

Inflammation targeting through collagen affinity improves antibody therapeutic ability.
Abstract
Enhancing the therapeutic efficacy of drugs for inflammatory diseases is of high demand. One possible approach is targeting drugs to the extracellular matrix of the inflamed area. Here, we target collagens in the matrix, which are inaccessible in most tissues yet are exposed to the bloodstream in the inflamed area because of vascular hyperpermeability. We conferred collagen affinity to anti–tumor necrosis factor-α (α-TNF) antibody by conjugating a collagen-binding peptide (CBP) derived from the sequence of decorin. CBP–α-TNF accumulated in the inflamed paw of the arthritis model, and arthritis development was significantly suppressed by treatment with CBP–α-TNF compared with the unmodified antibody. Similarly, CBP–anti-transforming growth factor-β (α–TGF-β) accumulated in the inflamed lung of pulmonary fibrosis model and significantly suppressed pulmonary fibrosis compared with the unmodified antibody. Together, collagen affinity enables the anticytokine antibodies to target arthritis and pulmonary fibrosis accompanied by inflammation, demonstrating a clinically translational approach to treat inflammatory diseases.
INTRODUCTION
Biological therapies to block cytokine signals in the human body are a powerful approach for treating inflammatory and autoimmune diseases. Therapeutic benefit has been shown for anti–tumor necrosis factor-α (α–TNF) therapy for rheumatoid arthritis (RA), which is an autoimmune inflammatory disorder that mainly damages joints (1). However, currently approved medications do not completely cure most patients and have the possibility of significant side effects by suppressing systemic immunity (2–4). Transforming growth factor-β (TGF-β) is a key inducer of collagen expression from mesenchymal cells such as fibroblasts and thus of fibrosis (5, 6). Fibrosis is a pathological condition characterized by excess deposition of extracellular matrix (ECM) components, mainly collagen, accompanied by inflammatory reactions (7). Although therapeutic interventions that block TGF-β action including anti–TGF-β (α–TGF-β) antibody have been reported to suppress fibrosis progression in multiple organs and tissues of animals (8), clinical trials have thus far been unsuccessful because of insufficient efficacy (9, 10). Thus, improving the efficacy of anti-inflammatory antibodies including α-TNF and α–TGF-β is a crucial challenge.
Inflammatory tissues release a range of mediators that induce the enhanced permeability and retention (EPR) effect (11, 12). The EPR effect results from loose endothelial junctions allowing extravasation of macromolecules and nonfunctional lymphatics, resulting in prolonged retention of macromolecules within the solid tumors and inflamed tissues (11, 12). Unlike tumor tissue, inflammatory tissue has a functional lymphatic system that drains agents from the inflamed site (13). Although the strategy of exploiting the abnormal vasculature of a diseased organ as a therapeutic target is currently being tested (14), rapid clearance of molecules from the inflamed tissues makes targeting inflamed tissues difficult (15). In addition, fibrosis that is often accompanied by inflammation may make drug penetration into the inflamed tissues more difficult because of the development of a dense ECM network (16). Therefore, active retention within the inflammatory site may be a promising approach, rather than a simple EPR effect–dependent passive targeting based on molecular size control.
Collagen is the most abundant protein in the mammalian body and exists in almost all tissues (17). While collagen is richly present in the blood vessel subendothelial space, it barely exists within the blood because of its insolubility under physiological conditions (18, 19). Vasculature in chronically inflamed tissues is reported to have an abnormal structure and be hyperpermeable to supply nutrients to accumulations of inflammatory cells (13, 15). Thus, with its leaky vasculature, collagen is exposed to the soluble elements of the blood in the inflamed tissues. Exploiting this corresponding leaky nature of tumor vessels, we have recently reported that a collagen-binding protein domain derived from the von Willebrand factor (VWF) A3 domain could be conjugated to checkpoint inhibitor antibodies or fused to cytokines to enable accumulation in the tumor (20). The VWF A3 domain binds to collagen types I and III, but not to type II, which is a major component of articular cartilage (21). Decorin is a small proteoglycan that interacts with collagen fibrils in all connective tissues (22) and binds to several types of collagen with strong affinity (23). On the basis of the analysis of decorin’s crystal structure, the peptide with the highest binding affinity for type I collagen has been identified (24).
Here, we have hypothesized that conjugation of the collagen-binding peptide (CBP) derived from decorin with anti-inflammatory antibodies can enhance therapeutic benefit. We engineered collagen-binding α–TNF-α and collagen-binding α–TGF-β using decorin’s CBP to achieve targeted therapy for arthritis and pulmonary fibrosis, respectively, via systemic injection.
RESULTS
CBP conjugation confers collagen affinity to α-TNF
After mixing α-TNF with sulfosuccinimidyl-4-(N-maleimidomethyl) cyclohexane-1-carboxylate (sulfo-SMCC), the CBP (LRELHLNNNC) was covalently conjugated via its C-terminal cysteine residue to α-TNF. Under the stoichiometric conditions used, the α-TNF bound up to five CBPs per one antibody as measured by matrix-assisted laser desorption/ionization–time-of-flight (MALDI-TOF) mass spectrometry (MS) (fig. S1). To examine the capacities of CBP-conjugated α-TNF (CBP–α-TNF) to bind collagens, the binding activities of CBP–α-TNF and unmodified α-TNF against human types I, II, and III collagen were determined by enzyme-linked immunosorbent assay (ELISA). CBP–α-TNF bound to all three tested types of collagen, whereas binding signal of unmodified α-TNF to collagens was undetectable (Fig. 1A). Dissociation constant (KD) values [95% confidence interval (CI)] of CBP–α-TNF were 4.5 × 10−7 (3.8 × 10−7 to 5.3 × 10−7) M for human type I collagen, 7.7 × 10−7 (5.9 × 10−7 to 10 × 10−7) M for human type II collagen, and 11 × 10−7 (7.5 × 10−7 to 18 × 10−7) M for human type III collagen. To examine the target specificity of CBP–α-TNF, the binding activities of CBP–α-TNF were determined against other ECM proteins, such as mouse types I and III collagen, bovine serum albumin (BSA), fibronectin, fibrinogen, elastin, and human type IV collagen (fig. S2A). CBP–α-TNF strongly bound mouse collagens, while the binding signals against BSA, fibronectin, fibrinogen, elastin, and human type IV collagen were limited. To examine the amino acid sequence specificity of CBP for collagen binding, we conjugated α-TNF with scrambled sequence peptides (fig. S2, B and C). The binding activities of all the tested scrambled peptide–conjugated α-TNF were much less than CBP–α-TNF against types I and III mouse collagens, demonstrating that CBP–α-TNF binds to collagen in an amino acid sequence–specific manner (fig. S2D). CBP–α-TNF also neutralized its target antigen with similar activity to unmodified α-TNF (Fig. 1B). The 50% inhibitory concentrations against binding between TNF-α and TNF receptor 2 (95% CI) of unmodified α-TNF and CBP–α-TNF were 3.2 × 10−6 (2.8 × 10−6 to 3.6 × 10−6) M and 3.9 × 10−6 (3.1 × 10−6 to 4.9 × 10−6) M, respectively. In addition, CBP–α-TNF bound to collagen in human tendon specimens from a patient with RA. The specimen was probed with either unmodified α-TNF or CBP–α-TNF, together with anti–type I collagen antibody and anti-CD31 antibody. CBP–α-TNF bound intensely around blood vessels, where type I collagen is enriched, in the human specimen, whereas unmodified α-TNF was not detected (Fig. 1C). Similarly, CBP–α-TNF binding to cartilage in human osteoarthritis specimens was evaluated, showing binding to regions that are rich in type II collagen (Fig. 1D); unmodified α-TNF was not detected. These data indicate that CBP conjugation provided α-TNF with affinity against types I, II, and III collagen, without impairment of its neutralizing activity against target antigen.
