

Abstract
Rationale:
Norepinephrine plays a critical role in the stress response. Clarifying the psychopharmacological effects of norepinephrine manipulation on stress reactivity in humans has important implications for basic neuroscience and treatment of stress-related psychiatric disorders, such as posttraumatic stress disorder and alcohol use disorders. Preclinical research implicates the norepinephrine alpha-1 receptor in responses to stressors. The No Shock, Predictable Shock, Unpredictable Shock (NPU) task is a human laboratory paradigm that is well positioned to test cross-species neurobiological stress mechanisms and advance experimental therapeutic approaches to clinical trials testing novel treatments for psychiatric disorders.
Objectives:
We hypothesized that acute administration of prazosin, a noradrenergic alpha-1 antagonist, would have a larger effect on reducing stress reactivity during unpredictable, compared to predictable, stressors in the NPU task.
Methods:
We conducted a double-blind, placebo-controlled, crossover randomized controlled trial in which sixty-four healthy adults (32 female) completed the NPU task at two visits (2mg prazosin vs. placebo).
Results:
A single acute dose of 2mg prazosin did not reduce stress reactivity in a healthy adult sample. Neither NPU startle potentiation nor self-reported anxiety was reduced by prazosin (vs. placebo) during unpredictable (vs. predictable) stressors.
Conclusions:
Further research is needed to determine whether this failure to translate preclinical neuroscience to human laboratory models is due to methodological factors (e.g., acute vs. chronic drug administration, brain penetration, study population) and/or suggests limited clinical utility of noradrenergic alpha-1 antagonists for treating stress-related psychiatric disorders.
Keywords: Stress, prazosin, noradrenaline, startle response, startle potentiation, posttraumatic stress disorder, alcohol use disorder
Introduction
Initial excitement for prazosin as a promising treatment for posttraumatic stress disorder (PTSD) and alcohol use disorder (AUD) has recently been tempered by larger clinical trials, which failed to show improvement in clinical outcomes (Petrakis et al. 2016; Raskind et al. 2018; Simpson et al. 2018). Prazosin is a norepinephrine alpha-1 (NE-α1) antagonist originally developed as an antihypertensive medication that has widespread actions in both the peripheral and central nervous systems. Prazosin’s ability to penetrate the blood-brain barrier and the well documented role of NE in arousal, sleep, and stress spurred researchers to test prazosin as a novel treatment for PTSD (for review see Hendrickson and Raskind 2016). Early studies demonstrated positive clinical outcomes related to nightmares, sleep disturbance, and patients’ overall functioning in PTSD (Raskind et al. 2003, 2007, 2013). Follow up studies suggested prazosin may reduce relapse in AUD (Simpson et al. 2009; Fox et al. 2012), which is highly comorbid and shares stress-related etiology with PTSD (McCarthy and Petrakis 2010; Gilpin and Weiner 2017). Understanding why the findings of most early small trials failed to replicate and examining if prazosin may improve other symptom targets remain pressing scientific and clinical questions (Krystal et al. 2017; Haass-Koffler et al. 2018).
Efforts to repurpose prazosin grew from robust animal neuroscience literature that clearly demonstrated NE’s broad role in coordinating the body’s response to stress (Berridge and Waterhouse 2003; Arnsten 2009). In rodents, brain NE levels are elevated in response to discrete stressors (Pacák et al. 1995; Galvez et al. 1996). Similarly, manipulations that increase NE release or NE-receptor binding elicit arousal and stress-related behaviors (Varty et al. 1999; Berridge 2008). The NE system and neural circuits can develop sensitized responses to acute stressors following exposure to prolonged or intense stressors or chronic alcohol/drug use (Smith and Aston-Jones 2008; Koob 2009; Rajbhandari et al. 2015). Although dysregulation in these NE neural systems may occur via multiple pathways, the resulting exaggerated stress reactivity may represent a transdiagnostic feature and viable treatment target for both PTSD and addiction. Indeed, preclinical rodent models of stress-induced reinstatement of alcohol seeking behavior have found promising effects of prazosin (Lê et al. 2011; Funk et al. 2016). Despite this neuroscientific foundation, repurposing prazosin has proceeded largely without basic psychopharmacology research in humans and has failed to include mechanism-relevant clinical outcomes such as exaggerated stress reactivity (but see Fox et al. 2012; Verplaetse et al. 2017).
Research is rapidly accruing to suggest that stress reactivity, and more specifically, acute response to a subset of stressors that are unpredictable (vs. predictable), may provide a critical mechanism to account for many maladaptive outcomes among stress-related psychiatric disorders (e.g., relapse, Kaye et al. 2017; Koob 2009). These unpredictable (i.e., ambiguous, ill-defined) stressors appear to produce phenomenologically distinct responses via overlapping yet partially separable neural mechanisms relative to predictable (i.e., well-defined, imminent) stressors (Davis et al. 2010). Unpredictable stressors and NE manipulations are ubiquitous in behavioral neuroscience animal models that probe anxiety-like and drug-seeking behaviors. For instance, unpredictable footshock and yohimbine challenge are widely used to instigate reinstatement of previously extinguished drug-seeking behavior, a model of stress-induced relapse (for review see Mantsch et al. 2016). Moreover, unpredictability is a cardinal feature of the typical stressors that humans experience in their daily lives (e.g., financial security, interpersonal conflicts); these types of stressors often exacerbate PTSD symptoms and precede relapse in addiction. As such, examination of NE mechanisms in stress reactivity in humans may benefit from evaluation of tasks that can parse unpredictable vs. predictable stressors.
To parse the neural mechanisms involved in response to unpredictable vs. predictable stressors, affective neuroscience has relied heavily on startle potentiation, an important animal-human translational bridge (Davis et al. 2010). As such, we have detailed knowledge of the neurobiology of the startle response and its potentiation. In preclinical rodent models, startle potentiation during unpredictable stressors has implicated NE- and corticotropin-releasing factor (CRF)-sensitive pathways through the lateral divisions of the central amygdala and bed nucleus of the stria terminalis (Walker et al. 2009; Davis et al. 2010). In contrast, distinct pathways through the medial division of the central amygdala appear responsible for startle potentiation during predictable stressors (Walker and Davis 1997; Davis et al. 2010). NE is a powerful modulator of extrahypothalamic CRF and many stress-related behaviors (Berridge and Dunn 1989; Gresack and Risbrough 2011). In rodents, acute prazosin pretreatment reduces startle potentiation elicited by direct administration of CRF, suggesting that CRF-enhanced startle is NE-α1 dependent (Gresack and Risbrough 2011). Prazosin administration prior to unpredictable stressors (e.g., restraint and inescapable tail-shock) reduces subsequent increases in startle response in rodents (Manion et al. 2007). In humans, the startle response is potentiated by pharmacological challenge that elevates NE levels via yohimbine in healthy adults and patients with PTSD or alcohol/drug addiction (Morgan et al. 1993, 1995; Stine et al. 2001). Thus, startle potentiation during unpredictable stressors 1) represents a psychophysiological index of heightened response to stressors; 2) has well known neurobiological substrates in rodents; and 3) can be assessed across species, positioning it as an attractive translational measure. However, the effect of an NE-α1 antagonist on startle potentiation has not been examined in humans to date.
Grillon and colleagues developed the No Shock, Predictable Shock, Unpredictable Shock (NPU) task to contrast responses to unpredictable vs. predictable stressors (Schmitz and Grillon 2012). Predictable shock conditions involve administration of 100% cue-contingent, imminent electric shock. Unpredictable shock conditions involve temporally and probabilistically uncertain administration of shock. Startle potentiation during unpredictable shock (relative to no-shock blocks) provides the primary measure of stressor reactivity. This task represents a direct translation of preclinical methods and measures to parse the neural mechanisms involved in response to unpredictable vs. predictable stressors (Davis et al. 2010).
