

Abstract
The microbiome's capacity to shape the host phenotype and its mutability underlie theorization that the microbiome might facilitate host acclimation to rapid environmental change. However, when environmental change occurs, it is unclear whether resultant microbiome restructuring is proximately driven by this changing external environment or by the host's physiological response to this change. We leveraged urbanization to compare the ability of host environment (urban or forest) versus multi-scale biological measures of host hypothalamic–pituitary–adrenal (HPA) axis physiology (neutrophil : lymphocyte ratio, faecal glucocorticoid metabolites, hair cortisol) to explain variation in the eastern grey squirrel (Sciurus carolinensis) faecal microbiome. Urban and forest squirrels differed across all three of the interpretations of HPA axis activity we measured. Direct consideration of these physiological measures better explained greater phylogenetic turnover between squirrels than environment. This pattern was strongly driven by trade-offs between bacteria which specialize on metabolizing digesta versus host-derived nutrient sources. Drawing on ecological theory to explain patterns in intestinal bacterial communities, we conclude that although environmental change can affect the microbiome, it might primarily do so indirectly by altering host physiology. We demonstrate that the inclusion and careful consideration of dynamic, rather than fixed (e.g. sex), dimensions of host physiology are essential for the study of host–microbe symbioses at the micro-evolutionary scale.
Keywords: bacterial ecology, ecophysiology, ecosystem on a leash, comparative endocrinology, holobiont, cortisol
1. Introduction
Our growing understanding of host–microbe symbioses has begun to blur the line between hosts and their inhabitant microorganisms [1]. In vertebrates, the most intimate host–microbe symbioses occur in the intestinal tract, which harbours a diverse bacterial community collectively known as the bacterial microbiome. Far from passive, bacterial microbiomes can actively shape hosts' phenotypes [2–4], leading to theorization that the microbiome might confer rapid ecological adaptation to wildlife threatened by environmental change [1,5]. While intriguing, this mechanism necessitates that the microbiome be plastic in response to environmental stimuli, and capable of concerted change across populations. However, in cases where analogous environmental conditions drive microbiome convergence, it is unclear whether convergence is a result of direct bacterial response to external environmental stimuli or the host's physiological reaction to external stimuli. Although at first glance a trivial distinction, clarifying the proximate mechanism is consequential for our understanding of the host–microbiome relationship.
Studies of wildlife microbiomes often centre their discussions on extrinsic mechanisms of diet and environment, with only cursory consideration of host physiology. For example, although diet is thought to drive patterns of microbiome convergence [6], recent analyses suggest shared ancestry and convergent digestive physiology better explain these patterns than diet [7,8]. While environmental effects are important, focusing on extrinsic factors alone offers no insight into how stable host–microbiome relationships are maintained (phylosymbiosis). Although some studies consider host physiological variation, scrutiny is often limited to coarse physiological measures like age [9], sex [10] or reproductive condition [11]. These coarse physiological dimensions often explain variation in the microbiome; however, they are also largely immutable to external stimuli, and are thus unlikely to explain patterns of microbiome divergence between environments. Furthermore, these immutable physiological attributes are not heritable from a quantitative genetics perspective [12]. Like emphasis placed on extrinsic factors, focus on non-quantitative physiological traits offers little insight into how phylosymbioses evolve and are maintained. However, we note that immutable traits (e.g. sex, age, reproductive condition) can play a role in explaining microbiome dynamics, especially when considering the underlying proximate physiological traits (e.g. hormonal, metabolic and immunological differences). Importantly, unlike sex, age or reproductive conditions, these more proximate physiological facets are both mutable in response to environmental conditions and heritable quantitative traits [13,14]. Directly measuring dynamic components of host physiology will provide greater mechanistic understanding of how the host–microbiome relationship evolves than the study of extrinsic factors alone or immutable dimensions of host physiology.
The hypothalamic–pituitary–adrenal (HPA) axis is a dynamic hormonal system that acts to maintain or restore host homeostasis through the release of glucocorticoids [15]. As the mediator between a vertebrate's perception of its environment and a suite of physiological responses, the HPA axis provides a useful universal measure of how host responses to environmental stimuli could shape the microbiome. Change in the HPA axis has been demonstrated to affect the microbiome in laboratory models [16–18] and in free-living wildlife [19–22]. However, the laboratory approach of comparing control and stressor-exposed animals under highly controlled conditions that lack ecological reality makes extrapolating results to the free-living organisms difficult. In contrast, while studying free-living wildlife is more firmly grounded in ecological reality, discretely delineating stressed versus non-stressed individuals under these conditions is somewhat arbitrary. Instead, field studies treat stress as a continuous metric by assaying glucocorticoids, but researchers often quantify only a single measure of HPA axis activity (e.g. plasma cortisol, hair/feather cortisol or faecal glucocorticoid metabolites), which is problematic since indices differ in the timescale of HPA axis activity they capture, and thus are limited in physiological scope [23,24].
The context of timing is important, as stress responses of differing durations can manifest a myriad of physiological outcomes. The microbiome will probably be affected very differently by acute stress that mobilizes an organism's immune response [25] and stimulates intestinal mucus secretion [26] than by the attenuated immunity [27] and reduced mucus production [28–30] typified by prolonged HPA upregulation. Furthermore, it is unclear whether only an organism's immediate physiological state matters, or if previous stress-based perturbation of the microbiome has lingering effects. The effect that environmental stimuli have on the microbiome may depend not only on whether an HPA axis response is elicited, but the duration, frequency and severity of the stressor.
We hypothesize that many environmental factors reported to affect the microbiome do so indirectly, their influence translated to the microbiome by an intermediary of host physiology. For the first time, we directly compare whether a host's environment or HPA axis physiology is a stronger determinant of intestinal bacterial communities by using urbanization as an opportunistic experiment which exposes free-living wildlife to an array of novel environmental stimuli. In doing so, we considered three measures of HPA axis activity in eastern grey squirrels (Sciurus carolinensis) to determine how the timing of HPA activation affects the bacterial microbiome. Upon a multi-environment and multi-stress measure backdrop, we predicted that measures of HPA axis activity will better explain bacterial microbiome variation than host environment and demonstrate the necessity of accounting for host physiology when parsing ecological patterns in the microbiome of free-living wildlife.
2. Methods
(a). Study species
Eastern grey squirrels are common in both deciduous forests and cities, their centuries-long colonization of which are uniquely well documented from a historical perspective [31]. This colonization of urban environments has undoubtedly been aided by their broad dietary niche, which has allowed grey squirrels to capitalize on anthropogenic food subsidies [32], which we predict would affect their microbiomes. This dietary shift and a well-described ecology and stress physiology [33] make eastern grey squirrels useful for investigating interactions between the HPA axis and microbiome within free-living wildlife. Furthermore, biomedical findings from laboratory mice, from which much of our understanding of the microbiome stems, may help inform patterns we observe in grey squirrels.
