

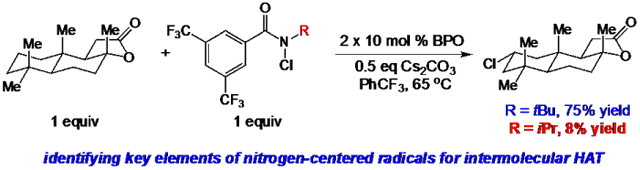
Abstract
Recent studies have demonstrated the capabilities of amidyl radicals to facilitate a range of intermolecular functionalizations of unactivated, aliphatic C─H bonds. Relatively little information is known regarding the important structural and electronic features of amidyl and related radicals that impart efficient reactivity. Herein, we evaluate a diverse range of nitrogen-centered radicals in unactivated, aliphatic C─H chlorinations. These studies establish the salient features of nitrogen-centered radicals critical to these reactions in order to expedite the future development of new site-selective, intermolecular C─H functionalizations.
Graphical Abstract
INTRODUCTION
The functionalization of aliphatic C─H bonds using a variety of strategies has become a valuable tool in chemical synthesis.1-3 For example, C─H functionalization provides facile access to molecular analogs of drugs with modified physicochemical and biological properties.4 A classical approach to C─H functionalization involves the use of heteroatom-centered radicals in intramolecular hydrogen-atom transfer (HAT) processes5-8 (Figure 1). Recent efforts by Suarez9, Du Bois10,11, Nagib12, and others13-23 have utilized this intramolecular HAT strategy to unlock practical C─H functionalizations of diverse substrates.
Figure 1.
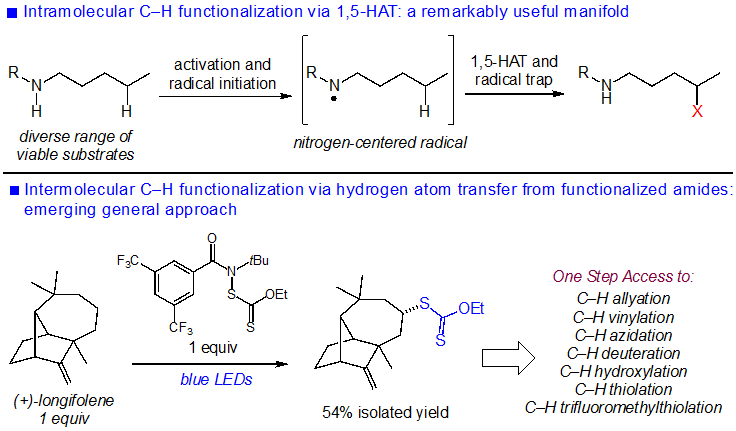
Intra- and intermolecular aliphatic C─H functionalization using nitrogen–centered radicals.
Pioneering work by Minisci24, Greene25, and Deno26 demonstrated the potential for intermolecular aliphatic C─H functionalizations using nitrogen-centered radicals. The development of practical synthetic reactions of this type, however, involves several difficult challenges that must be addressed. For example, these processes do not benefit from the kinetic advantages of intramolecular HAT. Furthermore, the highly reactive nature of heteroatom-centered radicals could lead to poor reaction site- and chemoselectivity.
We have recently demonstrated the ability of N-functionalized amides to achieve site-selective, intermolecular radical-mediated C─H functionalizations27-30. Both N-halo and N-xanthylamides are useful precursors of amidyl radicals, which using either photochemical or radical initiation undergo HAT with a diverse array of organic substrates (Figure 1). These aliphatic C─H functionalizations facilitate the introduction of a range of molecular functionality in concise fashion. Our initial efforts have used reagents derived from bulky N-t-Bu amides, which combined with the electrophilicity of the amidyl radical favor functionalization at sterically accessible, electron-rich C─H sites.
The modularity of the amide scaffold is amenable to the development of alternative reagents in this class. Indeed, we have begun work in this area via the modification of the aromatic substituent to increase the sterically-dictated selectivity of our system30. There is little known concerning the requirements and limitations of the N-functionalized amide (or related) reagents in promoting efficient intermolecular C─H functionalization, however (Figure 2). Herein, we report a detailed analysis of amidyl radical reactivity for intermolecular HAT, focusing on aliphatic C─H chlorination. These experimental and computational studies outline several key components of the amide scaffold important in developing intermolecular HAT processes, and identify several possible side reactions that are problematic with unsuccessful reactions of nitrogen-centered radicals.
Figure 2.
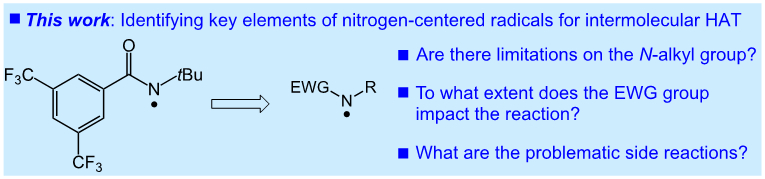
Elements of nitrogen-centered radicals addressed in these studies.
RESULTS AND DISCUSSION
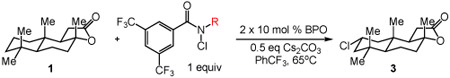
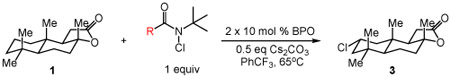
We commenced our studies by evaluating the C─H chlorination of (+)–sclareolide as a test substrate with a range of N-chloroamides containing varied N-alkyl substitution (Table 1). Our previously reported chlorination of this substrate using N-tBu amide 2a proceeded in good yield (entry 1, 75%) with excellent site selectivity and served as our baseline result. Surprisingly, only a slight variation in the chloroamide structure—using N-iPr amide 2b—led to a far inferior reaction providing only 8% of the chlorination product 3 (entry 2). Similar primary, branched, and non-branched N-alkyl chloroamides also displayed poor reactivity (entries 3-6). The N-trifluoroethyl amide 2g—which we previously reported was capable of functionalizing cyclohexane27—also provided very little product (entry 7). Attempts using an ester derivative containing a fully substituted α-carbon similar to reagent 2a provided 3 in good yield (entry 8). However, once again removing full alkyl substitution from the α-substituent completely shut down reactivity (entry 9). We note that N-aryl amides were not investigated owing to the low N─H bond strength, preventing an energetically favorable C─H abstraction.31
Table 1.
C─H chlorination of (+)–sclareolide varying chloroamide N–alkyl substitution.
 | ||
|---|---|---|
| entry | N-alkyl chloroamide | yield (%)a |
| 1 | R = tBu (2a) | 75 |
| 2 | iPr (2b) | 8 |
| 3 | Et (2c) | <2 |
| 4 | Me (2d) | 8 |
| 5 | nPr (2e) | <2b |
| 6 | iBu (2f) | <2b |
| 7 | 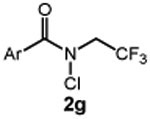 |
5 |
| 8 | 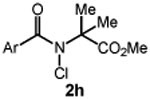 |
65 |
| 9 | 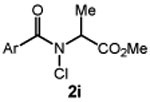 |
<2 |
Reactions were performed with [1]0 = 0.5 M.
Yields determined by 1H NMR spectroscopy of crude reaction mixture using an internal standard.
Minor chlorination of the N-alkyl group was observed by 1H NMR.
We next studied the effect of electronic modification of the aryl substituent of N-tBu chloroamide 2a on the C─H chlorination (Table 2). A range of aryl substituents were permitted, with the exception of p-OMe amide (2n) likely owing to the abstractable C─H bonds of the methoxy group. There is no clear electronic preference for the aryl substituent, as the electron poor aryl substituted 2k and 2l (66% and 67% yield, respectively) performed similarly to unsubstituted reagent 2m (68% yield, entry 5). Furthermore, the aryl group is not required, as a reagent containing a perfluoroalkyl groups (2p) also provided 3 in moderate yield (39% yield, entry 8). We note that PhCF3 is the solvent of choice, as non–aromatic solvents with abstractable C─H bonds are not tolerated (See Table S1). For example, the use of common organic solvents such as 1,2-dichloroethane revealed significant amounts of 1,1,2-trichloroethane, confirming C─H chlorination of the solvent.
Table 2.
