis and a substantial preclinical literature. Importantly, under some dose conditions, dronabinol attenuated oxycodone analgesia. For example, the low dose of dronabinol (2.5 mg) decreased the analgesic effect of oxycodone (10 mg) on the pressure algometer and oxycodone (5mg) on the cold pressor test (Fig 1). While the discrepancy between prior preclinical studies and the current findings may be attributable to species, dose and preparation differences, they are consistent with at least one other clinical study. Patients undergoing a surgical procedure were randomized to receive nabilone (0.5, 1 mg, p.o.) or placebo 1 hr prior to anesthesia and every 8 hrs through 24 hrs post-surgery along with allowed post-surgical opioid treatment. There were no significant differences in post-surgical morphine doses administered (via PCA pump) across the groups, indicating that nabilone was not opioid-sparing. In addition, patients receiving nabilone (2 mg) exhibited greater post-surgical pain (instead of less) on both outcomes measured – pain at rest and pain during movement – relative to those who received placebo (Beaulieu, 2006).
Healthy volunteers without current pain or chronic pain histories and without substance use histories were enrolled in the present study. It is, therefore, possible that the current results may not translate to individuals with 1) long histories of pain (e.g., chronic pain syndromes), which can induce dysregulation of endocannabinoid and/or endogenous opioid receptor systems (e.g., Greco et al., 2018; Schrepf et al., 2016); and/or 2) long histories of opioid/cannabinoid use, which can produce similar neurophysiological changes (e.g., Varrassi et al., 2018; Bloomfield et al, 2018). Three studies have reported that cannabinoid/opioid dose combinations produce a signal for increased analgesia in these populations. Two studies examined pain patients, all of whom had extensive opioid histories (but limited or no cannabinoid use), and administered cannabinoid agonists along with patients’ ongoing opioid analgesic regimens (no experimental pain tests were conducted; subjective pain ratings were the primary outcome). The first, which did not implement a placebo control, administered vaporized cannabis (3.6% THC) and reported decreased pain scores (Abrams et al., 2011). The second administered dronabinol (10, 20 mg, p.o.) or placebo to pain patients and reported that both doses decreased pain ratings (Narang et al., 2008). The third study enrolled heavy/daily cannabis smokers and assessed cold pressor response after smoked cannabis (0, 5.6% THC) and oxycodone (2.5, 5 mg, p.o.). The 2.5 mg oxycodone dose + active (5.6%) cannabis increased cold pressor threshold and tolerance (Cooper et al. 2018). These data suggest that chronic exposure to either drug class may alter subsequent response; however, when combined with the present results, suggest that cannabinoid agonists including THC/dronabinol may yield unpredictable analgesic results (and cannabinoid dose and route of administration [oral vs. smoked]/pharmacokinetic differences may contribute to these mixed findings). This coupled with the psychoactive effects and abuse potential of cannabinoids, warrant careful consideration for their widespread use an adjuncts for opioid therapy.
It is well known that CB1 agonists have abuse potential; thus, the potential therapeutic benefit of cannabinoid agonists must be weighed against their potential risks. In the present study, dronabinol robustly increased the subjective effects of oxycodone related to its abuse potential, including peak ratings of feeling a drug effect, drug liking and feeling high (Fig. 2–3; Table 1). Similar findings were reported in two of the studies mentioned above (Cooper et al., 2018; Abrams, 2011). A combination of active smoked cannabis (5.6% THC) + oral oxycodone (2.5 mg) produced significant increases in oxycodone abuse liability (relative to oxycodone alone) in cannabis smokers (Cooper et al., 2018). Increased ratings of feeling high were reported upon active cannabis administration (3.6%) in pain patients taking opioids, although these effects dissipated after several consecutive days of use (Abrams et al., 2011). These data have important implications for the clinical use of opioid/cannabinoid combinations. Even when the doses of both compounds are within a therapeutic dose range, as in the present study, cannabinoids may increase the abuse potential of opioids. In the present study, dronabinol also increased subjective ratings of nodding, nauseous, tired/sleepiness, heavy/sluggish, unmotivated/lazy, and impaired; ratings of bad drug effects were also increased (Table 1; Fig. 3). These findings suggest that acute use of these drugs in combination may be impairing and intoxicating and could pose a safety risk (e.g., driving).