(b). Capture protocol
We trapped eastern grey squirrel adults (14 forest, 15 urban) over 32 consecutive days in the summer of 2016. The University of Guelph campus served as our urban site (43°31′52.33″ N, 80°13′36.80″ W, Guelph, Ontario, Canada), while a 0.7 km2 forest at the rare Charitable Research Reserve served as our non-urban forest reference site (43°22′52.17″ N, 80°20′52.46″ W, Cambridge, Ontario, Canada). Notably, the rare Charitable Research Reserve is composed of forest fragments bordered by agricultural property and sits in close proximity to the cities of Kitchener and Cambridge. This site should not be considered a pristine natural habitat; however, it differs markedly in biotic and abiotic factors from urban landscapes. Furthermore, it is likely to be representative of the habitat to which eastern grey squirrels are now relegated throughout much of their native range [31]. We captured squirrels using tomahawk Model 102 traps (Tomahawk Live Trap Co., WI, USA) between 06.00 and 16.00, checking traps at 1 h intervals. Upon capture, we transferred caged squirrels to a cloth bag and checked at 15 min intervals for faecal pellets (usually present after 15 min). Using forceps ethanol-washed between use, we collected fresh faecal pellets which we stored on ice in the field before transfer to −20°C for storage within 4 h of collection. After faecal pellet collection, we transferred squirrels to a handling bag, and within 5 min drew 1 ml of blood from the saphenous vein via heparin rinsed syringe and created duplicate blood smears. We recorded general physiological measures (mass, sex, reproductive condition) and collected an approximately 3 × 3 cm patch of hair from the outer thigh using an electric razor. Squirrels were given a pair of unique alpha-numeric ear tags upon release to identify re-captures and all procedures were completed within 20 min of transferring a squirrel from the trap to the handling bag. All cloth holding bags were used only once within a trapping day and washed between trapping days to prevent cross contamination between squirrels.
(c). Quantification of HPA axis activity
We quantified three measures of HPA axis activity that differ temporally in the physiological snapshot they capture [24]. Our first and most acute measure, neutrophil : lymphocyte ratio (N : L), is correlated with circulating glucocorticoids approximately 30 min prior to sample collection [34,35]. Because this measure becomes elevated soon after capture, it cannot be interpreted as a baseline measure, but rather an immunological response to trapping stress [34]. We posit that stressor-induced innate immunity is a primary proximate mechanism by which an acute stress response drives change in the microbiome. As a direct immunological measure of stressor induced innate immunity, N : L is a more direct measure of this mechanism than plasma cortisol, a more commonly used measure of acute HPA axis activity. Limitations imposed by trapping live wildlife prevented time of capture standardization of blood draws, and therefore interpreting the causal mechanism underlying N : L differences between individuals is inadvisable. Regardless of the cause of N : L variation, N : L offers a useful glimpse into grey squirrel physiological state at the time of faecal pellet collection. Due to improper fixing of blood smears, two urban squirrel samples were excluded from analyses which included N : L as a fixed effect. Our second measure, faecal glucocorticoid metabolites (FGM), provides an integrated picture into HPA axis activity on the order of hours prior to capture but is not affected by trapping stress [33]. We extracted and assayed FGM using an enzyme immunoassay (EIA) protocol validated for use in eastern grey squirrels [33]. Our third measure, hair cortisol, is reflective of circulating glucocorticoids integrated over longer periods (days to weeks [36]) and thus should be best in reflecting baseline HPA activity. We extracted hair cortisol as per Davenport et al. [36] and assayed cortisol using a sensitive (0.007 µg dl−1 detection limit) commercially available salivary cortisol EIA kit commonly used for hair cortisol quantification (Salimetrics, Carlsbad, USA).
(d). Sequencing and bioinformatics
Using QIAamp DNA Stool Mini Kits (Qiagen, Hilden Germany), we extracted total DNA from 0.2 g subsamples of faecal samples, which in other rodents are indicative of bacterial communities in the large intestines [37]. Extracts underwent triplicate PCR amplification of the 16S rRNA gene v3-v4 region (515F-806R primers) at MetaGenomBio Inc. (Toronto, Canada). Triplicate PCR products were pooled for sequencing on an Illumina MiSeq platform to a depth of 30 000 reads per sample. We then processed pair-end reads in Mothur using a standardized amplicon processing pipeline [38], aligned sequences to the silva v132 reference database [39] and assigned taxonomy to operational taxonomic units (OTUs) clustered using OptiClust [40] based on a 97% cut-off. To remove potential contaminants or sequencing errors, all non-bacterial OTUs and OTUs which were not represented by at least 1 read in 10% of the samples were removed prior to analysis, similar to Knowles et al. [41]. OTUs were selected rather than exact sequence variants (ESVs) as we were interested primarily in identifying putative functional mechanisms underlying microbiome variation. The finer phylogenetic resolution offered by ESVs over OTUs does not further our understanding of these patterns. We rarefied OTU count tables to 4794 sequences per sample (rarefaction curve: electronic supplementary material, figure S1) for alpha diversity (see electronic supplementary material), non-Euclidean distance matrix and nearest taxon index computation. An unrarefied centred log ratio (CLR)-transformed dataset was used for all other analyses [42].
(e). Statistical methods
We used general linear models to determine whether urban and forest squirrels differed in indices of HPA axis activity. We included sex as a fixed effect, and duration of time in trap (estimated as the time between the last trap check and capture) in the N : L model. N : L and FGM were log-transformed and hair cortisol was Box–Cox transformed (λ = −1) to achieve normality.
To determine whether beta-diversity differed between urban and forest squirrels, we performed univariate PERMANOVAs on OTU matrices calculated using Jaccard [43], Bray–Curtis [44], unweighted UniFrac [45] and weighted UniFrac [46] distances. Next, to determine whether host physiology better explained variation in phylogenetically weighted beta-diversity than environment we used a distance-based redundancy analysis (dbRDA) to compare the marginal effects of environment, sex and HPA axis measures on weighted UniFrac-computed matrices. The weighted UniFrac measure balances both phylogenetic relatedness and differences in OTU relative abundance. Here, we emphasize the weighted UniFrac distance measures over a phylogeny agnostic measure as it more closely signals possible functional differences in the microbiome and is less likely to be biased by bacterial dispersal limitation between study sites. To refine our understanding of the taxonomic scales at which host environment and physiology act, we performed additional post hoc dbRDAs of Euclidean distance computed matrices of CLR-transformed datasets after successive binning of sequences to OTU, genus, family, order, class and phylum.
To explore how dominant components of the microbiome differed between environments and sexes, or covaried with measures of host physiology, we next restricted our focus to the five most abundant bacterial families (those representing greater than 5% of all sequences). To determine which predictors most parsimoniously explained variation in the relative abundances of these families, we used a multi-model inference approach [47], starting from a global model parametrized by environment (urban versus forest) and physiological (sex, N : L, FGM, hair cortisol) terms. Within a multi-model inference framework, Akaike's information criterion (AICc) scores of models composed of all independent variable combinations are compared. These contrasts allow us to determine whether host physiology explains variation in abundant bacterial families above and beyond what is explained by environment, and vice versa. All model combinations were tested using the ‘MuMIn’ package in R [48], and statistical significance was determined from full averaging of AICc scores.
