

Abstract
Background: Obesity and diabetes are associated with increased risk and worse outcomes for endometrial cancer. Metformin is a widely prescribed generic drug for the treatment of type II diabetes and metabolic syndrome and may also have anti-tumorigenic effects. Thus, we assessed the metabolic anti-tumorigenic effects of metformin in (1) human endometrial cancer cell lines under varying glucose concentrations, and (2) a novel genetically engineered mouse model of endometrioid endometrial cancer under obese and lean conditions. Methods: The effects of metformin on cytotoxicity, apoptosis, cell cycle progression, and the AMPK/mTOR/S6 and MAPK pathways were assessed in ECC-1 and Ishikawa cells under low, normal and high glucose conditions. The impact of metformin treatment on tumor growth under obese and lean conditions was evaluated using a novel LKB1fl/fl p53fl/fl mouse model of endometrial cancer. Global, untargeted metabolomics was used to identify (1) obesity-associated differences between endometrial tumors and (2) the obesity-dependent effects of metformin in the endometrial tumors. Results: Hypoglycemic conditions significantly enhanced the sensitivity of the cells to metformin in regards to its anti-proliferative and apoptotic effects, as compared to hyperglycemic and normal glucose conditions. Metformin inhibited tumor growth in both the obese and lean mice, which metformin-induced inhibition of tumor progression in obese mice was significantly greater than in lean mice. Metabolomic profiling in endometrial cancer tissues revealed significant differences between obese- and lean-mice. Enhanced energy metabolism was seen in obese- versus lean-mice as evidenced by increases in glycolytic and oxidative phosphorylation intermediates. In addition, dramatic increases in lipid biosynthesis and lipid peroxidation were found in the obese- versus lean-mice, whereas metformin obviously reversed the obesity-driven upregulation of lipid and protein biosynthesis in the obese mice. Conclusions: The obese state promoted tumor aggressiveness in the LKB1fl/fl p53fl/fl mouse model, accompanied by increases in energy metabolism, lipid biosynthesis, and markers of lipid peroxidation. Metformin had increased efficacy against endometrial cancer in obese versus lean mice and reversed the detrimental metabolic effects of obesity in the endometrial tumors. Taken together, it is likely that the unique metabolic milieu underlies metformin’s improved efficacy in treating endometrial cancer which develop in an obese host environment.
Keywords: Metformin, endometrial cancer, obesity, metabolomics, proliferation
Introduction
Endometrial cancer (EC) is the fourth most common cancer among women in the United States and has been increasing in frequency, with the rising rates of obesity as the potential major culprit. In 2019, approximately 61,880 new cases of EC will be diagnosed in the US, and 12,160 women will succumb to this disease [1]. Women with early-stage EC have a good prognosis; however, 30% of EC patients are diagnosed with advanced disease and have poor 5-year overall survival rates (15-40%). Obesity, diabetes, and insulin resistance are well-known factors associated with both increased risk of developing EC [2-4] and increased risk of death [5-9]. Among all obesity-related cancers, increasing body mass index is most strongly related to endometrial cancer risk, with at least 50-60% of all endometrial cancers attributable to obesity [10,11].
It has been postulated that hyperglycemia and hyperinsulinemia in obese patients provide abundant nutrients and growth factors to cancer cells, resulting in an ideal environment for pro-tumor signaling cascades, such as the insulin/insulin-like growth factor-1 (IGF-1) and PI3K/Akt/mTOR pathways [12]. Hyperinsulinemia, IGF-1, and IGF-1 receptor (IGF1R) levels are well-known to be important in EC development and progression [13,14]. Signaling through IGF-1R leads to activation of the PI3K/Akt/mTOR pathway, and components of this pathway are often mutated, amplified, or aberrantly expressed in EC [15-18]. Activation of the PI3K/Akt/mTOR pathway, through PIK3CA amplifications, PIK3CA/PIK3R1/PIK3R2 mutations and phosphatase and tensin homolog (PTEN) mutations and loss of function, is common in EC and has been linked to more aggressive tumor behavior [16-19].
In obese individuals [20,21], there is excess white adipose tissue (WAT) due to both individual adipocyte hypertrophy and stimulated adipocyte proliferation (hyperplasia). WAT is responsible for the storage of triglycerides as a fuel for energy [20,21]. Increased WAT, specifically visceral WAT, is associated with insulin resistance and alterations in metabolic signaling pathways leading to increased adipokine and inflammatory cytokine production, induction of lipolysis and greater release of free fatty acids into the circulation [20,21]. There is high demand for fatty acids by tumor cells as these are critical for building the lipid membrane bilayers in rapidly proliferating cells [20,21]. Thus, increased production of fatty acids is another mechanism by which obesity fuels tumor growth.
In essence, obesity is a high-energy, pro-inflammatory condition that culminates in increased growth factor signaling via the insulin/IGF-1 axis, as well as a nutrient-saturated environment via increased glucose, lipids, fatty acids (and other nutrients), all ultimately resulting in heightened cell proliferation, activation of AMP-activated protein kinase (AMPK) and excessive stimulation of the PI3K/AKT/mTOR pathway that mediates EC growth [22-25]. Therefore, obesity may create a unique tumor-enhancing environment that should be strategically and therapeutically targeted to improve outcomes for obese EC patients.
Metformin, an AMPK activator, is a classic biguanide drug that is widely used for treatment of type 2 diabetes and metabolic syndrome. Epidemiological evidence suggests that metformin lowers cancer risk and reduces cancer incidence and deaths among diabetic patients [26-28], including endometrial cancer [29-31]. This has led to the hypothesis that metformin has a role in cancer treatment and prevention for EC and many other cancers. Metformin is thought to exert anti-tumorigenic activity through indirect effects on the metabolic milieu and direct effects on the tumor through AMPK activation/mTOR inhibition and decreased fatty acid/lipid biosynthesis [32]. Its indirect effects are postulated to be due to a reduction in circulating glucose and insulin levels via inhibition of gluconeogenesis in the liver, and subsequent decreased insulin/IGF-1 growth factor stimulation in tumor cells [33]. AMPK is a central metabolic sensor involved in cellular energy homeostasis in energy deplete conditions including hypoxia, low glucose conditions, or oxidative stress and is activated by liver-kinase b1 (LKB1) and calcium/calmodulin-dependent protein-kinase (CaMKK) through phosphorylating the residue equivalent to Thr172. The LKB1-AMPK axis controls the mTOR pathway that regulates nutrient and growth factor inputs and controls cancer cell growth [34]. For its cellular or direct effects, metformin enters cells through transporters; inhibits mitochondrial respiratory complex 1, leading to suppression of tricarboxylic acid cycle flux; interrupts oxidative phosphorylation; and decreases mitochondrial ATP production [33,35-37]. The resulting cellular energetic stress from inhibition of complex 1 raises the AMP/ATP ratio, culminating in increased AMPK signaling and stimulated glycolysis and fatty acid beta-oxidation. Furthermore, activation of AMPK by metformin leads to inhibition of acetyl-CoA carboxylase (ACC) which in turn decreases fatty acid synthesis [38]. Lastly, the key lipogenic transcription factor, sterol regulatory element-binding protein-1 (SREBP-1), is a direct AMPK substrate, and metformin, through AMPK activation, suppresses SREBP-1 expression, blunting both lipogenesis and lipid accumulation [38].
We and others have shown that metformin-mediated AMPK activation decreases EC cell growth via inhibition of mTOR signaling [39-41]. In addition, metformin in combination with paclitaxel or cisplatin has synergistic anti-proliferative effects in EC cells [42,43]. Furthermore, in a mouse model of endometrial hyperplasia, metformin-induced anti-proliferative effects on the endometrium coincided with inhibition of targets of the mTOR pathway [44]. Thus, it has been hypothesized that a drug like metformin, which decreases circulating glucose, insulin, and free fatty acid levels and specifically disrupts the insulin/IGF-1, PI3K/AKT/mTOR and fatty acid/lipid biosynthetic pathways, may break the link between obesity and cancer, and emerge as a metabolically targeted therapy for obesity-driven cancers, such as EC. Only a few pre-clinical studies to date have evaluated host metabolic status as a potential biomarker or mediator for improved efficacy of metformin in the treatment of cancer [45-48]. In an effort to assess the impact of obesity on metformin’s effective action, we first created a transgenic mouse model of endometrial cancer bred on an FVB background with concurrent conditional loss of LKB1 and p53 (LKB1fl/fl p53fl/fl) in the mouse endometrium by local injection of the Adeno-Cre virus. After establishment of this mouse model, we aimed to assess the effects of metformin on cell proliferation in human endometrial cancer cells under varying glucose conditions and most importantly, endometrial tumor growth in LKB1fl/fl p53fl/fl mice under both obese and lean conditions.
Materials and methods
Cell culture and reagents
The human endometrial cancer cell lines, ECC-1 and Ishikawa, were used. The ECC-1 cells were maintained in 1640 medium with 5% fetal bovine serum (FBS), and the Ishikawa cells were cultured in DMEM medium supplemented with 5% FBS. All media was supplemented with 100 U/ml of penicillin and 100 ug/ml of streptomycin. The cells were cultured in a humidified 5% CO2 at 37°C. The glucose solution, MTT, and DMSO were purchased from Sigma-Aldrich (St. Louis, MO). The Cleaved Caspase-3 Activity Assay kit was bought from AAT Bioquest (Sunnyvale, CA). For the glucose-related studies, the cells were cultured in RPMI-1640 medium or DMEM medium (Cat #11879-020 and 11966-025, Gibco) containing 5% FBS in the presence or absence of various concentrations of glucose. All the primary antibodies for Ki-67, phosphorylated-S6, pan-S6, cyclin D1, CDK4, CDK6, Bcl-2, Mcl-1, cleaved caspase 3, PERK and α-tubulin were obtained from Cell Signaling Technology (Danvers, MA). The anti-p53 and LKB1 antibodies were from Santa Cruz (Dallas, TX). The enhanced chemiluminescence (ECL) detection reagents were purchased from GE Health care (Piscataway, NJ).
Cell proliferation assay
The ECC-1 and Ishikawa cell lines were seeded at 4000 cells/well in 96-well plates in their standard media for 24 hours. Cells were subsequently treated with varying concentrations of metformin (from 0. 1 to 5 mM) for 72 hours. MTT (5 mg/ml) was added to the 96-well plates at 5 μl/well for 1 hour. The MTT reaction was terminated through the addition of 100 μl of DMSO. Viable cell densities were determined by measuring absorbance of metabolic conversion of the colorimetric dye at 570 nm. For the glucose-related experiments, the ECC-1 and Ishikawa cells were seeded at 4000 cells/well in 96-well plates in standard media for 24 hours and then treated with different doses of metformin (from 0.1 to 5 mM) for 48 hours in the media supplied with 1, 5 and 25 mM glucose as well as without glucose. Cell proliferation was measured by MTT assay. Each experiment was performed in triplicate with N = 3 replicates per assay.
Colony formation assay
The ECC-1 and Ishikawa cells growing in log phase were seeded (600 cells/well in a 6-well plate) in their standard growth media. Cells were allowed to adhere for 24 hours, and then treated with metformin (0, 1 and 5 mM) for 24 hours. Cells were cultured at 37°C under different concentrations of glucose solution for 14 days, with media changes every third or fourth day. Cells were stained with 0.5% crystal violet, and colonies were counted under the microscope. Colony formation assays were performed in duplicate with three replicates per assay.
Cell cycle analysis
The ECC-1 and Ishikawa cells were seeded at 2.5 × 105 cells/well into 6-well plates and incubated overnight, and then treated with metformin in standard medium for 48 hours to measure cell cycle. The cells were then harvested and washed with phosphate buffered saline (PBS). The pellet was re-suspended and fixed in 90% pre-chilled methanol and stored overnight at -20°C. The cells were then washed with PBS again and re-suspended in 50 μl RNase A solution (250 μg/ml, 10 mM EDTA) for 30 minutes and then stained with 50 μl of staining solution [containing 2 mg/ml propidium iodide (Hayward, MA), 0.1 mg/ml Azide and 0.05% Triton X-100]. The final mixture was incubated for 15 minutes in the dark before analyzing with Cellometer (Nexelom, Lawrence, MA). The results were evaluated using FCS4 express software (Molecular Devices, Sunnyvale, CA). For the glucose-related experiments, the ECC-1 and Ishikawa cells were seeded at 2.5 × 105 cells/well into 6-well plates and incubated overnight in standard media, and then treated with 0.5 mM metformin for 36 hours in media with varying concentrations of glucose. Cell cycle progression was assessed by Cellometer. All experiments were performed in triplicate and repeated three times to ensure consistency of results.