Some recent reports describing correlational, not causative, outcomes have received widespread attention, particularly in the lay press, and suggest that the presence of medical cannabis laws (i.e., availability of medical marijuana) decrease the total number of opioid doses prescribed (Bradford & Bradford, 2017). Another study reports that opioid overdose deaths (as measured from a CDC multiple-cause death database) declined following the passage of medical cannabis legislation (Bachhuber et al., 2014). However, in both studies, the data reported does not examine effects on an individual level; for example, there is no evidence that patients taking medical cannabis received fewer opioid prescriptions or were protected from overdose – thus, numerous other (non-pharmacological) factors could account for these coinciding changes. Although there are several survey-type studies in which patients report decreasing their opioid doses after initiating medical cannabis use (e.g., Boehnke et al., 2016; Lucas et al., 2016), this relationship has not been examined in a controlled study. The strongest data to date on this topic suggests the exact opposite finding – that medical cannabis use is detrimental to chronic pain patients taking opioid analgesics. The POINT study conducted in Australia (Campbell et al., 2018) prospectively enrolled 1514 patients with chronic non-cancer pain who were prescribed opioids and collected data on the cohort annually for 4 years. The study examined pain, anxiety, depression and opioid use outcomes in those who elected to use cannabis for pain, compared to those who did not. The authors found that cannabis worsened patient outcomes – patients who were using cannabis had greater pain severity, greater pain interference in their daily activities, and greater anxiety scores than the patients who were not using cannabis. Cannabis did not reduce opioid doses (no opioid-sparing effects were detected) and it did not change the number of patients who were able to stop taking opioids (Campbell et al., 2018). Similarly, an analysis of the 2015 National Survey on Drug Use and Health data (Caputi & Humphries, 2018) found that medical cannabis users were more likely to use and misuse psychoactive prescription medications in general, and specifically, misuse opioid medications, relative to those not using medical cannabis. A recent report indicates that cannabis use increases the risk of developing opioid use disorder as well (see Olfson et al., 2018).
Taken together, there are mixed results on the efficacy and overall risk profile of combining cannabinoid agonists with prescription opioids for pain relief. Additional controlled data are needed to provide information on the safety and pharmacological interactions of these agents, and the types of pain that should be targeted by opioid/cannabinoid combinations. The data collected in the current study with healthy participants suggest that dronabinol may not be an effective or suitable opioid adjunct due to an unfavorable profile of effects (no increase in analgesic effects, robust increase in psychoactive effects). Importantly, these data, combined with findings from other studies with different participant samples (chronic pain patients, frequent cannabis users), suggest that cannabinoid agonists have the potential to increase the abuse potential of opioid agonists. Although more controlled data are needed, this finding should not be overlooked, particularly in the context of the current opioid epidemic. Until there is strong evidence that cannabinoid treatment can ultimately result in opioid dose reduction/opioid-sparing – which has not been demonstrated to date – the risk/benefit profile of the medical use of cannabinoids as opioid adjuncts cannot be considered clearly favorable.
Acknowledgments
Grants from the National Center for Research Resources and National Center for Advancing of Translational Sciences (KL2TR000116-04 [SB]; UL1TR001998 [UK CTSA]) and the University of Kentucky Center for Clinical and Translation Science provided support for this research. We thank Anne Estrup Olesen (Aalborg University Hospital, Denmark) for feedback on the experimental pain protocols; the staff at the University of Kentucky (UK) Center on Drug and Alcohol Research for research support: Amanda Kopca, Matthew Taylor and Victoria Vessels; the UK Investigational Pharmacy for preparing study medication; and Dr. Samy-Claude Elayi for patient support.
This study was funded by grants from the National Center for Research Resources and National Center for Advancing of Translational Sciences (KL2TR000116-04 [SB]; UL1TR001998 [UK CTSA]).
Footnotes
Publisher's Disclaimer: This Author Accepted Manuscript is a PDF file of a an unedited peer-reviewed manuscript that has been accepted for publication but has not been copyedited or corrected. The official version of record that is published in the journal is kept up to date and so may therefore differ from this version.
The authors have no conflicts of interest to declare related to this project.
REFERENCES
- Abrams DI, Couey P, Shade SB, Kelly ME, Benowitz NL. Cannabinoid-opioid interaction in chronic pain. Clin Pharmacol Thera 2011;90(6):844–851. [DOI] [PubMed] [Google Scholar]
- Altis K, Schmidtko A, Angioni C, Kuczka K, Schmidt H, Geisslinger G, Lötsch J, Tegeder I. Analgesic efficacy of tramadol, pregabalin and ibuprofen in menthol-evoked cold hyperalgesia. PAIN 2009;147(1–3):116–21. [DOI] [PubMed] [Google Scholar]
- American Pain Society. Pain Management and Dosing Guide. 2016 http://americanpainsociety.org/uploads/education/PAMI_Pain_Mangement_and_Dosing_Guide_02282017.pdf.