The NPU and related tasks have been used to identify common phenotypic characteristics of stress-related disorders and to probe pharmacological effects of anxiolytic agents (Schmitz and Grillon 2012; Shankman et al. 2013; Kaye et al. 2017). Individuals with PTSD, AUD and panic disorder display elevated startle potentiation to unpredictable stressors but not predictable stressors (Grillon et al. 2009b, 2008; Gorka et al. 2013; Moberg et al. 2017). This hypersensitivity to unpredictable stressors is not indicative of psychopathology broadly, as it is not observed in major depressive disorder or generalized anxiety disorder (Grillon et al. 2009b; Shankman et al. 2013). Pharmacological manipulations with expected anxiolytic effects (i.e., acute benzodiazepines and alcohol, chronic SSRIs) selectively reduce startle potentiation during unpredictable (vs. predictable) stressors (Grillon et al. 2006, 2009a; Moberg and Curtin 2009; Bradford et al. 2013). These studies support the utility of startle potentiation during unpredictable stress in the NPU task as a sensitive testbed to detect transdiagnostic perturbations in stress-related disorders and screen potential novel medications to target these processes. Examining prazosin’s effects on startle potentiation in the NPU task would be particularly informative considering conflicting reports on the efficacy of prazosin as a novel treatment for PTSD/AUD and the paucity of basic psychopharmacology research on how prazosin affects stress reactivity in humans.
The current double-blind, placebo-controlled, crossover randomized controlled trial (N = 64) examined the effects of acute prazosin administration on stress reactivity during unpredictable and predictable stressors in the NPU task in healthy adults. This is the first study, to our knowledge, to examine the impact of prazosin on the startle response in humans, a physiological measure of stress reactivity. We examined startle potentiation and self-reported anxiety during the NPU task to include translational and subjective markers of stress reactivity. We hypothesized that prazosin (vs. placebo) would have a larger effect on reducing stress reactivity during unpredictable (vs. predictable) stressors. Positive results would suggest greater NE-α1 receptor involvement in acute responses to unpredictable relative to predictable stressors. Further, if NE-α1 antagonism reduces stress reactivity in humans, this could provide guidance for prioritizing outcomes in future clinical research (e.g., exaggerated startle in PTSD, stress-related relapse in addiction).
Materials and Methods
Open science and preregistration
We took several steps to follow emerging open science guidelines to promote transparency and reproducibility. We preregistered the study design and data analysis plan prior to the start of data collection (Open Science Framework: https://osf.io/m8jmp/, ClinicalTrials.gov ). We have reported how we determined our sample size, all data exclusions, all manipulations, and all measures in the study (see Supplement, Simmons et al. 2012). Finally, we have made the data, analysis code, and other study materials publicly available (https://osf.io/un6h6/).
Participants
We recruited sixty-four participants (32 female) from November 2016 to March 2018 from the greater community (see Supplement for CONSORT diagram and a priori power calculations).1 Participants were 18 to 46 years old (mean age = 23 years, SD = 5.3 years). The racial composition of the sample was 64% White, 19% Asian, 6% Black, and 11% Other Race (8% Hispanic/Latino). We excluded those who self-reported: uncorrected auditory or visual problems; colorblindness; pregnancy, breastfeeding, or unreliable contraception in women; current medication with direct noradrenergic action (e.g., NE beta blockers, NE alpha2 agonists, NE alpha1 agonists, psychostimulants, SNRIs); current medication with acute anxiolytic or sedative properties (e.g., benzodiazepines, zolpidem); current medications with interactions with prazosin that increase side effect potential (e.g., sildenafil, trazadone); medical or psychiatric conditions that would contraindicate electric shock exposure or prazosin administration; substance use disorder other than tobacco; or severe, persistent mental illness. We excluded those with a blood alcohol concentration >0.00%, non-negative urine pregnancy test (female only), heart rate <56 or >100 bpm, systolic blood pressure <100 or >160 mmHg, orthostatic hypotension, or symptoms upon standing (e.g., dizziness, lightheadedness, etc.) at any study visit. We compensated participants $390 for completing the study ($15 screening visit, $150 per study visit, $75 completion bonus).
General procedures
University of Wisconsin (UW) Madison Health Sciences Institutional Review Board approved all procedures. We determined preliminary eligibility during a phone screening and screening visit. At this visit at UW-Madison, we explained the study purpose and procedures and obtained written informed consent. Eligible participants completed two subsequent overnight study visits at the UW Hospital separated by approximately two weeks (Mean = 12.6 days, Range = 4-35 days, Median = 8 days). At each study visit, we reassessed the self-reported and objectively-measured eligibility criteria, and the study physician completed a medical history and physical exam. The procedures were identical at both study visits except where noted (see Figure 1a for Study Procedures Flowchart).
Figure 1. Study Procedures Flowchart and No Shock, Predictable Shock, Unpredictable Shock (NPU) Task.


1a: This figure displays the procedures completed at each visit in this within-subjects crossover design. Screening visit procedures included obtaining informed consent, preliminary eligibility determination, and shock sensitivity assessment. At study visit 1 we randomly assigned participants to drug administration order (between-subjects). All participants receive both prazosin and placebo (within-subjects), one at each study visit. Participants randomized to order A (n = 34) received 2mg prazosin at study visit 1 and placebo at study visit 2. Participants randomized to order B (n = 30) received placebo at study visit 1 and 2mg prazosin at study visit 2. At study visits 1 and 2 participants were orally administered a pill and completed the General Startle Reactivity Task (75 minutes post-dose) and NPU Task (90 minutes post-dose).
1b: In the NPU task, participants viewed a series of colored square “cues” displayed briefly on a computer screen. We presented cues in a blocked design with three conditions: No Shock (N), Predictable Shock (P), and Unpredictable Shock (U). The upper panel displays counterbalanced conditions both within- and between-subjects. Participants completed the same condition order at both study visits. The lower panel displays examples of each condition. All blocks included 6 cues presented sequentially for 5 seconds separated by a variable inter-trial interval (ITI; 14-20 seconds). In No Shock, we instruct participants that no electric shocks will be administered at any time. In Predictable Shock, we instructed participants that they would receive a shock at the end of every cue, but never during the ITI, so that the cue ‘predicted’ that the shock would occur in several seconds. In Unpredictable Shock, we instructed participants that they could receive a shock at any time, during both the cues and ITIs, so that the occurrence of the shock was unpredictable to the participant. We measured the eye-blink startle response elicited by “startle probes” (5ms acoustic white noise) presented binaurally over headphones. We calculated startle potentiation during cues separately in Predictable and Unpredictable Shock conditions as the differences between response to startle probes during the shock conditions and no-shock conditions (i.e., predictable startle potentiation = predictable cue – no shock cue). After the NPU task, participants retrospectively reported their subjective anxiety/fear during each condition cue. A figure legend is displayed in the left panel.
Figure 1b modified with permission from Schmitz & Grillon (2012). Used with permission of Springer Nature.
Participants were administered prazosin or placebo (see Prazosin Dosing below) and 60 minutes post-dose were seated in a dimly lit room approximately 45 inches in front of a 20-inch CRT computer monitor. Participants completed the General Startle Reactivity Task (75 minutes post-dose) and NPU Task (90 minutes post-dose). At the first study visit only, participants completed a battery of questionnaires on an iPad (Apple Inc.) using Qualtrics software (Provo, UT, USA) to assess demographics, trait affect, and broadband personality traits. Participants were admitted overnight to the hospital for safety monitoring and discharged the following morning after medical assessment. Participants were debriefed at the final study visit.
Prazosin dosing
Participants were orally administered 2mg prazosin at one study visit and placebo at the other visit (randomly-assigned order was counterbalanced between subjects). Participants and study staff were blind to drug administration order.2 Participant blinding was assessed after the NPU Task; participants reported which pill they believed they received that day on a 5-point Likert scale (1 = “No Medication”; 5 = “Study Medication”).
Shock sensitivity assessment
We coded our experimental tasks in MATLAB using the Psychophysics Toolbox extensions (Kleiner et al. 2007). At the screening visit, we measured participants’ subjective tolerance using standard procedures from our laboratory (Kaye et al. 2016). Participants rated a series of 200ms electric shocks of increasing intensity (7 mA maximum) administered to the distal phalanges of the 2nd and 4th fingers of one hand. We used participants’ subjective maximum tolerated shock from this procedure during the NPU Task to control for individual differences in subjective shock tolerance. We used the same shock level in the NPU Task at both study visits.
