

Abstract
Only a minority of cancer patients respond to anti PD-1 immunotherapy. A recent study demonstrates that PD-1 therapy-resistant melanoma patients present distinct signatures of upregulated genes involved in immunosuppression, angiogenesis, monocyte and macrophage chemotaxis, extracellular matrix remodeling, and epithelial-mesenchymal transition (EMT). Combination-targeting of these pathways with PD-1 may help overcome PD-1 resistance, thus producing effective antitumor immunity.
Blockade of either the PD-1 receptor or its ligand PD-L1 has improved overall survival in Phase III trials in patients with melanoma, non-small cell lung cancer and kidney cancer. Early studies suggest that PD-1 pathway blockade may benefit a subset of patients in many other types of cancer. Nevertheless, the majority of patients fail to respond to PD-1 pathway blockade and insights into improving response rates are critically needed. In a recent report published in Cell, Hugo, et al. analyzed the genomic and transcriptomic features of pretreatment tumor specimens to determine molecular signatures distinguishing response from resistance in metastatic melanoma patients treated with PD-1 blockade immunotherapy [1].
Studies have supported the hypothesis that response to PD-1 blockade is associated with a smoldering, but ineffective immune response, sometimes referred to as a “hot” tumor, characterized by PD-L1 expression on tumor or infiltrating immune cells, CD8+ T cell infiltration, and high neoantigen load. Somewhat surprisingly, Hugo et al. found no difference between primary responders and progressors in the expression of genes encoding PD-L1, PD-L2, PD-1, or genes associated with activated T lymphocytes. This could be partly attributed to the fact that immune checkpoints such as PD-L1, are expressed on infiltrating immune cells associated with both a good prognosis (dendritic cells) [2] and a poor prognosis (myeloid derived suppressor cells). In addition, lack of differences in gene expression between groups could also be partly attributed to multiple strategies of immune evasion accumulated by tumor cells and infiltrating immune cells [3]. Using large-scale genomic analysis, several groups have found that the mutational burden or neoantigen load in tumors positively associates with a clinical benefit derived from therapeutic blockade using immune checkpoint inhibitors [4]. However, there has been a broad overlap in the number of mutations observed in responders and nonresponders. Here, the analysis provided by Roger Lo’s group suggests that high neoantigen load does not predict a response to PD-1 immunotherapy, but rather, that it is associated with improved patient survival. Some genes were found to be more frequently mutated in the responder cohort. In particular, mutations in BRCA2 were more frequent in tumors from the responding cohort. Loss of BRCA2-mediated DNA breakpoint repair function would not be expected to result in as large a number of neoantigens as tumors exhibiting mismatch repair deficiency. As such, BRCA2 is an example of a protein harboring mutations that can lead to potential neoantigens as well as loss of function, possibly driving tumorigenesis.
While the authors did not report a predictive up-regulated gene signature associated with response to PD-1 immunotherapy, they did report a transcriptional signature associated with resistance to PD-1 immunotherapy, termed Innate PD-1 RESistance (IPRES). As illustrated in Figure 1, the IPRES signature includes genes involved in immunosuppression (IL10) and angiogenesis (VEGFA, VEGFC, FLT1, and ANGPT2), monocyte and macrophage chemotaxis (CCL2, CCL7, CCL8, CCL13) and epithelial-mesenchymal transition (EMT) (AXL, ROR2, WNT5A, LOXL2, TWIST2, TAGLN, FAP). Consistent with this pattern, nonresponders presented lower expression of the CDH1 (E-cadherin) gene, a marker of differentiated epithelia. Alternate analyses by gene ontology enrichment and gene set variant analysis (GSVA) scores also showed that non-responding tumors were enriched for expressed genes associated with wound healing, angiogenesis, hypoxia, TGF-beta signaling, EMT, as well as cell adhesion and extracellular matrix organization. However, future studies are required to examine whether these pathways represent parallel patterns of response failure, and/or whether they are interconnected in any way.
Figure 1. The Innate PD-1 Resistance (IPRES) Signature.