Cleaved caspase 3 assay
Activity of cleaved caspase 3 was assessed with the Cleaved Caspase 3 Activity Assay kit to measure apoptosis. After cells were treated with metformin under varying concentrations of glucose in 96-well plate (6000 cells/well), 10 ml of caspase-3 assay loading buffer was added into each well, mixed gently and then the plates were incubated for 60 minutes at 37°C, 5% CO2. The fluorescence intensity was measured at an excitation wavelength of 350 nm and an emission wavelength of 450 nm using a plate reader from Tecan (Morrisville, NC).
Western immunoblot analysis
Total protein was extracted from the ECC-1 and Ishikawa cells using RIPA buffer (Boston Bioproducts, Ashland, MA). Protein samples with equal loading (30 µg) were separated by 10-12% SDS-PAGE and transferred onto polyvinylidene fluoride (PVDF) membranes. The membranes were blocked with 5% nonfat milk and then were incubated with a 1:2000 dilution of primary antibodies overnight at 4°C. The membranes were washed and incubated with a secondary peroxidase-conjugated antibody for 1 hour at room temperature. The membranes were developed using an enhanced ECL and the ChemiDoc Imaging System (Bio-Rad, Hercules, CA). After developing, the membranes were re-probed using antibody against α-tubulin or β-actin to confirm equal loading. The bands’ intensity were measured and normalized to α-tubulin or β-actin. Each experiment was repeated at least twice.
Generation of LKB1fl/fl p53fl/fl transgenic mice
The mouse floxed (fl) alleles of LKB1 and p53 were used in this study. LKB1loxP/loxP (FVB, 129S6-Stk11 tm1Rdp) conditional knockout mice with LoxP sites flanking exons 3 and 6 of the LKB1 gene, and p53loxP/loxP (FVB, 129P2-Trp53 tm1Brn) conditional mutation in the endogenous p53 gene (Trp53) with LoxP sites inserted into intron 1 and intron 10 of the p53 gene were obtained from the Mouse Models of Human Cancers Consortium Mouse Repository (NCI, Rockville, MD) [49,50]. LKB1loxP/loxP and p53loxP/loxP mice were intercrossed through multiple generations to create colonies of homozygous LKB1fl/fl p53fl/fl double conditional knockout mice. Mouse genomic DNA was extracted from distal toe clips as previously described [51]. LKB1fl/fl mice were genotyped using primers PCRS5: 5’-tct aac aat gcg ctc atc gtc atc ctc ggc-3’, LKB36: 5’-ggg ctt cc acct ggt gcc agc ctg t-3’ and LKB39: 5’-gag atg ggt acc agg agt tgg ggc t-3’ to yield a 220 bp band for wild type or a 300 bp band for floxed sequences [50]. p53fl/fl mice were genotyped using primers T008: 5’-CAC AAA AAC AGG TTA AAC CAG-3’ and T009: 5’-AGC ACA TAG GAG GCA GAG-3’. PCR results from wild type mice yielded a 288 bp band, while floxed mice yielded a 370 bp band [52]. Animal experiments were approved by our Institutional Animal Care and Usage Committee in accordance with NIH guidelines.
AdCre administration and treatments
For the evaluation of metformin in the LKB1fl/fl p53fl/fl genetically engineered endometrial cancer mouse model, 30 female mice were provided a HFD (obese group, 60% kcal from fat, Research Diets, New Brunswick, NJ) to induce obesity, and 30 female littermate control mice were provided a LFD (lean group; 10% kcal from fat) ad libitum, beginning at 3 weeks of age. Recombinant adenovirus Ad5-CMV-Cre (AdCre) was purchased from the University of Iowa Transfer Vector Core at a titer of 1011-1012 infectious particles/ml. Intrauterine Ad-Cre injections of LKB1fl/fl p53fl/fl mice were performed at 6-8 weeks of age to induce endometrial cancer as described with modifications [53,54]. In brief, mice were anesthetized with a mixture of ketamine (75 mg/kg) and medetomidine (1 mg/kg) by intraperitoneal injection. Under aseptic conditions, a laparotomy with a 1 cm longitudinal incision of the left side abdomen was performed to allow exposure of the left uterine horn with adjoining ovary. The ligatures were tied around the proximal (near uterine corpus) and distal (near oviduct) left uterine horn with a 4-0 Vicryl (Ethicon, New Brunswick, NJ). AdCre (5 µl) was injected into the left uterine cavity under a microscope using a syringe with a 30-gauge needle. The incisions were closed with clips. The body temperature of the mice was maintained using a heating pad throughout surgery until the mice regained consciousness. At 8 weeks after AdCre infection, each group was randomly divided with equal allocation into control (saline) and metformin treatment groups (obese + placebo, obese + metformin, lean + placebo, lean + metformin). The HFD- and LFD-fed mice were treated with metformin (250 mg/kg/day, oral gavage) or control saline gavage for 4 weeks. All mice were euthanized after 4 weeks of metformin or control treatment.
Immunohistochemical analysis
The mouse tumor tissue was formalin-fixed and paraffin-embedded. Slides (5 μm) were first incubated with protein block solution (Dako) for 1 hour and then with the primary antibodies for Ki-67 (1:400), phosphorylated-S6 (1:300), phosphorylated-AMPK (1:100), cyclin D1 (1:50) cleaved caspase 3 (1:300), LBK1 (1:200) and p53 (1:300) for 2 hours at room temperature. The slides were then washed and incubated with appropriate secondary antibodies at room temperature for 1 hour. The slides were washed, and the specific staining was visualized using the Signal Stain Boost Immunohistochemical Detection Reagent (Cell Signaling Technology), according to the manufacturer’s instructions. Individual slides were scanned using the Aperio™ ScanScope (Aperio Technologies, Vista, CA), and digital images were analyzed for target protein expression using Aperio™.
Biochemical analysis
Serum insulin, TNF-α, PAI-1, IL-6, and leptin were assessed using the Luminex technique. The multiplex ELISA kits were obtained from Milliplex Map (Millipore CA). Assays were run in triplicate according to the manufacturer’s protocols. Data was collected using the Luminex-200 system (Austin, TX).
Metabolomic profiling
Metabolomic profiling was performed on the endometrial tumors from control and metformin treated lean and obese mice. Samples were analyzed by Metabolon (Research Triangle Park, NC) according to their standard protocols (N = 5 mice/group) [55-58], and per our previous publications [59-62]. Briefly, unbiased global metabolomic profiling was achieved using methanol extracts of tumor tissues normalized to serum volume or tissue weight. Analysis of extracts consisted of either ultrahigh performance liquid chromatography (Waters Corporation, Milford, MA) coupled with tandem mass spectrometry (UHPLC/MS/MS; Thermo-Finnigan, San Jose, CA) in positive and negative ionization modes, or via gas chromatography/MS analysis (Thermo-Finnigan). Metabolites in tumor tissues were positively identified by matching chromatographic retention time, mass and MS/MS fragmentation patterns to a reference library of over 2500 purified, authenticated biochemicals. Data are presented as relative measures of “scaled intensity” and median scaling to 1. Missing values were imputed with the minimum.
Endometrial tumors from lean and obese LKB1fl/fl p53fl/fl mice have biochemical differences reflecting baseline differences in their endometrial tumor metabolism. To compare metformin’s effects between lean and obese groups, while accounting for their baseline metabolic differences, T-tests analyzed the biochemical ratio in metformin-treated groups, normalized by the control groups (For example, (Obese + Metformin)/average (Obese + Vehicle) vs (Lean + Metformin)/average (Lean + Vehicle).
Statistical analysis
Statistical differences between experimental groups were determined using statistics software within GraphPad Prism (GraphPad Software, Inc., La Jolla, CA). The non-parametric ANOVA was used to test if there were differences in metformin’s effects in obese versus lean LKB1 mice. Non-paired Student’s t test was then used to determine p values. Statistical significance was defined to be P < 0.05.
Results
Effect of glucose concentration on inhibition of cell proliferation by metformin
We have previously found that metformin significantly inhibited endometrial cancer cell growth whereas high glucose concentrations promoted endometrial cancer cell proliferation in a dose-dependent manner [39,42,63]. To define whether glucose concentrations modulate the anti-proliferative effects of metformin, two endometrial cancer cell lines, ECC-1 and Ishikawa, were treated in their standard culture media with three varying concentrations of glucose: low glucose (hypoglycemia, LG, 1 mM), normal glucose (euglycemia, NG, 5 mM) and high glucose (hyperglycemia, HG, 25 mM) for 48 hours. Cell proliferation was assessed by MTT assay. Metformin inhibited cell growth in both cell lines at all three different glucose concentrations. However, metformin was most effective under hypoglycemic conditions as compared to euglycemic conditions, with a mean IC50 between 1.3 mM to 2.1 mM. Metformin was least effective under hyperglycemic conditions for both cell lines (Figure 1A and 1B).
Figure 1.
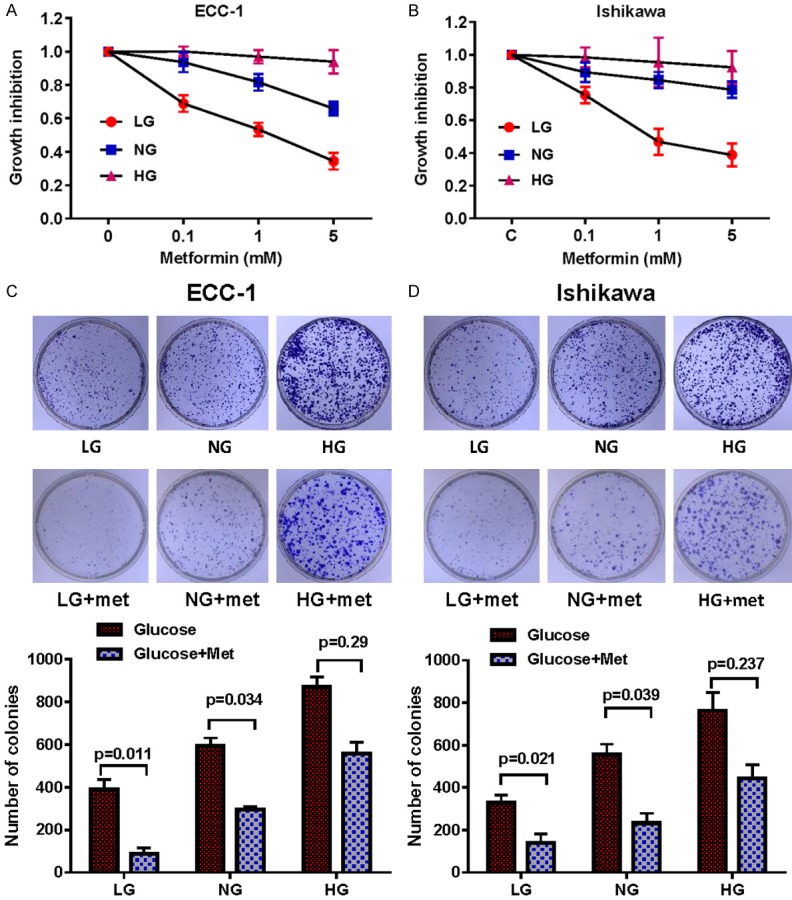
Metformin suppressed cell proliferation and colony formation, with the most pronounced inhibitory effects seen under low glucose conditions. The endometrial cancer cell lines, ECC-1 and Ishikawa, were cultured in glucose-free media supplemented with 25 mM glucose (HG), 5 mM glucose (NG) and 1 mM glucose (LG) for 24 h and then treated with the indicated concentration of metformin in 96-well plates for 48 h. Cell proliferation was assessed by MTT assay. LG increased sensitivity to metformin in the inhibition of cell proliferation in both cell lines (A and B). The effect of metformin on the long term growth in ECC-1 and Ishikawa was assessed through a colony-forming assay. LG increased sensitivity to metformin in the inhibition of colony formation in both cell lines (C and D). The results are shown as the mean of ± SE of triplicate samples and are representative of three independent experiments. *P < 0.05, **P < 0.01.