General startle reactivity
We measured participants’ resting startle response prior to initiating the NPU task at both study visits to assess their general startle reactivity (75 minutes post-dose). Participants viewed a white fixation cross in the center of the black screen while 9 acoustic startle probes were presented, separated by 13-20s (task length: 2.5min). No other images were displayed on the screen, and no shocks were delivered. General startle reactivity was calculated as the mean raw startle response during this procedure (excluding first 3 habituation probes). We assessed general startle reactivity to evaluate individual differences in startle response and to determine if prazosin (vs. placebo) affects the startle response prior to the threat context.3
No shock, predictable shock, unpredictable shock (NPU) task
Participants completed a version of the NPU Task with demonstrated adequate psychometric properties for repeated administrations (see Figure 1b; Kaye et al. 2016). During the NPU Task, participants viewed a series of colored square “cues” displayed in the center of a computer screen with a black background. We presented cues in a blocked design with three conditions: No Shock (N), Predictable Shock (P), and Unpredictable Shock (U). Each shock condition was presented twice and separated by no shock conditions. Condition order was counterbalanced both within- and between-subjects (i.e., 2 condition orders: PNUNUNP, UNPNPNU), and participants completed the same order at both study visits. All blocks included 6 cues presented for 5s separated by a variable inter-trial-interval (ITI; mean 17s, range 14-20s). A white fixation cross remained in the center of the monitor during the cues and ITI. We administered a 200ms electric shock 200ms prior to cue offset during every cue in the Predictable shock conditions, so that the cue ‘predicted’ that the shock would occur in several seconds. We administered electric shock at pseudo-random times during both cues and ITIs in the Unpredictable shock condition (2 or 4.8s post-cue onset and 4, 8, or 12s post-cue offset), so that the occurrence of the shock was unpredictable by the participant. Twelve electric shocks were administered in each Predictable and Unpredictable shock condition. No electric shock occurred during the No Shock condition. We took several steps to ensure participants clearly understood the differences between task conditions based on our previously published methods (see Supplement and Kaye et al. 2016). Each block lasted approximately 2.5m and the entire task lasted approximately 20m. After the NPU task, participants retrospectively reported their anxiety/fear during each condition on a 5-point Likert scale (1 = ‘Not at all anxious/fearful’, 5 = ‘Very anxious/fearful’).
Startle probes occurred at 4.5s post cue-onset on a pseudo-random subset of 8 cues and 13, 14, or 15s post-cue offset during 4 ITIs in both shock conditions (no shock condition: 12 cues and 6 ITIs). Startle probes occurred a minimum of 12.5s after another startle eliciting event (e.g., shock or startle probe). Serial position of startle probes across the three conditions for both cues and ITIs was counterbalanced within-subjects to account for habituation. We used two different orders of the serial position of startle probe, counterbalanced between-subjects.
Startle response measurement and quantification
We recorded eyeblink electromyogram (EMG) activity to the acoustic startle probes (50ms, 102dB) according to published guidelines (Blumenthal et al. 2005). We conducted data acquisition, offline processing, and artifact rejection using our previously published (Kaye et al. 2016) and preregistered criteria (see Supplement for details). We quantified the startle response as the peak amplitude 20-100ms post-startle probe onset relative to a 50ms pre-probe baseline. We calculated startle magnitude as the mean startle response during cues for each condition in the NPU task. We calculated startle potentiation during cues separately for unpredictable and predictable blocks as the difference between response to probes during the shock and no-shock blocks (i.e., predictable startle potentiation = predictable cue - no shock cue)4
Preregistered analysis plan
We preregistered our a priori analysis plan prior to initiating data collection. We analyzed startle potentiation and self-reported anxiety in the NPU Task in separate General Linear Models (GLMs) with repeated measures for Drug (Prazosin vs. Placebo) and NPU Task Condition (Unpredictable vs. Predictable). We report partial eta squared (ηp2) and raw GLM parameter estimates (b) to document effect sizes. We evaluated additive covariates to increase power and report covariates included in the final models5. We used the standard p < .05 criteria for determining that results from all tests are significantly different from those expected if the null hypothesis were correct. We removed any model outliers identified as Bonferroni-corrected studentized residuals of p < .05.
Our preregistered hypothesis was that prazosin (vs. placebo) would have a larger effect on startle potentiation (primary outcome) and self-reported fear/anxiety (secondary outcome) during unpredictable vs. predictable stressors. We tested these hypotheses with separate models for each outcome with a two-way interaction of Drug X NPU Task Condition. We report data analysis of our a priori pre-registered hypothesis tests separately from all subsequent analyses for manipulation checks, robustness, and exploratory analyses. We accomplished data analysis and figure preparation with R within R-Studio.
RESULTS
NPU task preregistered analyses
NPU startle potentiation:
We analyzed startle potentiation in a GLM with repeated measures for Drug (prazosin vs placebo) and NPU Condition (unpredictable vs predictable shock), see Figure 2a and Table S1. Test of our primary preregistered hypothesis showed that there was not a significant Drug X NPU Condition interaction, ηp2 = .006, b = 2.4μV, t(63) = 0.64, p = .526, indicating that prazosin (vs. placebo) did not have a larger effect on reducing startle potentiation during unpredictable (vs. predictable) threat.6
Figure 2. Startle Potentiation and Self-Reported Anxiety by Drug and NPU Condition.
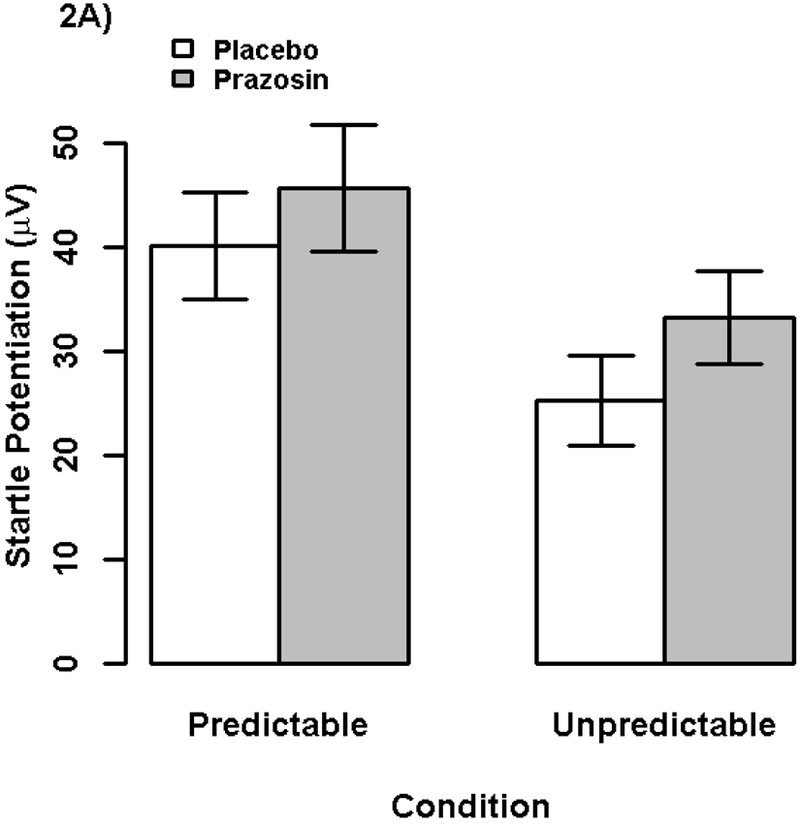
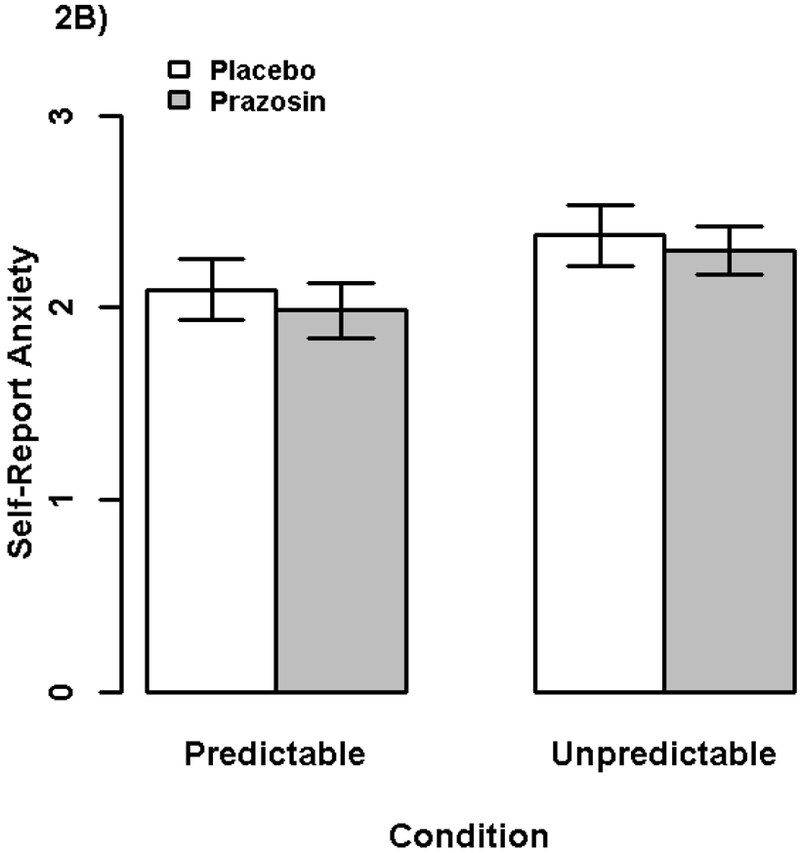
Bars display (2A) startle potentiation and (2B) self-reported anxiety/fear to predictable and unpredictable shock (vs. no shock) following prazosin (gray) and placebo (white) administration. Confidence bars represent ± one standard error for point estimates of startle potentiation from the general linear models.