- Babalonis S, Hampson AJ, Lofwall MR, Nuzzo PA, Walsh SL. Quinine as a potential tracer for medication adherence: A pharmacokinetic and pharmacodynamic assessment of quinine alone and in combination with oxycodone in humans. J Clin Pharmacol 2015;55(12):1332–1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babalonis S, Lofwall MR, Nuzzo PA, Walsh SL. Pharmacodynamic effects of oral oxymorphone: abuse liability, analgesic profile and direct physiologic effects in humans. Addiction Biol 2016; 21(1), 146–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachhuber MA, Saloner B, Cunningham CO, Barry CL. Medical cannabis laws and opioid analgesic overdose mortality in the United States, 1999–2010. JAMA Int Med 2014;174(10), 1668–1673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaulieu P Effects of nabilone, a synthetic cannabinoid, on postoperative pain. Can J Anaesth 2006; 53(8), 769–775. [DOI] [PubMed] [Google Scholar]
- Binder A, Stengel M, Klebe O, Wasner G, Baron R. Topical high-concentration (40%) menthol-somatosensory profile of a human surrogate pain model. J Pain 2011; 12(7), 764–773. [DOI] [PubMed] [Google Scholar]
- Bloomfield MA, Hindocha C, Green SF, Wall MB, Lees R, Petrilli K, Costello H, Ogunbiyi MO, Bossong MG, Freeman TP. The neuropsychopharmacology of cannabis: a review of human imaging studies. Pharm Thera; 2018. [DOI] [PMC free article] [PubMed]
- Bradford AC, Bradford WD. Medical marijuana laws may be associated with a decline in the number of prescriptions for Medicaid enrollees. Health Affairs 2017; 36(5), 945–951. [DOI] [PubMed] [Google Scholar]
- Boehnke KF, Litinas E, Clauw DJ. Medical Cannabis Use Is Associated With Decreased Opiate Medication Use in a Retrospective Cross-Sectional Survey of Patients With Chronic Pain. J Pain 2016;17(6):739–744. [DOI] [PubMed] [Google Scholar]
- Califf RM, Woodcock J, Ostroff S. A proactive response to prescription opioid abuse. NEJM 2016; 374(15):1480–1485. [DOI] [PubMed] [Google Scholar]
- Campbell G, Hall WD, Peacock A, Lintzeris N, Bruno R, Larance B, Nielsen S, Cohen M, Chan G, Mattick RP, Blyth F. Effect of cannabis use in people with chronic non-cancer pain prescribed opioids: findings from a 4-year prospective cohort study. Lancet Public Health 2018;3(7):e341–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caputi TL, Humphreys K. Medical marijuana users are more likely to use prescription drugs medically and nonmedically. J Addict Med 2018; July 1;12(4):295–9. [DOI] [PubMed] [Google Scholar]
- Cichewicz DL, Martin ZL, Smith FL, Welch SP. Enhancement mu opioid antinociception by oral delta9-tetrahydrocannabinol: dose-response analysis and receptor identification. J Pharmacol Exp Ther 1999; 289(2), 859–867. [PubMed] [Google Scholar]
- Cichewicz DL, McCarthy EA. Antinociceptive synergy between delta(9)-tetrahydrocannabinol and opioids after oral administration. J Pharmacol Exp Ther 2003;304(3):1010–1015. [DOI] [PubMed] [Google Scholar]
- Cichewicz DL. Synergistic interactions between cannabinoid and opioid analgesics. Life Sci 2004;74(11):1317–1324. [DOI] [PubMed] [Google Scholar]
- Compton P, Kehoe P, Sinha K, Torrington MA, Ling W. Gabapentin improves cold-pressor pain responses in methadone-maintained patients. Drug Alcohol Depend 2010; 109(1–3), 213–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Congressional Research Service (2018). The Opioid Epidemic and the Food and Drug Administration: Legal Authorities and Recent Agency Action Updated December 27, 2018 https://fas.org/sgp/crs/misc/R45218.pdf
- Cooper ZD, Bedi G, Ramesh D, Balter R, Comer SD, Haney M. Impact of co-administration of oxycodone and smoked cannabis on analgesia and abuse liability. Neuropsychopharmacology; 2018. [DOI] [PMC free article] [PubMed]
- Greco R, Demartini C, Zanaboni AM, Piomelli D, Tassorelli C. Endocannabinoid System and Migraine Pain: An Update. Frontiers in neuroscience; 2018; 12, 172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dowell D, Haegerich TM, Chou R. CDC Guideline for Prescribing Opioids for Chronic Pain — United States, 2016. MMWR Recomm Rep 2016;65(No. RR-1):1–49. 10.15585/mmwr.rr6501e1. [DOI] [PubMed] [Google Scholar]