C─H chlorination of (+)–sclareolide varying N-tBu chloroamide electronics.
 | ||
|---|---|---|
| entry | N-tBu chloroamide | yield (%)a |
| 1 | 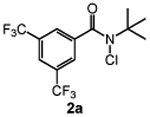 |
75 |
| 2 | 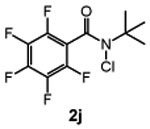 |
75 |
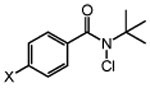 |
||
| 3 | X = NO2 (2k) | 66 |
| 4 | F (2l) | 67 |
| 5 | H (2m) | 68 |
| 6 | OMe (2n) | 21b |
| 7 | tBu (2o) | 58 |
| 8 | 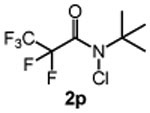 |
39 |
Reactions were performed with [1]0 = 0.5 M.
Isolated yields.
Chlorination on the methoxy substituent observed by 1H NMR.
Lastly, we investigated the efficiency of C─H chlorination using several other classes of N-chloro reagents (Figure 3). In addition to N-chloroamide 2a, N-chlorosulfonamide 2q provided product 3 in moderate yield (53%). Reactions involving the N-chlorophosphoramide 2r and N-chlorocarbamate 2s also delivered product, although both reactions proceeded with incomplete conversion. The use of common chlorinating agent N-chlorophthalimide provided no product. These results are consistent with the requirement of strong parent N─H bonds (i.e., >100 kcal/mol) for the C─H chlorination reagents. Our calculated N─H BDE for the parent amide of 2a is 110.7 kcal/mol, and those for reagents 2r and 2s are estimated to be within a ∼3 kcal/mol range of this value.32 According to our calculations, the N─H BDE of the parent sulfonamide for 2q is 7.2 kcal/mol weaker than that of 2a but still relatively strong (103.5 kcal/mol). The N─H BDE of phthalimide is only ∼89 kcal/mol making C─H abstraction significantly endergonic.33
Figure 3.
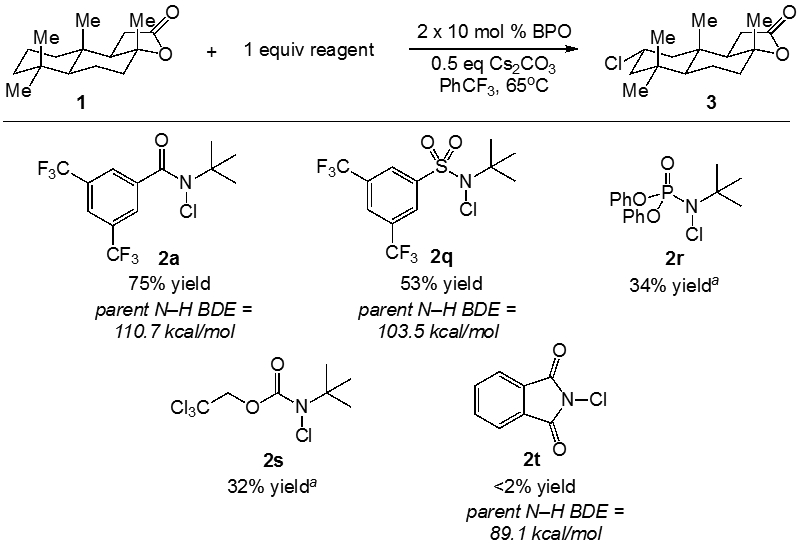
Intermolecular C─H chlorination using alternative electron-poor nitrogen–centered radicals. aIncomplete conversion of the reagent observed, see the Supporting Information for more details.
We next examined the structures of the N-tBu and N-iPr amidyl radicals computationally to glean any insights into the remarkable difference in reaction efficiency–85% yield versus 8% yield, respectively–involving these species (Figure 4). While the C–O and C–N bond lengths of the amidyl radicals are almost identical (<0.001 Å difference), the bulkier t-Bu group induces a more pronounced “twisting” effect of the N-tBu amidyl. The nitrogen substituent is markedly displaced out of the plane of the carbonyl group, with a –49° OC7NC10 dihedral in the case of the N-t-Bu amidyl radical. On the other hand, two different conformers (α and β; see Figure S4 for the complete potential energy surface scan) with an almost identical energy content. Nevertheless, both conformers of the N-iPr analogue show a less pronounced twisting (−32° and −49° OC7NC10 dihedral angle, for α and β, respectively). This disparity affects the distribution of spin density of the radicals, with a larger share of radical character localized on the nitrogen atom in the N-tBu amidyl radical, with more spin density located on oxygen in the N-iPr analogue.34
Figure 4.
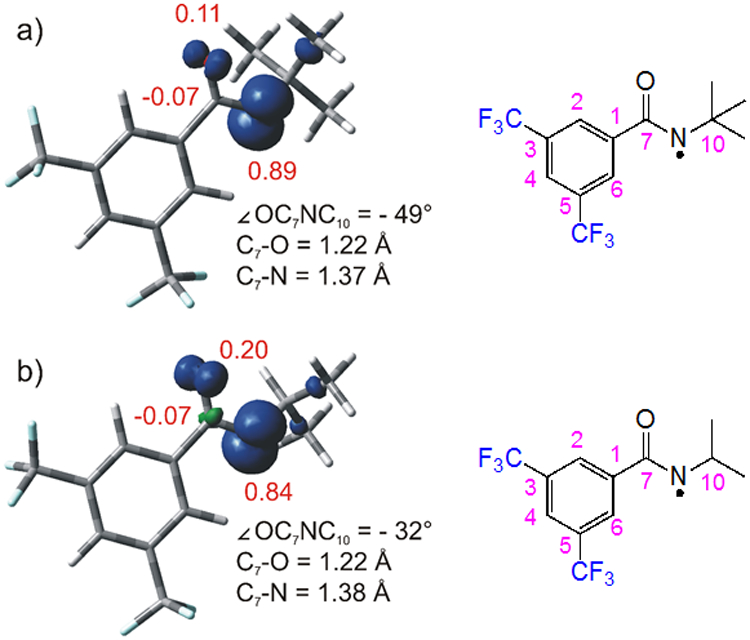
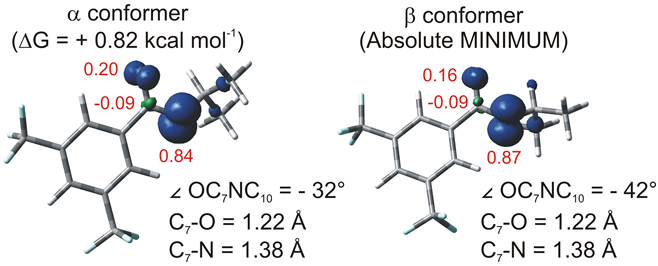
Spin density plots for: a) N-t-Bu and b) α and β conformers of N-i-Pr amidyl radicals, along with some relevant geometric parameters. Spin densities for O, C7 and N have been reported in red. These data have been obtained from calculations at the UwB97XD/def2svp level in the gas phase.
In order to study the electronics of the two amidyl radicals in further detail, we performed a dedicated NBO (natural bond order) analysis.35 This approach determines the contribution of the second-order perturbation stabilization terms (E(2)) and the orbitals involved in them, shedding light on specific stereochemical interactions. It has recently been demonstrated to provide reliable estimates in predicting the reactivity of species with diradical transition state character.36 As depicted in Figure 5, the N-i-Pr amidyl radical achieves more stabilization via both n→π* and π→n interactions with the C═O group than the N-t-Bu amidyl radical. This behavior can also be appreciated comparing the percentage of σ character of the orbital of the N radical.
Figure 5.
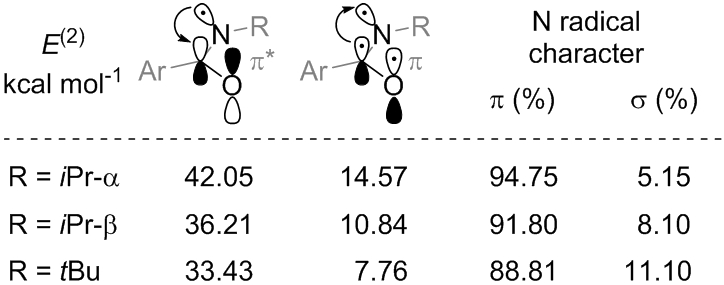
Stabilization energies and orbital analysis obtained from the interactions of the N-iPr (the analysis of both conformers has been reported) and N-tBu amidyl radicals with π and π* orbitals of the neighbouring C═O calculated with the NBO6 software at the ωB97X-D/def2-SVP level of theory.