Finally, to infer changes in the strengths of deterministic versus stochastic processes underlying community assembly in response to changes in host physiology, we completed a nearest taxon index (NTI) analysis using the ‘picante’ R package [49]. Positive values of NTI indicate that community membership is more phylogenetically clustered (indicating ecological filtering) than would be expected by chance alone. Conversely, negative NTI values indicate the communities are phylogenetically over-dispersed, and thus strongly structured by competition [50]. The magnitude of |NTI| is therefore expected to be proportional to the strength of deterministic processes [51]. We tested for a relationship between NTI and N : L, FGM and hair cortisol, fitting general linear models to each separately, as well as to the first principal component of a PCA of HPA measures which represented major variation in HPA axis activity. All statistical analyses presented were performed in R v. 3.4.4 (R Core Team 2018) using the package ‘phyloseq’ [52] unless otherwise stated.
3. Results
(a). HPA-axis activity
Our haematological proxy of acute HPA axis activity (N : L) was higher in forest squirrels (2.9 ± 0.47; mean ± s.e.) than their urban counterparts (1.4 ± 0.32, t1,23 = −2.07, p = 0.05; electronic supplementary material, figure S2A), after controlling for both sex and time captive in a trap. In contrast, our intermediate measure (FGM) was greater in urban squirrels (784 ± 129 ng g−1 of dried faeces) than forest residents (349 ± 57 ng g−1, t1,26 = 2.77, p = 0.01; electronic supplementary material, figure S2B) after controlling for sex. Finally, after controlling for sex, our furthest back-casting measure (hair cortisol) was higher in forest squirrels (127 ± 34 ng g−1 of hair) than urban squirrels (54 ± 7 ng g−1, t1,26 = −2.27, p = 0.03; electronic supplementary material, figure S2C).
(b). Beta diversity
Urban and forest squirrel microbiomes differed in the OTUs present (Jaccard distance: R2 = 0.07, p < 0.001; figure 1a), the differential abundance of OTUs (Bray–Curtis distance: R2 = 0.10, p < 0.001; figure 1b), the phylogenetic weighting of unshared OTUs (UniFrac: R2 = 0.12, p < 0.001; figure 1c) and the relative abundance weighted phylogenetic beta-diversity (weighted UniFrac: R2 = 0.09, p < 0.001; figure 1d), based on univariate PERMANOVAs. However, when the marginal effects of environment, sex and stress measures were concurrently compared in a distance-based redundancy analysis (dbRDA), only N : L (R2 = 0.07, p = 0.01) and hair cortisol (R2 = 0.07, p < 0.01) were found to explain variation in weighted UniFrac beta diversity (environment: p = 0.17; sex: p = 0.29; FGM: p = 0.25). To determine the taxonomic scale at which these effects were acting, we performed successive dbRDAs after binning bacterial sequences to increasingly broad taxonomic levels. Environment explained significant variation only among closely related taxa (OTUs and genus), while hair cortisol explained variation at higher taxonomic groupings (genus to phylum; electronic supplementary material, table S1).
Figure 1.

Non-multidimensional scaling ordinations of urban and forest eastern grey squirrel faecal bacterial microbiomes. Distances computed using (a) Jaccard (OTU; stress 0.19), (b) Bray–Curtis (OTU; stress = 0.19), (c) unweighted UniFrac (stress = 0.18) and (d) weighted UniFrac (stress = 0.17) measures. Dashed lines denote 95% confidence intervals. (Online version in colour.)
To generate hypotheses as to what might underlie beta-diversity in the microbiome, we focused our attention on the five dominant bacterial families found in the grey squirrel microbiome: Lachnospiraceae (44% of all reads on average), Ruminococcacae (17%), Muribaculaceae (12%), Akkermansiaceae (8%) and Prevotellaceae (6%) (electronic supplementary material, figure S3). When environment was the only term fitted, the relative abundance of some of these families appeared to differ between urban and forest environments. However, a full multi-model inference analyses revealed that physiological terms more parsimoniously explained this variation (electronic supplementary material, table S2). Namely, hair cortisol was observed to negatively correlate with Akkermansiaceae (t1,27 = −4.32, R2 = 0.39, p < 0.001; figure 2a) but positively correlate with Lachnospiraceae (t1,27 = 3.39, R2 = 0.27, p < 0.01; figure 2b). Additionally, N : L was weakly positively correlated with Prevotellaceae (t1,25 = 2.44, R2 = 0.16, p = 0.02; electronic supplementary material, figure S4), however the model fit was greatly improved after removal of 2 outlying data points (t1,24 = 4.82, R2 = 0.48, p < 0.001; figure 3), corresponding to squirrels in which Prevotellaceae was not detected.
Figure 2.
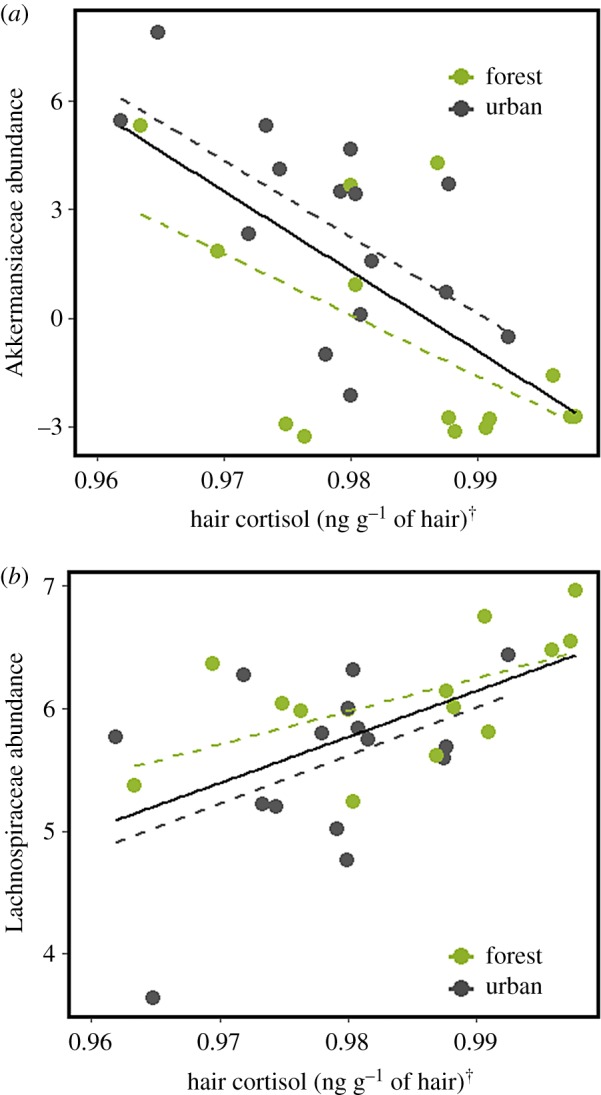
Hair cortisol is (a) negatively correlated with CLR-transformed Akkermansiaceae relative abundance, but (b) positively correlated with CLR-transformed Lachnospiraceae relative abundance. Hair cortisol values Box–Cox transformed (λ = −1). Dashed lines are fitted separately to both environments but do not differ statistically in their slope or intercept. (Online version in colour.)
Figure 3.