Given that in vitro colony formation assays are excellent measures of long term tumor cell survival, we then assessed whether metformin under low, normal and high glucose conditions had an effect on the colonization ability of the ECC-1 and Ishikawa cells. Long term exposure to hyperglycemia over 14 days significantly increased the clonogenicity of the ECC-1 and Ishikawa cells by 1.3-1.4-fold and 2.0-2.3-fold, respectively, as compared with the euglycemic and hypoglycemic groups. Metformin was least effective in decreasing colony formations in hyperglycemic media compared to euglycemic and was most effective in hypoglycemic conditions. For ECC-1 and Ishikawa cells, treatment with metformin under hyperglycemic conditions caused a reduction of 36% and 43% in colony formation, while in hypoglycemic conditions metformin had approximatively 78% and 60% reduced clonogenicity in ECC-1 and Ishikawa cells, respectively (Figure 1C and 1D). Together, these results confirm that glucose concentration has the ability to modulate sensitivity to metformin in EC cells in vitro.
Effect of glucose concentrations on induction of apoptosis by metformin
To evaluate the underlying mechanism of growth inhibition by metformin in the presence of high and low glucose concentrations, the activity of cleaved caspase 3 was assessed as a measure of apoptosis. Incubation of the EC cells lines under hypoglycemic conditions increased cleaved caspase 3 activity by approximately 20% in both cell lines compared with the normal glucose groups. Metformin significantly increased cleaved caspase 3 activity under hypoglycemic conditions in both cell lines (P < 0.05) (Figure 2A and 2B). In the ECC-1 cell line, metformin also increased cleaved caspase 3 activity under normal glucose and hyperglycemic conditions (P < 0.05), but failed to do this in the Ishikawa cell line. Western blotting analysis found that metformin reduced expression of the anti-apoptotic proteins BCL-2 and MCL-1 in a dose-dependent manner after treatment for 14 hours. Treatment of the cells with metformin caused greater reduction of BCL-2 and MCL-1 expression in the hypoglycemic groups as compared to the hyperglycemic groups in both cell lines (Figure 2C and 2D). These results suggest that glucose levels modulate mitochondrial apoptosis induced by metformin in EC cells.
Figure 2.
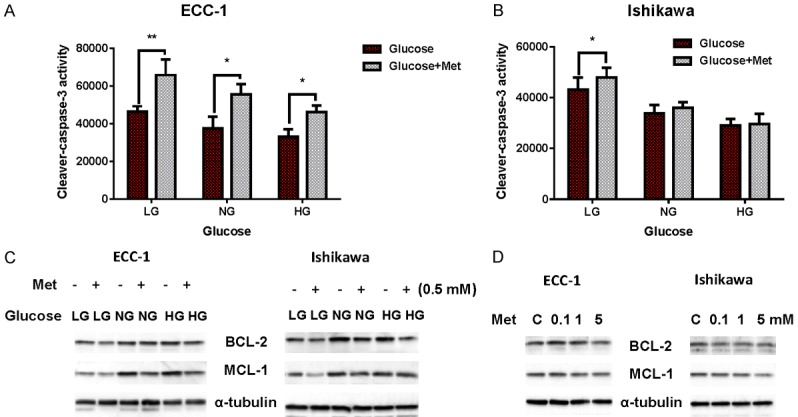
Induction of apoptosis by metformin is glucose-dependent. The ECC-1 and Ishikawa cells were treated with metformin for 12 h in 25 (HG), 5 (NG) and 1 (LG) mM glucose containing media. Cleaved caspase 3 activity was assayed by ELISA assay. Metformin increased cleaved caspase 3 activity under LG conditions in both cell lines (A and B); metformin increased cleaved caspase 3 activity under NG and HG conditions for ECC-1 but not Ishikawa. Western blotting was used to evaluate the expression of BCL-2 and MCL-2 in both cell lines after treatment with metformin under varying glucose conditions for 18 h. Metformin decreased expression of the BCL-2 and MCL-2 proteins under all glucose conditions (C). Metformin decreased BCL-2 and MCL-2 expression in a dose-dependent manner in both cell lines (D). The results are shown as the mean ± SD and are representative of three independent experiments. *P < 0.05, **P < 0.01.
Effect of glucose concentrations on induction of cell cycle arrest by metformin
To assess the impact of glucose levels on the underlying mechanism of growth inhibition induced by metformin in the endometrial cancer cells, the cell cycle profile was analyzed by Cellometer. Metformin treatment resulted in cell cycle G1 phase arrest in a dose-dependent manner in both the endometrial cancer cell lines when compared with the control groups (Figure 3A and 3B). Hypoglycemia increased the G1 population from 48.1% to 55.8% in ECC-1 cells, and from 45.1% to 56.3% in Ishikawa cells, respectively, compared with euglycemic groups. A combination of metformin and hypoglycemia strongly induced cell cycle G1 arrest compared with either the hyperglycemic groups or normal groups in the both cell lines (Figure 3C and 3D).
Figure 3.

Induction of cell cycle G1 arrest by metformin is glucose-dependent. The ECC-1 and Ishikawa cells were cultured in low glucose (LG), normal glucose (NG) and high glucose (HG) conditions and then treated with metformin for 24 h. Cell cycle was assessed by Cellometer. Metformin induced cell cycle G1 arrest in a dose-dependent manner in both cell lines (A and B). Treatment with metformin under LG conditions led to greater cell cycle G1 arrest compared with NG and HG condition in both cell lines (C and D). Western blotting showed that metformin decreased the expression of CDK4, CDK6 and cyclin D in a dose-dependent manner in both cell lines after 18 h of exposure (E). The ECC-1 and Ishikawa cells were cultured for 24 h and then treated with metformin with varying concentrations of glucose for 18 h. HG induced an increase in expression of cyclin D1 and CDK6 as compared to NG and LG. Metformin inhibited expression of cyclin D1 and CDK6 under all glucose conditions with greater effects seen under LG conditions (F). The results shown are one of three independent experiments.
To further understand the molecular events underlying the observed G1 arrest by metformin under varying glucose concentrations, the expression of CDK4, CDK6 and Cyclin D1 were measured by Western blotting. Under normal glucose conditions, metformin reduced the expression of CDK4, CDK6 and Cyclin D in a dose-dependent manner in both endometrial cancer cell lines after 24 hours of treatment (Figure 3E). In addition, hypoglycemia alone also reduced the expression of CDK4, CDK6 and Cyclin D1. ECC-1 and Ishikawa cells under hypoglycemic conditions were more responsive to metformin in the reduction of CDK6 and Cyclin D1 expression as compared with normal glucose and hyperglycemic conditions (Figure 3F). These results indicate that both hypoglycemia and metformin treatment leads to G1 phase arrest, and that hyperglycemia partially antagonizes induction of G1 arrest by metformin in the EC cells.
Effect of metformin on AMPK/mTOR/S6 pathways in different glucose conditions
Given that AMPK and mTOR pathways are central regulators of the energy status and glucose metabolism of the cell [63-65], we next investigated the effect of metformin on AMPK and mTOR pathways under high, euglycemic, and low glucose conditions in the EC cell lines. The ECC-1 and Ishikawa cells were treated with various doses of metformin for 24 hours. Phosphorylation of AMPK was significantly increased and phosphorylation of S6 was decreased by metformin treatment in a dose-dependent fashion in both cell lines, suggesting inhibition of downstream signaling targets of mTOR by metformin, as expected (Figure 4A). Furthermore, we examined the effects of metformin on AMPK and S6 protein expression under different concentrations of glucose. Hypoglycemia caused a slight increase in AMPK phosphorylation and a significant decrease in S6 phosphorylation in both cell lines compared with normal and high glucose conditions for 24 hours. Hyperglycemia decreased phosphorylation of AMPK and increased phosphorylation of S6 in both cells as compared with normal glucose and hypoglycemic conditions. Metformin more effectively activated phosphorylation of AMPK in both cell lines after 24 hours of treatment under hypoglycemic versus normal glucose and hyperglycemic conditions. In particular, metformin resulted in a 2.5 fold increase in phosphorylated AMPK in ECC-1 cells and a 2.1 fold increase in Ishikawa cells under hypoglycemic compared to hyperglycemic conditions for 24 hours of treatment. In contrast, phosphorylation of S6 was more affected by metformin under euglycemic and hyperglycemic conditions versus hypoglycemic conditions after 24 hours of treatment. Metformin reduced 45% and 67% phosphorylated S6 expression in normal glucose conditions in ECC-1 and Ishikawa cells respectively, while in low glucose conditions, metformin only decreased approximatively 20 to 25% expression of phosphorylation of S6 in both cells. These results suggest that phosphorylation of AMPK and S6 may be involved in sensitivity to metformin under varying glucose conditions in EC cells (Figure 4B).
Figure 4.
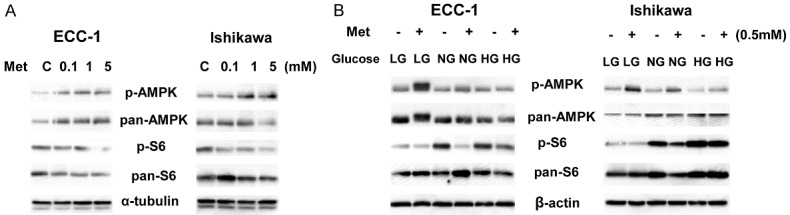
Effect of glucose concentrations on AMPK/mTOR pathways by metformin. The ECC-1 and Ishikawa cells were treated with metformin in regular media for 24 h. The expression of phosphorylated AMPK and pS6 was analyzed using western blotting. Metformin increased expression of AMPK phosphorylation and decreased phosphorylation of S6 in a dose-dependent fashion in both cells (A). Metformin was more effective in activation of AMPK phosphorylation and less effective in inhibition of phosphorylation of S6 under LG conditions than NG and HG conditions in both cell lines (B). The results shown are one of three independent experiments.
Generation of a mouse model of LKB1-deficient and p53 inactivation invasive endometrial cancer
Deficiency of LKB1 has been implicated in the carcinogenesis of endometrial cancer, and loss of LKB1 in mice has been found to lead to invasive endometrial cancer [54]. However, mice with the LKB1 homozygous floxed allele (LKB1fl/fl) have a long latency for tumor growth and only 65% penetrance by 9 months of age [54]. Although p53 mutations occur in 10-20% of endometrial adenocarcinoma, it is associated with aggressive behavior and increased risk of recurrence [66]. Therefore, we hypothesized that intercrossing LKB1fl/fl and p53fl/fl mice would result in a shorter latency and increased penetrance of endometrial cancer (i.e. LKB1fl/fl p53fl/fl mouse model). In addition, this mouse model is most reflective of high grade endometrioid ECs in women in that LKB1 loss is most commonly seen in grade 3 versus grade 1/2 ECs and is accompanied by p53 loss about 20% of the time [67].
We created the homozygous LKB1fl/fl p53fl/fl double conditional knockout mice through multiple breeding by genotype (Figure 5A). To inactivate the LKB1fl/fl p53fl/fl alleles, we injected AdCre into the unilateral uterine cavity of LKB1fl/fl p53fl/fl mice, and the contralateral uterus served as a control (Figure 5B). It has been previously found that by 55 weeks of age 53% of LKB1f/+ mice had uterine neoplasms and by 36 weeks of age 65% of LKB1f/f mice developed uterine tumors [54]. In the LKB1fl/fl p53fl/fl mouse model, we found complex atypical hyperplasia (CAH) at 4-5 weeks and invasive cancer at 6-7 weeks post AdCre injections in the left uterine horn injected with AdCre (Figure 5C). Analysis of H&E stained sections of the left uterine horns revealed well-differentiated adenocarcinoma (Figure 5D). Within 8 weeks post induction, tumors of endometrioid histology develop in the affected uterine horn in 93% of the mice, with a median survival of 144 days after AdeCre injection, while the un-injected uterine horn remains normal (Figure 5E). Immunohiostochemical analysis confirmed that LKB1 and wild type p53 were positive in the normal endometrium and markedly reduced expression of LBK1 and p53 was observed in the majority of the tumor tissues (Figure 5F).
Figure 5.