NPU self-report anxiety:
We analyzed self-reported fear/anxiety potentiation in a GLM with repeated measures for Drug and NPU Condition, see Figure 2b and Table S1. Test of our secondary preregistered hypothesis showed that there was not a significant Drug X NPU Condition interaction, ηp2 < .001, b = 0.03, t(63) = 0.18, p = .857, indicating that prazosin (vs. placebo) did not have a larger effect on reducing retrospective self-reported anxiety/fear during unpredictable (vs. predictable) threat.
NPU task exploratory analyses
We report the following non-preregistered exploratory analysis to characterize the data more fully and provide insights for future research.
NPU startle drug main effect:
We examined whether prazosin had an overall effect of reducing stress reactivity irrespective of stressor predictability. There was a significant main effect of Drug on startle potentiation, ηp2 = .107, b = −8.0μV, t(61) = −2.71, p = .009, indicating that startle potentiation was larger following prazosin than placebo administration (see Figure 2a).7 Following our preregistered analysis plan for our primary analysis we included general startle reactivity as a covariate and removed one model outlier.8
NPU self-report anxiety drug main effect:
There was not a significant main effect of Drug on overall self-reported fear/anxiety, ηp2 = .001, b = 0.03, t(62) = 0.27, p = .792, indicating that prazosin did not affect overall self-reported fear/anxiety.9
General startle reactivity
We analyzed general startle reactivity in a GLM with repeated measures for Drug. There was not a significant effect of Drug on general startle reactivity (Prazosin Mean = 79.2μV, Placebo Mean = 75.4μV), ηp2 = .010, b = −3.8μV, t(63) = −0.81, p = .421, indicating that prazosin did not affect overall startle response prior to stressor exposure (i.e., NPU Task).
Manipulation checks, robustness, post hoc power, and exploratory analyses
We conducted follow-up analyses to evaluate the robustness, reliability, and internal validity of the NPU task, placebo blind, and peripheral effects of prazosin (see Supplement). These analyses support the effectiveness of the stressor and drug manipulations and evaluate alternative explanations for the primary results. We confirmed that 2mg prazosin reduced blood pressure overall, with significant effect on diastolic by 1-hour post-administration (Figure 3), suggesting prazosin was physiologically active during the time window of the NPU Task (~1.5-2hr). We also report results from post hoc Monte Carlo power simulation that indicate we had high power (> 99.9%) to detect a medium effect size for prazosin (see Supplement).
Figure 3. Blood Pressure by Drug and Time.
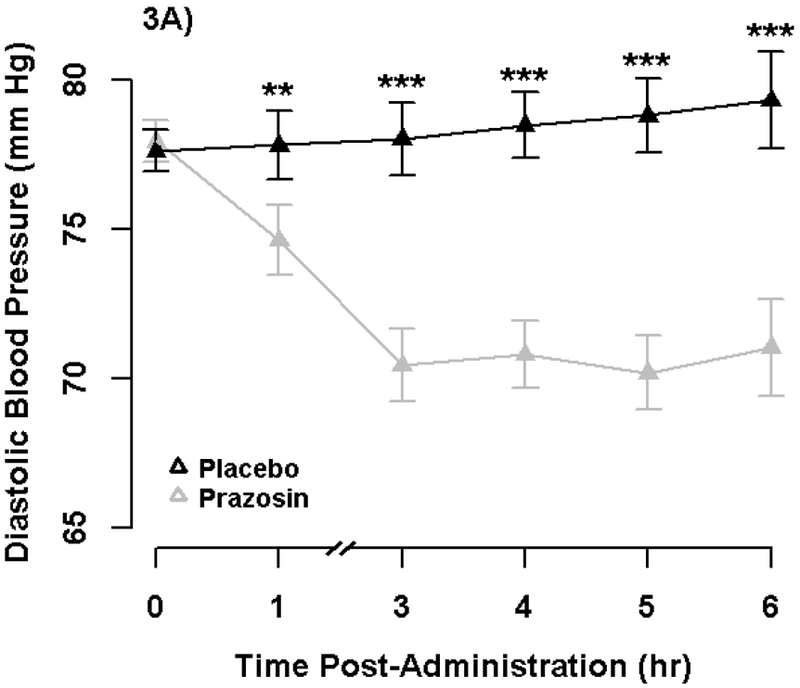
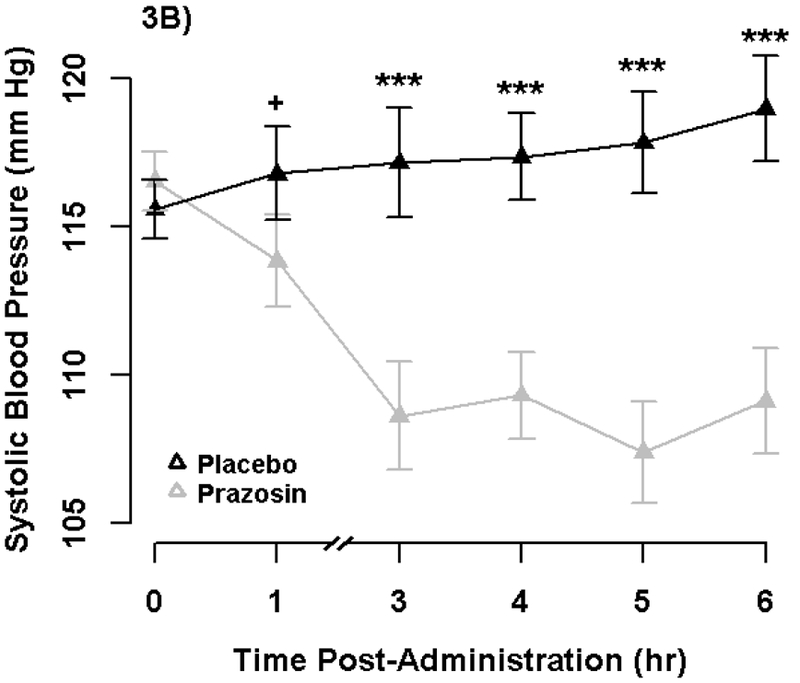
Points display standing diastolic (3A) and systolic (3B) blood pressure (BP) by Drug and Time. Error bars represent +−1 SE of the Drug effect from separate covariate adjusted General Linear Models at each time point. We analyzed standing diastolic and systolic BP in separate General Linear Models with repeated measures for Drug and Time and baseline BP (mean-centered averaged across visits) as a between-subjects regressor. Prazosin produced a significant reduction in both diastolic and systolic BP overall (p’s < .001). Furthermore, prazosin produced a significant reduction in diastolic BP at both 1-hour (p =.008) and 3-hour (p < .001). Prazosin produced a significant reduction in systolic BP by 3-hours (p < .001), but non-significant change at 1-hour (p = .067), consistent with the known greater effects on diastolic than systolic BP. See Supplement for additional analyses.