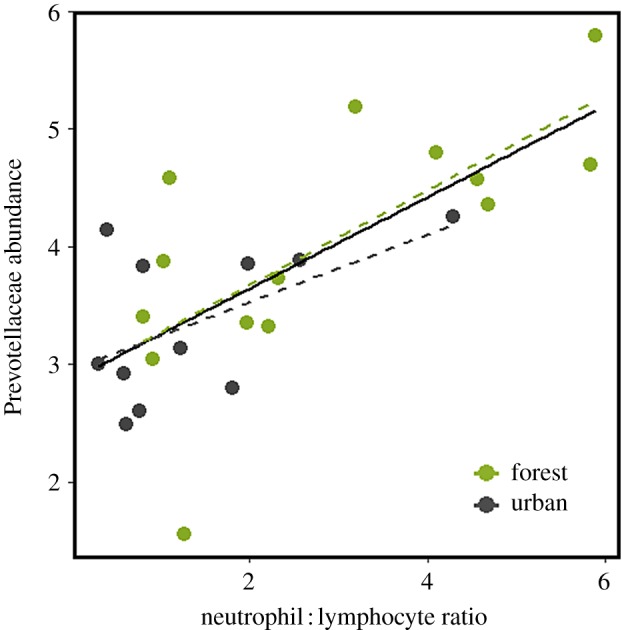
Neutrophil : lymphocyte ratio is positively correlated with CLR-transformed Prevotellaceae relative abundance. Dashed lines are fitted separately to both environments but do not differ statistically in their slope or intercept. (Online version in colour.)
Finally, to determine how host HPA axis activity affected the form and strength of bacterial community assembly, we completed an NTI analysis. NTI was negatively correlated with FGM (t1,27 = −2.632, R2 = 0.17, p = 0.01), but not N : L (t1,25 = 1.714, p = 0.10) or hair cortisol (t1,27 = 1.497, p = 0.15). However, a strong positive relationship was observed between NTI and the first principal component of a HPA axis measure PCA (electronic supplementary material, figure S5), such that extremes in HPA axis PC scores were characterized by stronger evidence for environmental filtering (t1,25 = 3.644, R2 = 0.32, p < 0.01; figure 4).
Figure 4.
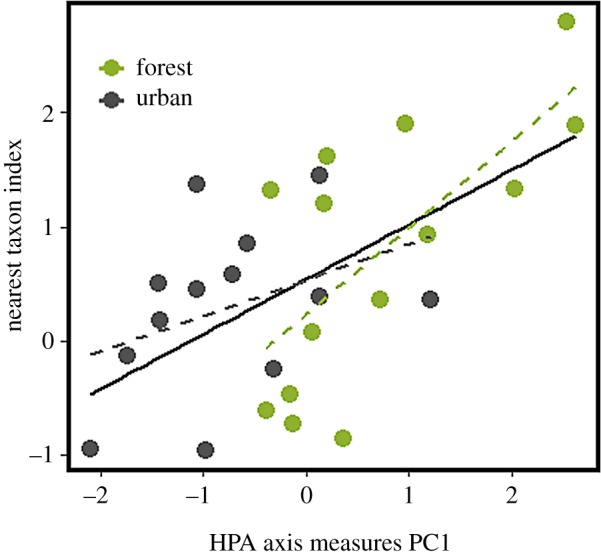
The first component of a principal component analysis of hypothalamic–pituitary–adrenal axis measures (neutrophil : lymphocyte ratio, faecal glucocorticoid metabolite concentration and hair cortisol) is positively correlated with eastern grey squirrel faecal bacterial microbiome nearest taxon index. Dashed lines are fitted separately to both environments but do not differ statistically in their slope or intercept. (Online version in colour.)
4. Discussion
We sought to determine whether host environment or physiology was a stronger predictor of bacterial microbiome structure. The eastern grey squirrel microbiome differed between urban and forest environments however HPA axis measures better explained variation among more distantly related taxa than urbanization. Similarly, HPA axis measures more parsimoniously explained microbiome variation than host sex. FGM had little explanatory power, leading us to suggest that while HPA axis upregulation might remodel the intestinal microbiome, resiliency of bacterial communities makes stress experienced in the recent past less consequential for deterministic assembly of microbiota communities than an organism's immediate physiological state. In contrast, the strongest relationships we observed were between the microbiome and hair cortisol, which represents the most stable baseline measure of HPA axis activity, and thus may be most representative of the intestinal physiological landscape.
The close relatedness (OTU and genus) of bacteria distinguishing urban from forest squirrel microbiomes suggests that microbiome functional capacity does not differ markedly between these two environments and that variable selection has not played a substantial role in structuring inter-environment differences [51]. Instead, microbiome differences observed between urban and forest squirrels not accounted for by host physiology might have resulted from bacterial dispersal limitation [53] or exposure to different environmental pools of microbial diversity. These results suggest that ecological processes of drift and dispersal, not just selection, might play an important role in driving microbiome divergence between host environments. In contrast, the stronger phylogenetic signal we detected within bacterial communities in response to HPA axis measures indicates that changes in host physiology may reshape the ecological filters imposed on resident microbial communities. Incidentally, our nearest taxon index analysis also indicates that changes in host physiology might affect the strength, not just the direction of deterministic assembly. FGM concentration, which did not correlate with beta-diversity in the microbiome, best explained variation in NTI values. Consideration of how changes in host physiology might modulate the relative strengths of deterministic versus stochastic processes of community assembly is an important next step for understanding the maintenance or collapse of host–microbe symbioses, particularly within the Anna Karenina paradigm of microbiome dysbiosis [54].
To explain disparities in the taxonomic scale at which physiology and environment act, we draw on community assembly theory, which posits that communities form as the result of hierarchically structured ecological filters [55,56]. We assert that host physiology is a higher-order filter than that of the host's external environment or diet. Before occupying an intestinal niche and becoming a stable resident of the microbiome, a bacterium must first be able to colonize a host and persist despite dynamics in host physiological state. Evolutionary familiarity between vertebrates and bacteria, and a deep rooting of the gut–brain axis in vertebrate phylogeny [57], might mean that the combinations of traits adapted to conditions of reduced or elevated host HPA axis activity may likewise be deeply rooted in bacterial phylogeny. Although dietary differences between urban and forest squirrels might favour different bacterial metabolic traits, and thereby drive microbiome divergence as we predicted, the bacteria able to exploit this new dietary niche are most likely to be drawn from among those taxa able to surpass host physiological filters. This perspective helps to reconcile the conflict between hypotheses that function (not bacterial identity) matters for host–microbiome relationships [58,59], with widespread evidence for phylosymbiosis [60].
Mucin glycan production is one conserved facet of vertebrate physiology, which probably acts as a primary environmental filter during microbiome assembly [61]. Mucin glycan subsidies are an important host-derived nutrient source for bacterial communities. However, the enzymes required to metabolize mucin glycans are not universal among bacteria, which often specialize on either dietary or host-derived energy channels [62]. Furthermore, the availability of host-derived nutrients is dynamic; prolonged HPA axis upregulation, like that indicated by elevated hair cortisol, depletes intestinal mucus [28–30]. We speculate that this mechanism might underlie the negative relationship we observed between hair cortisol and the mucin glycan specialist, Akkermansiaceae [63,64], in contrast to the positive relationship observed between hair cortisol and Lachnospiraceae, a plant fibre specialist [65] (figure 2). Interestingly, these findings echo patterns observed in a dynamic in vitro gut model in response to experimental mucin glycan supplementation [66]. A comparable in vivo mucin glycan supplementation in free-living wildlife would be effective follow-up manipulation for parsing causality in the correlative patterns we report. Notably, Akkermansiaceae and Lachnospiraceae relative abundances were higher and lower in urban squirrels, respectively. However, environment was not identified as a significant term in multi-model inference analyses when considered alongside hair cortisol. This suggests that although these bacterial families differ in relative abundance between urban and forest environments, an altered host physiological state is the underlying cause.