Given the overall structural and electronic similarities of the N-tBu and N-iPr amidyl radicals, combined with the similar parent amide N─H bond strengths,31 we considered other possibilities to explain the much greater efficiency of the N-tBu reagents in C─H functionalization. It is clear from Table 1 that the presence of α-C─H bonds on the N-alkyl group leads to a dramatic decrease in reaction yield. A hypothesis that is consistent with this trend is undesired C─H abstraction on the N-alkyl group occurring in preference to reaction with the substrate (Scheme 1). This side product pathway would consume N-chloroamide while providing no C─H chlorination product. The α-C─H abstraction of amines is well-documented,37 while that of amides is much less known. The reported α-C─H BDE of N-iPr acetamide is ∼93 kcal/mol, making the HAT from the radicals studied herein exergonic.28
Scheme 1.
Proposed undesired α-C─H abstraction pathways of N-alkyl chloroamides.
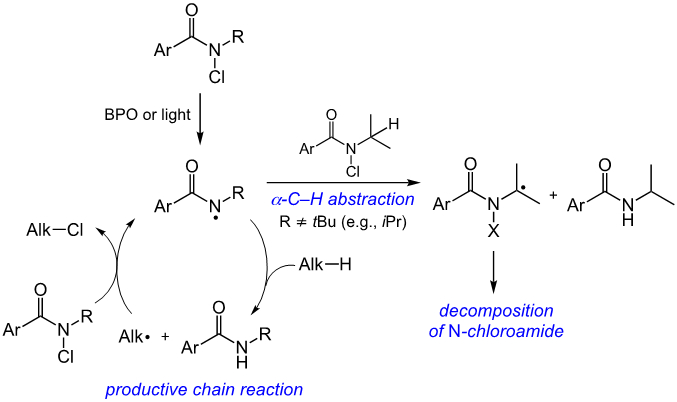
In order to observe potential products of N-alkyl group functionalization, we performed reactions in the absence of substrate (Figure 6). The reaction of N-chloroamide 2b with benzoyl peroxide yielded products with a strong high-resolution mass spectrometry signal at m/z = 258.03461 (exact mass calculated for C9H6F6N1O1 [M+H]+ = 258.034789) indicating the formation of primary amide 4, which could result from chlorination of the iPr group followed by cleavage. We also subjected N-methyl chloroamide 2d to standard photochemical reaction conditions, to remove the possibility of HAT involving the radical initiator. In this case, we observed the production of presumed α-chloro N-methyl amide 5, albeit in low yield (10%). While the chemical yields of products 4 and 5 are modest, they could still play a significant role by decreasing the effective length of the radical chain process involved in the C─H functionalization.
Figure 6.
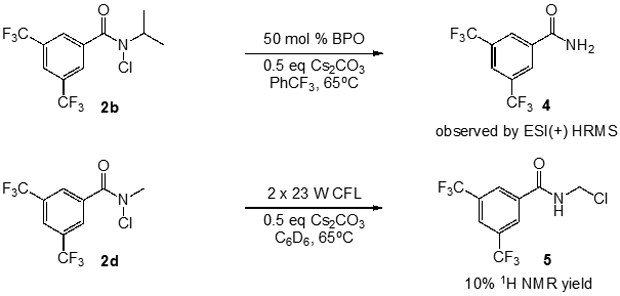
Evidence of α-C─H abstraction of the N-alkyl substituent.
We next studied the kinetics of the C─H chlorination of cyclohexane using N-tBu chloroamide 2a and N-iPr chloroamide 2b (Figure 7a). In these experiments, cyclohexane was chosen as substrate owing to the significantly higher yield with 2b. These studies revealed significant differences in reaction rates, with N-tBu reagent 2a reacting faster at each time point. For example, at 7 h the conversion of 2a is 93%, whereas that of 2b is only 23%. This is consistent with a much more efficient chain process using reagent 2a versus 2b. At longer reaction times and additional peroxide initiator, conversion of N-iPr chloroamide 2b is significant, while the yield of chlorocyclohexane is moderately lower than N-tBu reagent 2a (Figure 7b). We note that the large difference in reaction yields between (+)-sclareolide (8%) and cyclohexane (48%) using 2b is consistent with the well-documented entropic component of HAT processes.38 The C─H chlorination of (+)-sclareolide requires HAT at a single methylene site, in direct contrast to cyclohexane. This difference in the kinetic profile enables side reactions to become much more problematic in the functionalization of complex substrates, requiring judicious reagent selection.
Figure 7.
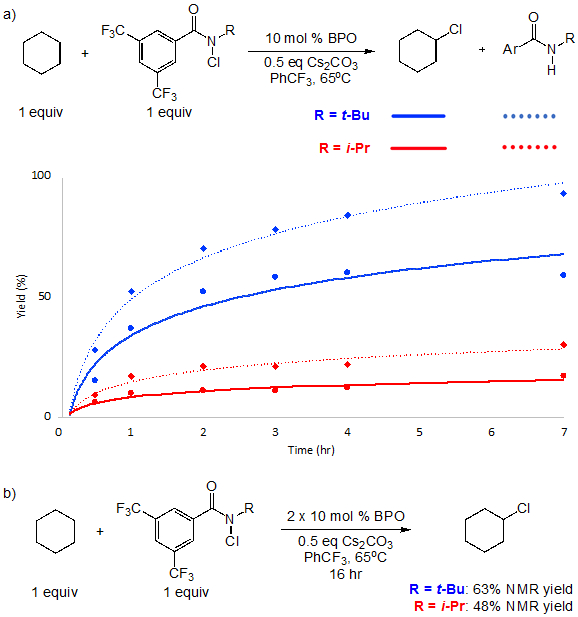
a) Conversion of the C─H chlorination of cyclohexane and N-chloroamide using reagents 2a and 2b at different time points. b) C─H chlorination of cyclohexane with N-chloroamide 2a and 2b at 16 hours with sequential additions of peroxide initiator
Finally, we investigated the effect of either the N-t-Bu or the N-i-Pr parent amide on the efficiency of the (+)-sclareolide chlorination using 2a (Figure 8). As the C─H chlorination is stoichiometric in N-chloroamide, these amides are present in significant amounts throughout the course of the reaction. When the successful reaction using 2a (75%, Table 1, entry 1) is performed in the presence of 0.5 equiv of the parent N-t-Bu amide, only a slight decrease in yield is observed (70%). On the other hand, the addition of 0.5 equiv of the N-i-Pr amide significantly lowers the reactions efficiency (16%). We also observed a 35% yield of N-chloroamide 2b in this reaction from chlorine atom transfer from 2a. Additionally, the rate of C─H functionalization decreased when 0.5 equivalents of the N-i-Pr amide were added to reaction of cyclohexane with 2b. These results are consistent with the N-i-Pr amide participating in the same non-productive pathways previously described, significantly lowering the yield of 3.
Figure 8.
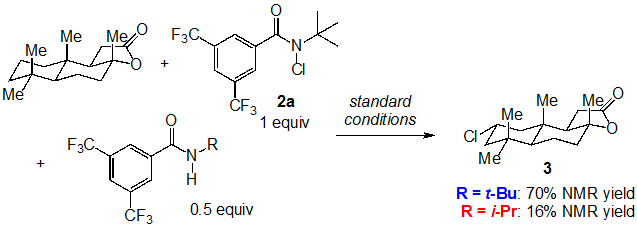
C─H chlorination of (+)-sclareolide in the presence of N-t-Bu and N-i-Pr amides.
The requirement for fully substituted α-positions on the N-alkyl sidechain of functionalized amides reduces the possibilities to diversify these reagents for C─H functionalization. In order to address this issue, we examined the use of a more hindered aromatic scaffold. We have recently reported the enhanced steric selectivity of C─H functionalizations using 2,6-bis-trifluoromethyl N-t-Bu amides.30 We hypothesized that the significantly increased steric profile of this derivative could mitigate undesired side reactions (i.e., α-C─H abstraction). The reaction of 2,6-bis-trifluoromethyl N-i-Pr chloroamide 2u with (+)-sclareolide did provide product 3 in 36% yield (Figure 9), with up to a 75% yield using excess substrate (3 equiv). For comparison, the reaction with N-i-Pr reagent 2b under the same conditions produced only 8% of product. These results suggest that tuning the steric profile of the amidyl radical can permit the use of N-substituents which otherwise would have little chance of success.
Figure 9.
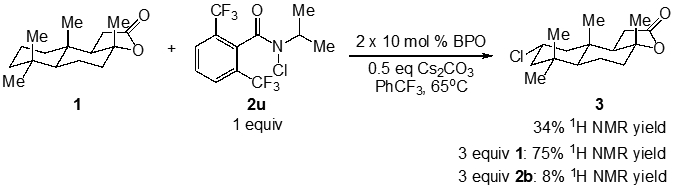
C─H chlorination of (+)-sclareolide with an N-i-Pr chloroamide containing sterically demanding aryl substitution.