+p < .10; *p < .05; **p < .01; ***p < .001
DISCUSSION
Results of the current study indicate that a single acute dose of prazosin does not reduce stress reactivity in healthy adults. We did not find evidence to support our a priori hypothesis that acute administration of 2mg prazosin (vs placebo) would decrease startle potentiation or self-reported anxiety to a greater degree during unpredictable than predictable stressors. Prazosin did not have differential effects on either measure of stress reactivity as a function of predictability. Robustness analyses (see Supplement) suggest these null results are not attributable to individual difference moderators (e.g., age, sex, baseline blood pressure, trait affect measures) or methodological factors (e.g., prazosin-placebo order, expectancy effects, shock intensity). Following a rigorous preregistered analysis plan in a well-powered efficient within-subjects cross-over study design increases confidence in our null results.
Exploratory follow up analysis suggests prazosin may have acutely increased, rather than decreased, overall startle potentiation during threat of shock. This effect was robust to numerous analytic checks (see Supplement, e.g., no drug order moderation, between-subject drug effect observed at study visit 1 only). We did not, however, see this effect for self-reported anxiety potentiation. Given the exploratory nature of these tests, the conclusion that acute prazosin increases overall startle potentiation should be interpreted cautiously and awaits replication to bolster confidence in its reliability. Regardless, contrary to our hypothesis, prazosin did not decrease startle potentiation or self-report measures in the NPU task. Furthermore, prazosin did not affect startle response at baseline prior to stressor exposure in the NPU task (i.e., general startle reactivity) nor during the No Shock condition in the NPU task.
Current study strengths and limitations
Prior to initiating the current study, we comprehensively evaluated the psychometric properties of the NPU task in a large sample (n=128) to confirm it was well-suited for repeated administration (Kaye et al. 2016). The current study confirmed that the NPU task was effective at eliciting robust stress reactivity across measures. Startle potentiation displayed good internal consistency in the NPU task (split-half reliability correlations >.8, see Supplement), bolstering our confidence in the reliability of this task-measure pairing. To maximize statistical power, we utilized a fully within-subjects design in a large sample size. We followed emerging open science recommendations by preregistering our a priori hypotheses to strengthen the validity of our results and we performed exploratory analyses to guide future research.
We conducted the NPU task when prazosin was most likely to be maximally active based on its pharmacokinetics and pharmacodynamics (Vincent et al. 1985). It remains possible that prazosin was not sufficiently active in the brain during this time due to insufficient dose or individual differences in first-pass metabolism, bioavailability, or blood-brain barrier penetrance.10 This concern is reduced by our observation that prazosin lowered participants’ blood pressure, indicating that the dose was physiologically relevant at least in the periphery when the NPU task occurred (see Figure 2 and Supplement). We administered 2mg, double the typical initial dose, to maximize our ability to detect acute effects while ensuring safety. However, this single dose may have been insufficient to impact stress reactivity robustly. Clinical doses to treat PTSD are typically higher (i.e., ≥10mg), though the optimal therapeutic dose range (if any) remains unclear. However, these higher chronic doses may only be required when studying noisy clinical outcomes (e.g., AUD heavy drinking, PTSD hyperarousal symptoms), which arise from many mechanistic pathways. In contrast, we used a physiological measure (e.g., startle potentiation) that is tightly linked to putative NE stress mechanism affected by prazosin. As such, our use of this stress-mechanism focused measure in a controlled laboratory setting likely provided greater sensitivity to detect much smaller reductions in stressor reactivity. Further, it is possible that prazosin differentially affects stress reactivity following acute dosing versus chronic dosing used in clinical practice; similar to how two weeks of SSRI administration (but not acute administration) selectively reduces startle potentiation to unpredictable stress in the NPU task, mirroring their anxiolytic clinical profile in humans (Grillon et al. 2007, 2009a). Again, higher doses may be necessary to achieve long-lasting suppression of the stress system clinically. However, our study was designed to detect even the expected short-term suppression of stress reactivity at peak prazosin activity following an acute dose, consistent with the time course of peripheral prazosin administration on brain activity and behavioral responses in rodents (Darracq et al. 1998).
The placebo blind was not completely effective, which can constrain interpreting the study results. However, follow-up tests confirmed the NPU task results did not differ by drug administration order (see Supplement). The results were also comparable when examining between-subject drug manipulation at the first study visit only. Furthermore, participants’ self-reported expectancy of which drug they received did not moderate the effects of drug on NPU task results. These analyses support the robustness of the conclusions from the NPU task but do not rule out the potential impact of inadequate drug blind or expectancy effects.
Future directions and conclusions
To address some of the concerns and limitations from the current study, our research team is conducting a larger randomized controlled trial of another NE-α1 antagonist, doxazosin, on NPU task stress reactivity (ClinicalTrials.gov ). This experimental medicine approach incorporates the NPU task into a traditional double-blind, placebo-controlled trial of AUD to examine clinical outcomes (e.g., heavy drinking days) as well as potential stress mechanisms. Doxazosin has a similar chemical structure to prazosin but has a more favorable clinical profile for use in psychiatric practice (e.g., longer half-life, once daily dosing). In this trial, participants complete the NPU task after titrating up to a therapeutic dose of doxazosin (8 mg) over several weeks. This could help clarify if the null effects in the current study may be due to an acute, single-dose administration or insufficient dose (vs. prolonged, higher-dose administration). Furthermore, this trial may be more likely to detect the effects of NE-α1 blockade in AUD patients who as a group show sensitized responses to unpredictable (vs predictable) stressors relative to healthy controls (as in the current study) (Gorka et al. 2013; Moberg et al. 2017). However, failure to detect effects of doxazosin on either NPU stress reactivity or clinical outcomes would cast serious doubt on the utility of NE-α1 antagonists as treatments for AUD and the translational nature of the NPU task.
We and others have proposed that the NPU task may be a viable surrogate endpoint to efficiently screen novel or repurposed pharmacotherapies targeting stress mechanisms in addiction and PTSD (Davis et al. 2010; Kaye et al. 2017). Acute administration of CRF1 and NE-α1 antagonists in humans have not reduced startle potentiation during unpredictable stressors in the NPU task (current study; Grillon et al. 2015). This runs counter to predictions based on influential theories in behavioral neuroscience and raises questions regarding the utility of startle potentiation during unpredictable stress to identify cross-species neural mechanisms and/or candidate drug targets that successfully translate from rodent to human models (Davis et al. 2010; for critique see Shackman and Fox 2016).11 While CRF1 and NE-α1 antagonist effects on the NPU have not confirmed predictions from rodent models, they do in fact appear to more closely align with emerging results from failed clinical trials for PTSD and AUD (Dunlop et al. 2017; Raskind et al. 2018; Simpson et al. 2018). The NPU task has been sensitive to effects of other medications (e.g., benzodiazepines) that do have anxiolytic clinical benefit in humans (Grillon et al. 2006, 2015). Thus, it remains possible that the NPU task is working as a surrogate endpoint should, correctly identifying effective versus ineffective treatments. Utilizing human laboratory measures to screen novel or repurposed pharmacotherapies earlier in the drug development process has potential to increase “fast-fails” at the Phase 2a stage and save critical downstream resources (Grillon et al. 2015; Schwandt et al. 2016). However, there remain many important unanswered questions the field must rigorously address (e.g., meaningful prediction of clinical outcomes) for these laboratory measurement approaches (be it the NPU or other paradigms) to prove valuable as surrogate endpoints for clinical trials.