Regulating microbial access to mucin glycan resources is a conserved example of how vertebrates select for microbiome gene content, rather than specific bacterial identity, when interacting with the microbiome [58,59]. Since the HPA axis is deeply rooted in vertebrate phylogeny [57], similar re-balancing of digesta versus mucin glycan energy channels in response to stressors experienced by the host could be a conserved mechanism by which vertebrates regulate their microbiomes. Trade-offs between mucin glycan and digesta metabolizing bacteria are reported in laboratory mice exposed to chronic stressors [67–69], HIV patients experiencing mucosal barrier disruption [70], fasting vertebrates [71,72] and mammals undergoing torpor or hibernation [73–77]. Although these studies typically discuss the bacterial patterns observed within the context of diet or specific stressors, homologous HPA axis responses across these biological groups might provide an alternate or complimentary mechanism. We suggest that mucin glycan production should be considered of equal importance as the host's diet when discussing proximate causes of patterns in the microbiome. Whenever possible, researchers might consider quantifying mucin glycan production in the intestinal microbiome, as exemplified by Dill-McFarland et al. [73].
Alongside bottom-up regulation of bacterial communities through host subsidies (e.g. mucin glycans), host immunity exerts strong top-down control [78]. While not overtly pathogenic, Prevotellaceae is a family that contains members which appear strongly mediated by host immunity [79,80]. This may underlie the positive correlation we observed between Prevotellaceae and N : L. Importantly, the reverse causality cannot be discounted since bacterial translocation has been demonstrated under laboratory conditions to prime host innate immunity [17,81], and Prevotellaceae has been shown to trigger strong host immune responses [82]. If a similar mechanism operates under wild conditions, then our results and the well characterized correlation between neutrophil/heterophil : lymphocyte ratios and acute stress in vertebrates [34] might have a bacterial basis. Understanding the bacterial basis for this and other well characterized eco-physiological phenomena is an exciting frontier for wildlife eco-physiologists [83].
Ultimately, parsing causality in the correlative patterns we report will require in-field manipulations. In this study we have generated specific predictions for future directed experimental testing. Nevertheless, we argue that urbanization itself may be viewed as an opportunistic experiment which can inform our interpretation of causality. Both the microbiome and HPA axis could covary independent of one another in response to urban environmental cues, however current understanding of the gut–brain axis makes complete independence unlikely [84]. Similarly, it is unlikely that forest–urban microbiome differences wholly underlie adult HPA axis differences between such disparate environments in free-living wildlife, especially as the microbiome's capacity to programme the mammalian HPA axis is attenuated after an early developmental window [85]. Therefore, our suggestion that differences in stressors between environments affect host physiology, which in turn affects resident bacterial communities, appears to be the most parsimonious interpretation. However, future experimental manipulations present exciting possibilities to directly test these relationships.
Our findings do not indicate that environmental change (urbanization) only affects the microbiome at a fine taxonomic scale. Rather, large phylogenetic turnover observed between environments may be driven by changes in host physiology. When investigating underlying physiological mechanisms of microbiome variation, we caution researchers to carefully select their physiological indices of interest and, when feasible, entertain multiple metrics. Comprehensive physiological measures appear to better explain variation in bacterial community structure than indicators of an organism's immediate physiological state, or physiological state in the recent past. However, the latter might better capture stochasticity in the microbiome, an important but heretofore overlooked feature of host associated microbial communities [54]. Finally, although we focus heavily on the importance of understanding proximate mechanisms, change in HPA axis activity is an ultimate physiological cause of microbiome variation that links external environmental change to more direct physiological mechanisms (e.g. immunity and host-derived nutrients). Characterizing ultimate and proximate physiological causes of microbiome variation, rather than relying on external mechanisms, is necessary for advancing our knowledge of host–microbe relationships, whether in pursuit of health biomarkers, therapeutic targets or more complete fundamental knowledge of host–microbiome phylosymbiosis.
Supplementary Material
Acknowledgements
We thank S. Taylor and L. McCaw for their aid in data collection and the rare Charitable Research Reserve and the City of Guelph for logistical support. Thank you also to Drs E. Allen-Vercoe and A. McAdam for their comments on early manuscript drafts.
Ethics
Animal utilization protocol was approved by the University of Guelph Animal Care Committee (AUP no. 3506). Trapping authorization was granted by the Ontario Ministry of Natural Resources (WSCA no. 1087323).
Data accessibility
All data and scripts have been made publicly available (Raw sequences: 10.6084/m9.figshare.8264003; meta-data: 10.6084/m9.figshare.8264054; mothur pipeline: 10.6084/m9.figshare.9916193; R-script: 10.6084/m9.figshare.9916220).
Authors' contributions
A.E.M.N. and M.R.S. participated in study design, M.R.S. completed data collection and bioinformatics, M.R.S. and R.P. performed the hormone assays, M.R.S. wrote the first manuscript draft. All authors contributed critically to the drafts and gave final approval of publication.
Competing interests
We declare we have no competing interests.
Funding
M.R.S. was funded by a NSERC CGS-M and Vanier scholarship while completing this work. A.E.M.N. funded the research costs using University of Guelph research funds.