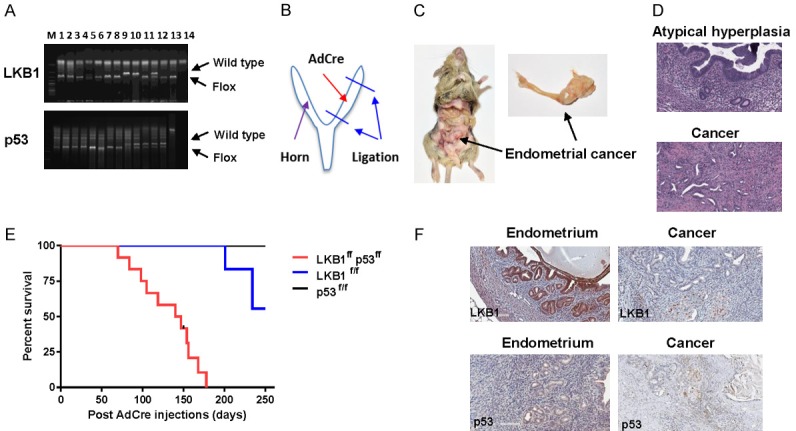
Concurrent loss of LKB1 and p53 in the mouse endometrium led to the development of invasive endometrial tumors. Genotyping of generated LKB1fl/fl p53fl/fl mice in mouse toes. The genotypes were confirmed by polymerase chain reaction using specific deigned primers (A). AdCre injection procedure: After laparotomy under general anesthesia, the left side of uterus was closed by suture ligation as shown in (B). Adcre (5 ul) was injected by puncturing the uterine wall with a 30 gauge needle. Gross pictures of the intact uterus at 12 weeks after injection. An endometrial tumor is located in the left side of uterus (C). Histopathology of normal uterus (top) and LKB1 p53-deficient endometrial carcinomas (bottom, D). Kaplan-Meier survival curves for LKB1fl/fl, p53fl/fl and LKB1fl/fl p53fl/fl mice of indicated genotypes as a function of days after Ade-cre administration (E). IHC results showed that expression of LKB1 and p53 was decreased in endometrial tumor tissues of LKB1fl/fl p53fl/fl (F).
Metformin inhibits tumor growth in the LKB1fl/fl/p53fl/fl mouse model of endometrial cancer
To determine whether obesity could increase the anti-tumorigenic potential of metformin in vivo, we fed LKB1fl/fl p53fl/fl mice with either a HFD (obese) at 3 weeks of age to induce obesity or fed them LFD to remain as lean controls [59]. The initial average body weight of the obese mice when starting treatment was 41.98 gm, while that of lean mice was 31.86 gm (P < 0.01, Figure 6A). The levels of serum cholesterol (non-fasting) in obese mice was 31% higher than those of lean mice (P = 0.014), prior to metformin treatment (Figure 6B). There was no significant difference in random blood glucose levels between obese and lean mice over the course of the diet treatment (data not shown). The obese and lean mice were treated with metformin (250 mg/kg, oral gavage, 4 weeks) at 8 weeks after AdCre injection to induce endometrial tumor growth. During treatment, tumor growth was monitored by palpation twice a week. Regular twice-weekly measurements yielded no changes in body weight during metformin or placebo treatment. Obesity accelerated tumor growth with a 1.9-fold increase in tumor weight at sacrifice compared to mice fed a LFD. Both obese and lean mice treated with metformin had a significant reduction in tumor weight (Figure 6C and 6D, P < 0.05). Metformin had a more pronounced impact on the tumor growth of obese mice with a 77% reduction in tumor weight in obese mice (1.51 g in control versus 0.34 g in metformin mice, P < 0.01) compared to a 62% reduction in tumor weight with metformin treatment in lean mice (0.79 g in control mice versus 0.30 g in metformin mice, P < 0.05). These data suggest that obesity promotes endometrial tumor growth and metformin effectively suppresses the endometrial tumor growth in both obese and lean LKB1fl/fl p53fl/fl mice, with greater anti-tumorigenic effects in the setting of obesity.
Figure 6.

Metformin inhibited tumor growth in the LKB1fl/fl p53fl/fl endometrial cancer mouse model. LKB1fl/fl p53fl/fl mice were fed high fat diet (HFD) or low fat diet (LFD) at 3 weeks of age to induce obesity. The mice were divided into four groups: obese, obese + metformin, lean and lean + metformin. HFD induced an increase in body weight in the LKB1fl/fl p53fl/fl mice (A). The levels of cholesterol were significantly increased in obese mice compared with lean mice (B). The obese and lean mice in both groups were treated with metformin (250 mg/kg, oral gavage) or placebo for 4 weeks. Obesity promoted tumor growth in obese mice versus lean mice. Metformin significantly reduced tumor weight in the obese and lean mice, with a great impact on tumor weight in obese mice (C and D). The changes of Ki-67, phosphorylated-S6, phosphorylated-AMPK, cyclin D and cleaved caspase 3 were assessed by immunohistochemistry in the endometrial cancer tissues. The expression of Ki-67, cyclin D and phosphorylated-S6 was significantly reduced and cleaved caspase 3 and phosphorylated-AMPK was increased in both groups after metformin treatment (E). *P < 0.05, **P < 0.01.
To further investigate the anti-tumorigenic mechanism of metformin in vivo, the expression of Ki-67, phosphorylated S6, phosphorylated AMPK, cyclin D1 and cleaved caspase-3 in the endometrial tumor tissues was evaluated by immunohistochemistry (Figure 6E). As expected, the expression of the proliferation marker Ki-67 was significantly reduced in the endometrial tumors following metformin treatment, quantified as a 65% decrease in tumors of obese mice and 18% in tumors of lean mice compared to controls (P < 0.05). Metformin treatment increased the cleaved caspase-3 positive index by 2.3-fold in obese mice and 2.1-fold in lean mice, respectively (P < 0.05). Consistent with our results in vitro, metformin significantly reduced the expression of phosphorylated S6 and increased the expression of phosphorylated AMPK in obese and lean mice compared with the untreated mice, suggesting that metformin inhibited tumor growth through the AMPK/mTORC1 pathway in vivo (P < 0.05). Given that cyclin D1 severs as a cell cycle protein that promotes cellular proliferation in endometrial cancer, we then analyzed effect of metformin on cyclin D1 in vivo. Metformin dramatically decreased the expression of cyclin D1 in only obese mice, resulting in a 42% decrease in mean expression (P < 0.05). Together, these results further confirm that metformin inhibits tumor growth of EC potentially via targeting of the AMPK/mTOR/S6 pathway and activation of apoptosis in vivo in both obese and lean mice.
Effect of metformin on inflammatory factors in the LKB1fl/fl p53fl/fl mouse model of endometrial cancer
Given that obesity-associated inflammatory adipokines contribute to insulin resistance and promote carcinogenesis in EC [68], serum leptin, IL-6, TNF-α, PAI-1, and insulin were determined by multiplex ELISA in obese and lean LKB1fl/fl p53fl/fl mice. Metformin significantly increased insulin levels in the obese mice (P < 0.05) but did not change concentrations in lean mice. Similarly, metformin increased serum leptin in obese mice but decreased serum leptin in lean mice (P < 0.05), suggesting that metformin had differing effects on insulin and leptin pathways in obese versus lean mice (Figure 7A and 7B). In addition, obesity significantly elevated serum cytokines IL-6 and TNF-α. Serum IL-6 and TNF-α levels were significantly reduced by metformin in both obese and lean mice groups compared to control groups (P < 0.05) (Figure 7C and 7D). However, metformin only decreased PAI-1 levels in obese mice and not lean mice (Figure 7E).
Figure 7.
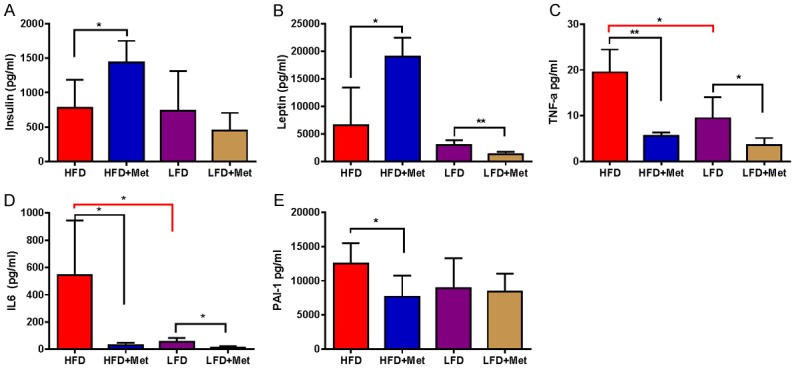
Effect of metformin on inflammatory factors in the LKB1fl/fl p53fl/fl mouse model of endometrial cancer. Serum leptin, IL-6, TNF-α, PAI-1 and insulin were determined by multiplex ELISA in LKB1fl/fl p53fl/fl mice after completion of metformin treatment. In obese mice, metformin increased serum insulin and leptin (A and B) and decreased IL-6, TNF-α and PAI-1 production (C-E). In lean mice, metformin only reduced serum leptin, IL-6 and PAI-1 compared with controls. *P < 0.05, **P < 0.01.
Metabolic effects of obesity and metformin in the endometrial tumors from LBK1fl/fl/p53fl/fl mice
Given that obesity is attendant to profound metabolic changes that promote tumor growth [65], we compared the metabolic profiles of endometrial tumors from obese and lean mice. Overall, 251 up- or down-regulated metabolites differentiated endometrial tumors in obese versus lean LKB1fl/fl p53fl/fl mice (Figure 8A and Supplementary File). The most striking difference between obese versus lean endometrial tumors was the dramatic stimulation of lipid biosynthesis and lipid peroxidation in obesity as indicated by increases in free fatty acids (FFAs up to 38 fold), plasmologens (up to 3 fold), phospholipids (up to 11 fold), lysolipids (up to 8 fold), diacylglycerols (DAGs up to 27 fold), monacylglycerols (MAGS up to 250 fold), and sphingolipids (up to 3 fold). Enhanced energy metabolism was also detected in obese versus lean endometrial tumors as evidenced by increases in glycolytic (i.e. glucose-6-phosphate and fructose-6-phosphate) and oxidative phosphorylation (i.e isocitrate, fumarate and malate) intermediates. Protein biosynthesis in the endometrial tumors was also heightened in obesity as demonstrated by increases in amino acids and dipeptides (up to 10 fold).
Figure 8.

Metabolic effects of obesity and metformin on endometrial tumors in the LBKfl/flp53fl/fl mouse model. Metabolomic profiling indicated that obesity significantly increased energy metabolism and protein/lipid biosynthesis, as demonstrated by increases in glycolytic and oxidative phosphorylation intermediates, amino acids, peptides, nucleotides, fatty acids, and phospholipids in endometrial tumor tissues of obese compared with lean mice (Red = increased, Green = decreased, P < 0.05) (A). Metformin treatment decreased free fatty acids, phospholipids, lysolipids and sphingolipids in the obese- versus lean-endometrial tumors (B), and increased protein degradation markers (C), indicative of reversal of obesity-driven upregulation of lipid and protein biosynthesis. Schematic of the effects of obesity and metformin in obese versus lean endometrial tumors (A and C).
We next investigated the effects of metformin in the endometrial tumors of lean and obese mice using metabolomic profiling to determine specific pathways that may mediate the improved benefit of metformin in the obese state. The metabolic profiles of endometrial tumors from obese and lean LKB1fl/fl p53fl/fl mice treated with either vehicle or metformin were compared. Free fatty acids, phospholipids, lysolipids, MAGS, DAGs and sphingolipids were decreased and 3-hydroxybutyrate was increased by metformin treatment in tumors derived from obese mice compared to lean, suggesting that lipids were being degraded and/or oxidized (Figure 8B). Protein degradation markers were also decreased in obese endometrial tumors with metformin treatment, suggesting reduced protein turnover and biosynthesis. Thus, metabolomic profiling of the endometrial tumors revealed that metformin reversed obesity-driven upregulation of lipid and protein biosynthesis. We hypothesize that this metabolic obese signature partially underlies metformin’s improved efficacy in treating tumors. A proposed schematic of the impact of obesity on the endometrial tumors in LKB1fl/fl p53fl/fl mice and resulting effect of metformin treatment is summarized in Figure 8C.
Discussion
Obesity, insulin resistance and hyperglycemia/hyperinsulinemia are all thought to contribute to the pathogenesis of EC, potentially through activation of insulin/IGF-1 and PI3K/Akt/mTOR signaling pathways [68,69]. Our previous studies have found that metformin inhibited EC cell proliferation through activation of AMPK and inhibition of mTOR pathways in vitro [39]. In this study, we investigated the impact of varying glucose levels on the anti-tumorigenic activity of metformin in human endometrial cancer cell lines as well as the efficacy of metformin on tumor inhibition in LKB1fl/fl p53fl/fl mice under obese and lean conditions. We found that metformin inhibited EC cell proliferation via activation of the AMPK pathway, as we previously demonstrated. In addition, metformin induced G1 phase cell cycle arrest and apoptosis under both hypoglycemic and hyperglycemic conditions. However, the sensitivity of EC cells to metformin was greatly enhanced under low versus normal and high glucose conditions. Obesity promoted endometrial tumor growth in LKB1fl/fl p53fl/fl mice, and the metabolic profiles of the endometrial tumors were distinct between obese and lean mice, with striking up-regulation of lipid biosynthesis in the setting of obesity. Metformin significantly reduced tumor growth in both obese and lean LKB1fl/fl p53fl/fl mice. Most importantly, metformin had greater anti-tumor efficacy in obese as compared to lean mice that aligned with distinct metabolic effects of metformin that were dependent on obesity status [59,70].