In conclusion, these studies provide a set of guidelines for the design of reagents for intermolecular C─H functionalization using N-functionalized amides and derivatives. High effective N─H BDEs (>100 kcal/mol) are necessary to promote HAT of unactivated C─H bonds, and amides are desirable in this regard. Most importantly, reagents containing α-C─H bonds in the N-substituent can lead to sharply decreased efficiencies of intermolecular C─H functionalization. Increasing the steric profile of the aryl substituents can overcome this issue and permit further reagent diversification. We expect this work will support further reagent development in intermolecular radical-mediated aliphatic C─H functionalization in the future.
EXPERIMENTAL SECTION
General Information.
Unless otherwise noted, all reagents were obtained from commercial sources and used without further purification unless otherwise noted. Proton and carbon magnetic resonance spectra (1H NMR and 13C NMR) were recorded on a Bruker model DRX 400, or a Bruker AVANCE III 600 CryoProbe (1H NMR at 400 or 600 MHz and 13C NMR at 100 or 151 MHz) spectrometer with solvent resonance as the internal standard (1H NMR: CDCl3 at 7.26 ppm; 13C NMR: CDCl3 at 77.16 ppm). 1H NMR data are reported as follows: chemical shift, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, dd = doublet of doublets, ddd = doublet of doublet of doublets, td = triplet of doublets, tdd = triplet of doublet of doublets, qd = quartet of doublets, m = multiplet, br. s. = broad singlet, app = apparent), coupling constants (Hz), and integration. Mass spectra were obtained using a ThermoScientific Q Exactive HF-X Hybrid Quadrapole-Orbitrap with electrospray introduction and external calibration. Thin layer chromatography (TLC) was performed on SiliaPlate 250μm thick silica gel plates provided by Silicycle. Visualization was accomplished with short wave UV light (254 nm), iodine, aqueous basic potassium permanganate solution, or aqueous acidic ceric ammonium molybdate solution followed by heating. Flash chromatography was performed using SiliaFlash P60 silica gel (40-63 μm) purchased from Silicycle. Tetrahydrofuran, diethyl ether, and dichloromethane were dried by passage through a column of neutral alumina under nitrogen prior to use.
General Procedure for C─H chlorination of (+)–sclareolide using N-chloro reagents 2a – 2u:
A flame-dried 1 dram vial was charged with a stir bar and brought into the glove where substrate (0.15 mmol, 1 equiv), BPO (0.015 mmol, 10 mol %), cesium carbonate (0.075 mmol, 0.5 equiv), PhCF3 (0.3mL, 0.5 M), and N-chloro reagent (0.15 mmol, 1.0 equiv) were added. The reaction was capped with a PTFE lined screw cap, sealed with Teflon tape, taken out of the glovebox, and stirred at 65 °C on a heating mantle with a pie block for 7 hours. After cooling to room temperature, the reaction was brought back into the glovebox where BPO (0.015 mmol, 10 mol %) was added. The reaction was capped and resealed with Teflon tape, taken out of the glovebox, heated to 65 °C on the pie block, and stirred for an additional 17 hours. Upon completion, the reaction mixture was then filtered through a silica plug, concentrated down in vacuo, and analyzed by NMR using 10.0 μL hexamethyldisiloxane as an internal standard.
General Procedure A for amide synthesis:
Prepared similarly to previous reports from our lab.24 To a solution of benzoic acid (10 mmol, 1 equiv) in CH2Cl2 and a few drops of DMF at 0 °C was added oxalyl chloride (20 mmol, 2 equiv) dropwise, and the resulting solution was allowed to warm to rt overnight. The mixture was concentrated in vacuo, resuspended in THF, and chilled to 0 °C. Amine (20 mmol, 2 equiv) was added, and the mixture was warmed to room temperature and stirred overnight. The mixture was concentrated in vacuo and the residue suspended in Et2O and washed with 3M NaOH, 1M HCl, brine, dried with MgSO4 and concentrated to afford the corresponding amide which was used without purification.
N-(tert-butyl)-3,5-bis(trifluoromethyl)benzamide.
Obtained as a white powder according to General Procedure A. Spectral data matched literature values.24
N-isopropyl-3,5-bis(trifluoromethyl)benzamide.
Obtained as a white powder according to General Procedure A. Spectral data matched literature values39
N-ethyl-3,5-bis(trifluoromethyl)benzamide.
Obtained as a white powder according to General Procedure A. 1H NMR (600 MHz, Chloroform-d): δ 8.24 (d, J = 1.5 Hz, 2H), 8.01 (s, 1H), 6.45 (s, 1H), 3.61 – 3.51 (m, 2H), 1.32 (td, J = 7.3, 1.3 Hz, 3H). 13C{1H} NMR (151 MHz, Chloroform-d): δ 164.5, 132.2 (q, J = 33.3, 32.9 Hz), 127.3, 125.4 – 123.8 (m), 122.0, 35.4, 14.8. HRMS (ESI+): Exact mass calcd for C11H10N1O1F6 [M+H]+, 286.0661. Found 286.0658.
N-methyl-3,5-bis(trifluoromethyl)benzamide.
Obtained as a white powder according to General Procedure A. 1H NMR (600 MHz, Chloroform-d): δ 8.07 (s, 2H), 8.01 (s, 1H), 3.51 (s, 3H). 13C{1H} NMR (151 MHz, Chloroform-d): δ 169.5, 135.5, 131.9 (q, J = 34.1 Hz), 128.4 (d, J = 4.1 Hz), 124.6, 122.8 (d, J = 272.9 Hz), 42.5. HRMS (ESI+): Exact mass calcd for C10H8N1O1F6 [M+H]+, 272.0504. Found 272.0507.
N-propyl-3,5-bis(trifluoromethyl)benzamide
Obtained as a white powder according to General Procedure A. 1H NMR (600 MHz, Chloroform-d): δ 8.23 (s, 2H), 8.02 (s, 1H), 6.37 (s, 1H), 3.49 (dt, J = 7.4, 5.9 Hz, 2H), 1.71 (m, J = 7.3 Hz, 2H), 1.03 (t, J = 7.4 Hz, 3H). 13C{1H} NMR (151 MHz, Chloroform-d): δ 164.7, 136.8, 132.1 (q, J = 34.0 Hz), 127.3 (q, J = 3.7 Hz), 125.4 – 124.3 (m), 126.3 – 115.5 (m), 42.2, 22.8, 11.4. HRMS (ESI+): Exact mass calcd for C12H12N1O1F6 [M+H]+, 300.0817. Found 300.0819.
N-isobutyl-3,5-bis(trifluoromethyl)benzamide
Obtained as a white powder according to General Procedure A. 1H NMR (500 MHz, Chloroform-d): δ 8.01 (s, 2H), 8.00 – 7.99 (s, 1H), 3.63 (d, J = 7.3 Hz, 2H), 2.30 (dp, J = 13.7, 6.9 Hz, 1H), 1.00 (d, J = 6.7 Hz, 5H). 13C{1H} NMR (101 MHz, Chloroform-d): δ 164.7, 136.9, 132.4 (t, J = 33.9 Hz), 127.2, 125.4 – 123.0 (m), 121.6, 47.8, 28.6, 20.2. HRMS (ESI+): Exact mass calcd for C13H14N1O1F6 [M+H]+, 314.0974. Found 314.0976.
N-(2,2,2-trifluoroethyl)-3,5-bis(trifluoromethyl)benzamide.
Obtained as a white powder according to General Procedure A. Spectral data matched the reported literature values27
Methyl 2-(3,5-bis(trifluoromethyl)benzamido)-2-methylpropanoate.
Obtained as a white powder according to General Procedure A. 1H NMR (500 MHz, Chloroform-d): δ 8.25 (s, 1H), 8.04 (s, 1H), 6.95 (s, 1H), 3.84 (s, 3H), 1.75 (s, 6H). HRMS (ESI+): Exact mass calcd for C14H14F6N1O3, 358.0872. Found 358.0865.
Methyl (3,5-bis(trifluoromethyl)benzoyl)alaninate.
Obtained as a white powder according to General Procedure A. Spectral data matched the reported literature values40
N-(tert-butyl)-2,3,4,5,6-pentafluorobenzamide.
Obtained as a white powder according to General Procedure A. 1H NMR (600 MHz, Chloroform-d): δ 5.70 (s, 1H), 1.49 (s, 9H). 13C{1H} NMR (151 MHz, Chloroform-d): δ 156.3, 145.9 – 140.6 (m), 139.3 – 134.4 (m), 112.7 (t, J = 20.4 Hz), 53.2, 28.9. 19F NMR (564 MHz, Chloroform-d): δ −139.1 – −143.2 (m), −151.8 (d, J = 20.4 Hz), −158.0 – −161.5 (m). HRMS (ESI+): Exact mass calcd for C11H11N1O1F5 [M+H]+, 268.0755. Found 268.0755.