Fig. 1. CBP conjugation provided collagen affinity to α-TNF.
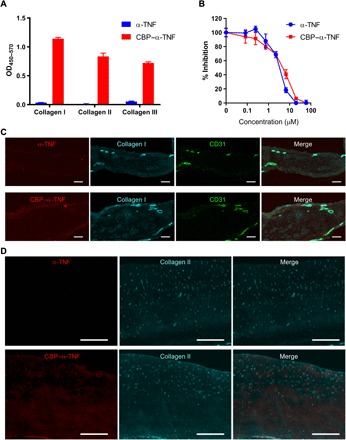
(A) Unmodified α-TNF and CBP–α-TNF binding affinities to types I, II, and III human collagen are analyzed by ELISA (n = 3, means + SD). OD450–570, optical density at 450 nm with subtraction of optical density at 570 nm. (B) Blocking activity of unmodified α-TNF and CBP–α-TNF against the binding between TNF-α and TNF receptor 2 was analyzed by ELISA (n = 3, means ± SD). (C) Representative images of human RA specimen probed with either unmodified α-TNF or CBP–α-TNF, anti-CD31 antibody, and anti–type I collagen antibody. Scale bars, 200 μm. (D) Representative images of human osteoarthritis specimen probed with either unmodified α-TNF or CBP–α-TNF and anti–type II collagen antibody. Scale bars, 500 μm.
CBP conjugation enabled α-TNF to localize in the inflamed paw of the arthritis model
Localization of CBP–α-TNF in the inflamed paw of the collagen antibody–induced arthritis (CAIA) model through binding to endogenous collagen was determined by in vivo biodistribution analysis. Arthritis was selectively induced in the right hind paw by systemic passive immunization with anticollagen antibodies, followed by subcutaneous injection of lipopolysaccharide (LPS) at the right hind footpad. The local LPS injection induced severe arthritis at the right hind paw compared with the other paws. On the day following LPS injection, fluorescently labeled CBP–α-TNF or unmodified α-TNF was intravenously injected into the CAIA and naïve mice. One hour after the injection, major organs and tissues including paws were collected, and their fluorescence levels were measured. The fluorescence level of CBP–α-TNF in the arthritic paw was markedly increased compared with the nonarthritic paw, and there was no significant difference in distribution to nonpathogenic organs, such as the liver and kidney, between CBP–α-TNF and unmodified α-TNF (fig. S3). To examine the accumulation of CBP–α-TNF in the arthritic paw over time, the whole-body fluorescence level was measured before the antibody injection and at 0.5, 1, 2, 4, 6, 24, and 48 hours after the injection. The fluorescence level in the right hind arthritic paw of the CAIA mice injected with CBP–α-TNF and unmodified α-TNF increased immediately after the injection, whereas that of naïve mice was almost the same in both hind paws (Fig. 2, A and B). The ratio of the level in the arthritic paw to the nonarthritic paw was significantly higher in mice injected with CBP–α-TNF than with unmodified α-TNF (Fig. 2C). The injected CBP–α-TNF was distributed in the synovium and pannus, the principal inflamed areas of arthritis, as determined by immunohistochemistry (Fig. 2D). These data indicate that CBD–α-TNF preferentially localizes to the inflamed tissue (i.e., the arthritic paw) after systemic injection more than its unmodified form.
Fig. 2. CBP–α-TNF accumulated in the inflamed paw.
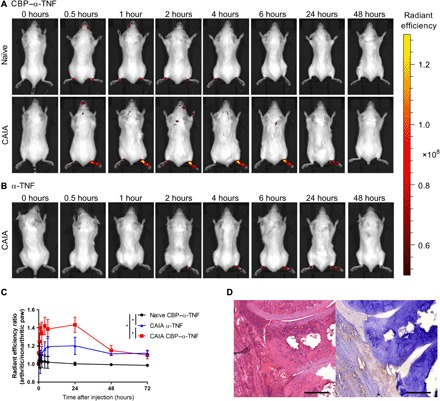
Arthritis (CAIA) was induced selectively in the right hind paw by passive immunization of anticollagen antibodies, followed by subcutaneous injection of LPS at the right hind footpad and phosphate-buffered saline at the left hind footpad. On the day following LPS injection, Cy7-labeled CBP–α-TNF and Cy7-labeled α-TNF were intravenously injected into naïve and CAIA mice. Representative images of accumulation in arthritic or nonarthritic paws of mice injected with CBP–α-TNF (A) and α-TNF (B) are shown. (C) Changes in radiant efficiency ratio of the arthritic paw (right hind) to the nonarthritic paw (left hind) in naïve and CAIA mice (n = 3 to 4, means ± SD). *P < 0.05 compared with the area under the radiant efficiency ratio-time curve from 0 to 72 hours of each treatment group (Tukey’s multiple comparison test). (D) Representative histology images of joints in CBP–α-TNF–injected CAIA mouse [left, hematoxylin and eosin (H&E) staining; right, immunohistochemistry staining against anti-rat immunoglobulin G (IgG)]. Scale bars, 200 μm.