Most cancer cells use aerobic glycolysis as a means of energy production, regardless of whether they are under normoxic or hypoxic condition. Consumption of glucose by cancer cells is typically much higher than that of normal cells [59,63]. Heavy consumption of glucose and increased glycolysis are essential to generate both catabolic and anabolic precursors for the synthesis of DNA, RNA, proteins, and lipids for cancer cell growth. Our previous studies found that glucose is essential for ovarian and endometrial cancer cell growth and survival, and metformin can induce a metabolic shift in ovarian and endometrial cancer cells through reduced oxidative phosphorylation [63,64,70,71]. In addition, the combination of metformin and 2-deoxyglucose (2DG), a compound that blocks glycolysis, exerts a more deleterious effect on cancer cell viability than treatment with metformin or 2DG alone [72]. Low glucose conditions or glucose deprivation potentiates the cytotoxicity of metformin in breast, thyroid and ovarian cancer cells by reducing the capacity for metformin to stimulate glycolysis in vitro and in vivo [73-75]. High glucose conditions protect against metformin cytotoxicity by providing a fuel source for glycolysis, which maintains cellular ATP levels even when metformin blocks mitochondrial oxidative phosphorylation [73]. Consistent with these results, we found that metformin effectively inhibits cell proliferation in a dose-dependent manner in EC cells, such that the greatest repression was observed under hypoglycemic conditions. The precise mechanisms to explain this difference in in vitro sensitivity to metformin remains unknown. Several groups have found that the effect of glucose on metformin activity was dependent on inhibition of the mTOR pathway and potentially independent of the AMPK pathway [73,75,76]. Herein, our data showed that metformin, under normal or high versus low glucose conditions, caused a dramatic decrease in phosphorylation of S6, a downstream target of the mTOR pathway, while at the same time metformin significantly increased the phosphorylation of AMPK in both cell lines under low glucose conditions to a greater extent than in normal or high glucose conditions. This suggests that the AMPK/mTOR pathways are involved in inhibition of cell proliferation by metformin under varying glucose conditions in EC cells.
Exposure to metformin has been shown to induce cell cycle arrest in G1 phase and increase cell apoptosis in different types of cancers including EC cells, culminating in decreased cell viability [39,77-79]. Given that metformin increases glucose uptake and stimulates glycolysis, the increased rate of glucose consumption in the presence of metformin in low glucose medium results in an earlier onset of glucose starvation and more cell death in cancer cells in vitro [80]. Treatment of thyroid cancer cells with metformin under low glucose conditions was associated with induction of endoplasmic reticulum stress, autophagy, and oncosis, but not induction of caspase cascade expression [75]. In addition, the combination of metformin and 2DG had a significant effect on cell cycle G2 arrest in p53-deficient prostate cells as well as induction of apoptosis in doxorubicin-resistant breast cancer cells [72,81]. Menendez et. al. found that metformin mostly caused cell cycle arrest without signs of apoptotic cell death under high glucose conditions, while under glucose withdrawal stress, metformin circumvented the ability of oncogenes such as HER2 to protect cancer cells from glucose-deprivation apoptosis in breast cancer cells [74]. These findings suggest that glucose is a critical component in determining sensitivity to metformin-induced cytotoxicity, and genetic background may also play a role in cellular response to metformin under hypoglycemic conditions in breast cancer cells [74]. In our study, we evaluated the effect of metformin on apoptosis and the cell cycle under varying glucose conditions, including hyperglycemia, normal glucose and hypoglycemia. We found different patterns of change in caspase 3 activity between the Ishikawa and ECC-1 cell lines. We observed metformin induced caspase 3 activity under low, normal and high glucose conditions in the ECC-1 cells, whereas caspase 3 activity was only increased under low glucose conditions in the Ishikawa cells. Interestingly, metformin induced cell cycle G1 arrest under low and normal glucose conditions in both cell lines, whereas cycle G1 arrest was observed under high glucose conditions in the Ishikawa but not in the ECC-1 cell line. These results highlight the importance of glucose levels in modulating the regulation of cell cycle progression and apoptosis in EC cells treated with metformin.
Loss of LKB1 is observed in 20% of primary endometrial adenocarcinomas, most of which are high grade tumors, and transgenic mouse models have revealed a uniquely potent role of LKB1 as an endometrial cancer tumor suppressor [67,82]. Lack of LKB1 leads to hyperactivation of the mTOR pathway and elevated levels of IGF-I and is thought to contribute to carcinogenesis and progression of EC [83,84]. Inactivation of LKB1 by AdCre in the uterus is sufficient to induce endometrial adenocarcinoma with incomplete penetrance and long latency, even though LKB1 has remarkably tissue specific attributes [84]. Both long latency and incomplete penetrance limits the use of inactivation of LKB1 as a pre-clinical mouse model for EC. For this reason, conditional LKB1 mouse models must be crossed with other mouse models with additional cooperating oncogenic mutations in order to overcome these limitations. LKB1 mice either crossed with Sprr2f-Cre or PTEN floxed mice demonstrate a synergistic effect in shortening tumor latency and increasing penetrance for endometrioid adenocarcinomas that arise [83,85]. Although p53 mutations are often detected in serous endometrial carcinomas, p53 mutations are also associated with high grade endometrioid endometrial adenocarcinomas which also bear an unfavorable prognosis among endometrioid endometrial cancers [82]. Mice with a combined deletion of endometrial p53 and PTEN had a shorter lifespan with an exacerbated disease state [82]. More importantly, sequence analysis revealed four potential binding sites for p53 in the LKB1 promoter region along with a strong correlation between p53 and LKB1 protein expression levels in high grade endometrial cancers [67], indicating that p53 may regulate LKB1 activity and possibly contribute to the more aggressive phenotype seen in ECs with both LKB1 loss and p53 mutations [67]. In contrast to LKB1fl/fl mice, we found that the LKB1fl/fl p53fl/fl mouse model resulted in a shorter latency (8 weeks) and lifespan (median of 144 days) and an increase in penetrance (93%) compared to LKB1-deficient mice (65% penetrance at 36 weeks of age). The mice started to develop invasive adenocarcinomas by 14 to 16 weeks of age (8 weeks after AdCre injections), and these tumors were confined to the uterus with no evidence of extrauterine spread, suggesting p53 loss may increase sensitivity of the uterus to LKB1 loss [86]. Since loss of p53 does not cause endometrial cancer in p53fl/fl mice, it will be of interest to further investigate functional interactions between p53 and LKB1 in EC [82,86].
Given that obesity is a major risk factor for the development of EC and is associated with worse prognosis, we evaluated the effects of metformin after first inducing obesity through dietary changes (HFD versus LFD) in the LKB1fl/fl p53fl/fl mouse model. Similar to our previous studies in the K18-gT121 +/-; p53fl/fl Brca1fl/fl (KpB) genetically engineered mouse model of high grade serous epithelial ovarian cancer [70], exposure to a HFD significantly increased tumor size and weight in the LKB1fl/flp53fl/fl mouse model. Metformin reduced tumor growth in both obese and lean mice, however, metformin showed a more potent inhibitory effect on tumor weights in obese as compared to lean mice, suggesting that metformin may be a more beneficial therapeutic strategy in an obese versus lean host. Similar results have been reported in mouse models of breast and lung cancer, as well as our own work in ovarian cancer in that metformin was more effective in decreasing tumor growth in animals fed a HFD compared to a LFD or standard diet [45,87-89]. Furthermore, in a randomized, placebo-controlled preoperative window study in patients with breast cancer, women with both higher body mass index (BMI) and Homeostatic Model Assessment of Insulin Resistance (HOMA) indexes had a greater response to metformin as evidenced by a decrease in Ki-67 staining [90]. However, in striking contrast, metformin treatment was found to elicit greater reductions in tumor growth in normoglycemic versus hyperglycemic conditions in a syngeneic ovarian cancer mouse model [91], suggesting the opposite effect in that metformin may have greater anti-tumorigenic efficacy in non-diabetic as opposed to diabetic patients. Taken together, while most data point to a synergistic effect of metformin in obesity, not every cancer study supports that. It confirms that hyperglycemia and obesity may not be interchangeable in their impact on modifying metformin response for cancer treatment and that other factors such as cytokines and inflammation etc. may be important in EC.
Obesity activates numerous oncogenic signaling pathways that induce a variety of systemic changes including altered levels of insulin, IGF-1, leptin, adiponectin, steroid hormones and cytokines that ultimately promote tumor cell proliferation. Each of these factors has the potential to create an environment that favors tumor initiation and progression, including that of EC [68,92]. Insulin resistance and hyperinsulinemia are commonly observed in EC, which potentiate IGF-1 biologic activity and promote hyperactivity of MAPK and PI3K/AKT/mTOR signaling which are also frequently observed in EC [68,93]. Inflammatory cytokines, such as tumor necrosis factor (TNF)-α, interleukin (IL)-1β and IL-6, produced by a tumor and/or activated by immune cells, have been shown to stimulate cancer cell growth and influence prognosis [94]. Recent studies showed that metformin reduced secretion of IGF-1 and expression of IGF-1R and inhibited cell proliferation stimulated by insulin in EC cells [95,96]. Pre-operative metformin treatment has been found to decrease circulating metabolic factors, including insulin, glucose, IGF-1, and leptin, in patients with EC [78,97]. In addition, pre-operative metformin treatment in endometrial cancer patient led to a reduction in the production of inflammatory cytokines, such as TNF and IL-6 through the inactivation of nuclear-factor kappa-B (NF-B) and hypoxia-inducible factor (HIF)-1 [98]. In this study, we found that metformin significantly reduced serum IL-6, TNF-α and PA1-A levels in obese and lean mice, suggesting it exerts anti-inflammatory effects in addition to its anti-tumorigenic effects in vivo.
Knowledge of metabolic profiling can enable identification of novel therapeutic targets and metabolic pathways as possible targets of intervention in cancer treatment [99]. Though this approach, lipid, kynurenine and endocannabinoid signaling pathways as well as RNA editing pathways were found to be dysregulated in human endometrial cancer tissues [99,100]. In this study, metabolomic profiling analysis found that energy metabolism, protein biosynthesis, and most strikingly lipid biosynthesis/lipid peroxidation were upregulated in obese versus lean endometrial tumors. These results further confirmed that the aggressive phenotype of EC in obese LKB1fl/fl p53fl/fl mice was accompanied by distinct alterations in metabolism to fuel the cancer cellular machinery. In addition, metabolomic profiling revealed that that lipids were being degraded and oxidized and excess free fatty acids in obesity were being shunted to beta-oxidation by metformin rather than being used for lipid biosynthesis to fuel tumor growth. We have previously reported elevated obesity-driven acylcarnitines as evidence of increased yet incomplete fatty acid oxidation associated with elevated FA metabolism and obesity [101,102]. In addition, protein degradation markers were decreased in obese ECs with metformin treatment, suggesting reduced protein turnover and biosynthesis. Thus, metabolomics profiling of the endometrial tumors revealed that metformin may reverse obesity-driven upregulation of lipid and protein biosynthesis. We hypothesize that this metabolic obese signature partially underlies metformin’s improved efficacy in treating obesity-driven tumors.
Interestingly, when comparing our previous obesity and metformin studies in the KpB ovarian cancer mouse model to those in the LKB1fl/fl p53fl/fl endometrial cancer mouse model, similarities and differences were noted. Both our ovarian cancer and endometrial cancer mouse studies found that obesity results in metabolically different tumors under obese and lean conditions, and that metformin had differing metabolic effects on the tumors that aligned with obesity status. Despite these similarities, the obesity- and metformin-driven effects in the transgenic KpB ovarian cancer mouse model were remarkably distinct from the LKB1fl/fl p53fl/fl endometrial cancer mouse model. For the KpB mouse model, the ovarian tumors from obese mice had evidence of impaired mitochondrial complex 2 function and energy supplied by omega fatty acid oxidation rather than glycolysis as compared to lean mice [70]. The improved efficacy of metformin in the obese ovarian tumors corresponded with inhibition of mitochondrial complex 1 and fatty acid oxidation, and stimulation of glycolysis [70]. In contrast, the endometrial tumors from LKB1fl/fl p53fl/fl obese mice had evidence of upregulation of glycolysis, lipid and protein metabolism, and metformin reversed these obesity-driven effects via shunting of free fatty acids to beta-oxidation as opposed to lipid biosynthesis. We hypothesize that these differences in obesity- and metformin-driven effects in ovarian versus endometrial cancer mouse models may be due to the organ of origin of the cancer (ovarian versus endometrial) versus the genetic alterations in the corresponding mouse models (Rb/Brca1/p53 versus LKB1/p53). For example, LKB1 and p53 are key regulators of glycolysis in cancer and may account for some of the metabolic differences found between the ovarian and endometrial cancer mouse models [103].