N-(tert-butyl)-4-nitrobenzamide.
Obtained as a white powder according to General Procedure A. Spectral data matched the reported literature values41
N-(tert-butyl)-4-fluorobenzamide.
Obtained as a white powder according to General Procedure A. Spectral data matched the reported literature values41
N-(tert-butyl)-benzamide.
Obtained as a white powder according to General Procedure A. Spectral data matched the reported literature values42
N-(tert-butyl)-4-methoxybenzamide.
Obtained as a white powder according to General Procedure A. Spectral data matched the reported literature values41
N,4-di-tert-butylbenzamide.
Obtained as a white powder according to General Procedure A. Spectral data matched the reported literature values41
N-isopropyl-2,6-bis(trifluoromethyl)benzamide.
Obtained as colorless crystals according to General Procedure A. 1H NMR (600 MHz, Chloroform-d): δ 7.93 (d, J = 8.0 Hz, 2H), 7.68 (t, J = 8.0 Hz, 1H), 5.58 (s, 1H), 4.37 (m, J = 6.6 Hz, 1H), 1.28 (d, J = 6.6, 1.7 Hz, 6H). 13C{1H} NMR (151 MHz, Chloroform-d): δ 163.1, 134.5 (d, J = 1.8 Hz), 129.9 (q, J = 4.9 Hz), 129.6, 129.2 (q, J = 32.0 Hz), 123.1 (q, J = 274.6 Hz), 42.4, 22.2. HRMS (ESI+): Exact mass calcd for C12H12N1O1F6 [M+H]+, 300.0817. Found 300.0815.
Procedure for synthesis of diphenyl tert-butylphosphoramidate:
Prepared similarly to previous reports in the literature. A solution of diphenyl chlorophosphate (10 mmol, 1 equiv) in CH2Cl2 (25 mL) was cooled in an ice bath. t-Butylamine (25 mmol, 2.5 equiv) was added dropwise, and the mixture was warmed to rt and stirred overnight. The mixture was washed with sat. NaHCO3, 1M HCl, brine, dried with MgSO4 and concentrated to afford the corresponding phosphoramidate as a white solid (3.0 g, 98% yield), which was used without purification. 1H NMR (600 MHz, Chloroform-d): δ 7.35 (t, J = 7.4 Hz, 4H), 7.30 – 7.24 (m, 6H), 7.17 (t, J = 7.3 Hz, 4H), 3.01 (s, 1H), 1.38 (d, J = 1.9 Hz, 9H). 13C{1H} NMR (151 MHz, Chloroform-d): δ 151.1 (d, J = 7.1 Hz), 129.6, 124.7, 120.2 (d, J = 5.1 Hz), 51.8, 31.3 (d, J = 4.9 Hz). HRMS (ESI+): Exact mass calcd for C16H21N1O3P1 [M+H]+, 306.1254. Found 306.1247.
Procedure for synthesis of N-(tert-butyl)-3,5-bis(trifluoromethyl)benzenesulfonamide:
Prepared similarly to previous reports in the literature. A solution of 3,5-bis(trifluoromethyl)benzenesulfonyl chloride (15 mmol, 1 equiv) in CH2Cl2 was cooled in an ice bath. t-Butylamine (38 mmol, 2.5 equiv) was added dropwise, and the mixture was warmed to rt and stirred overnight. The mixture was washed with sat. NaHCO3, 1M HCl, brine, dried with MgSO4 and concentrated to afford the corresponding sulfonamide as a white solid (5.0 g, 95% yield) which was used without purification. 1H NMR (600 MHz, Chloroform-d): δ 8.36 (s, 2H), 8.07 (s, 1H), 4.81 (s, 1H), 1.30 (s, 9H). 13C{1H} NMR (151 MHz, Chloroform-d): δ 146.1, 132.8 (q, J = 34.3 Hz), 127.2 (d, J = 3.9 Hz), 122.5 (q, J = 273.3 Hz), 55.8, 30.2. HRMS (ESI−): Exact mass calcd for C12H12F6N1O2S [M–H]−, 348.0487. Found 348.0487.
Procedure for synthesis of 2,2,2-trichloroethyl tert-butylcarbamate:
Prepared similarly to previous reports in the literature. A solution of 2,2,2-trichloroethyl chloroformate (10 mmol, 1 equiv) in CH2Cl2 was cooled in an ice bath. t-Butylamine (25 mmol, 2.5 equiv) was added dropwise, and the mixture was warmed to rt and stirred overnight. The mixture was washed with sat. NaHCO3, 1M HCl, brine, dried with MgSO4 and concentrated to afford the corresponding carbamate as a colorless amorphous solid (2.2 g, 89% yield), which was used without purification. 1H NMR (600 MHz, Chloroform-d): δ 4.94 (s, 1H), 4.69 (s, 2H), 1.38 (s, 9H). 13C{1H} NMR (151 MHz, Chloroform-d): δ 152.6, 95.9, 73.9, 50.9, 28.7. HRMS (ESI+): Exact mass calcd for C7H12Cl3N1O1 [M+H]+, 248.0006. Found 248.0007.
Procedure for synthesis of N-(tert-butyl)-2,2,3,3,3-pentafluoropropanamide:
A solution of 2,2,3,3,3-pentafluoropropionic anhydride (11 mmol, 1.1 equiv) in CH2Cl2 (75 mL) was cooled in an ice bath. t-Butylamine (10 mmol, 1 equiv) and triethylamine (12 mmol, 1.2 equiv) were carefully added dropwise [caution: reaction is very exothermic]. The mixture was then warmed to rt and stirred overnight. The mixture was washed with water, sat. NH4Cl, brine, dried with MgSO4 and concentrated to afford the corresponding amide as a yellow liquid (1.9 g, 89% yield), which was used without purification. 1H NMR (600 MHz, Chloroform-d): δ 6.18 (s, 1H), 1.44 (s, 9H). 13C{1H} NMR (151 MHz, Chloroform-d): δ 156.6 (t, J = 24.6 Hz), 121.4 – 115.2 (m), 105.6 (dd, J = 266.7, 38.5 Hz), 53.0, 28.2. 19F NMR (564 MHz, Chloroform-d) δ −82.9, −122.7. HRMS (ESI−): Exact mass calcd for C7H9F5N1O1 [M], 218.0610. Found 218.0603.
General Procedure B for synthesis of N-chloro reagents.
Synthesized according to the literature procedure28. To a foil wrapped flask under N2, amide (5 mmol, 1 equiv) was added and dissolved in a mixture of ethyl acetate (10 mL) and tert-butanol (5 mmol, 1 equiv). 10 equivalents of a sodium hypochlorite solution was then added (~1.5M in H2O Sigma-Aldrich), followed by acetic acid (50 mmol, 10 equiv). The reaction was stirred at room temperature for 3-4 hours. When the reaction was complete as judged by TLC analysis (3 hours usually sufficient) the reaction was diluted with Et2O, washed three times with saturated sodium bicarbonate solution, once with water, and once with brine. The organic layer was dried with magnesium sulfate and concentrated under reduced pressure. The crude material usually contains traces of chlorinated ethyl acetate. The crude product was purified via column chromatography (EtOAc/Hexane) to give the desired product. General storage: N-Chloro reagents were stored in foil-wrapped vials in the freezer when not in use. The reagents can be weighed out on the bench top without risk of decomposition.
General Procedure C for synthesis of N-chloro reagents.
To a foil wrapped flask under N2, amide (5 mmol, 1 equiv) was added and dissolved in a mixture of ethyl acetate (10 mL) and tert-butanol (5 mmol, 1 equiv). 1 equivalent of a sodium hypochlorite solution was then added (~1.5M in H2O Sigma-Aldrich), followed by acetic acid (10 mmol, 2 equiv). The reaction was monitored by TLC until all starting material was consumed. When the reaction was complete the solution was diluted with Et2O, washed three times with saturated sodium bicarbonate solution, once with water, and once with brine. The organic layer was dried with magnesium sulfate and concentrated under reduced pressure. The crude material usually contains traces of chlorinated ethyl acetate. The crude product was purified via column chromatography (EtOAc/Hexane) to give the desired product.