- Fattore L, Spano M, Melis V, Fadda P, Fratta W. Differential effect of opioid and cannabinoid receptor blockade on heroin-seeking reinstatement and cannabinoid substitution in heroin-abstinent rats. Br J Pharmacol 2011;163(7):1550–1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser HF, Van Horn GD, Martin WR, Wolbach AB, Isbell H. Methods for evaluating addiction liability. (A) “Attitude” of opiate addicts toward opiate-like drugs. (B) a short-term “direct” addiction test. J Pharmacol Exp Ther 1961;133: 371–387. [PubMed] [Google Scholar]
- Greenwald MK, & Stitzer ML Antinociceptive, subjective and behavioral effects of smoked marijuana in humans. Drug Alcohol Depend 2000; 59(3), 261–275. [DOI] [PubMed] [Google Scholar]
- Haney M, Malcolm RJ, Babalonis S, et al. Oral Cannabidiol Does Not Alter the Subjective, Reinforcing or Cardiovascular Effects of Smoked Cannabis. Neuropsychopharmacology 2016;41(8):1974–1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haghparast A, Shamsizadeh A, Samandari R, Omranifard A, Vaziri A, Razavi Y. Cannabinoid receptors in the basolateral amygdala are involved in the potentiation of morphine rewarding properties in the acquisition, but not expression of conditioned place preference in rats. Brain Res 2014;1565:28–36. [DOI] [PubMed] [Google Scholar]
- Hill SY, Schwin R, Goodwin DW, Powell BJ. Marihuana and pain. J Pharmacol Exp Thera 1974; 188(2), 415–418. [PubMed] [Google Scholar]
- Kinsey SG, Long JZ, Cravatt BF, Lichtman AH. Fatty acid amide hydrolase and monoacylglycerol lipase inhibitors produce anti-allodynic effects in mice through distinct cannabinoid receptor mechanisms. Pain 2010; 11(12), 1420–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan MIA, Walsh D, Brito-Dellan N. Opioid and adjuvant analgesics: compared and contrasted. Am J Hosp Palliat Care 2011; 28(5), 378–383. [DOI] [PubMed] [Google Scholar]
- Kraft B, Frickey NA, Kaufmann RM, Reif M, Frey R, Gustorff B, Kress HG. Lack of analgesia by oral standardized cannabis extract on acute inflammatory pain and hyperalgesia in volunteers. Anesthesiology 2008; 109(1), 101–110. [DOI] [PubMed] [Google Scholar]
- Li JX, Koek W, France CP. Interactions between Delta(9)-tetrahydrocannabinol and heroin: self-administration in rhesus monkeys. Behav Pharmacol 2012;23(8):754–761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lile JA, Kelly TH, Charnigo RJ, Stinchcomb AL, Hays LR. Pharmacokinetic and Pharmacodynamic Profile of Supratherapeutic Oral Doses of Δ9‐ THC in Cannabis Users. J Clin Pharmacol 2013; 53(7), 680–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucas P, Walsh Z, Crosby K. Substituting cannabis for prescription drugs, alcohol and other substances among medical cannabis patients: The impact of contextual factors. Drug Alcohol Rev 2016;35(3):326–333. [DOI] [PubMed] [Google Scholar]
- Maguire DR, Yang W, France CP. Interactions between μ-opioid receptor agonists and cannabinoid receptor agonists in rhesus monkeys: antinociception, drug discrimination, and drug self-administration. Journal of Pharmacology and Experimental Therapeutics 2013; 345(3):354–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maguire DR, France CP. Effects of daily delta-9-tetrahydrocannabinol treatment on heroin self-administration in rhesus monkeys. Behav Pharmacol 2016;27(2–3 Spec Issue):249–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maguire DR, France CP. Reinforcing effects of opioid/cannabinoid mixtures in rhesus monkeys responding under a food/drug choice procedure. Psychopharmacology 2018; 3:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marinol® (dronabinol) Package Insert. Banner Pharmacaps, Inc. Package Insert and Prescribing Information for Marinol® (dronabinol) capsules 2013; AbbVie Inc., Chicago, IL. [Google Scholar]