There remains an urgent need to develop treatments that target stress-related processes such as hyperarousal symptom cluster in PTSD and stress-induced relapse in addiction. Repurposing available NE medications held initial promise based on hypothesized stress mechanisms identified in preclinical behavioral neuroscience (Hendrickson and Raskind 2016; Krystal et al. 2017; Haass-Koffler et al. 2018). In spite of the burgeoning clinical trials literature of prazosin as a treatment for PTSD and AUD and increasing off-label prescribing, the current study is among the first experimental psychopharmacology study to investigate the effects of prazosin on stress reactivity in humans (Fox et al. 2012; Homan et al. 2017; also see Verplaetse et al. 2017). We failed to detect any indication that prazosin acutely reduces stress reactivity, measured via startle response and self-reported anxiety. These findings join with recent failures to replicate the treatment effects of prazosin for PTSD and AUD, suggesting the possibility that prazosin is a far less promising intervention for stress-related psychiatric disorders than originally believed (Petrakis et al. 2016; Raskind et al. 2018; Simpson et al. 2018; Kleinman and Ostacher 2019).
Supplementary Material
Acknowledgements:
We thank Heather M. Williams for assistance with data collection and the University of Wisconsin (UW) Clinical Research Unit and UW Pharmaceutical Research Center for study support. Portions of this research have previously been presented at the annual conferences of the Society for Psychophysiology Research. This manuscript has been published on the preprint server PsyArXiv.
Funding: Research reported in this publication was supported by the National Institute of Alcohol Abuse and Alcoholism (NIAAA) of the National Institutes of Health (NIH) under award numbers R01 AA024388 (J Curtin) and F31 AA022845 (J Kaye), NIH National Center for Advancing Translational Science under the Clinical and Translational Science Award number UL1TR000427 to the UW Institute for Clinical and Translational Research, and by intramural research awards from the UW Office of the Vice Chancellor for Research and Graduate Education (J Curtin).
Footnotes
Publisher's Disclaimer: This Author Accepted Manuscript is a PDF file of a an unedited peer-reviewed manuscript that has been accepted for publication but has not been copyedited or corrected. The official version of record that is published in the journal is kept up to date and so may therefore differ from this version.
Conflicts of Interest: The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH, VA, or UW. The authors have no biomedical financial interests or potential conflicts of interest.
We preregistered a sequential recruitment plan to enroll an equal number of participants with AUD in early recover if we confirmed our hypothesis that prazosin reduced stress reactivity to unpredictable (vs. predictable) stressors in health adults (https://clinicaltrials.gov/ct2/show/NCT02966340).
The University of Iowa Pharmaceuticals prepared and over-encapsulated study drug and matching placebos. The University of Wisconsin Pharmaceutical Research Center implemented and maintained the randomization and blind. At visit 1 participants were randomized 1:1 to Drug Order (A: visit 1 prazosin and visit 2 placebo; B: visit 1 placebo and visit 2 prazosin) and NPU Task Order (four condition and startle probe counterbalancing orders), stratified by Sex.
In accordance with our pre-registration we excluded and replaced 2 participants with general startle reactivity at their first study visit of <5μV (non-responders).
We analyze raw startle potentiation consistent with our preregistered analysis plan and numerous previous studies with this and related tasks (Moberg and Curtin 2009; Bradford et al. 2013, 2014; Kaye et al. 2016; Moberg et al. 2017). We report analyses of startle response during the no-shock blocks to confirm that observed effects result from shock threat rather than control condition (no-shock block) differences (see footnote 6 and 8). We do not standardize startle potentiation as it yields lower internal consistency and temporal stability than raw startle potentiation in the NPU task (Bradford et al. 2015; Kaye et al. 2016). Consistent with our previous studies, we also limit analyses to the cue period in predictable and unpredictable blocks to control for (i.e., match) the attentional demands associated with the visual foreground across these blocks (Lang et al. 1990).
We collected a battery of other measures that were available to be used as either covariates or moderators in the analysis of the primary and secondary dependent variables (see Supplement). We utilize covariates to increase power to detect the focal effect in our analytic models. We preregistered to select covariates if we confirmed that the specific covariate (e.g., general startle reactivity, drug order, intolerance of uncertainty) significantly predicted the test of the primary hypothesis (i.e., two-way interaction between Drug and NPU Task Condition). Any categorical between-subject factors were coded as unit-weighted, centered, orthogonal regressors (e.g., Sex: male = −0.5, female = 0.5). Any continuous/quantitative individual difference covariates were mean-centered. We conducted analyses separately for each dependent variable (e.g., startle potentiation, self-reported fear/anxiety potentiation) with only one covariate in the model at a time to determine covariate selection. We only used the covariate if it was a significant predictor of the Drug X NPU Condition interaction for each dependent variable separately (e.g. startle potentiation or self-reported fear/anxiety potentiation).
We did not include any covariates in models predicting the 2-way interaction on startle potentiation (primary outcome) or self-reported anxiety (secondary outcome) as none met our preregistered decision threshold. We did not identify or remove any model outliers (i.e., Bonferroni-corrected studentized residuals, p < .05).
There was not a significant effect of Drug on startle response only during the No Shock condition, ηp2 = .039, b = 4.2 μV, t(61) = 1.57, p = .121, suggesting that the main effect of prazosin on startle potentiation (i.e., shock cues minus no-shock cues) was not driven by a reduction in startle during No Shock.
In the unadjusted model there was not a significant main effect of Drug on overall startle potentiation, ηp2 = .107, b = −6.8 μV, t(63) = −1.97, p = .053, with no covariates included or outliers removed.
We removed one model outlier, but there was still not a significant main effect of Drug on overall self-reported anxiety/fear, ηp2 = .008, b = 0.09, t(63) = 0.70, p = .484, with no outliers removed. We did not include any covariates in either model predicting the Drug main effect on self-reported anxiety as none met our preregistered decision threshold. We also confirmed that was not a significant effect of Drug on startle response during the No Shock condition, ηp2 = .013, b = 0.06, t(62) = 0.89, p = 0.375.
Previous literature suggests that prazosin’s peak effects on peripheral physiology and plasma concentration occur 1-4 hours post-administration (Jaillon 1980). Unfortunately, there is limited research in humans to confirm the time course of effects in the brain (but see Rutland et al. 1980). Although prazosin is still widely used in rodent behavioral neuroscience research today to study the central nervous system, human studies have primarily examined peripheral physiology as prazosin was originally developed as an antihypertensive agent. Indeed, very few studies have examined basic acute effects of prazosin in humans since the 1970s.
Considerable preclinical research supports our study hypothesis that acute prazosin would selectively reduce startle potentiation during unpredictable (relative to predictable) shock. However, the most direct translational design of our current study in humans (i.e., acute prazosin effects on startle potentiation to unpredictable vs predictable shock), has not been performed in rodent models to date. We believe reverse-translation of our current study design in rodents, using parallel pharmacological manipulation (acute prazosin), stressor manipulation (unpredictable vs. predictable shock), and measurement (startle potentiation) is essential to clarify convergent or divergent results across species. Furthermore, additional psychopharmacology studies in both human and rodent models should address differences in acute vs chronic prazosin administration. Only recently have preclinical labs begun to examine the effects of chronic prazosin on relevant anxiety-like behaviors and alcohol use/seeking behaviors (Froehlich et al. 2013; Skelly and Weiner 2014; Rasmussen et al. 2017). However, no studies have examined chronic prazosin administration effects on startle response as the primary outcome measure.