N-(tert-butyl)-N-chloro-3,5-bis(trifluoromethyl)benzamide (2a)
was obtained as a pale yellow oil (0.75 g, 86% yield) according to General Procedure B (2.5 mmol). Purified using 1:20 EtOAc/hexanes. Spectral data are in accordance with the literature28
N-chloro-N-isopropyl-3,5-bis(trifluoromethyl)benzamide (2b)
was obtained as a pale yellow oil (1.5 g, 90% yield) according to General Procedure B (5.0 mmol). Purified using 1:20 EtOAc/hexanes. 1H NMR (600 MHz, Chloroform-d): δ 8.07 – 7.96 (m, 3H), 4.77 (m, 1H), 1.36 (d, J = 8.7, 6H). 13C{1H} NMR (151 MHz, Chloroform-d): δ 168.8, 136.8, 132.8 – 131.0 (m), 128.0, 124.3, 122.8 (q, J = 273.1 Hz), 52.7, 19.7. HRMS (ES−): Exact mass calcd for C12H11Cl1F6N1O1 [M+H]+, 334.0439. Found 334.0430.
N-chloro-N-ethyl-3,5-bis(trifluoromethyl)benzamide (2c)
was obtained as a pale yellow oil (0.60 g, 75% yield) according to General Procedure B (2.5 mmol). Purified using 1:10 EtOAc/hexanes. 1H NMR (600 MHz, Chloroform-d): δ 8.05 (s, 2H), 8.01 (s, 1H), 3.86 (q, J = 1.4 Hz, 2H), 1.39 (t J = 1.4 Hz, 3H). 13C{1H} NMR (151 MHz, Chloroform-d): δ 168.8, 136.0, 132.0 (q, J = 34.2 Hz), 128.2, 125.51, 124.1 (d, J = 118.1 Hz), 121.9, 49.5, 12.5. HRMS (ES−): Exact mass calcd for C11H9Cl1F6N1O1 [M+H]+, 320.0282. Found 320.0270.
N-chloro-N-methyl-3,5-bis(trifluoromethyl)benzamide (2d)
was obtained as an off-white solid (0.78 g, 51% yield) according to General Procedure B (5.0 mmol). Purified using 1:10 EtOAc/hexanes. 1H NMR (600 MHz, Chloroform-d): δ 8.07 (s, 2H), 8.01 (s, 1H), 3.51 (s, 3H). 13C{1H} NMR (151 MHz, Chloroform-d): δ 169.5, 135.5, 131.9 (q, J = 34.1 Hz), 128.4 (d, J = 4.1 Hz), 124.6, 122.8 (q, J = 272.9), 42.5. HRMS (ES−): Exact mass calcd for C10H7Cl1F6N1O1 [M+H]+, 306.0126. Found 306.0116.
N-chloro-N-propyl-3,5-bis(trifluoromethyl)benzamide (2e)
was obtained as a pale yellow oil (1.68 g, 98% yield) according to General Procedure B (5.0 mmol). Purified using 1:20 EtOAc/hexanes. 1H NMR (500 MHz, Chloroform-d): δ 8.02 (s, 2H), 8.00 (s, 1H), 3.81 – 3.72 (m, 2H), 1.85 (h, J = 7.4 Hz, 2H), 0.99 (t, J = 7.4 Hz, 3H). 13C{1H} NMR (151 MHz, Chloroform-d): δ 169.0, 136.3, 131.9 (q, J = 33.9 Hz), 128.1 (t, J = 3.8 Hz), 126.1 – 117.0 (m), 60.6, 26.7, 19.4. HRMS (ES−): Exact mass calcd for C12H11Cl1F6N1O1 [M+H]+, 334.0439. Found 334.0429.
N-chloro-N-isobutyl-3,5-bis(trifluoromethyl)benzamide (2f)
was obtained as a pale yellow oil (0.70 g, 40% yield) according to General Procedure B (5.0 mmol). Purified using 1:20 EtOAc/hexanes. 1H NMR (500 MHz, Chloroform-d): δ 8.01 (s, 2H), 8.00 – 7.99 (s, 1H), 3.63 (d, J = 7.3 Hz, 2H), 2.30 (m, 1H), 1.00 (d, J = 6.7 Hz, 6H). 13C{1H} NMR (151 MHz, Chloroform-d): δ 169.0, 136.3, 131.9 (q, J = 33.9 Hz), 128.1 (t, J = 3.8 Hz), 126.1 – 117.0 (m), 60.6, 26.7, 19.4. HRMS (ES−): Exact mass calcd for C13H13Cl1N1O1F6 [M+H]+, 348.0584. Found 348.0588.
N-chloro-N-(2,2,2-trifluoroethyl)-3,5-bis(trifluoromethyl)benzamide (2g)
was obtained as an off-white solid (1.7 g, 91% yield) according to General Procedure B (5.0 mmol). Purified using 1:20 EtOAc/hexanes. 1H NMR (600 MHz, Chloroform-d) δ 8.11 (s, 2H), 8.09 (s, 1H), 4.49 (q, J = 8.1 Hz, 2H). 13C NMR{1H} (151 MHz, Chloroform-d) δ 169.3, 133.9, 132.1 (q, J = 34.3 Hz), 128.5 (q, J = 3.8 Hz), 126.1 – 118.9 (m), 52.8 (q, J = 35.6 Hz). 19F NMR (564 MHz, Chloroform-d): δ −63.1, −69.7. HRMS (ES−): Exact mass calcd for C11H6Cl1F9N1O1 [M+H]+, 373.9999. Found 373.9989.
Methyl 2-(N-chloro-3,5-bis(trifluoromethyl)benzamido)-2-methylpropanoate (2h)
was obtained as an off-white solid (0.31 g, 63% yield) according to General Procedure B (1.25 mmol). Purified using 1:20 EtOAc/hexanes. 1H NMR (600 MHz, Chloroform-d): δ 8.13 (s, 2H), 8.05 – 7.98 (s, 1H), 3.79 (s, 3H), 1.69 (s, 6H). 13C{1H} NMR (151 MHz, Chloroform-d): δ 172.0, 170.9, 135.9, 131.8 (d, J = 34.2 Hz), 127.1 – 119.2 (m), 66.5, 53.0, 23.4. Exact mass calcd for C14H13Cl1F6N1O3 [M+H]+, 392.0493. Found 392.0482.
Methyl N-(3,5-bis(trifluoromethyl)benzoyl)-N-chloroalaninate (2i)
was obtained as an off-white solid (0.95 g, 50% yield) according to General Procedure B (5.0 mmol). Purified using 1:20 EtOAc/hexanes. 1H NMR (500 MHz, Chloroform-d): δ 8.09 (d, J = 1.7 Hz, 2H), 8.03 (s, 1H), 5.27 (br s, 1H), 3.85 (s, 3H), 1.68 (d, J = 7.0 Hz, 3H). 13C{1H} NMR (151 MHz, Chloroform-d): δ 169.9, 169.5, 135.5, 132.0 (q, J = 34.1 Hz), 128.3 (q, J = 3.5 Hz), 125.3 – 124.6 (m), 122.8 (d, J = 273.0 Hz), 57.9, 53.0, 14.7. HRMS (ES−): Exact mass calcd for C13H11Cl1F6N1O3 [M+H]+, 378.0337. Found 378.0326.
N-(tert-butyl)-N-chloro-2,3,4,5,6-pentafluorobenzamide (2j)
was obtained as a white solid (1.0 g, 66% yield) according to General Procedure B (5.0 mmol). Purified using 1:20 EtOAc/hexanes. 1H NMR (600 MHz, Chloroform-d): δ 1.64 (s, 9H). 13C{1H} NMR (151 MHz, Chloroform-d): δ 156.3, 145.9 – 140.6 (m), 139.3 – 134.4 (m), 112.7 (t, J = 20.4 Hz), 53.2, 28.9. 19F NMR (564 MHz, Chloroform-d) δ −141.7 (d, J = 21.8 Hz), −152.5 (t, J = 20.9 Hz), −158.8 – −161.8 (m). HRMS (ES−): Exact mass calcd for C11H10Cl1F5N1O1 [M+H]+, 302.0376. Found 302.0365.
N-(tert-butyl)-N-chloro-4-nitrobenzamide (2k)
was obtained as a white solid (0.31 g, 61% yield) according to General Procedure B. Purified using 1:20 EtOAc/hexanes (2.0 mmol). 1H NMR (600 MHz, Chloroform-d): δ 8.28 (d, J = 8.8 Hz, 2H), 7.78 (d, J = 8.8 Hz, 2H), 1.62 (s, 9H). 13C{1H} NMR (151 MHz, Chloroform-d): δ 129.1, 123.4, 64.6, 28.0. HRMS (ES+): Exact mass calcd for C11H14Cl1N2O3 [M+H]+, 257.0698. Found 257.0694.