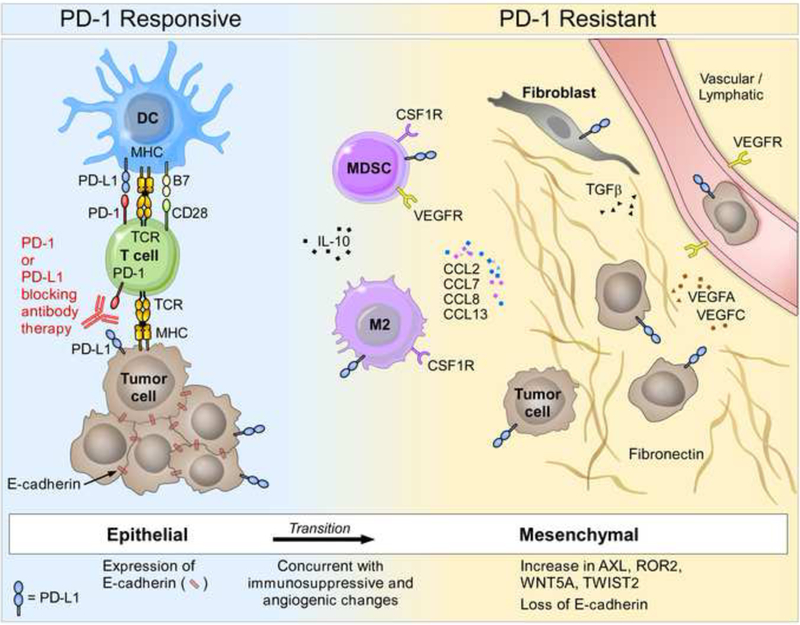
Data from the Hugo et al. study reveal upregulated expression of gene sets associated with wound healing, angiogenesis, hypoxia, TGF-beta signaling, epithelial-mesenchymal transition (EMT), as well as cell adhesion and extracellular matrix organization in nonresponding melanoma tumors from patients undergoing anti-PD-1 treatment. Nonresponder tumors express less CDH1 (E-cadherin, a marker of differentiated epithelia) than responder tumors. The IPRES signature includes genes involved in immunosuppression (IL10) and angiogenesis (VEGFA, VEGFC, FLT1, and ANGPT2), monocyte and macrophage chemotaxis (CCL2, CCL7, CCL8, CCL13) and EMT (AXL, ROR2, WNT5A, LOXL2, TWIST2, TAGLN, FAP). DC; dendritic cells; MDSC, myeloid derived suppressor cells; VEGFR, vascular endothelial growth factor receptor.
EMT is a normal process in embryonic development and wound healing. In wounds, the early acute inflammatory process produces a red, warm, inflamed injury, followed by scarring and repair of the tissue architecture, which are tightly regulated processes. These include the induction of angiogenesis, the suppression of potentially destructive inflammatory responses and the resolution of tissue injury (wound closure). Tumors may utilize this process to increase tumor survival by increasing metastatic features and immunosuppression. Interestingly, Spranger et al. have shown that increased β-catenin signaling leads to poor T cell infiltration of T cells into melanoma tumors [5]. Moreover, increased Wnt-GSK3β-β-catenin signaling can also promote EMT [5], perhaps connecting these mechanisms to tumor resistance. In a prior study, Lo’s group compared the transcriptomic signatures of melanomas prior to treatment with MAPK inhibitors (BRAF monotherapy or dual BRAF and MEK inhibition) [6]. They found that at the time of resistance to MAPK inhibition, the majority of tumors developed gain-of–function changes in NRAS and KRAS, as well as upregulated EMT-gene associated changes. Some of these tumors harbored high intratumoral T lymphocytes prior to treatment, but presented decreased infiltrating lymphocytes and increased M2 macrophage markers (CSF1R and CD163) after developing resistance to treatment. In the present study, Hugo et al. found that the signature of MAPK inhibition-resistance bore many features in common with the IPRES signature. Furthermore, the IPRES signature could also be found in the TCGA database in a subset of tumors, including lung adenocarcinoma, renal clear cell carcinoma, pancreatic adenocarcinoma, and colon adenocarcinoma. Whether the IPRES signature is associated with resistance to PD-1 monotherapy in these cancers remains to be tested.
The link between activation in the RAS–RAF–MEK–ERK–MAPK pathway and reduced lymphocyte infiltration in tumors is not unique to MAPK inhibitor therapy. For instance, in breast cancer patients treated with standard chemotherapy, increased MAPK signaling has been associated with fewer tumor-infiltrating lymphocytes, while the presence of tumor infiltrating lymphocytes has been associated with a better clinical outcome [7]. Since PD-1 blockade is an approved therapy for lung and kidney cancer, determining whether the IPRES signature is associated with resistance to PD-1 blockade in these patients is an important next step. The analysis of other tumor types is of particular importance since EMT status has been linked to an inflammatory tumor microenvironment in lung adenocarcinoma [8]. And, this microenvironment has been characterized by the expression of PD-L1/2, PD-1, TIM-3, LAG-3, B7-H3, BTLA and CTLA-4, as evidenced by mRNA expression, protein array and immunohistochemistry [8].