CBP conjugation enhances the efficacy of α-TNF in the CAIA model
We next examined the anti-inflammatory efficacy of CBP–α-TNF in the CAIA model. Arthritis was induced in all paws by passive immunization of anticollagen antibodies, followed by intraperitoneal injection of LPS. On the day of LPS injection, control immunoglobulin G (IgG), unmodified α-TNF, or CBP–α-TNF was intravenously injected. The arthritis score was increased in control mice, and the score was reduced by unmodified α-TNF and CBP–α-TNF (Fig. 3A). The score reduction in CBP–α-TNF–treated mice was significantly greater than in unmodified α-TNF–treated mice. Histological observation revealed that joint destruction was significantly suppressed by CBP–α-TNF (Fig. 3B). The joint condition in CBP–α-TNF–treated mice was comparable to that in naïve mice. Unmodified α-TNF did not significantly suppress the histological score compared to control IgG–treated mice, since α-TNF–treated mice showed wide variability in pathology scores (e.g., two of six mice showed score 4, the severest score, while four of six mice showed score 2 or less). On the contrary, CBP–α-TNF treatment showed small variability in pathology scores (e.g., six of six mice showed either score 0 or 1). The injected CBP–α-TNF was detected in synovium and pannus by immunohistochemistry (Fig. 2D). CBP–α-TNF effectively suppressed infiltration of proinflammatory cells into the paws. CBP–α-TNF reduced neutrophils and macrophages within the paws, whereas unmodified α-TNF did not (fig. S4). These data demonstrate that effective delivery of CBP–α-TNF to the arthritic paws suppressed joint destruction. Furthermore, a similar tendency in the accumulation and the inhibitory efficacy was observed even with subcutaneous injection of CBP–α-TNF (fig. S5). To investigate whether the CBP conjugate showed toxicity, blood cell counts and histological examination of the major organs were assessed after injection of control IgG, unmodified α-TNF, or CBP–α-TNF into CAIA mice. Although reduction of cell counts in white blood cells and platelets by arthritis induction was observed, there was no significant difference in the tested blood parameters and histology between the groups (fig. S6). These data indicate that CBP modification of α-TNF provides superior anti-inflammatory efficacy in the CAIA model of RA compared to its unmodified form without any enhancement in toxicity.
Fig. 3. CBP–α-TNF suppressed arthritis development more effectively than unmodified α-TNF.
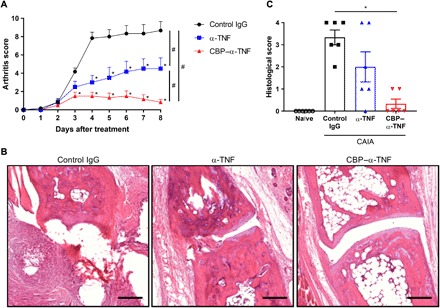
Arthritis (CAIA) was induced by passive immunization of anticollagen antibodies, followed by intraperitoneal injection of LPS. On the day of LPS injection, control IgG, unmodified α-TNF, or CBP–α-TNF was injected intravenously into the arthritic mice. (A) Arthritis scores represent the means + SE from six mice. *P < 0.05, compared with control (Dunnett’s multiple comparison test). #P < 0.05 compared with the scores on day 8 of each treatment group (Tukey’s multiple comparison test). (B) Representative H&E image of joints on day 8 in each treatment group. Scale bars, 200 μm. (C) The severity of synovial hyperplasia and bone resorption was scored 0 to 4 as described in Materials and Methods. *P < 0.05 compared with control (Dunnett’s multiple comparison test).
CBP conjugation enhances the efficacy of α–TGF-β in a pulmonary fibrosis model
To explore whether the collagen-binding antibody engineering approach has versatile application for inflammatory diseases, we next conjugated CBP with α–TGF-β, which neutralizes all three isoforms of TGF-β. The collagen affinity was installed to α–TGF-β as with α-TNF (fig. S7). Accumulation of CBP–α–TGF-β in the inflamed tissue was determined in the bleomycin (BLM)–induced pulmonary fibrosis model by fluorescence imaging. When the lung was inflamed, fluorescently labeled α–TGF-β and CBP–α–TGF-β were intravenously injected. CBP–α–TGF-β was detected in the lung of the fibrosis model, whereas α–TGF-β was not (Fig. 4A). To investigate the efficacy of CBP–α–TGF-β in modulating fibrosis, control IgG, unmodified α–TGF-β, or CBP–α–TGF-β was injected into mice three times a week from 1 week after the BLM instillation, and the histological fibrosis severity in the lung was determined 3 weeks after the BLM instillation. The lung sections were stained with hematoxylin and eosin (H&E) and Masson’s trichrome staining, which visualizes collagen fibers in blue, and then the extent of fibrosis was quantified by Ashcroft scoring (25). Severe fibrosis was observed in control IgG-treated mice; the disease score was significantly reduced by CBP–α–TGF-β but not by unmodified α–TGF-β (Fig. 4, B and C). These results suggest that CBP conjugation is applicable to other antibodies and that installing collagen affinity enables anti-inflammatory antibodies to target inflamed tissues and enhance its efficacy in inflammatory diseases and in fibrosis accompanied by inflammation.
Fig. 4. Localization in lung and efficacy of CBP–α–TGF-β in the BLM-induced pulmonary fibrosis model.
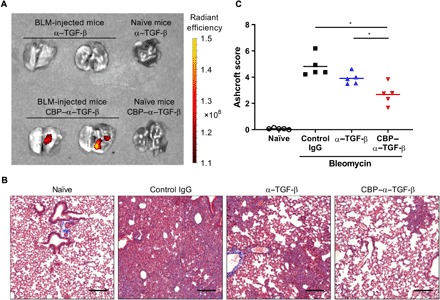
Mice were intranasally instilled with 50 μg of BLM sulfate on day 0. (A) Cy7-labeled CBP–α–TGF-β or Cy7-labeled α–TGF-β was intravenously injected into naïve mice or BLM-injected mice on day 7. The lung was harvested 4 hours after the fluorescence injection, and fluorescence intensity was measured. (B and C) Control IgG, unmodified α–TGF-β, or CBP–α–TGF-β at a dose of 50 μg per mouse three times a week from day 7. On day 21, the left lung lobe was fixed and provided to histological analysis. (B) Representative image of Masson’s trichrome staining of the lung on day 21 in each treatment group. Scale bars, 200 μm (C) Lung fibrosis was scored 0 to 8 by Ashcroft scoring as described in Materials and Methods. *P < 0.05 compared with the scores of each treatment group (Tukey’s multiple comparison test).