N-(tert-butyl)-N-chloro-4-fluorobenzamide (2l)
was obtained as a white solid (0.50 g, 48% yield) without purification according to General Procedure B (5.0 mmol). 1H NMR (600 MHz, Chloroform-d): δ 7.73 – 7.65 (m, 2H), 7.13 – 7.06 (m, 2H), 1.58 (s, 9H). 13C{1H} NMR (151 MHz, Chloroform-d): δ 174.6, 165.1, 147.8 (d, J = 4721.3 Hz), 131.0 (d, J = 8.8 Hz), 115.0 (d, J = 21.8 Hz), 78.7 – 74.5 (m), 63.8, 27.9.19F NMR (564 MHz, Chloroform-d): δ −108.4 (d, J = 10.0 Hz). HRMS (ES+): Exact mass calcd for C11H14F1Cl1N1O1 [M+H]+, 230.0742. Found 230.0740.
N-(tert-butyl)-N-chlorobenzamide (2m)
was obtained as a white solid (0.55 g, 52% yield) according to General Procedure C. Purified using 1:10 EtOAc/hexanes. 1H NMR (600 MHz, Chloroform-d): δ 7.68 – 7.64 (m, 2H), 7.50 – 7.46 (m, 1H), 7.42 (dd, J = 8.2, 6.7 Hz, 2H), 1.60 (s, 9H). 13C{1H} NMR (151 MHz, Chloroform-d): δ 175.6, 136.3, 130.9, 128.42, 128.0, 78.3 – 75.4 (m), 63.7, 28.0. HRMS (ES+): Exact mass calcd for C11H15Cl1N1O1 [M+H]+, 212.0837. Found 212.0834.
N-(tert-butyl)-N-chloro-4-methoxybenzamide (2n)
was obtained as a white solid (0.40 g, 34% yield) according to General Procedure C. Purified using 1:10 EtOAc/hexanes. 1H NMR (500 MHz, Chloroform-d): δ 7.69 (d, J = 8.8 Hz, 2H), 6.91 (d, J = 8.8 Hz, 2H), 3.87 (s, 3H), 1.57 (s, 9H). HRMS (ES+): Exact mass calcd for C12H17Cl1N1O2 [M+H]+, 244.0904. Found 244.0921.
N,4-di-tert-butyl-N-chlorobenzamide (2o)
was obtained as a white solid (0.39 g, 24% yield) according to General Procedure C. Purified using 1:10 EtOAc/hexanes. 1H NMR (600 MHz, Chloroform-d): δ 7.62 (d, J = 8.5 Hz, 2H), 7.42 (d, J = 8.6 Hz, 2H), 1.58 (s, 9H), 1.35 (s, 9H). 13C{1H} NMR (151 MHz, Chloroform-d): δ 175.7, 154.5, 133.2, 128.5, 124.9, 63.5, 34.9, 31.2, 28.0. HRMS (ES+): Exact mass calcd for C15H23Cl1N1O1 [M+H]+, 268.1474. Found 268.1459.
N-(tert-butyl)-N-chloro-2,2,3,3,3-pentafluoropropanamide (2p)
obtained as a yellow oil (70 mg, 15%) according to General Procedure B (2.5 mmol). Purified using 1:10 EtOAc/hexanes. 1H NMR (600 MHz, Chloroform-d): δ 1.56 (s, 9H). 13C{1H} NMR (151 MHz, Chloroform-d): δ 159.9, 118.1 (qt, J = 286.6, 34.1 Hz), 107.7 (tq, J = 274.4, 36.4 Hz), 67.2, 27.7. 19F NMR (564 MHz, Chloroform-d): δ −81.5, −112.1 – −118.0 (m). HRMS (ES−): Exact mass calcd for C7H10Cl1F5N1O1 [M+H]+, 254.0365. Found 254.0370.
N-(tert-butyl)-N-chloro-3,5-bis(trifluoromethyl)benzenesulfonamide (2q)
was obtained as a colorless solid (0.82 g, 85% yield) without purification according to General Procedure B (2.5 mmol). 1H NMR (600 MHz, Chloroform-d) δ 8.46 (s, 2H), 8.14 (s, 1H), 1.60 (s, 9H). 13C{1H} NMR (151 MHz, Chloroform-d): δ 141.1, 132.8 (q, J = 34.8 Hz), 129.1 (q, J = 3.8 Hz), 127.1 (p, J = 3.6 Hz), 122.4 (q, J = 273.5 Hz), 69.5, 29.3. HRMS (ES+): Exact mass calcd for C12H13Cl1N1O2S1F6 [M+H]+, 384.0247. Found 384.0254.
Diphenyl tert-butylchlorophosphoramidate (2r)
was obtained as a pale yellow oil (1.7 g, 98% yield) without purification according to General Procedure C (5.0 mmol). 1H NMR (600 MHz, Chloroform-d): δ 7.42 – 7.32 (m, 8H), 7.22 (t, J = 8.2 Hz, 2H), 1.45 (s, J = 1.0 Hz, 9H). 13C{1H} NMR (151 MHz, Chloroform-d): δ 150.6, 129.7, 125.3, 120.4, 63.4, 29.3. 31P NMR (243 MHz, Chloroform-d): δ −4.97. HRMS (ES+): Exact mass calcd for C16H20Cl1P1N1O3 [M+H]+, 340.0864. Found 340.0862.
2,2,2-trichloroethyl tert-butylchlorocarbamate (2s)
was obtained as a colorless oil (0.46 g, 65% yield) according to General Procedure B (2.5 mmol). Purified using 1:20 EtOAc/hexanes. 1H NMR (600 MHz, Chloroform-d): δ 4.79 (s, 2H), 1.55 (s, 9H). 13C{1H} NMR (151 MHz, Chloroform-d): δ 154.5, 94.9, 76.0, 64.5, 28.7. HRMS (ES−): Exact mass calcd for C7H12Cl4N1O2 [M+H]+, 281.9628. Found 281.9611.
N-chloro-N-isopropyl-2,6-bis(trifluoromethyl)benzamide (2u)
was obtained as a white solid (0.30 g, 45% yield) according to General Procedure B (2.5 mmol). Purified using 1:10 EtOAc/hexanes. 1H NMR (600 MHz, Chloroform-d): δ 7.94 (d, J = 8.0 Hz, 2H), 7.74 – 7.67 (m, 1H), 5.21 (p, J = 6.5 Hz, 1H), 1.30 (d, J = 6.5 Hz, 6H). 13C{1H} NMR (151 MHz, Chloroform-d): δ 166.2, 132.8 – 132.1 (m), 130.5 – 129.5 (m), 128.3 (q, J = 32.5 Hz), 123.0 (q, J = 274.7 Hz), 49.9, 19.0. HRMS (ES−): Exact mass calcd for C12H11Cl1F6N1O1 [M+H]+, 334.0439. Found 334.0426.
Supplementary Material
ACKNOWLEDGMENT
This work was supported by Award R01 GM 120163 from the National Institute of General Medical Sciences. M.M.T. thanks the NSF for a Graduate Research Fellowship. We would also like to thank Christina Na for performing N–H BDE calculations. Amide radical and structure calculations were carried out at the CINECA Supercomputer Center (Italy), with computer time granted by ISCRA projects (code: HP10C30YAJ).