References
- Arnsten AFT (2009) Stress signalling pathways that impair prefrontal cortex structure and function. Nat Rev Neurosci 10:410–422. doi: 10.1038/nrn2648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berridge CW (2008) Noradrenergic modulation of arousal. Brain Res Rev 58:1–17. doi: 10.1016/j.brainresrev.2007.10.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berridge CW, Dunn AJ (1989) Restraint-stress-induced changes in exploratory behavior appear to be mediated by norepinephrine-stimulated release of CRF. The Journal of Neuroscience 9:3513–3521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berridge CW, Waterhouse BD (2003) The locus coeruleus-noradrenergic system: modulation of behavioral state and state-dependent cognitive processes. Brain Res Brain Res Rev 42:33–84 [DOI] [PubMed] [Google Scholar]
- Blumenthal TD, Cuthbert BN, Filion DL, et al. (2005) Committee report: Guidelines for human startle eyeblink electromyographic studies. Psychophysiology 42:1–15. doi: 10.1111/j.1469-8986.2005.00271.x [DOI] [PubMed] [Google Scholar]
- Bradford DE, Kaye JT, Curtin JJ (2014) Not just noise: individual differences in general startle reactivity predict startle response to uncertain and certain threat. Psychophysiology 51:407–411. doi: 10.1111/psyp.12193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradford DE, Shapiro BL, Curtin JJ (2013) How bad could it be? Alcohol dampens stress responses to threat of uncertain intensity. Psychol Sci 24:2541–2549. doi: 10.1177/0956797613499923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradford DE, Starr MJ, Shackman AJ, Curtin JJ (2015) Empirically based comparisons of the reliability and validity of common quantification approaches for eyeblink startle potentiation in humans. Psychophysiol 52:1669–1681. doi: 10.1111/psyp.12545 https://www.ncbi.nlm.nih.gov/pubmed/26372120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darracq L, Blanc G, Glowinski J, Tassin JP (1998) Importance of the noradrenaline-dopamine coupling in the locomotor activating effects of D-amphetamine. J Neurosci 18:2729–2739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis M, Walker DL, Miles L, Grillon C (2010) Phasic vs sustained fear in rats and humans: Role of the extended amygdala in fear vs anxiety. Neuropsychopharmacology Reviews 35:105–135. doi: doi: 10.1037/npp.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunlop BW, Binder EB, Iosifescu D, et al. (2017) Corticotropin-Releasing Factor Receptor 1 Antagonism Is Ineffective for Women With Posttraumatic Stress Disorder. Biol Psychiatry 82:866–874. doi: 10.1016/j.biopsych.2017.06.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox HC, Anderson GM, Tuit K, et al. (2012) Prazosin effects on stress- and cue-induced craving and stress response in alcohol-dependent individuals: preliminary findings. Alcohol Clin Exp Res 36:351–360. doi: 10.1111/j.1530-0277.2011.01628.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Froehlich JC, Hausauer BJ, Federoff DL, et al. (2013) Prazosin Reduces Alcohol Drinking Throughout Prolonged Treatment and Blocks the Initiation of Drinking in Rats Selectively Bred for High Alcohol Intake. Alcohol Clin Exp Res. doi: 10.1111/acer.12116 PMID: 23731093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funk D, Coen K, Tamadon S, et al. (2016) Effects of prazosin and doxazosin on yohimbine-induced reinstatement of alcohol seeking in rats. Psychopharmacology (Berl). doi: 10.1007/s00213-016-4273-2 [DOI] [PubMed] [Google Scholar]
- Galvez R, Mesches MH, McGaugh JL (1996) Norepinephrine release in the amygdala in response to footshock stimulation. Neurobiol Learn Mem 66:253–257. doi: 10.1006/nlme.1996.0067 [DOI] [PubMed] [Google Scholar]
- Gilpin NW, Weiner JL (2017) Neurobiology of comorbid post-traumatic stress disorder and alcohol-use disorder. Genes Brain Behav 16:15–43. doi: 10.1111/gbb.12349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorka SM, Nelson BD, Shankman SA (2013) Startle response to unpredictable threat in comorbid panic disorder and alcohol dependence. Drug and Alcohol Dependence 132:216–222. doi: 10.1016/j.drugalcdep.2013.02.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gresack J, Risbrough V (2011) Corticotropin-releasing factor and noradrenergic signalling exert reciprocal control over startle reactivity. Int J Neuropsychopharm 14:1179–94. doi: 10.1017/S1461145710001409 PMID: 21205416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grillon C, Baas JM, Pine DS, et al. (2006) The benzodiazepine alprazolam dissociates contextual fear from cued fear in humans as assessed by fear-potentiated startle. Biological Psychiatry 60:760–766. doi: 10/1016/j.biopsych.2005.11.027 [DOI] [PubMed] [Google Scholar]
- Grillon C, Chavis C, Covington MF, Pine DS (2009a) Two-week treatment with the selective serotonin reuptake inhibitor citalopram reduces contextual anxiety but not cued fear in healthy volunteers: A fear-potentiated startle study. Neuropsychopharmacology 34:964–971. doi: 10.1038/npp.2008.141 PMID 18800069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grillon C, Hale E, Lieberman L, et al. (2015) The CRH1 Antagonist GSK561679 Increases Human Fear But Not Anxiety as Assessed by Startle. Neuropsychopharmacology 40:1064–1071. doi: 10.1038/npp.2014.316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grillon C, Levenson J, Pine DS (2007) A single dose of the selective serotonin reuptake inhibitor citalopram exacerbates anxiety in humans: a fear-potentiated startle study. Neuropsychopharmacology 32:225–231. doi: 10.1038/sj.npp.1301204 [DOI] [PubMed] [Google Scholar]
- Grillon C, Lissek S, Rabin S, et al. (2008) Increased anxiety during anticipation of unpredictable but not predictable aversive stimuli as a psychophysiologic marker of panic disorder. American Journal of Psychiatry 165:898–904. doi: 10.1176/appi.ajp.2007.07101581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grillon C, Pine DS, Lissek S, et al. (2009b) Increased anxiety during anticipation of unpredictable aversive stimuli in posttraumatic stress disorder but not in generalized anxiety disorder. Biological Psychiatry 66:47–53. doi: 10.1016/j.biopsych.2008.12.028 PMID 19217076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haass-Koffler CL, Swift RM, Leggio L (2018) Noradrenergic targets for the treatment of alcohol use disorder. Psychopharmacology (Berl) 235:1625–1634. doi: 10.1007/s00213-018-4843-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendrickson R, Raskind M (2016) Noradrenergic dysregulation in the pathophysiology of PTSD. Exp Neurol 284:181–195. doi: 10.1016/j.expneurol.2016.05.014 [DOI] [PubMed] [Google Scholar]
- Homan P, Lin Q, Murrough JW, et al. (2017) Prazosin during threat discrimination boosts memory of the safe stimulus. Cold Spring Harbor Laboratory Press 24:597–601. doi: 10.1101/lm.045898.117 PMID 29038221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaillon P (1980) Clinical pharmacokinetics of prazosin. Clin Pharmacokinet 5:365–376 [DOI] [PubMed] [Google Scholar]
- Kaye JT, Bradford DE, Curtin JJ (2016) Psychometric properties of startle and corrugator response in NPU, affective picture viewing, and resting state tasks. Psychophysiology 53:1241–1255. doi: 10.1111/psyp.12663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaye JT, Bradford DE, Magruder KP, Curtin JJ (2017) Probing for Neuroadaptations to Unpredictable Stressors in Addiction: Translational Methods and Emerging Evidence. J Stud Alcohol Drugs 78:353–371. doi: 10.15288/jsad.2017.78.353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleiner M, Brainard D, Pelli D, et al. (2007) What’s new in Psychtoolbox-3. Perception 36:1 [Google Scholar]
- Kleinman RA, Ostacher MJ (2019) Prazosin and Alcohol Use Disorder. Am J Psychiatry 176:165. doi: 10.1176/appi.ajp.2018.18101143 https://www.ncbi.nlm.nih.gov/pubmed/30704280 [DOI] [PubMed] [Google Scholar]
- Koob GF (2009) Brain stress systems in the amygdala and addiction. Brain Research 1293:61–75. doi: 10.1016/j.brainres.2009.03.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krystal JH, Davis LL, Neylan TC, et al. (2017) It Is Time to Address the Crisis in the Pharmacotherapy of Posttraumatic Stress Disorder: A Consensus Statement of the PTSD Psychopharmacology Working Group. Biol Psychiatry 82:e51–e59. doi: 10.1016/j.biopsych.2017.03.007 [DOI] [PubMed] [Google Scholar]