References
- 1.Rosenberg E, Zilber-Rosenberg I. 2018. The hologenome concept of evolution after 10 years. Microbiome 6, 1–14. ( 10.1186/s40168-018-0457-9) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Moloney RD, Desbonnet L, Clarke G, Dinan TG, Cryan JF. 2014. The microbiome: stress, health and disease. Mamm. Genome 25, 49–74. ( 10.1007/s00335-013-9488-5) [DOI] [PubMed] [Google Scholar]
- 3.Rooks MG, Garrett WS. 2016. Gut microbiota, metabolites and host immunity. Nat. Rev. Immunol. 16, 341–352. ( 10.1038/nri.2016.42) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kohl KD, Denise Dearing M. 2016. The woodrat gut microbiota as an experimental system for understanding microbial metabolism of dietary toxins. Front. Microbiol. 7, 1–9. ( 10.3389/fmicb.2016.01165) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Alberdi A, Aizpurua O, Bohmann K, Zepeda-Mendoza ML, Gilbert MTP. 2016. Do vertebrate gut metagenomes confer rapid ecological adaptation? Trends Ecol. Evol. 31, 689–699. ( 10.1016/j.tree.2016.06.008) [DOI] [PubMed] [Google Scholar]
- 6.Ley RE, et al. 2008. Evolution of mammals and their gut microbes. Science 320, 1647–1651. ( 10.1126/science.1155725) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Amato KR, et al. 2018. Evolutionary trends in host physiology outweigh dietary niche in structuring primate gut microbiomes. ISME J. 13, 576–587. ( 10.1038/s41396-018-0175-0) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nishida AH, Ochman H. 2018. Rates of gut microbiome divergence in mammals. Mol. Ecol. 27, 1884–1897. ( 10.1111/mec.14473) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Heitlinger E, Ferreira SCM, Thierer D, Hofer H, East ML. 2017. The intestinal eukaryotic and bacterial biome of spotted hyenas: the impact of social status and age on diversity and composition. Front. Cell. Infect. Microbiol. 7, 1–17. ( 10.3389/fcimb.2017.00262) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Menke S, Meier M, Mfune JKE, Melzheimer J, Wachter B, Sommer S. 2017. Effects of host traits and land-use changes on the gut microbiota of the Namibian black-backed jackal (Canis mesomelas). FEMS Microbiol. Ecol. 93, 1–16. ( 10.1093/femsec/fix123) [DOI] [PubMed] [Google Scholar]
- 11.Phillips CD, et al. 2012. Microbiome analysis among bats describes influences of host phylogeny, life history, physiology and geography. Mol. Ecol. 21, 2617–2627. ( 10.1111/j.1365-294X.2012.05568.x) [DOI] [PubMed] [Google Scholar]
- 12.Lynch M, Walsh B. 1998. Genetics and analysis of quantitative traits, 1st edn Sunderland, MA: Sinauer Associates; See http://www.invemar.org.co/redcostera1/invemar/docs/RinconLiterario/2011/febrero/AG_8.pdf. [Google Scholar]
- 13.Burnham KP, Anderson DR. 2002. Model selection and multimodel inference: a practical information-theoretic approach. 2nd edition. New York, NY: Springer-Verlag. [Google Scholar]
- 14.Jenkins BR, Vitousek MN, Hubbard JK, Safran RJ. 2014. An experimental analysis of the heritability of variation in glucocorticoid concentrations in a wild avian population. Proc. R. Soc. B 281, 1–8. ( 10.1098/rspb.2014.1302) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McEwen BS. 1998. Stress, adaptation, and disease: allostasis and allostatic load. Ann. N. Y. Acad. Sci. 840, 33–44. ( 10.1111/j.1749-6632.1998.tb09546.x) [DOI] [PubMed] [Google Scholar]
- 16.Galley JD, Yu Z, Kumar P, Dowd SE, Lyte M, Bailey MT. 2014. The structures of the colonic mucosa-associated and luminal microbial communities are distinct and differentially affected by a prolonged murine stressor. Gut Microbes 5, 748–760. ( 10.4161/19490976.2014.972241) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bailey MT, Dowd SE, Galley JD, Hufnagle AR, Allen RG, Lyte M. 2011. Exposure to a social stressor alters the structure of the intestinal microbiota: implications for stressor-induced immunomodulation. Brain Behav. Immun. 25, 397–407. ( 10.1016/J.BBI.2010.10.023) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bailey MT, Dowd SE, Parry NM, Galley JD, Schauer DB, Lyte M. 2010. Stressor exposure disrupts commensal microbial populations in the intestines and leads to increased colonization by Citrobacter rodentium. Infect. Immun. 78, 1509–1519. ( 10.1128/IAI.00862-09) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stothart MR, Bobbie CB, Schulte-hostedde AI, Boonstra R, Palme R, Mykytczuk NCS, Newman AEM. 2016. Stress and the microbiome: linking glucocorticoids to bacterial community dynamics in wild red squirrels. Biol. Lett. 12, 14–17. ( 10.1098/rsbl.2015.0875) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vlckova K, et al. 2018. Impact of stress on the gut microbiome of free-ranging western lowland gorillas. Microbiology 164, 40–44. ( 10.1099/mic.0.000587) [DOI] [PubMed] [Google Scholar]
- 21.Noguera JC, Aira M, Pérez-Losada M, Domínguez J, Velando A. 2018. Glucocorticoids modulate gastrointestinal microbiome in a wild bird. R. Soc. open sci. 5, 171743 ( 10.1098/rsos.171743) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Levin II, Zonana DM, Fosdick BK, Song SJ, Knight R, Safran RJ. 2016. Stress response, gut microbial diversity and sexual signals correlate with social interactions. Biol. Lett. 12, 739–745. ( 10.1098/rsbl.2016.0352) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sheriff MJ, Dantzer B, Delehanty B, Palme R, Boonstra R. 2011. Measuring stress in wildlife: techniques for quantifying glucocorticoids. Oecologia 166, 869–887. ( 10.1007/s00442-011-1943-y) [DOI] [PubMed] [Google Scholar]
- 24.Palme R. 2019. Non-invasive measurement of glucocorticoids: advances and problems. Physiol. Behav. 199, 229–243. ( 10.1016/j.physbeh.2018.11.021) [DOI] [PubMed] [Google Scholar]
- 25.Martin LB. 2009. Stress and immunity in wild vertebrates: timing is everything. Gen. Comp. Endocrinol. 163, 70–76. ( 10.1016/j.ygcen.2009.03.008) [DOI] [PubMed] [Google Scholar]
- 26.Castagliuolo I, Lamont JT, Qiu B, Fleming SM, Bhaskar KR, Nikulasson ST, Kornetsky C, Pothoulakis C. 1996. Acute stress causes mucin release from rat colon: role of corticotropin releasing factor and mast cells. Am. J. Physiol. 271, G884–G892. ( 10.1152/ajpgi.1996.271.5.G884) [DOI] [PubMed] [Google Scholar]
- 27.Brooks KC, Mateo JM. 2013. Chronically raised glucocorticoids reduce innate immune function in Belding's ground squirrels (Urocitellus beldingi) after an immune challenge. Gen. Comp. Endocrinol. 193, 149–157. ( 10.1016/j.ygcen.2013.07.019) [DOI] [PubMed] [Google Scholar]
- 28.Shigeshiro M, Tanabe S, Suzuki T. 2012. Repeated exposure to water immersion stress reduces the Muc2 gene level in the rat colon via two distinct mechanisms. Brain Behav. Immun. 26, 1061–1065. ( 10.1016/J.BBI.2012.05.016) [DOI] [PubMed] [Google Scholar]
- 29.Söderholm JD, Yang PC, Ceponis P, Vohra A, Riddell R, Sherman PM, Perdue MH. 2002. Chronic stress induces mast cell-dependent bacterial adherence and initiates mucosal inflammation in rat intestine. Gastroenterology 123, 1099–1108. ( 10.1053/gast.2002.36019) [DOI] [PubMed] [Google Scholar]