DISCUSSION
Various drugs are in development based on passive targeting strategies to noncancer diseases using accumulation of macromolecular prodrugs, liposomes, and nanoparticles administered through the systemic circulation, although they are still in preclinical stages or suspended in early clinical stages (26–28). While these techniques are useful for delivering drugs to inflammatory sites, inflammation site targeting is challenging because of biological barriers such as ECM proteins and rapid clearance from the inflamed tissues via lymphatic drainage (15, 29). Immunocytokines have been targeted to the ECM spice variants fibronectin extra domain A (EDA) and extra domain B (EDB), which are expressed in sites of chronic inflammation (30). These targets are principally present in the inflamed subendothelium. Here, we target a majority component, collagen, which is present throughout the matrix of the inflamed tissue.
In this study, we have demonstrated that installing collagen-binding affinity to anti-inflammatory antibodies enhances their retention at the inflamed tissue site and thereby their therapeutic efficacy. CBP conjugation increased the amount of antibodies detected in the inflamed tissues in the CAIA model and the BLM-induced pulmonary fibrosis model after systemic injection. This demonstrates that antibodies with collagen affinity can target and be retained at inflamed sites and that this technology is widely applicable to various inflammatory diseases. This is because collagen is universally and abundantly present around the vasculature and within the inflamed tissue but is exposed to the soluble components of the bloodstream only when hyperpermeability of the vasculature occurs, as in inflammatory tissue. Thus, collagen affinity as a drug retention method is neither tissue nor molecular expression specific and may be applied to chronic inflammation generally.
Fibrosis is a pathological condition of inflammatory diseases where collagen expression is greatly up-regulated (31–34). Thus, collagen targeting is a reasonable approach to deliver therapeutics to sites of fibrosis. A number of preclinical studies indicate that TGF-β inhibition is a promising strategy for fibrosis treatment (35–38). However, clinical translation has not progressed well because of the failure of showing efficacy in clinical trials (9, 10). Here, we have shown that the disease site targeting approach markedly improved the efficacy of α–TGF-β. Considering that there are few studies focusing on improving the efficacy of α–TGF-β itself, our molecular engineering is an innovative approach.
CBP-conjugated antibody reduced the arthritis score and the fibrosis score more effectively than unmodified antibody in the two models that we examined. Unmodified α-TNF treatment reduced the arthritis score, as expected, but did not show a significant reduction in histological score. Current α-TNF therapy for RA does not show a complete response in terms of joint damage by persistent synovitis even in patients in clinical remission (2–4). Similarly, CBP–α–TGF-β significantly reduced the fibrosis score, whereas unmodified α–TGF-β did not. That is because increased ECM protein deposition in the lung and rapid clearance of antibody induced by inflammation might prevent α–TGF-β from blocking the up-regulated TGF-β signal that induces fibrosis in the tissue, suggesting that CBP conjugation to α–TGF-β enables the antibody to accumulate and be retained in the microenvironment to thoroughly block TGF-β signal.
Biological therapies such as adalimumab are often used via a subcutaneous route in clinical settings. The feasibility of targeting inflammatory sites using CBP antibody by subcutaneous injection could be a possible concern because the subcutaneous tissue contains collagens. Therefore, we have tested subcutaneous injection of CBP–α-TNF. Even when injected subcutaneously, CBP–α-TNF was accumulated in the arthritic paw and was more effective against arthritis development than unmodified α-TNF. The avidity of the CBP conjugates to each type of collagen is relatively moderate (KD, approximately 0.5 to 1 μM), which enables antibody from the subcutaneous tissue to be drained and enter the bloodstream presumably via the lymphatics, with subsequent accumulation of CBP–α-TNF at the inflammatory sites. Thus, it may be that the moderate collagen affinity of the CBP from decorin is advantageous when the CBP antibodies are administered subcutaneously.
For clinical translation of collagen-binding anti-inflammatory drugs, an advantage also lies in the use of a CBP that naturally exists in the body, limiting the possibility of immune system recognition. It was confirmed that the CBP-conjugated antibody was capable of binding with human collagens both in vitro and in human specimens. Also, the CBP can be conjugated to antibodies with a simple chemical reaction. The advantage of this feature is in the simplicity in production, in that it is possible to work with antibodies for which production has already been optimized. The CBP conjugation synthesis reaction for antibodies can be done in only 90 min using chemistry that is analogous to PEGylation of proteins. The same reaction is used in antibody-drug conjugates, such as in the production of trastuzumab emtansine (39, 40). These features may facilitate development of collagen-binding drug therapy to overcome the barriers to clinical translation.
In conclusion, we have found that installing collagen affinity to an anticytokine antibody enables it to target and be retained in inflammatory sites. Moreover, collagen-binding anti-inflammatory antibodies showed higher therapeutic efficacy than their unmodified forms. This simple approach of generating an engineered collagen-binding biologic may hold potential for clinical translation as an inflammation-targeted therapeutic.
MATERIALS AND METHODS
Study design
This study was designed to test the strategy of targeting anti-inflammatory therapeutic antibodies to inflammation sites through engineered affinity for collagen. Specifically, we tested whether inflammatory site-targeted delivery of anti-inflammatory therapeutic antibodies by CBP conjugation is superior to current nontargeted delivery using two examples: α-TNF in the model of RA and α–TGF-β in the model of pulmonary fibrosis. To evaluate the efficacy of α-TNF, α–TGF-β, and their CBP-conjugated forms, we scored arthritic symptom and joint histology in a CAIA model and fibrosis development in a BLM-induced pulmonary fibrosis model. Statistical methods were not used to predetermine necessary sample size, but sample sizes were chosen on the basis of estimates from pilot experiments so that appropriate statistical tests could yield statistically significant results. Production of CBP–α–TNF and CBP–α–TGF-β was performed by multiple individuals to ensure reproducibility. All experiments were replicated at least twice. For animal studies, mice were randomized into treatment groups within a cage immediately before the first drug injection and treated in the same manner. The n values used to calculate statistics are indicated in the figure legends. Drug administration and pathological analyses were performed in a blinded fashion. Statistical methods are described in the “Statistical analysis” section. Original data are located in data file S1.