- McLeod DR, Griffiths RR, Bigelow GE, Yingling JE. An automated version of the digit symbol substitution test (DSST). Behav Res Methods Instrum Comput 1982; 14, 463–466. [Google Scholar]
- Moulin DE, Clark AJ, Gilron I, Ware MA, Watson CP, Sessle BJ. Pharmacological management of chronic neuropathic pain—consensus statement and guidelines from the Canadian Pain Society. Pain Res Manag 2007;12(1):13–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naef M, Curatolo M, Petersen-Felix S, Arendt-Nielsen L, Zbinden A, Brenneisen R. The analgesic effect of oral delta-9-tetrahydrocannabinol (THC), morphine, and a THC-morphine combination in healthy subjects under experimental pain conditions. Pain 2003;105(1–2):79–88. [DOI] [PubMed] [Google Scholar]
- Narang S, Gibson D, Wasan AD, et al. Efficacy of dronabinol as an adjuvant treatment for chronic pain patients on opioid therapy. J Pain 2008;9(3):254–264. [DOI] [PubMed] [Google Scholar]
- Olesen AE, Andresen T, Staahl C, Drewes AM. Human experimental pain models for assessing the therapeutic efficacy of analgesic drugs. Pharmacol Rev 2012; 64(3), 722–779. [DOI] [PubMed] [Google Scholar]
- Olfson M, Wall MM, Liu SM, Blanco C. Cannabis use and risk of prescription opioid use disorder in the United States. Am J Psychiatry 2017;175(1), 47–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redmond WJ, Goffaux P, Potvin S, Marchand S. Analgesic and antihyperalgesic effects of nabilone on experimental heat pain. Curr Med Res Opin 2008; 24(4), 1017–1024. [DOI] [PubMed] [Google Scholar]
- Roberts JD, Gennings C, Shih M. Synergistic affective analgesic interaction between delta-9-tetrahydrocannabinol and morphine. Eur J Pharmacol 2006;530(1–2):54–58. [DOI] [PubMed] [Google Scholar]
- Schrepf A, Harper DE, Harte SE, Wang H, Ichesco E, Hampson JP, Zubieta JK, Clauw DJ, Harris RE. Endogenous opioidergic dysregulation of pain in fibromyalgia: a PET and fMRI study. Pain 2016; 157(10), 2217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solinas M, Panlilio LV, Tanda G, Makriyannis A, Matthews SA, Goldberg SR. Cannabinoid agonists but not inhibitors of endogenous cannabinoid transport or metabolism enhance the reinforcing efficacy of heroin in rats. Neuropsychopharmacology 2005;30(11):2046–2057. [DOI] [PubMed] [Google Scholar]
- Staahl C, Olesen AE, Andresen T, Arendt-Nielsen L, Drewes AM. Assessing analgesic actions of opioids by experimental pain models in healthy volunteers - an updated review. Br J Clin Pharmacol 2009; 68(2), 149–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- United States Food and Drug Administration. Statement by FDA Commissioner Scott Gottlieb, M.D., on the agency’s ongoing work to forcefully address the opioid crisis. 2018 https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm618831.htm.
- Varrassi G, Fusco M, Skaper SD, Battelli D, Zis P, Coaccioli S, Pace MC, Paladini A. A Pharmacological Rationale to Reduce the Incidence of Opioid Induced Tolerance and Hyperalgesia: A Review. Pain Thera 2018:1–7. [DOI] [PMC free article] [PubMed]
- Wallace M, Schulteis G, Atkinson JH, Wolfson T, Lazzaretto D, Bentley H, Gouaux B, Abramson I. Dose-dependent effects of smoked cannabis on capsaicin-induced pain and hyperalgesia in healthy volunteers. Anesthesiology 2007; 107(5), 785–796. [DOI] [PubMed] [Google Scholar]
- Walsh SL, Nuzzo PA, Lofwall MR, Holtman JR Jr. The relative abuse liability of oral oxycodone, hydrocodone and hydromorphone assessed in prescription opioid abusers. Drug Alcohol Depend 2008; 98:191–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter C, Oertel BG, Lötsch J. THC may reproducibly induce electrical hyperalgesia in healthy volunteers. Euro J Pain 2015;19(4):516–8. [DOI] [PubMed] [Google Scholar]
- Welch SP. Interaction of the cannabinoid and opioid systems in the modulation of nociception. Int Rev Psychiatry 2009; 21(2), 143–151. [DOI] [PubMed] [Google Scholar]
- Welch SP, Eads M. Synergistic interactions of endogenous opioids and cannabinoid systems. Brain Res 1999; 848(1–2), 183–190. [DOI] [PubMed] [Google Scholar]