- 30.Estienne M, Claustre J, Clain-Gardechaux G, Paquet A, Taché Y, Fioramonti J, Plaisancié P. 2010. Maternal deprivation alters epithelial secretory cell lineages in rat duodenum: role of CRF-related peptides. Gut 59, 744–751. ( 10.1136/gut.2009.190728) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Benson E. 2013. The urbanization of the eastern gray squirrel in the United States. J. Am. Hist. 100, 691–710. ( 10.1093/jahist/jat353) [DOI] [Google Scholar]
- 32.Koprowski JL. 1994. Sciurus carolinensis. Mamm. Species 1, 1–9. [Google Scholar]
- 33.Bosson CO, Palme R, Boonstra R. 2013. Assessing the impact of live-capture, confinement, and translocation on stress and fate in eastern gray squirrels. J. Mammal. 94, 1401–1411. ( 10.1644/13-MAMM-A-046.1) [DOI] [Google Scholar]
- 34.Davis AK, Maney DL, Maerz JC. 2008. The use of leukocyte profiles to measure stress in vertebrates: a review for ecologists. Funct. Ecol. 22, 760–772. ( 10.1111/j.1365-2435.2008.01467.x) [DOI] [Google Scholar]
- 35.Johnstone CP, Lill A, Reina RD. 2012. Does habitat fragmentation cause stress in the agile antechinus? A haematological approach. J. Comp. Physiol. B Biochem. Syst. Environ. Physiol. 182, 139–155. ( 10.1007/s00360-011-0598-7) [DOI] [PubMed] [Google Scholar]
- 36.Davenport MD, Tiefenbacher S, Lutz CK, Novak MA, Meyer JS. 2006. Analysis of endogenous cortisol concentrations in the hair of rhesus macaques. Gen. Comp. Endocrinol. 147, 255–261. ( 10.1016/j.ygcen.2006.01.005) [DOI] [PubMed] [Google Scholar]
- 37.Gu S, Chen D, Zhang J-N, Lv X, Wang K, Duan L-P, Nie Y, Wu X-L. 2013. Bacterial community mapping of the mouse gastrointestinal tract. PLoS ONE 8, e74957 ( 10.1371/journal.pone.0074957) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kozich JJ, Westcott SL, Baxter NT, Highlander SK, Schloss PD. 2013. Development of a dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the MiSeq Illumina sequencing platform. Appl. Environ. Microbiol. 79, 5112–5120. ( 10.1128/AEM.01043-13) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yilmaz P, et al. 2014. The SILVA and ‘All-species Living Tree Project (LTP)’ taxonomic frameworks. Nucleic Acids Res. 42, 643–648. ( 10.1093/nar/gkt1209) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Westcott SL, Schloss PD. 2017. OptiClust, an improved method for assigning amplicon-based sequence data to operational taxonomic units. mSphere 2, 1–11. ( 10.1128/mSphereDirect.00073-17) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Knowles SCL, Eccles RM, Baltrūnaitė L. 2019. Species identity dominates over environment in shaping the microbiota of small mammals. Ecol. Lett. 22, 826–837. ( 10.1111/ele.13240) [DOI] [PubMed] [Google Scholar]
- 42.Aitchison J. 1982. The statistical analysis of compositional data. J. R. Stat. Soc. Ser. B. 44, 139–160. [Google Scholar]
- 43.Jaccard P. 1912. The distribution of the flora in the alpine zone. New Phytol. 11, 37–50. ( 10.1111/j.1469-8137.1912.tb05611.x) [DOI] [Google Scholar]
- 44.Bray JR, Curtis JT. 1957. An ordination of the upland forest communities of southern Wisconsin. Ecol. Monogr. 27, 325–349. ( 10.2307/1942268) [DOI] [Google Scholar]
- 45.Lozupone C, Knight R. 2005. UniFrac: a new phylogenetic method for comparing microbial communities. Appl. Environ. Microbiol. 71, 8228–8235. ( 10.1128/AEM.71.12.8228-8235.2005) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lozupone CA, Hamady M, Kelley ST, Knight R. 2007. Quantitative and qualitative beta diversity measures lead to different insights into factors that structure microbial communities. Appl. Environ. Microbiol. 73, 1576–1585. ( 10.1128/AEM.01996-06) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Grueber CE, Nakagawa S, Laws RJ, Jamieson IG. 2011. Multimodel inference in ecology and evolution: challenges and solutions. J. Evol. Biol. 24, 699–711. ( 10.1111/j.1420-9101.2010.02210.x) [DOI] [PubMed] [Google Scholar]
- 48.Fairbanks LA, Jorgensen MJ, Bailey JN, Breidenthal SE, Grzywa R, Laudenslager ML. 2011. Heritability and genetic correlation of hair cortisol in vervet monkeys in low and higher stress environments. Psychoneuroendocrinology 36, 1201–1208. ( 10.1016/J.PSYNEUEN.2011.02.013) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kembel SW, Cowan PD, Helmus MR, Cornwell WK, Morlon H, Ackerly DD, Blomberg SP, Webb CO. 2010. Picante: R tools for integrating phylogenies and ecology. Bioinformatics 26, 1463–1464. ( 10.1093/bioinformatics/btq166) [DOI] [PubMed] [Google Scholar]
- 50.Horner-Devine MC, Bohannan BJM. 2006. Phylogenetic clustering and overdispersion in bacterial communities. Ecology 87, S100–S108. ( 10.1890/0012-9658(2006)87[100:PCAOIB]2.0.CO;2) [DOI] [PubMed] [Google Scholar]
- 51.Stegen JC, Lin X, Konopka AE, Fredrickson JK. 2012. Stochastic and deterministic assembly processes in subsurface microbial communities. ISME J. 6, 1653–1664. ( 10.1038/ismej.2012.22) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.McMurdie PJ, Holmes S. 2013. phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE 8, e61217 ( 10.1371/journal.pone.0061217) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Linnenbrink M, Wang J, Hardouin EA, Künzel S, Metzler D, Baines JF. 2013. The role of biogeography in shaping diversity of the intestinal microbiota in house mice. Mol. Ecol. 22, 1904–1916. ( 10.1111/mec.12206) [DOI] [PubMed] [Google Scholar]
- 54.Zaneveld JR, McMinds R, Vega Thurber R. 2017. Stress and stability: applying the Anna Karenina principle to animal microbiomes. Nat. Microbiol. 2, 17121 ( 10.1038/nmicrobiol.2017.121) [DOI] [PubMed] [Google Scholar]
- 55.Hillerislambers J, Adler PB, Harpole WS, Levine JM, Mayfield MM. 2012. Rethinking community assembly through the lens of coexistence theory. Annu. Rev. Ecol. Evol. Syst. 43, 227–275. ( 10.1146/annurev-ecolsys-110411-160411) [DOI] [Google Scholar]
- 56.Pearson DE, Ortega YK, Eren Ö, Hierro JL. 2018. Community assembly theory as a framework for biological invasions. Trends Ecol. Evol. 33, 313–325. ( 10.1016/j.tree.2018.03.002) [DOI] [PubMed] [Google Scholar]
- 57.Denver RJ. 2009. Structural and functional evolution of vertebrate neuroendocrine stress systems. Ann. N. Y. Acad. Sci. 1163, 1–16. ( 10.1111/j.1749-6632.2009.04433.x) [DOI] [PubMed] [Google Scholar]
- 58.Foster KR, Schluter J, Coyte KZ, Rakoff-Nahoum S. 2017. The evolution of the host microbiome as an ecosystem on a leash. Nature 548, 43–51. ( 10.1038/nature23292) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Doolittle WF, Booth A. 2017. It's the song, not the singer: an exploration of holobiosis and evolutionary theory. Biol. Philos. 32, 5–24. ( 10.1007/s10539-016-9542-2) [DOI] [Google Scholar]