Conclusions
Overall, our results suggest that the efficacy of metformin could be modulated by glucose levels in vitro and obesity status in vivo. In human EC cell lines, metformin had increased antiproliferative effects under conditions of glucose deprivation as opposed to glucose excess, mostly likely due to metformin’s known effects on increasing glycolysis as seen in other studies. These findings suggest that inhibition of glucose uptake or glycolysis could be an effective adjunctive treatment option to enhance the inhibitory effect of metformin on tumor growth in EC patients. In our LKB1fl/fl p53fl/fl mouse model of endometrioid EC, obesity resulted in a doubling of tumor size and increased EC lipid and protein biosynthesis. Metformin had increased efficacy against ECs in obese versus lean mice, reversing the detrimental metabolic effects of obesity in the tumors via shunting fatty acids to beta-oxidation as opposed to lipid biosynthesis. Clinical trials are already underway for metformin in EC patients [104], and our work suggests that evaluating the metabolic milieu of the patient and their corresponding tumor as potential biomarkers of metformin response warrants further investigation in therapeutic clinical trials in endometrial cancer.
Acknowledgements
This work is supported by: (1) Bae-Jump: American Cancer Society (ACS) Research Scholar Grant - RSG CCE 128826. (2) Bae-Jump: NIH/NCI - R37CA226969. (3) Bae-Jump: V Foundation Translational Award.
Disclosure of conflict of interest
None.
Abbreviations
- EC
endometrial cancer
- IGF-1
insulin-like growth factor-1
- IGF1R
IGF-1 receptor
- PTEN
phosphatase and tensin homolog
- WAT
white adipose tissue
- AMPK
AMP-activated protein kinase
- SREBP-1
sterol regulatory element-binding protein-1
- LKB1
liver-kinase b1
- CaMKK
calmodulin-dependent protein-kinase
- mTOR
mammalian target-of-rapamycin
- ACC
acetyl-CoA carboxylase
- SREBP-1
sterol regulatory element-binding protein-1
- FBS
fetal bovine serum
- ECL
enhanced chemiluminescence
- PBS
phosphate buffered saline
- AdCre
Ad5-CMV-Cre
- LG
hypoglycemia
- NG
euglycemia
- HG
hyperglycemia
- CAH
complex atypical hyperplasia
- HFD
high-fat diet
- LFD
Low-fat diet
- HOMA
Homeostatic Model Assessment of Insulin Resistance
- BMI
body mass index
- TNF-α
tumor necrosis factor-α
- IL
interleukin
- NF-κB
nuclear-factor kappa-B
- HIF-1
hypoxia-inducible factor-1
Supporting Information
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin. 2019;69:7–34. doi: 10.3322/caac.21551. [DOI] [PubMed] [Google Scholar]
- 2.Ko EM, Walter P, Clark L, Jackson A, Franasiak J, Bolac C, Havrilesky L, Secord AA, Moore DT, Gehrig PA, Bae-Jump VL. The complex triad of obesity, diabetes and race in Type I and II endometrial cancers: prevalence and prognostic significance. Gynecol Oncol. 2014;133:28–32. doi: 10.1016/j.ygyno.2014.01.032. [DOI] [PubMed] [Google Scholar]
- 3.Setiawan VW, Yang HP, Pike MC, McCann SE, Yu H, Xiang YB, Wolk A, Wentzensen N, Weiss NS, Webb PM, van den Brandt PA, van de Vijver K, Thompson PJ, Strom BL, Spurdle AB, Soslow RA, Shu XO, Schairer C, Sacerdote C, Rohan TE, Robien K, Risch HA, Ricceri F, Rebbeck TR, Rastogi R, Prescott J, Polidoro S, Park Y, Olson SH, Moysich KB, Miller AB, McCullough ML, Matsuno RK, Magliocco AM, Lurie G, Lu L, Lissowska J, Liang X, Lacey JV Jr, Kolonel LN, Henderson BE, Hankinson SE, Hakansson N, Goodman MT, Gaudet MM, Garcia-Closas M, Friedenreich CM, Freudenheim JL, Doherty J, De Vivo I, Courneya KS, Cook LS, Chen C, Cerhan JR, Cai H, Brinton LA, Bernstein L, Anderson KE, Anton-Culver H, Schouten LJ, Horn-Ross PL. Type I and II endometrial cancers: have they different risk factors? J. Clin. Oncol. 2013;31:2607–2618. doi: 10.1200/JCO.2012.48.2596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schmandt RE, Iglesias DA, Co NN, Lu KH. Understanding obesity and endometrial cancer risk: opportunities for prevention. Am J Obstet Gynecol. 2011;205:518–525. doi: 10.1016/j.ajog.2011.05.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chia VM, Newcomb PA, Trentham-Dietz A, Hampton JM. Obesity, diabetes, and other factors in relation to survival after endometrial cancer diagnosis. Int J Gynecol Cancer. 2007;17:441–446. doi: 10.1111/j.1525-1438.2007.00790.x. [DOI] [PubMed] [Google Scholar]
- 6.Calle EE, Rodriguez C, Walker-Thurmond K, Thun MJ. Overweight, obesity, and mortality from cancer in a prospectively studied cohort of U.S. adults. N Engl J Med. 2003;348:1625–1638. doi: 10.1056/NEJMoa021423. [DOI] [PubMed] [Google Scholar]
- 7.Steiner E, Plata K, Interthal C, Schmidt M, Faldum A, Hengstler JG, Sakuragi N, Watari H, Yamamoto R, Kolbl H. Diabetes mellitus is a multivariate independent prognostic factor in endometrial carcinoma: a clinicopathologic study on 313 patients. Eur J Gynaecol Oncol. 2007;28:95–97. [PubMed] [Google Scholar]
- 8.Arem H, Park Y, Pelser C, Ballard-Barbash R, Irwin ML, Hollenbeck A, Gierach GL, Brinton LA, Pfeiffer RM, Matthews CE. Prediagnosis body mass index, physical activity, and mortality in endometrial cancer patients. J Natl Cancer Inst. 2013;105:342–349. doi: 10.1093/jnci/djs530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Secord AA, Hasselblad V, Von Gruenigen VE, Gehrig PA, Modesitt SC, Bae-Jump V, Havrilesky LJ. Body mass index and mortality in endometrial cancer: a systematic review and meta-analysis. Gynecol Oncol. 2016;140:184–190. doi: 10.1016/j.ygyno.2015.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Felix AS, Yang HP, Bell DW, Sherman ME. Epidemiology of endometrial carcinoma: etiologic importance of hormonal and metabolic influences. Adv Exp Med Biol. 2017;943:3–46. doi: 10.1007/978-3-319-43139-0_1. [DOI] [PubMed] [Google Scholar]
- 11.Kitson SJ, Evans DG, Crosbie EJ. Identifying high-risk women for endometrial cancer prevention strategies: proposal of an endometrial cancer risk prediction model. Cancer Prev Res (Phila) 2017;10:1–13. doi: 10.1158/1940-6207.CAPR-16-0224. [DOI] [PubMed] [Google Scholar]
- 12.Khandekar MJ, Cohen P, Spiegelman BM. Molecular mechanisms of cancer development in obesity. Nat Rev Cancer. 2011;11:886–895. doi: 10.1038/nrc3174. [DOI] [PubMed] [Google Scholar]
- 13.Gunter MJ, Hoover DR, Yu H, Wassertheil-Smoller S, Manson JE, Li J, Harris TG, Rohan TE, Xue X, Ho GY, Einstein MH, Kaplan RC, Burk RD, Wylie-Rosett J, Pollak MN, Anderson G, Howard BV, Strickler HD. A prospective evaluation of insulin and insulin-like growth factor-I as risk factors for endometrial cancer. Cancer Epidemiol Biomarkers Prev. 2008;17:921–929. doi: 10.1158/1055-9965.EPI-07-2686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McCampbell AS, Broaddus RR, Loose DS, Davies PJ. Overexpression of the insulin-like growth factor I receptor and activation of the AKT pathway in hyperplastic endometrium. Clin Cancer Res. 2006;12:6373–6378. doi: 10.1158/1078-0432.CCR-06-0912. [DOI] [PubMed] [Google Scholar]
- 15.Gehrig PA, Bae-Jump VL. Promising novel therapies for the treatment of endometrial cancer. Gynecol Oncol. 2010;116:187–194. doi: 10.1016/j.ygyno.2009.10.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dedes KJ, Wetterskog D, Ashworth A, Kaye SB, Reis-Filho JS. Emerging therapeutic targets in endometrial cancer. Nat Rev Clin Oncol. 2011;8:261–271. doi: 10.1038/nrclinonc.2010.216. [DOI] [PubMed] [Google Scholar]
- 17.Cheung LW, Hennessy BT, Li J, Yu S, Myers AP, Djordjevic B, Lu Y, Stemke-Hale K, Dyer MD, Zhang F, Ju Z, Cantley LC, Scherer SE, Liang H, Lu KH, Broaddus RR, Mills GB. High frequency of PIK3R1 and PIK3R2 mutations in endometrial cancer elucidates a novel mechanism for regulation of PTEN protein stability. Cancer Discov. 2011;1:170–185. doi: 10.1158/2159-8290.CD-11-0039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cancer Genome Atlas Research Network. Kandoth C, Schultz N, Cherniack AD, Akbani R, Liu Y, Shen H, Robertson AG, Pashtan I, Shen R, Benz CC, Yau C, Laird PW, Ding L, Zhang W, Mills GB, Kucherlapati R, Mardis ER, Levine DA. Integrated genomic characterization of endometrial carcinoma. Nature. 2013;497:67–73. doi: 10.1038/nature12113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Salvesen HB, Carter SL, Mannelqvist M, Dutt A, Getz G, Stefansson IM, Raeder MB, Sos ML, Engelsen IB, Trovik J, Wik E, Greulich H, Bo TH, Jonassen I, Thomas RK, Zander T, Garraway LA, Oyan AM, Sellers WR, Kalland KH, Meyerson M, Akslen LA, Beroukhim R. Integrated genomic profiling of endometrial carcinoma associates aggressive tumors with indicators of PI3 kinase activation. Proc Natl Acad Sci U S A. 2009;106:4834–4839. doi: 10.1073/pnas.0806514106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.O’Flanagan CH, Bowers LW, Hursting SD. A weighty problem: metabolic perturbations and the obesity-cancer link. Horm Mol Biol Clin Investig. 2015;23:47–57. doi: 10.1515/hmbci-2015-0022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.International Agency for Research on Cancer, Globocan. Cancer Epidemiology Database. 2002 [Google Scholar]
- 22.Hursting SD, Lashinger LM, Wheatley KW, Rogers CJ, Colbert LH, Nunez NP, Perkins SN. Reducing the weight of cancer: mechanistic targets for breaking the obesity-carcinogenesis link. Best Pract Res Clin Endocrinol Metab. 2008;22:659–669. doi: 10.1016/j.beem.2008.08.009. [DOI] [PubMed] [Google Scholar]
- 23.Dann SG, Selvaraj A, Thomas G. mTOR Complex1-S6K1 signaling: at the crossroads of obesity, diabetes and cancer. Trends Mol Med. 2007;13:252–259. doi: 10.1016/j.molmed.2007.04.002. [DOI] [PubMed] [Google Scholar]
- 24.Wysocki PJ, Wierusz-Wysocka B. Obesity, hyperinsulinemia and breast cancer: novel targets and a novel role for metformin. Expert Rev Mol Diagn. 2010;10:509–19. doi: 10.1586/erm.10.22. [DOI] [PubMed] [Google Scholar]
- 25.Gonzalez-Angulo AM, Meric-Bernstam F. Metformin: a therapeutic opportunity in breast cancer. Clin Cancer Res. 2010;16:1695–700. doi: 10.1158/1078-0432.CCR-09-1805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Evans JM, Donnelly LA, Emslie-Smith AM, Alessi DR, Morris AD. Metformin and reduced risk of cancer in diabetic patients. BMJ. 2005;330:1304–1305. doi: 10.1136/bmj.38415.708634.F7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bowker SL, Majumdar SR, Veugelers P, Johnson JA. Increased cancer-related mortality for patients with type 2 diabetes who use sulfonylureas or insulin. Diabetes Care. 2006;29:254–258. doi: 10.2337/diacare.29.02.06.dc05-1558. [DOI] [PubMed] [Google Scholar]