Synthesis of peptide-conjugated antibody
Rat anti-mouse TNF-α antibody (clone XT3.11, Bio X Cell) or mouse anti-mouse TGF-β antibody (clone 1D11.16.8, Bio X Cell) was incubated with 30-fold molar excess of sulfo-SMCC cross-linker (Thermo Fisher Scientific) for 30 min at room temperature. Unreacted cross-linker was removed using a Zeba spin desalting column (Thermo Fisher Scientific), and then 30-fold molar excess of CBP (LRELHLNNNC) or CBP sequence scrambled peptide [scramble 1 (LNNLRLHENC), scramble 2 (LEHNNRLLNC), or scramble 3 (RNNLHENLLC)] was added and reacted for 1 hour at room temperature for conjugation to the thiol moiety on the C residue. The peptide had been synthesized with >95% purity by GenScript.
Matrix-assisted laser desorption/ionization–time-of-flight mass spectroscopy
Antibodies were analyzed by MALDI-TOF MS using a Bruker UltrafleXtreme MALDI TOF/TOF instrument. All spectra were collected with acquisition software Bruker flexControl and processed with analysis software Bruker flexAnalysis. First, a saturated solution of the matrix, α-cyano-4-hydroxycinnamic acid (Sigma-Aldrich), was prepared in 50:50 (v/v) acetonitrile:(1% trifluoroacetic acid in water) as a solvent. The analyte in phosphate-buffered saline (PBS) (5 μl, 0.1 mg/ml) and the matrix solution (25 μl) were then mixed, and 1 μl of that mixture was deposited on the MTP 384 ground steel target plate. The drop was allowed to dry in a nitrogen gas flow, which resulted in the formation of uniform sample/matrix coprecipitate. All samples were analyzed using the high mass linear positive mode method with 5000 laser shots at a laser intensity of 75%. The measurements were externally calibrated at three points with a mix of carbonic anhydrase, phosphorylase B, and BSA.
Detection of CBP antibody binding to collagens and ECM proteins
Ninety-six–well ELISA plates (Greiner Bio-One) were coated with recombinant human type I, type II, type III, or type IV collagen (Millipore, Sigma); mouse types I and III collagen mixed (Bio-Rad Laboratories); BSA (Sigma-Aldrich), fibronectin (Sigma-Aldrich), fibrinogen (VWF- and fibronectin-depleted; Enzyme Research Laboratories), or elastin (Millipore, Sigma) at 10 μg/ml in PBS overnight at 37°C, then blocked with 2% BSA in 0.05% Tween 20 containing PBS (PBS-T) for 1 hour at room temperature. Then, wells were washed with PBS-T and further incubated with 15.6 to 1000 nM CBP-conjugated, sequence-scrambled CBP-conjugated, or unmodified antibody for 1 hour at room temperature. After three washes with PBS-T, antibody was detected by horseradish peroxidase (HRP)–conjugated antibody against rat IgG or mouse IgG and incubated 1 hour at room temperature (Jackson ImmunoResearch). After washes, bound proteins were detected with tetramethylbenzidine substrate by measurement of the absorbance at 450 nm with subtraction of the absorbance at 570 nm. The apparent KD values were obtained by nonlinear regression analysis in Prism software (GraphPad Software v8), assuming one site-specific binding.
CBP antibody binding to human specimens
Frozen sections of tendon from a patient with RA and cartilage from a patient with osteoarthritis were purchased from OriGene Technologies. The sections were blocked with 2% fetal bovine serum (FBS) in PBS for overnight at room temperature. The sections were incubated with primary antibodies for 3 hours at room temperature. For tendon sections, either rat α-TNF (50 μg/ml) or equimolar CBP–α-TNF, mouse anti-human CD31 antibody (5 μg/ml; Abcam), and rabbit anti-human type I collagen antibody (5 μg/ml; Abcam) were used as primary antibodies. For cartilage sections, either rat α-TNF or equimolar CBP–α-TNF (50 μg/ml) and rabbit anti-human type II collagen antibody (5 μg/ml; Abcam) were used as primary antibodies. After incubating with the fluorescently tagged secondary antibodies, slides were covered with ProLong Gold Antifade Mountant with 4′,6-diamidino-2-phenylindole (Thermo Fisher Scientific). The images were scanned with a Pannoramic Digital Slide Scanner (3DHISTECH) and analyzed using Pannoramic Viewer software (3DHISTECH).
Mice
Balb/c mice at 7 to 8 weeks of age were obtained from the Jackson laboratory. C57BL/6 mice at 7 to 12 weeks of age were obtained from Charles River Laboratories. Experiments were performed with approval from the Institutional Animal Care and Use Committee of the University of Chicago.
In vivo biodistribution study
To make fluorescently labeled CBP antibody, α-TNF and α–TGF-β were incubated with eightfold molar excess of SM(PEG)24 cross-linker (Thermo Fisher Scientific) for 30 min at room temperature. Unreacted cross-linker was removed using a Zeba spin desalting column, and then 30-fold molar excess of Cy7-labeled CBP ([Cy7]LRELHLNNNC[COOH]) was added and reacted for 30 min at room temperature for conjugation to the thiol moiety on the C residue. The peptide had been synthesized with >95% purity by GenScript. Unreacted dye was removed by dialysis against PBS. As to CBP-unconjugated antibodies, α-TNF and α–TGF-β were labeled using sulfo-Cy7 N-hydroxysuccinimide ester (Lumiprobe) according to the manufacturer’s instruction. When the target tissue of the mouse was inflamed, Cy7-labeled antibody at 50 μg/ml was intravenously injected. Mice and organs harvested from the disease model were imaged with the Xenogen IVIS Imaging System 100 (Xenogen) under the following conditions: f/stop, 2; optical filter excitation, 745 nm; excitation, 800 nm; exposure time, 5 s; small binning.