- Lang PJ, Bradley MM, Cuthbert BN (1990) Emotion, attention, and the startle reflex. Psychological Review 97:377–395. doi: 10.1037/0033-295X.97.3.377 [DOI] [PubMed] [Google Scholar]
- Lê AD, Funk D, Juzytsch W, et al. (2011) Effect of prazosin and guanfacine on stress-induced reinstatement of alcohol and food seeking in rats. Psychopharmacology 218:89–99. doi: 10.1007/s00213-011-2178-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manion S, Gamble E, Li H (2007) Prazosin administered prior to inescapable stressor blocks subsequent exaggeration of acoustic startle response in rats. Pharmacol Biochem Behav 86:559–65. doi: 10.1016/j.pbb.2007.01.019 [DOI] [PubMed] [Google Scholar]
- Mantsch JR, Baker DA, Funk D, et al. (2016) Stress-induced reinstatement of drug seeking: 20 years of progress. Neuropsychopharmacology 41:335–356. doi: 10.1038/npp.2015.142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy E, Petrakis I (2010) Epidemiology and management of alcohol dependence in individuals with post-traumatic stress disorder. CNS Drugs 24:997–1007. doi: 10.2165/11539710-000000000-00000 [DOI] [PubMed] [Google Scholar]
- Moberg CA, Bradford DE, Kaye JT, Curtin JJ (2017) Increased startle potentiation to unpredictable stressors in alcohol dependence: Possible stress neuroadaptation in humans. J Abnorm Psychol 126:441–453. doi: 10.1037/abn0000265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moberg CA, Curtin JJ (2009) Alcohol selectively reduces anxiety but not fear: startle response during unpredictable vs. predictable threat. J Abnorm Psychol 118:335–347. doi: 10.1037/a0015636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan C, Grillon C, Southwick S, et al. (1995) Yohimbine facilitated acoustic startle in combat veterans with post-traumatic stress disorder. Psychopharmacology 117:466–71. [DOI] [PubMed] [Google Scholar]
- Morgan C, Southwick S, Grillon C, et al. (1993) Yohimbine-facilitated acoustic startle reflex in humans. Psychopharmacology 110:342–6 [DOI] [PubMed] [Google Scholar]
- Pacák K, McCarty R, Palkovits M, et al. (1995) Effects of immobilization on in vivo release of norepinephrine in the bed nucleus of the stria terminalis in conscious rats. Brain Res 688:242–246 [DOI] [PubMed] [Google Scholar]
- Petrakis IL, Desai N, Gueorguieva R, et al. (2016) Prazosin for Veterans with Posttraumatic Stress Disorder and Comorbid Alcohol Dependence: A Clinical Trial. Alcohol Clin Exp Res 40:178–186. doi: 10.1111/acer.12926 [DOI] [PubMed] [Google Scholar]
- Rajbhandari AK, Baldo BA, Bakshi VP (2015) Predator stress-induced CRF release causes enduring sensitization of basolateral amygdala norepinephrine sstems that promote PTSD-like startle abnormalities. J Neurosci 35:14270–14285. doi: 10.1523/JNEUROSCI.5080-14.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raskind M, Peskind ER, Chow B, et al. (2018) Trial of Prazosin for Post-Traumatic Stress Disorder in Military Veterans. N Engl J Med 378:507–517. doi: 10.1056/NEJMoa1507598 [DOI] [PubMed] [Google Scholar]
- Raskind M, Peskind ER, Hoff DJ, et al. (2007) A parallel group placebo controlled study of prazosin for trauma nightmares and sleep disturbance in combat veterans with post-traumatic stress disorder. Biol Psychiatry 61:928–934. doi: 10.1016/j.biopsych.2006.06.032 [DOI] [PubMed] [Google Scholar]
- Raskind M, Peskind ER, Kanter ED, et al. (2003) Reduction of nightmares and other PTSD symptoms in combat veterans by prazosin: a placebo-controlled study. Am J Psychiatry 160:371–373 [DOI] [PubMed] [Google Scholar]
- Raskind M, Peterson K, Williams T, et al. (2013) A Trial of Prazosin for Combat Trauma PTSD With Nightmares in Active-Duty Soldiers Returned From Iraq and Afghanistan. Am J Psychiatry. doi: 10.1176/appi.ajp.2013.12081133 PMID 23846759 [DOI] [PubMed] [Google Scholar]
- Rasmussen DD, Kincaid CL, Froehlich JC (2017) Prazosin Prevents Increased Anxiety Behavior That Occurs in Response to Stress During Alcohol Deprivations. Alcohol Alcohol 52:5–11. doi: 10.1093/alcalc/agw082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutland MD, Lee TY, Nimmon CC, et al. (1980) Measurement of the effects of a single dose of prazosin on the cerebral blood flow in hypertensive patients. Postgraduate medical journal 56:818–822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz A, Grillon C (2012) Assessing fear and anxiety in humans using the threat of predictable and unpredictable aversive events (the NPU-threat test). Nat Protoc 7:527–532. doi: 10.1038/nprot.2012.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwandt ML, Cortes CR, Kwako LE, et al. (2016) The CRF1 antagonist verucerfont in anxious alcohol dependent women: Translation of neuroendocrine, but not of anti-craving effects. Neuropsychopharmacology, doi: 10.1038/npp.2016.61 PMID 27109623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shackman AJ, Fox AS (2016) Contributions of the central extended amygdala to fear and anxiety. J Neurosci 36:8050–8063. doi: 10.1523/JNEUROSCI.0982-16.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shankman SA, Nelson BD, Sarapas C, et al. (2013) A psychophysiological investigation of threat and reward sensitivity in individuals with panic disorder and/or major depressive disorder. J Abnorm Psychol 122:322–338. doi: 10.1037/a0030747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmons JP, Nelson LD, Simonsohn U (2012) A 21 Word Solution. 10.2139/ssrn.2160588. Accessed 21 Mar 2016 [DOI]
- Simpson TL, Saxon A, Meredith C, et al. (2009) A pilot trial of the alpha-1 adrenergic antagonist, prazosin, for alcohol dependence. Alcoholism: Clinical and Experimental Research 33:255–263. doi: 10.1111/j.1530-0277.2008.00807.X https://www.ncbi.nlm.nih.gov/pubmed/25827659 [DOI] [PubMed] [Google Scholar]
- Simpson TL, Saxon AJ, Stappenbeck C, et al. (2018) Double-Blind Randomized Clinical Trial of Prazosin for Alcohol Use Disorder. Am J Psychiatry appiajp201817080913. doi: 10.1176/appi.ajp.2018.17080913 https://www.ncbi.nlm.nih.gov/pubmed/30153753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skelly MJ, Weiner JL (2014) Chronic treatment with prazosin or duloxetine lessens concurrent anxietyldmg-ike behavior and alcohol intake: evidence of disrupted noradrenergic signaling in anxiety-related alcohol use. Brain Behav 4:468–483. doi: 10.1002/brb3.230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith R, Aston-Jones G (2008) Noradrenergic transmission in the extended amygdala: role in increased drug-seeking and relapse during protracted drug abstinence. Brain structure & function 213:43–61. doi: 10.1007/s00429-008-0191-3 https://www.ncbi.nlm.nih.gov/pubmed/18651175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stine S, Grillon C, Morgan CA III, et al. (2001) Methadone patients exhibit increased startle and cortisol response after intravenous yohimbine. Psychopharmacology 154:274–281. doi: 10.1007/s002130000644 [DOI] [PubMed] [Google Scholar]
- Varty GB, Bakshi VP, Geyer MA (1999) M100907, a serotonin 5-HT2A receptor antagonist and putative antipsychotic, blocks dizocilpine-induced prepulse inhibition deficits in Sprague-Dawley and Wistar rats. Neuropsychopharmacology 20:311–321. doi: 10.1016/S0893-133X(98)00072-4 [DOI] [PubMed] [Google Scholar]
- Verplaetse TL, Weinberger AH, Oberleitner LM, et al. (2017) Effect of doxazosin on stress reactivity and the ability to resist smoking. J Psychopharmacol (Oxford) 31:830–840. doi: 10.1177/0269881117699603 https://www.ncbi.nlm.nih.gov/pubmed/28440105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincent J, Meredith PA, Reid JL, et al. (1985) Clinical pharmacokinetics of prazosin--1985. Clin Pharmacokinet 10:144–154 [DOI] [PubMed] [Google Scholar]
- Walker D, Davis M (1997) Double dissociation between the involvement of the bed nucleus of the stria terminalis and the central nucleus of the amygdala in startle increases produced by conditioned versus unconditioned fear. J Neurosci 17:9375–9383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker D, Miles LA, Davis M (2009) Selective participation of the bed nucleus of the stria terminalis and CRF in sustained anxiety-like versus phasic fear-like responses. Progress in Neuro-Psychopharmacology & Biological Psychiatry 33:1291–1308. doi: 10.1016/j.pnpbp.2009.06.022 https://www.ncbi.nlm.nih.gov/pubmed/19595731 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.