- 28.Libby G, Donnelly LA, Donnan PT, Alessi DR, Morris AD, Evans JM. New users of metformin are at low risk of incident cancer: a cohort study among people with type 2 diabetes. Diabetes Care. 2009;32:1620–1625. doi: 10.2337/dc08-2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ko EM, Walter P, Jackson A, Clark L, Franasiak J, Bolac C, Havrilesky LJ, Secord AA, Moore DT, Gehrig PA, Bae-Jump V. Metformin is associated with improved survival in endometrial cancer. Gynecol Oncol. 2014;132:438–442. doi: 10.1016/j.ygyno.2013.11.021. [DOI] [PubMed] [Google Scholar]
- 30.Currie CJ, Poole CD, Jenkins-Jones S, Gale EA, Johnson JA, Morgan CL. Mortality after incident cancer in people with and without type 2 diabetes: impact of metformin on survival. Diabetes Care. 2012;35:299–304. doi: 10.2337/dc11-1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nevadunsky NS, Van Arsdale A, Strickler HD, Moadel A, Kaur G, Frimer M, Conroy E, Goldberg GL, Einstein MH. Metformin use and endometrial cancer survival. Gynecol Oncol. 2014;132:236–240. doi: 10.1016/j.ygyno.2013.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Daugan M, Dufay Wojcicki A, d’Hayer B, Boudy V. Metformin: an anti-diabetic drug to fight cancer. Pharmacol Res. 2016;113:675–685. doi: 10.1016/j.phrs.2016.10.006. [DOI] [PubMed] [Google Scholar]
- 33.Pollak MN. Investigating metformin for cancer prevention and treatment: the end of the beginning. Cancer Discov. 2012;2:778–790. doi: 10.1158/2159-8290.CD-12-0263. [DOI] [PubMed] [Google Scholar]
- 34.Shackelford DB, Shaw RJ. The LKB1-AMPK pathway: metabolism and growth control in tumour suppression. Nat Rev Cancer. 2009;9:563–575. doi: 10.1038/nrc2676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pernicova I, Korbonits M. Metformin--mode of action and clinical implications for diabetes and cancer. Nat Rev Endocrinol. 2014;10:143–156. doi: 10.1038/nrendo.2013.256. [DOI] [PubMed] [Google Scholar]
- 36.Morales DR, Morris AD. Metformin in cancer treatment and prevention. Annu Rev Med. 2015;66:17–29. doi: 10.1146/annurev-med-062613-093128. [DOI] [PubMed] [Google Scholar]
- 37.Wheaton WW, Weinberg SE, Hamanaka RB, Soberanes S, Sullivan LB, Anso E, Glasauer A, Dufour E, Mutlu GM, Budigner GS, Chandel NS. Metformin inhibits mitochondrial complex I of cancer cells to reduce tumorigenesis. Elife. 2014;3:e02242. doi: 10.7554/eLife.02242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Quinn BJ, Kitagawa H, Memmott RM, Gills JJ, Dennis PA. Repositioning metformin for cancer prevention and treatment. Trends Endocrinol Metab. 2013;24:469–480. doi: 10.1016/j.tem.2013.05.004. [DOI] [PubMed] [Google Scholar]
- 39.Cantrell LA, Zhou C, Mendivil A, Malloy KM, Gehrig PA, Bae-Jump VL. Metformin is a potent inhibitor of endometrial cancer cell proliferation--implications for a novel treatment strategy. Gynecol Oncol. 2010;116:92–98. doi: 10.1016/j.ygyno.2009.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Iglesias DA, Yates MS, van der Hoeven D, Rodkey TL, Zhang Q, Co NN, Burzawa J, Chigurupati S, Celestino J, Bowser J, Broaddus R, Hancock JF, Schmandt R, Lu KH. Another surprise from metformin: novel mechanism of action via k-ras influences endometrial cancer response to therapy. Mol Cancer Ther. 2013;12:2847–2856. doi: 10.1158/1535-7163.MCT-13-0439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sarfstein R, Friedman Y, Attias-Geva Z, Fishman A, Bruchim I, Werner H. Metformin downregulates the insulin/IGF-I signaling pathway and inhibits different uterine serous carcinoma (USC) cells proliferation and migration in p53-dependent or -independent manners. PLoS One. 2013;8:e61537. doi: 10.1371/journal.pone.0061537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hanna RK, Zhou C, Malloy KM, Sun L, Zhong Y, Gehrig PA, Bae-Jump VL. Metformin potentiates the effects of paclitaxel in endometrial cancer cells through inhibition of cell proliferation and modulation of the mTOR pathway. Gynecol Oncol. 2012;125:458–469. doi: 10.1016/j.ygyno.2012.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dong L, Zhou Q, Zhang Z, Zhu Y, Duan T, Feng Y. Metformin sensitizes endometrial cancer cells to chemotherapy by repressing glyoxalase I expression. J Obstet Gynaecol Res. 2012;38:1077–1085. doi: 10.1111/j.1447-0756.2011.01839.x. [DOI] [PubMed] [Google Scholar]
- 44.Erdemoglu E, Guney M, Giray SG, Take G, Mungan T. Effects of metformin on mammalian target of rapamycin in a mouse model of endometrial hyperplasia. Eur J Obstet Gynecol Reprod Biol. 2009;145:195–199. doi: 10.1016/j.ejogrb.2009.04.034. [DOI] [PubMed] [Google Scholar]
- 45.Algire C, Zakikhani M, Blouin MJ, Shuai JH, Pollak M. Metformin attenuates the stimulatory effect of a high-energy diet on in vivo LLC1 carcinoma growth. Endocr Relat Cancer. 2008;15:833–839. doi: 10.1677/ERC-08-0038. [DOI] [PubMed] [Google Scholar]
- 46.Phoenix KN, Vumbaca F, Fox MM, Evans R, Claffey KP. Dietary energy availability affects primary and metastatic breast cancer and metformin efficacy. Breast Cancer Res Treat. 2010;123:333–44. doi: 10.1007/s10549-009-0647-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Algire C, Amrein L, Zakikhani M, Panasci L, Pollak M. Metformin blocks the stimulative effect of a high-energy diet on colon carcinoma growth in vivo and is associated with reduced expression of fatty acid synthase. Endocr Relat Cancer. 2010;17:351–360. doi: 10.1677/ERC-09-0252. [DOI] [PubMed] [Google Scholar]
- 48.Checkley LA, Rho O, Angel JM, Cho J, Blando J, Beltran L, Hursting SD, DiGiovanni J. Metformin inhibits skin tumor promotion in overweight and obese mice. Cancer Prev Res (Phila) 2014;7:54–64. doi: 10.1158/1940-6207.CAPR-13-0110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Vooijs M, van der Valk M, te Riele H, Berns A. Flp-mediated tissue-specific inactivation of the retinoblastoma tumor suppressor gene in the mouse. Oncogene. 1998;17:1–12. doi: 10.1038/sj.onc.1202169. [DOI] [PubMed] [Google Scholar]
- 50.Bardeesy N, Sinha M, Hezel AF, Signoretti S, Hathaway NA, Sharpless NE, Loda M, Carrasco DR, DePinho RA. Loss of the Lkb1 tumour suppressor provokes intestinal polyposis but resistance to transformation. Nature. 2002;419:162–167. doi: 10.1038/nature01045. [DOI] [PubMed] [Google Scholar]
- 51.Vuong S, Delgado-Olguin P. Mouse genotyping. Methods Mol Biol. 2018;1752:1–9. doi: 10.1007/978-1-4939-7714-7_1. [DOI] [PubMed] [Google Scholar]
- 52.Marino S, Vooijs M, van Der Gulden H, Jonkers J, Berns A. Induction of medulloblastomas in p53-null mutant mice by somatic inactivation of Rb in the external granular layer cells of the cerebellum. Genes Dev. 2000;14:994–1004. [PMC free article] [PubMed] [Google Scholar]
- 53.Takahashi K, Saga Y, Mizukami H, Takei Y, Urabe M, Kume A, Suzuki M, Ozawa K. Development of a mouse model for lymph node metastasis with endometrial cancer. Cancer Sci. 2011;102:2272–2277. doi: 10.1111/j.1349-7006.2011.02099.x. [DOI] [PubMed] [Google Scholar]
- 54.Contreras CM, Gurumurthy S, Haynie JM, Shirley LJ, Akbay EA, Wingo SN, Schorge JO, Broaddus RR, Wong KK, Bardeesy N, Castrillon DH. Loss of Lkb1 provokes highly invasive endometrial adenocarcinomas. Cancer Res. 2008;68:759–766. doi: 10.1158/0008-5472.CAN-07-5014. [DOI] [PubMed] [Google Scholar]
- 55.Lawton KA, Berger A, Mitchell M, Milgram KE, Evans AM, Guo L, Hanson RW, Kalhan SC, Ryals JA, Milburn MV. Analysis of the adult human plasma metabolome. Pharmacogenomics. 2008;9:383–397. doi: 10.2217/14622416.9.4.383. [DOI] [PubMed] [Google Scholar]
- 56.Reitman ZJ, Jin G, Karoly ED, Spasojevic I, Yang J, Kinzler KW, He Y, Bigner DD, Vogelstein B, Yan H. Profiling the effects of isocitrate dehydrogenase 1 and 2 mutations on the cellular metabolome. Proc Natl Acad Sci U S A. 2011;108:3270–3275. doi: 10.1073/pnas.1019393108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Evans AM, DeHaven CD, Barrett T, Mitchell M, Milgram E. Integrated, nontargeted ultrahigh performance liquid chromatography/electrospray ionization tandem mass spectrometry platform for the identification and relative quantification of the small-molecule complement of biological systems. Anal Chem. 2009;81:6656–6667. doi: 10.1021/ac901536h. [DOI] [PubMed] [Google Scholar]
- 58.Freemerman AJ, Johnson AR, Sacks GN, Milner JJ, Kirk EL, Troester MA, Macintyre AN, Goraksha-Hicks P, Rathmell JC, Makowski L. Metabolic reprogramming of macrophages: glucose transporter (GLUT1)-mediated glucose metabolism drives a pro-inflammatory phenotype. J Biol Chem. 2014;289:7884. doi: 10.1074/jbc.M113.522037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Makowski L, Zhou C, Zhong Y, Kuan PF, Fan C, Sampey BP, Difurio M, Bae-Jump VL. Obesity increases tumor aggressiveness in a genetically engineered mouse model of serous ovarian cancer. Gynecol Oncol. 2014;133:90–97. doi: 10.1016/j.ygyno.2013.12.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Freemerman AJ, Zhao L, Pingili AK, Teng B, Cozzo AJ, Fuller AM, Johnson AR, Milner JJ, Lim MF, Galanko JA, Beck MA, Bear JE, Rotty JD, Bezavada L, Smallwood HS, Puchowicz MA, Liu J, Locasale JW, Lee DP, Bennett BJ, Abel ED, Rathmell JC, Makowski L. Myeloid Slc2a1-deficient murine model revealed macrophage activation and metabolic phenotype are fueled by GLUT1. J Immunol. 2019;202:1265–1286. doi: 10.4049/jimmunol.1800002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Johnson AR, Qin Y, Cozzo AJ, Freemerman AJ, Huang MJ, Zhao L, Sampey BP, Milner JJ, Beck MA, Damania B, Rashid N, Galanko JA, Lee DP, Edin ML, Zeldin DC, Fueger PT, Dietz B, Stahl A, Wu Y, Mohlke KL, Makowski L. Metabolic reprogramming through fatty acid transport protein 1 (FATP1) regulates macrophage inflammatory potential and adipose inflammation. Mol Metab. 2016;5:506–526. doi: 10.1016/j.molmet.2016.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Freemerman AJ, Johnson AR, Sacks GN, Milner JJ, Kirk EL, Troester MA, Macintyre AN, Goraksha-Hicks P, Rathmell JC, Makowski L. Metabolic reprogramming of macrophages: glucose transporter 1 (GLUT1)-mediated glucose metabolism drives a proinflammatory phenotype. J Biol Chem. 2014;289:7884–7896. doi: 10.1074/jbc.M113.522037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Han J, Zhang L, Guo H, Wysham WZ, Roque DR, Willson AK, Sheng X, Zhou C, Bae-Jump VL. Glucose promotes cell proliferation, glucose uptake and invasion in endometrial cancer cells via AMPK/mTOR/S6 and MAPK signaling. Gynecol Oncol. 2015;138:668–675. doi: 10.1016/j.ygyno.2015.06.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sun L, Yin Y, Clark LH, Sun W, Sullivan SA, Tran AQ, Han J, Zhang L, Guo H, Madugu E, Pan T, Jackson AL, Kilgore J, Jones HM, Gilliam TP, Zhou C, Bae-Jump VL. Dual inhibition of glycolysis and glutaminolysis as a therapeutic strategy in the treatment of ovarian cancer. Oncotarget. 2017;8:63551–63561. doi: 10.18632/oncotarget.18854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Doerstling SS, O’Flanagan CH, Hursting SD. Obesity and cancer metabolism: a perspective on interacting tumor-intrinsic and extrinsic factors. Front Oncol. 2017;7:216. doi: 10.3389/fonc.2017.00216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Piulats JM, Guerra E, Gil-Martin M, Roman-Canal B, Gatius S, Sanz-Pamplona R, Velasco A, Vidal A, Matias-Guiu X. Molecular approaches for classifying endometrial carcinoma. Gynecol Oncol. 2017;145:200–207. doi: 10.1016/j.ygyno.2016.12.015. [DOI] [PubMed] [Google Scholar]