CAIA model
Arthritis was induced in female Balb/c mice by intraperitoneal injection of anticollagen antibody cocktail (1.5 mg per mouse; Chondrex) on day −3, followed by intraperitoneal injection of LPS (50 μg per mouse; Chondrex) on day 0. On the day of LPS injection, mice were intravenously or subcutaneously injected with control IgG (rat IgG1 isotype control, Bio X Cell), unmodified α-TNF, or CBP–α-TNF at a dose of 200 μg per mouse. Joint swelling was scored every day according to the manufacturer’s protocol (Chondrex). On day 8, hind paws were fixed in 10% neutral formalin (Sigma-Aldrich), decalcified in Decalcifier II (Leica), and then provided to histological analysis. Paraffin-embedded paws were sliced at 5-μm thickness and stained with H&E. The images were scanned with a Pannoramic Digital Slide Scanner and analyzed using Pannoramic Viewer software. The severity of synovial hyperplasia and bone resorption for the arthritis model was scored by three-grade evaluation (0 to 2) according to the previously reported criteria with slight modifications as follows: 0, normal to minimal infiltration of pannus in cartilage and subchondral bone of marginal zone; 1, mild to moderate infiltration of marginal zone with minor cortical and medullary bone destruction; 2, severe infiltration associated with total or near total destruction of joint architecture. The scores in both hind paws were summed for each mouse (score per mouse total, 0 to 4). Immunohistostaining was performed according to standard procedures. Briefly, the sections were incubated in 0.3% H2O2 for 20 min, blocked with PBS-buffered 1% BSA for 1 hour, and incubated overnight at 4°C with HRP-conjugated anti-rat IgG (Jackson ImmunoResearch) for 1 hour at room temperature and visualized with diaminobenzidine. The histopathological analyses were performed in a blinded fashion.
Flow cytometry
CAIA mice were intravenously injected with control IgG, unmodified α-TNF, or CBP–α-TNF at a dose of 200 μg per mouse. On day 5, hind paws were harvested and digested in Dulbecco’s modified eagle medium supplemented with 2% FBS, collagenase D (2 mg/ml) and deoxyribonuclease I (40 μg/ml; Roche) for 30 min at 37°C. Single-cell suspensions were obtained by gently disrupting through a 70-μm cell strainer. Antibodies against the following molecules were used: anti-mouse CD45 (30-F11, BD Biosciences), F4/80 (T45-2342, BD Biosciences), Ly6G (1A8, BioLegend), Ly6C (HK1.4, BioLegend), and CD11b (M1/70, BioLegend). Fixable live/dead cell discrimination was performed using Fixable Viability Dye eFluor 455 (eBioscience) according to the manufacturer’s instructions. Following a washing step, cells were stained with specific antibodies for 20 min on ice. Flow cytometric analyses were done using a Fortessa (BD Biosciences) flow cytometer and analyzed using FlowJo software (Tree Star).
BLM-induced pulmonary fibrosis model
Mice (C57BL/6, female) were intranasally instilled with 50 μg of BLM sulfate (Cayman Chemical) under isoflurane anesthesia on day 0 and were intravenously injected with control IgG (mouse IgG1 isotype control, Bio X Cell), unmodified α–TGF-β, or CBP–α–TGF-β at a dose of 50 μg per mouse three times a week from day 7. On day 21, left lung lobe was fixed with 10% neutral formalin and then provided to histological analysis. Paraffin-embedded lungs were sliced at 5 μm thickness and stained with H&E and Masson’s trichrome. The images were scanned with a Pannoramic Digital Slide Scanner and analyzed using Pannoramic Viewer software. Lung fibrosis was scored 0 to 8 by Ashcroft scoring described previously (25).
Statistical analysis
Statistical analyses were performed using GraphPad Prism software, and P < 0.05 was considered statistically significant. To compare the ratio of fluorescence level in an arthritic paw to a nonarthritic paw in each treatment group, the area under the radiant efficiency ratio-time curve from 0 to 72 hours of each treatment group was assessed by Tukey’s multiple comparison test. Changes in arthritis scores over time were assessed by repeated measurement two-way analysis of variance (ANOVA), and when interactions were considered significant, data were assessed using Dunnett’s multiple comparison test at each time point of measurement. To compare the efficacy of CBP–α-TNF with unmodified α-TNF, the data on day 8 were analyzed again via Tukey’s multiple comparison test. For histological scores in the CAIA model, Dunnett’s multiple comparison test was used to compare the difference between the control IgG–injected group and the treatment groups. The Ashcroft scores in BLM-induced pulmonary fibrosis model were assessed by Tukey’s multiple comparison test to compare each treatment group.
Supplementary Material
Acknowledgments
We thank the Human Tissue Resource Center of the University of Chicago for histology analysis. We thank the Integrated Light Microscopy Core of the University of Chicago for Imaging. We thank A. Solanki for assistance in tail vein injections. Funding: This work was supported by the University of Chicago (to J.A.H.). Author contributions: (Note: K.K. is on leave from Astellas Pharma Inc., Tsukuba, Ibaraki 305-8585, Japan.) K.K., J.I., A.M., and J.A.H. designed the project. K.K., A.M., and E.Y. performed experiments. K.K., J.I., and J.A.H. analyzed data. K.K., J.I., and J.A.H. wrote the paper. A.I. performed the histopathological analysis. M.M.R. performed the protein MS. Competing interests: K.K., J.I., A.I., and J.A.H. are inventors on a patent related to this work filed by the University of Chicago (no. 62/809,988, filed 25 February 2019). The other authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/11/eaay1971/DC1
Fig. S1. The molecular weight of α-TNF was increased by CBP conjugation.
Fig. S2. The target and sequence specificity of CBP.
Fig. S3. Distribution of CBP–α-TNF to organs and tissues in the arthritis model.
Fig. S4. CBP–α-TNF reduces macrophages and neutrophils within the paws.
Fig. S5. Effect of subcutaneous injection of CBP–α-TNF in the arthritis model.
Fig. S6. Short-term safety study of CBP–α-TNF in the arthritis model.
Fig. S7. CBP conjugation provided collagen affinity to α–TGF-β.
Data file S1. Original data.