Many questions remain on how EMT, hypoxia, and angiogenic pathways affect the development of an antitumor T cell response. While 34% of melanoma patients treated with nivolumab (PD-1 blocking antibody) survive more than 5 years, targeting patterns of response failure may further improve patient outcomes [9]. Notably, over thirty percent of patients whose melanomas expressed the IPRES signature responded to CTLA-4 blockade (ipilimumab), indicating that the IPRES signature does not indicate resistance to every immunotherapy. Targeting other immunosuppressive components or pathways identified by this study may represent a means of overcoming resistance to PD-1 monotherapy, and allow combination therapy to benefit more patients. Moreover, targeting putative candidates of EMT might not only delay progression of a tumor, but could also augment the local immune milieu and shift the balance towards a productive antitumor response. In this rapidly moving field, the combination of a MEK inhibitor and PD-L1 blockade has already been tested. Since MEK is a critical signaling component for both normal T cells and Ras-pathway mutated tumors, there has been concern that targeting MEK could inhibit the immune response. While MEK inhibition blocked in vivo priming of murine naïve T cells, it potentiated previously activated antitumor T cells by impairing TCRdriven apoptosis [10]. Moreover, the combination of MEK inhibition and PD-L1 blockade yielded durable tumor remission in a mouse model of colorectal CT26 carcinoma. Accumulating evidence thus suggests that PD-1 therapy should be initiated before MEK inhibitor resistance can develop. Consequently, the design of PD-1 combinations that target immune escape features of the IPRES signature could represent a means to overcome resistance to PD-1 monotherapy across many tumor types, providing the clinical benefit of cancer immunotherapy to more patients.
Acknowledgments
Research Support: DF/HCC Kidney Cancer SPORE P50CA101942 (GJF). Research supported by Claudia Adams Barr Program for Innovative Cancer Research (KM).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hugo W, et al. (2016) Genomic and Transcriptomic Features of Response to Anti-PD-1 Therapy in Metastatic Melanoma. Cell 165, 35–4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Giraldo N, et al. (2015) Orchestration and Prognostic Significance of Immune Checkpoints in the Microenvironment of Primary and Metastatic Renal Cell Cancer. Clinical cancer research : an official journal of the American Association for Cancer Research 21, 3031–3040 [DOI] [PubMed] [Google Scholar]
- 3.Koyama S, et al. (2016) Adaptive resistance to therapeutic PD-1 blockade is associated with upregulation of alternative immune checkpoints. Nature communications 7, 10501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rizvi NA, et al. (2015) Mutational landscape determines sensitivity to PD-1 blockade in nonsmall cell lung cancer. Science 348, 124–128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Spranger S, et al. (2015) Melanoma-intrinsic beta-catenin signalling prevents anti-tumour immunity. Nature 523, 231–235 [DOI] [PubMed] [Google Scholar]
- 6.Hugo W, et al. (2015) Non-genomic and Immune Evolution of Melanoma Acquiring MAPKi Resistance. Cell 162, 1271–1285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Loi S, et al. (2016) RAS/MAPK Activation Is Associated with Reduced Tumor-Infiltrating Lymphocytes in Triple-Negative Breast Cancer: Therapeutic Cooperation Between MEK and PD-1/PD-L1 Immune Checkpoint Inhibitors. Clinical cancer research : an official journal of the American Association for Cancer Research 22, 1499–1509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lou Y, et al. (2016) Epithelial-mesenchymal transition is associated with a distinct tumor microenvironment including elevation of inflammatory signals and multiple immune checkpoints in lung adenocarcinoma. Clinical cancer research : an official journal of the American Association for Cancer Research [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hodi FS, et al. (2016) Five-year Survival Rates For Nivolumab-treated Metastatic Melanoma Patients Much Higher Than Historical Rates AACR, CT001 [Google Scholar]
- 10.Ebert PJ, et al. (2016) MAP Kinase Inhibition Promotes T Cell and Anti-tumor Activity in Combination with PD-L1 Checkpoint Blockade. Immunity 44, 609–621 [DOI] [PubMed] [Google Scholar]