Footnotes
Supporting Information
Computational data and spectral data for all new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
REFERENCES
- 1.White MC Adding Aliphatic C─H Bonds to Synthesis. Science 2012, 335, 807–809. [DOI] [PubMed] [Google Scholar]
- 2.Newhouse T; Baran PS If C─H Bonds Could Talk: Selective C─H Bond Oxidation. Angew. Chem. Int. Ed 2011, 50, 3362–3374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yamaguchi J; Yamaguchi AD; Itami K C─H Bond Functionalization: Emerging Synthetic Tools for Natural Products and Pharmaceuticals. Angew. Chem. Int. Ed 2012, 51, 8960–9009. [DOI] [PubMed] [Google Scholar]
- 4.Cernak T; Dykstra KD; Tyagarajan S; Vachal P; Krska SW The Medicinal Chemist's Toolbox for Late Stage Functionalization of Drug-Like Molecules. Chem. Soc. Rev 2016, 45, 546–576. [DOI] [PubMed] [Google Scholar]
- 5.Hofmann AW Zur Kenntniss des Piperidins und Pyridins. Berichte Dtsch. Chem. Ges 1879, 12, 984–990. [Google Scholar]
- 6.Zard SZ Recent Progress in the Generation and Use of Nitrogen-Centered Radicals. Chem. Soc. Rev 2008, 37, 1603–1618. [DOI] [PubMed] [Google Scholar]
- 7.Stateman LM; Nakafuku KM; Nagib D Remote C─H Functionalization via Selective Hydrogen Atom Transfer. A. Synthesis 2018, 50, 1569–1586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Majetich G; Wheless K Remote Intramolecular Free Radical Functionalizations: An Update. Tetrahedron 1995, 51, 7095–7129. [Google Scholar]
- 9.Hernández R; Rivera A; Salazar JA; Suárez E Nitroamine Radicals as Intermediates in the Functionalization of Non-activated Carbon Atoms. J. Chem. Soc. Chem. Commun 1980, 958–959. [Google Scholar]
- 10.Zalatan DN; Du Bois J Oxidative Cyclization of Sulfamate Esters Using NaOCl – A Metal-Mediated Hoffman–Löffler–Freytag Reaction. Synlett 2009, 1, 143–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Du Bois J Rhodium-Catalyzed C─H Amination. An Enabling Method for Chemical Synthesis. Org. Process Res. Dev 2011, 15, 758–762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wappes EA; Fosu SC; Chopko TC; Nagib DA Triiodide-Mediated δ–Amination of Secondary C─H Bonds. Angew. Chem. Int. Ed 2016, 55, 9974–9978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen K; Baran PS Total Synthesis of Eudesmane Terpenes by Site-Selective C─H Oxidations. Nature 2009, 459, 824–828. [DOI] [PubMed] [Google Scholar]
- 14.Petterson RC; Wambsgans A Photochemical Rearrangement of N-Chloroimides to 4-Chloroimides. A New Synthesis of γ-Lactones. J. Am. Chem. Soc 1964, 86, 1648–1649. [Google Scholar]
- 15.Kerwin JF; Wolff ME; Owings FF; Lewis BB; Blank B; Magnani A; Karash C; Georgian V The Synthesis of C-18 Functionalized Steroid Hormone Analogs. II. Preparation and Some Reactions of 18-Chloro Steroids. J. Org. Chem 1962, 27, 3628–3639. [Google Scholar]
- 16.Chow YL; Joseph TC Selectivity of intramolecular hydrogen transfer in the free amido-radical. J. Chem. Soc. Chem. Commun 1969, 9, 490–491. [Google Scholar]
- 17.Groendyke BJ; AbuSalim DI; Cook SP Iron-Catalyzed, Fluoroamide-Directed C─H Fluorination. J. Am. Chem. Soc 2016, 138, 12771–12774. [DOI] [PubMed] [Google Scholar]
- 18.Reddy LR; Reddy BVS; Corey EJ Efficient Method for Selective Introduction of Substituents as C(5) of Isoleucine and Other α-Amino Acids. Org. Lett 2006, 8, 2819–2821. [DOI] [PubMed] [Google Scholar]
- 19.Liu T; Mei T-S; Yu J-Q γ,δ,ε-C(sp3)–H Functionalization through Directed Radical H-Abstraction. J. Am. Chem. Soc 2015, 137, 5871–5874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Choi GJ; Zhu Q; Miller DC; Gu CJ; Knowles RR Catalytic Alkylation of Remote C─H Bonds Enabled by Proton-Coupled Electron Transfer. Nature 2016, 539, 268–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chu JCK; Rovis T Amide-Directed Photoredox-Catalysed C–C Bond Formation at Unactivated sp3 C─H Bonds. Nature 2016, 539, 272–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen D-F; Chu JCK; Rovis T Directed γ-C(sp3)–H Alkylation of Carboxylic Acid Derivatives through Visible Light Photoredox Catalysis. J. Am. Chem. Soc 2017, 139, 14897–14900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tang N; Wu X; Zhu C; Practical, Metal-free Remote Heteroarylation of Amides via Unactivated C(sp3)–H Bond Functionalization. Chem. Sci 2019, 10, 6915–6919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Minisci F; Vismara E; Fontana F; Platone E; Faraci GJ Selective Aromatic Chlorination and Bromination with N-Halogenoamines in Acidic Solution. Chem. Soc. Perkin Trans. 2 1989, 2, 123–126. [Google Scholar]
- 25.Johnson RA; Greene FD Chlorination with N-Chloro Amides. II. Selectivity of Hydrogen Abstraction by Amidyl Radicals. J. Org. Chem 1975, 40, 2192–2196. [Google Scholar]
- 26.Deno NC; Fishbein R; Wyckoff JC Cation Radicals. III. Sterically Hindered Chlorinating Agents. J. Am. Chem. Soc 1971, 93, 2065–2066. [Google Scholar]
- 27.Schmidt VA; Quinn RK; Brusoe AT; Alexanian EJ Site-Selective Aliphatic C─H Bromination Using N-Bromoamides and Visible Light. J. Am. Chem. Soc 2014, 136, 14389–14392. [DOI] [PubMed] [Google Scholar]
- 28.Quinn RK; Könst ZA; Michalak SE; Schmidt Y; Szklarski AR; Flores AR; Nam S; Horne DA; Vanderwal CD; Alexanian EJ Site-Selective Aliphatic C─H Chlorination Using N-Chloroamides Enables a Synthesis of Chlorolissoclimide. J. Am. Chem. Soc 2016, 138, 696–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Czaplyski WL; Na CG; Alexanian EJ C─H Xanthylation: A Synthetic Platform for Alkane Functionalization. J. Am. Chem. Soc 2016, 138, 13854–13857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Carestia AM; Ravelli D; Alexanian EJ Reagent-Dictated Site Selectivity in Intermolecular Aliphatic C─H Functionalizations Using Nitrogen-Centered Radicals. Chem. Sci 2018, 9, 5360–5365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hioe J; Sakic D; Vrcek V; Zipse H The Stability of Nitrogen-Centered Radicals. Org. Biomol. Chem 2015, 13, 157–169. [DOI] [PubMed] [Google Scholar]
- 32.Šakić D; Zipse H Radical Stability as a Guideline in C─H Amination Reactions. Adv. Synth. Catal 2016, 358, 3983–3991. [Google Scholar]
- 33.Luo Yu-Ran. Handbook of Bond Dissociation Energies in Organic Compounds. CRC Press, 2003, ISBN 0-8493-1589-1. [Google Scholar]
- 34.Sutcliffe R; Griller D; Lessard J; Ingold KU The Structure of Amidyl Radicals. Evidence for the π-Electronic Ground State and for Twist about the Acyl-Nitrogen Bond by Electron Paramagnetic Resonance Spectroscopy. J. Am. Chem. Soc 1981, 103, 624–628. [Google Scholar]
- 35.Glendening ED; Landis CR, Weinhold F Natural bond orbital methods. Wiley Interdiscip. Rev. Comput. Mol. Sci 2012, 2, 1–42. [Google Scholar]
- 36.Crespi S; Simeth NA; Bellisario A; Fagnoni M; König B Unraveling the Thermal Isomerization Mechanisms of Heteroaryl Azoswitches: Phenylazoindoles as Case Study. J. Phys. Chem. A 2019, 123, 1814–1823. [DOI] [PubMed] [Google Scholar]
- 37.Hu J; Wang J; Nguyen TH; Zheng N The Chemistry of Amine Radical Cations Produced by Visible Light Photoredox Catalysis. Beilstein J. Org. Chem 2013, 9, 1977–2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Finn M; Friedline R; Suleman NK; Wohl CJ; Tanko JM Chemistry of the t-Butoxyl Radical: Evidence that Most Hydrogen Abstractions from Carbon are Entropy-Controlled. J. Am. Chem. Soc 2004, 126, 7578–7584. [DOI] [PubMed] [Google Scholar]
- 39.Prosser AR, Banning JE, Rubina M, Rubin M Formal Nucleophilic Substitution of Bromocyclopropanes with Amides en route to Conformationally Constrained β-Amino Acid Derivatives. Org. Lett, 2010, 12, 3968–3971. [DOI] [PubMed] [Google Scholar]
- 40.Frébault F, Luparia M, Oliveira MT, Goddard R, Maulide N A Versatile and Stereoselective Synthesis of Functionalized Cyclobutenes. Angewandte Chemie International Edition, 2010, 49, 5672–5676. [DOI] [PubMed] [Google Scholar]
- 41.Jiang H, Liu B, Li Y, Wang A, Huang H Synthesis of Amides via Palladium-Catalyzed Amidation of Aryl Halides. Org. Lett, 2011, 13, 1028–1031. [DOI] [PubMed] [Google Scholar]
- 42.Yoo W-J, Li C-J Highly Efficient Oxidative Amidation of Aldehydes with Amine Hydrochloride Salts. J. Am. Chem. Soc, 2006, 128, 13064–13065. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.