REFERENCES AND NOTES
- 1.Weinblatt M. E., Keystone E. C., Furst D. E., Moreland L. W., Weisman M. H., Birbara C. A., Teoh L. A., Fischkoff S. A., Chartash E. K., Adalimumab, a fully human anti-tumor necrosis factor alpha monoclonal antibody, for the treatment of rheumatoid arthritis in patients taking concomitant methotrexate: The ARMADA trial. Arthritis Rheum. 48, 35–45 (2003). [DOI] [PubMed] [Google Scholar]
- 2.Bellis E., Scirè C. A., Carrara G., Adinolfi A., Batticciotto A., Bortoluzzi A., Cagnotto G., Caprioli M., Canzoni M., Cavatorta F. P., De Lucia O., Di Sabatino V., Draghessi A., Filippou G., Farina I., Focherini M. C., Gabba A., Gutierrez M., Idolazzi L., Luccioli F., Macchioni P., Massarotti M. S., Mastaglio C., Menza L., Muratore M., Parisi S., Picerno V., Piga M., Ramonda R., Raffeiner B., Rossi D., Rossi S., Rossini P., Sakellariou G., Scioscia C., Venditti C., Volpe A., Matucci-Cerinic M., Iagnocco A., Ultrasound-detected tenosynovitis independently associates with patient-reported flare in patients with rheumatoid arthritis in clinical remission: results from the observational study STARTER of the Italian Society for Rheumatology. Rheumatology 55, 1826–1836 (2016). [DOI] [PubMed] [Google Scholar]
- 3.Lisbona M. P., Solano A., Ares J., Almirall M., Salman-Monte T. C., Maymó J., ACR/EULAR definitions of remission are associated with lower residual inflammatory activity compared with DAS28 remission on hand MRI in rheumatoid arthritis. J. Rheumatol. 43, 1631–1636 (2016). [DOI] [PubMed] [Google Scholar]
- 4.Vreju F. A., Filippucci E., Gutierrez M., Di Geso L., Ciapetti A., Ciurea M. E., Salaffi F., Grassi W., Subclinical ultrasound synovitis in a particular joint is associated with ultrasound evidence of bone erosions in that same joint in rheumatoid patients in clinical remission. Clin. Exp. Rheumatol. 34, 673–678 (2016). [PubMed] [Google Scholar]
- 5.Biernacka A., Dobaczewski M., Frangogiannis N. G., TGF-β signaling in fibrosis. Growth Factors 29, 196–202 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Verrecchia F., Mauviel A., Transforming growth factor- β and fibrosis. World J. Gastroenterol. 13, 3056–3062 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Birbrair A., Zhang T., Files D. C., Mannava S., Smith T., Wang Z. M., Messi M. L., Mintz A., Delbono O., Type-1 pericytes accumulate after tissue injury and produce collagen in an organ-dependent manner. Stem Cell Res Ther 5, 122 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Akhurst R. J., Hata A., Targeting the TGFβ signalling pathway in disease. Nat. Rev. Drug Discov. 11, 790–811 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Voelker J., Berg P. H., Sheetz M., Duffin K., Shen T., Moser B., Greene T., Blumenthal S. S., Rychlik I., Yagil Y., Zaoui P., Lewis J. B., Anti–TGF-β1 antibody therapy in patients with diabetic nephropathy. J. Am. Soc. Nephrol. 28, 953–962 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Denton C. P., Merkel P. A., Furst D. E., Khanna D., Emery P., Hsu V. M., Silliman N., Streisand J., Powell J., Akesson A., Coppock J., Hoogen F., Herrick A., Mayes M. D., Veale D., Haas J., Ledbetter S., Korn J. H., Black C. M., Seibold J. R.; Cat-192 Study Group; Scleroderma Clinical Trials Consortium , Recombinant human anti-transforming growth factor β1 antibody therapy in systemic sclerosis: A multicenter, randomized, placebo-controlled phase I/II trial of CAT-192. Arthritis Rheum. 56, 323–333 (2007). [DOI] [PubMed] [Google Scholar]
- 11.Maeda H., Wu J., Sawa T., Matsumura Y., Hori K., Tumor vascular permeability and the EPR effect in macromolecular therapeutics: a review. J. Control. Release 65, 271–284 (2000). [DOI] [PubMed] [Google Scholar]
- 12.Fang J., Nakamura H., Maeda H., The EPR effect: Unique features of tumor blood vessels for drug delivery, factors involved, and limitations and augmentation of the effect. Adv. Drug Deliv. Rev. 63, 136–151 (2011). [DOI] [PubMed] [Google Scholar]
- 13.Nehoff H., Parayath N. N., Domanovitch L., Taurin S., Greish K., Nanomedicine for drug targeting: Strategies beyond the enhanced permeability and retention effect. Int. J. Nanomedicine 9, 2539–2555 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bajpayee A. G., Grodzinsky A. J., Cartilage-targeting drug delivery: Can electrostatic interactions help? Nat. Rev. Rheumatol. 13, 183–193 (2017). [DOI] [PubMed] [Google Scholar]
- 15.McDonald D. M., Angiogenesis and remodeling of airway vasculature in chronic inflammation. Am. J. Respir. Crit. Care Med. 164, S39–S45 (2001). [DOI] [PubMed] [Google Scholar]
- 16.Wynn T. A., Cellular and molecular mechanisms of fibrosis. J. Pathol. 214, 199–210 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ricard-Blum S., The collagen family. Cold Spring Harb. Perspect. Biol. 3, a004978 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dubois C., Panicot-Dubois L., Merrill-Skoloff G., Furie B., Furie B. C., Glycoprotein VI-dependent and -independent pathways of thrombus formation in vivo. Blood 107, 3902–3906 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bergmeier W., Hynes R. O., Extracellular matrix proteins in hemostasis and thrombosis. Cold Spring Harb. Perspect. Biol. 4, a005132 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ishihara J., Ishihara A., Sasaki K., Lee S. S., Williford J. M., Yasui M., Abe H., Potin L., Hosseinchi P., Fukunaga K., Raczy M. M., Gray L. T., Mansurov A., Katsumata K., Fukayama M., Kron S. J., Swartz M. A., Hubbell J. A., Targeted antibody and cytokine cancer immunotherapies through collagen affinity. Sci. Transl. Med. 11, eaau3259 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Aigner T., Stoss H., Weseloh G., Zeiler G., von der Mark K., Activation of collagen type II expression in osteoarthritic and rheumatoid cartilage. Virchows Arch. B 62, 337–345 (1992). [DOI] [PubMed] [Google Scholar]