- 60.Brooks AW, Kohl KD, Brucker RM, van Opstal EJ, Bordenstein SR.. 2016. Phylosymbiosis: relationships and functional effects of microbial communities across host evolutionary history. PLoS Biol. 14, 1–29. ( 10.1371/journal.pbio.2000225) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Crost EH, Tailford LE, Le Gall G, Fons M, Henrissat B, Juge N.. 2013. Utilisation of mucin glycans by the human gut symbiont Ruminococcus gnavus is strain-dependent. PLoS ONE 8, e76341 ( 10.1371/journal.pone.0076341) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tailford LE, Crost EH, Kavanaugh D, Juge N. 2015. Mucin glycan foraging in the human gut microbiome. Front. Genet. 6, 1–18. ( 10.3389/fgene.2015.00081) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.van Passel MWJ, et al. 2011. The genome of Akkermansia muciniphila, a dedicated intestinal mucin degrader, and its use in exploring intestinal metagenomes. PLoS ONE 6, 1–8. ( 10.1371/journal.pone.0016876) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ottman N, Geerlings SY, Aalvink S, De Vos WM, Belzer C.. 2017. Action and function of Akkermansia muciniphila in microbiome ecology, health and disease. Best Pract. Res. Clin. Gastroenterol. 31, 637–642. ( 10.1016/j.bpg.2017.10.001) [DOI] [PubMed] [Google Scholar]
- 65.Meehan CJ, Beiko RG. 2014. A phylogenomic view of ecological specialization in the Lachnospiraceae, a family of digestive tract-associated bacteria. Genome Biol. Evol. 6, 703–713. ( 10.1093/gbe/evu050) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Van Herreweghen F, De Paepe K, Roume H, Kerckhof F-M, Van de Wiele T.. 2018. Mucin degradation niche as a driver of microbiome composition and Akkermansia muciniphila abundance in a dynamic gut model is donor independent. FEMS Microbiol. Ecol. 94, 1–13. ( 10.1093/femsec/fiy186) [DOI] [PubMed] [Google Scholar]
- 67.Li S, et al. 2017. Lachnospiraceae shift in the microbial community of mice faecal sample effects on water immersion restraint stress. AMB Express 7, 1–11. ( 10.1186/s13568-017-0383-4) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Moussaoui N, Jacobs JP, Larauche M, Biraud M, Million M, Mayer E, Taché Y. 2017. Chronic early-life stress in rat pups alters basal corticosterone, intestinal permeability, and fecal microbiota at weaning: influence of sex. J. Neurogastroenterol. Motil. 23, 135–143. ( 10.5056/jnm16105) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gautam A, Kumar R, Chakraborty N, Muhie S, Hoke A, Hammamieh R, Jett M. 2018. Altered fecal microbiota composition in all male aggressor-exposed rodent model simulating features of post-traumatic stress disorder. J. Neurosci. Res. 96, 1311–1323. ( 10.1002/jnr.24229) [DOI] [PubMed] [Google Scholar]
- 70.San-Juan-Vergara H, et al. 2018. A Lachnospiraceae-dominated bacterial signature in the fecal microbiota of HIV-infected individuals from Colombia, South America. Sci. Rep. 8, 1–13. ( 10.1038/s41598-018-22629-7) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Costello EK, Gordon JI, Secor SM, Knight R. 2010. Postprandial remodeling of the gut microbiota in Burmese pythons. ISME J. 471, 1375–1385. ( 10.1038/ismej.2010.71) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sonoyama K, Fujiwara R, Takemura N, Ogasawara T, Watanabe J, Ito H, Morita T. 2009. Response of gut microbiota to fasting and hibernation in Syrian hamsters. Appl. Environ. Microbiol. 75, 6451–6456. ( 10.1128/AEM.00692-09) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Dill-McFarland KA, Neil KL, Zeng A, Sprenger RJ, Kurtz CC, Suen G, Carey HV. 2014. Hibernation alters the diversity and composition of mucosa-associated bacteria while enhancing antimicrobial defence in the gut of 13-lined ground squirrels. Mol. Ecol. 23, 4658–4669. ( 10.1111/mec.12884) [DOI] [PubMed] [Google Scholar]
- 74.Carey HV, Walters WA, Knight R. 2013. Seasonal restructuring of the ground squirrel gut microbiota over the annual hibernation cycle. Am. J. Physiol. Regul. Integr. Comp. Physiol. 304, 33–42. ( 10.1152/ajpregu.00387.2012) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Stevenson TJ, Duddleston KN, Buck CL. 2014. Effects of season and host physiological state on the diversity, density, and activity of the arctic ground squirrel cecal microbiota. Appl. Environ. Microbiol. 80, 5611–5622. ( 10.1128/AEM.01537-14) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sommer F, Ståhlman M, Ilkayeva O, Arnemo JM, Kindberg J, Josefsson J, Newgard CB, Fröbert O, Bäckhed F. 2016. The gut microbiota modulates energy metabolism in the hibernating brown bear Ursus arctos. Cell Rep. 14, 1655–1661. ( 10.1016/j.celrep.2016.01.026) [DOI] [PubMed] [Google Scholar]
- 77.Carey HV, Assadi-Porter FM. 2017. The hibernator microbiome: host-bacterial interactions in an extreme nutritional symbiosis. Annu. Rev. Nutr. 37, 477–500. ( 10.1146/annurev-nutr-071816) [DOI] [PubMed] [Google Scholar]
- 78.Van den Abbeele P, Van de Wiele T, Verstraete W, Possemiers S. 2011. The host selects mucosal and luminal associations of coevolved gut microorganisms: a novel concept. FEMS Microbiol. Rev. 35, 681–704. ( 10.1111/j.1574-6976.2011.00270.x) [DOI] [PubMed] [Google Scholar]
- 79.Lozupone CA, Li M, Campbell TB, Flores SC, Linderman D, Gebert MJ, Knight R, Fontenot AP, Palmer BE. 2013. Alterations in the gut microbiota associated with HIV-1 infection. Cell Host Microbe 14, 329–339. ( 10.1016/J.CHOM.2013.08.006) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Elinav E, et al. 2011. NLRP6 inflammasome regulates colonic microbial ecology and risk for colitis. Cell 145, 745–757. ( 10.1016/j.cell.2011.04.022) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Fagundes CT, Amaral FA, Teixeira AL, Souza DG, Teixeira MM. 2012. Adapting to environmental stresses: the role of the microbiota in controlling innate immunity and behavioral responses. Immunol. Rev. 245, 250–264. ( 10.1111/j.1600-065X.2011.01077.x) [DOI] [PubMed] [Google Scholar]
- 82.Palm NW, et al. 2014. Immunoglobulin a coating identifies colitogenic bacteria in inflammatory bowel disease. Cell 158, 1000–1010. ( 10.1016/J.CELL.2014.08.006) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kohl KD, Carey HV. 2016. A place for host-microbe symbiosis in the comparative physiologist's toolbox. J. Exp. Biol. 219, 3496–3504. ( 10.1242/jeb.136325) [DOI] [PubMed] [Google Scholar]
- 84.Carabotti M, Scirocco A, Maselli MA, Severi C. 2015. The gut-brain axis: interactions between enteric microbiota, central and enteric nervous systems. Ann. Gastroenterol. 28, 203–209. ( 10.1038/ajgsup.2012.3) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Sudo N, Chida Y, Aiba Y, Sonoda J, Oyama N, Yu XN, Kubo C, Koga Y. 2004. Postnatal microbial colonization programs the hypothalamic–pituitary–adrenal system for stress response in mice. J. Physiol. 558, 263–275. ( 10.1113/jphysiol.2004.063388) [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data and scripts have been made publicly available (Raw sequences: 10.6084/m9.figshare.8264003; meta-data: 10.6084/m9.figshare.8264054; mothur pipeline: 10.6084/m9.figshare.9916193; R-script: 10.6084/m9.figshare.9916220).