- 22.Svensson L., Heinegard D., Oldberg A., Decorin-binding sites for collagen type I are mainly located in leucine-rich repeats 4-5. J. Biol. Chem. 270, 20712–20716 (1995). [DOI] [PubMed] [Google Scholar]
- 23.Bidanset D. J., Guidry C., Rosenberg L. C., Choi H. U., Timpl R., Hook M., Binding of the proteoglycan decorin to collagen type VI. J. Biol. Chem. 267, 5250–5256 (1992). [PubMed] [Google Scholar]
- 24.Federico S., Pierce B. F., Piluso S., Wischke C., Lendlein A., Neffe A. T., Design of Decorin-Based Peptides That Bind to Collagen I and their Potential as Adhesion Moieties in Biomaterials. Angew. Chem. Int. Ed. Eng. 54, 10980–10984 (2015). [DOI] [PubMed] [Google Scholar]
- 25.Ashcroft T., Simpson J. M., Timbrell V., Simple method of estimating severity of pulmonary fibrosis on a numerical scale. J. Clin. Pathol. 41, 467–470 (1988). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Durymanov M., Kamaletdinova T., Lehmann S. E., Reineke J., Exploiting passive nanomedicine accumulation at sites of enhanced vascular permeability for non-cancerous applications. J. Control. Release 261, 10–22 (2017). [DOI] [PubMed] [Google Scholar]
- 27.Yuan F., Quan L. D., Cui L., Goldring S. R., Wang D., Development of macromolecular prodrug for rheumatoid arthritis. Adv. Drug Deliv. Rev. 64, 1205–1219 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Anderson R., Franch A., Castell M., Perez-Cano F. J., Brauer R., Pohlers D., Gajda M., Siskos A. P., Katsila T., Tamvakopoulos C., Rauchhaus U., Panzner S., Kinne R. W., Liposomal encapsulation enhances and prolongs the anti-inflammatory effects of water-soluble dexamethasone phosphate in experimental adjuvant arthritis. Arthritis Res. Ther. 12, R147 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sagnella S. M., McCarroll J. A., Kavallaris M., Drug delivery: Beyond active tumour targeting. Nanomedicine 10, 1131–1137 (2014). [DOI] [PubMed] [Google Scholar]
- 30.Pasche N., Neri D., Immunocytokines: a novel class of potent armed antibodies. Drug Discov. Today 17, 583–590 (2012). [DOI] [PubMed] [Google Scholar]
- 31.Emblom-Callahan M. C., Chhina M. K., Shlobin O. A., Ahmad S., Reese E. S., Iyer E. P., Cox D. N., Brenner R., Burton N. A., Grant G. M., Nathan S. D., Genomic phenotype of non-cultured pulmonary fibroblasts in idiopathic pulmonary fibrosis. Genomics 96, 134–145 (2010). [DOI] [PubMed] [Google Scholar]
- 32.Whitfield M. L., Finlay D. R., Murray J. I., Troyanskaya O. G., Chi J. T., Pergamenschikov A., McCalmont T. H., Brown P. O., Botstein D., Connolly M. K., Systemic and cell type-specific gene expression patterns in scleroderma skin. Proc. Natl. Acad. Sci. U.S.A. 100, 12319–12324 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Naitoh M., Hosokawa N., Kubota H., Tanaka T., Shirane H., Sawada M., Nishimura Y., Nagata K., Upregulation of HSP47 and collagen type III in the dermal fibrotic disease, keloid. Biochem. Biophys. Res. Commun. 280, 1316–1322 (2001). [DOI] [PubMed] [Google Scholar]
- 34.Trojanowska M., LeRoy E. C., Eckes B., Krieg T., Pathogenesis of fibrosis: type 1 collagen and the skin. J. Mol. Med. 76, 266–274 (1998). [DOI] [PubMed] [Google Scholar]
- 35.Herbertz S., Sawyer J. S., Stauber A. J., Gueorguieva I., Driscoll K. E., Estrem S. T., Cleverly A. L., Desaiah D., Guba S. C., Benhadji K. A., Slapak C. A., Lahn M. M., Clinical development of galunisertib (LY2157299 monohydrate), a small molecule inhibitor of transforming growth factor- β signaling pathway. Drug Des. Devel. Ther. 9, 4479–4499 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ruzek M. C., Jha S., Ledbetter S., Richards S. M., Garman R. D., A modified model of graft-versus-host-induced systemic sclerosis (scleroderma) exhibits all major aspects of the human disease. Arthritis Rheum. 50, 1319–1331 (2004). [DOI] [PubMed] [Google Scholar]
- 37.Ziyadeh F. N., Hoffman B. B., Han D. C., Iglesias-De La Cruz M. C., Hong S. W., Isono M., Chen S., McGowan T. A., Sharma K., Long-term prevention of renal insufficiency, excess matrix gene expression, and glomerular mesangial matrix expansion by treatment with monoclonal antitransforming growth factor- β antibody in db/db diabetic mice. Proc. Natl. Acad. Sci. U.S.A. 97, 8015–8020 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Santiago B., Gutierrez-Canas I., Dotor J., Palao G., Lasarte J. J., Ruiz J., Prieto J., Borras-Cuesta F., Pablos J. L., Topical application of a peptide inhibitor of transforming growth factor- β1 ameliorates bleomycin-induced skin fibrosis. J. Invest. Dermatol. 125, 450–455 (2005). [DOI] [PubMed] [Google Scholar]
- 39.Lambert J. M., Chari R. V., Ado-trastuzumab Emtansine (T-DM1): an antibody-drug conjugate (ADC) for HER2-positive breast cancer. J. Med. Chem. 57, 6949–6964 (2014). [DOI] [PubMed] [Google Scholar]
- 40.Peters C., Brown S., Antibody-drug conjugates as novel anti-cancer chemotherapeutics. Biosci. Rep. 35, e00225 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/11/eaay1971/DC1
Fig. S1. The molecular weight of α-TNF was increased by CBP conjugation.
Fig. S2. The target and sequence specificity of CBP.
Fig. S3. Distribution of CBP–α-TNF to organs and tissues in the arthritis model.
Fig. S4. CBP–α-TNF reduces macrophages and neutrophils within the paws.
Fig. S5. Effect of subcutaneous injection of CBP–α-TNF in the arthritis model.
Fig. S6. Short-term safety study of CBP–α-TNF in the arthritis model.
Fig. S7. CBP conjugation provided collagen affinity to α–TGF-β.
Data file S1. Original data.