- 67.Co NN, Iglesias D, Celestino J, Kwan SY, Mok SC, Schmandt R, Lu KH. Loss of LKB1 in high-grade endometrial carcinoma: LKB1 is a novel transcriptional target of p53. Cancer. 2014;120:3457–3468. doi: 10.1002/cncr.28854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Onstad MA, Schmandt RE, Lu KH. Addressing the role of obesity in endometrial cancer risk, prevention, and treatment. J. Clin. Oncol. 2016;34:4225–4230. doi: 10.1200/JCO.2016.69.4638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lambe M, Wigertz A, Garmo H, Walldius G, Jungner I, Hammar N. Impaired glucose metabolism and diabetes and the risk of breast, endometrial, and ovarian cancer. Cancer Causes Control. 2011;22:1163–1171. doi: 10.1007/s10552-011-9794-8. [DOI] [PubMed] [Google Scholar]
- 70.Han J, Wysham WZ, Zhong Y, Guo H, Zhang L, Malloy KM, Dickens HK, Huh G, Lee D, Makowski L, Zhou C, Bae-Jump VL. Increased efficacy of metformin corresponds to differential metabolic effects in the ovarian tumors from obese versus lean mice. Oncotarget. 2017;8:110965–110982. doi: 10.18632/oncotarget.20754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Yuan L, Sheng X, Willson AK, Roque DR, Stine JE, Guo H, Jones HM, Zhou C, Bae-Jump VL. Glutamine promotes ovarian cancer cell proliferation through the mTOR/S6 pathway. Endocr Relat Cancer. 2015;22:577–591. doi: 10.1530/ERC-15-0192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ben Sahra I, Laurent K, Giuliano S, Larbret F, Ponzio G, Gounon P, Le Marchand-Brustel Y, Giorgetti-Peraldi S, Cormont M, Bertolotto C, Deckert M, Auberger P, Tanti JF, Bost F. Targeting cancer cell metabolism: the combination of metformin and 2-deoxyglucose induces p53-dependent apoptosis in prostate cancer cells. Cancer Res. 2010;70:2465–2475. doi: 10.1158/0008-5472.CAN-09-2782. [DOI] [PubMed] [Google Scholar]
- 73.Zhuang Y, Chan DK, Haugrud AB, Miskimins WK. Mechanisms by which low glucose enhances the cytotoxicity of metformin to cancer cells both in vitro and in vivo. PLoS One. 2014;9:e108444. doi: 10.1371/journal.pone.0108444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Menendez JA, Oliveras-Ferraros C, Cufi S, Corominas-Faja B, Joven J, Martin-Castillo B, Vazquez-Martin A. Metformin is synthetically lethal with glucose withdrawal in cancer cells. Cell Cycle. 2012;11:2782–2792. doi: 10.4161/cc.20948. [DOI] [PubMed] [Google Scholar]
- 75.Bikas A, Jensen K, Patel A, Costello J Jr, McDaniel D, Klubo-Gwiezdzinska J, Larin O, Hoperia V, Burman KD, Boyle L, Wartofsky L, Vasko V. Glucose-deprivation increases thyroid cancer cells sensitivity to metformin. Endocr Relat Cancer. 2015;22:919–932. doi: 10.1530/ERC-15-0402. [DOI] [PubMed] [Google Scholar]
- 76.Matsuo J, Tsukumo Y, Saito S, Tsukahara S, Sakurai J, Sato S, Kondo H, Ushijima M, Matsuura M, Watanabe T, Tomida A. Hyperactivation of 4E-binding protein 1 as a mediator of biguanide-induced cytotoxicity during glucose deprivation. Mol Cancer Ther. 2012;11:1082–1091. doi: 10.1158/1535-7163.MCT-11-0871. [DOI] [PubMed] [Google Scholar]
- 77.Wallbillich JJ, Josyula S, Saini U, Zingarelli RA, Dorayappan KD, Riley MK, Wanner RA, Cohn DE, Selvendiran K. High glucose-mediated STAT3 activation in endometrial cancer is inhibited by metformin: therapeutic implications for endometrial cancer. PLoS One. 2017;12:e0170318. doi: 10.1371/journal.pone.0170318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Soliman PT, Zhang Q, Broaddus RR, Westin SN, Iglesias D, Munsell MF, Schmandt R, Yates M, Ramondetta L, Lu KH. Prospective evaluation of the molecular effects of metformin on the endometrium in women with newly diagnosed endometrial cancer: a window of opportunity study. Gynecol Oncol. 2016;143:466–471. doi: 10.1016/j.ygyno.2016.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Takahashi A, Kimura F, Yamanaka A, Takebayashi A, Kita N, Takahashi K, Murakami T. Metformin impairs growth of endometrial cancer cells via cell cycle arrest and concomitant autophagy and apoptosis. Cancer Cell Int. 2014;14:53. doi: 10.1186/1475-2867-14-53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ariaans G, Jalving M, Vries EG, Jong S. Anti-tumor effects of everolimus and metformin are complementary and glucose-dependent in breast cancer cells. BMC Cancer. 2017;17:232. doi: 10.1186/s12885-017-3230-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Xue C, Wang C, Sun Y, Meng Q, Liu Z, Huo X, Sun P, Sun H, Ma X, Ma X, Peng J, Liu K. Targeting P-glycoprotein function, p53 and energy metabolism: combination of metformin and 2-deoxyglucose reverses the multidrug resistance of MCF-7/Dox cells to doxorubicin. Oncotarget. 2017;8:8622–8632. doi: 10.18632/oncotarget.14373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Pena CG, Castrillon DH. LKB1 as a tumor suppressor in uterine cancer: mouse models and translational studies. Adv Exp Med Biol. 2017;943:211–241. doi: 10.1007/978-3-319-43139-0_7. [DOI] [PubMed] [Google Scholar]
- 83.Contreras CM, Akbay EA, Gallardo TD, Haynie JM, Sharma S, Tagao O, Bardeesy N, Takahashi M, Settleman J, Wong KK, Castrillon DH. Lkb1 inactivation is sufficient to drive endometrial cancers that are aggressive yet highly responsive to mTOR inhibitor monotherapy. Dis Model Mech. 2010;3:181–193. doi: 10.1242/dmm.004440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Troiano G, Mastrangelo F, Caponio VCA, Laino L, Cirillo N, Lo Muzio L. Predictive prognostic value of tissue-based microRNA expression in oral squamous cell carcinoma: a systematic review and meta-analysis. J Dent Res. 2018;97:759–766. doi: 10.1177/0022034518762090. [DOI] [PubMed] [Google Scholar]
- 85.Cheng H, Liu P, Zhang F, Xu E, Symonds L, Ohlson CE, Bronson RT, Maira SM, Di Tomaso E, Li J, Myers AP, Cantley LC, Mills GB, Zhao JJ. A genetic mouse model of invasive endometrial cancer driven by concurrent loss of Pten and Lkb1 Is highly responsive to mTOR inhibition. Cancer Res. 2014;74:15–23. doi: 10.1158/0008-5472.CAN-13-0544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Daikoku T, Hirota Y, Tranguch S, Joshi AR, DeMayo FJ, Lydon JP, Ellenson LH, Dey SK. Conditional loss of uterine Pten unfailingly and rapidly induces endometrial cancer in mice. Cancer Res. 2008;68:5619–5627. doi: 10.1158/0008-5472.CAN-08-1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Phoenix KN, Vumbaca F, Fox MM, Evans R, Claffey KP. Dietary energy availability affects primary and metastatic breast cancer and metformin efficacy. Breast Cancer Res Treat. 2010;123:333–344. doi: 10.1007/s10549-009-0647-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Hou M, Venier N, Sugar L, Musquera M, Pollak M, Kiss A, Fleshner N, Klotz L, Venkateswaran V. Protective effect of metformin in CD1 mice placed on a high carbohydrate-high fat diet. Biochem Biophys Res Commun. 2010;397:537–542. doi: 10.1016/j.bbrc.2010.05.152. [DOI] [PubMed] [Google Scholar]
- 89.Schneider MB, Matsuzaki H, Haorah J, Ulrich A, Standop J, Ding XZ, Adrian TE, Pour PM. Prevention of pancreatic cancer induction in hamsters by metformin. Gastroenterology. 2001;120:1263–1270. doi: 10.1053/gast.2001.23258. [DOI] [PubMed] [Google Scholar]
- 90.Bonanni B, Puntoni M, Cazzaniga M, Pruneri G, Serrano D, Guerrieri-Gonzaga A, Gennari A, Trabacca MS, Galimberti V, Veronesi P, Johansson H, Aristarco V, Bassi F, Luini A, Lazzeroni M, Varricchio C, Viale G, Bruzzi P, Decensi A. Dual effect of metformin on breast cancer proliferation in a randomized presurgical trial. J. Clin. Oncol. 2012;30:2593–2600. doi: 10.1200/JCO.2011.39.3769. [DOI] [PubMed] [Google Scholar]
- 91.Litchfield LM, Mukherjee A, Eckert MA, Johnson A, Mills KA, Pan S, Shridhar V, Lengyel E, Romero IL. Hyperglycemia-induced metabolic compensation inhibits metformin sensitivity in ovarian cancer. Oncotarget. 2015;6:23548–23560. doi: 10.18632/oncotarget.4556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Hopkins BD, Goncalves MD, Cantley LC. Obesity and cancer mechanisms: cancer metabolism. J. Clin. Oncol. 2016;34:4277–4283. doi: 10.1200/JCO.2016.67.9712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Wise MR, Jordan V, Lagas A, Showell M, Wong N, Lensen S, Farquhar CM. Obesity and endometrial hyperplasia and cancer in premenopausal women: a systematic review. Am J Obstet Gynecol. 2016;214:689, e1–e17. doi: 10.1016/j.ajog.2016.01.175. [DOI] [PubMed] [Google Scholar]
- 94.Deng T, Lyon CJ, Bergin S, Caligiuri MA, Hsueh WA. Obesity, Inflammation, and Cancer. Annu Rev Pathol. 2016;11:421–449. doi: 10.1146/annurev-pathol-012615-044359. [DOI] [PubMed] [Google Scholar]
- 95.de Barros Machado A, Dos Reis V, Weber S, Jauckus J, Brum IS, von Eye Corleta H, Strowitzki T, Capp E, Germeyer A. Proliferation and metastatic potential of endometrial cancer cells in response to metformin treatment in a high versus normal glucose environment. Oncol Lett. 2016;12:3626–3632. doi: 10.3892/ol.2016.5041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Zhang Y, Li MX, Wang H, Zeng Z, Li XM. Metformin down-regulates endometrial carcinoma cell secretion of IGF-1 and expression of IGF-1R. Asian Pac J Cancer Prev. 2015;16:221–225. doi: 10.7314/apjcp.2015.16.1.221. [DOI] [PubMed] [Google Scholar]
- 97.Mitsuhashi A, Kiyokawa T, Sato Y, Shozu M. Effects of metformin on endometrial cancer cell growth in vivo: a preoperative prospective trial. Cancer. 2014;120:2986–2995. doi: 10.1002/cncr.28853. [DOI] [PubMed] [Google Scholar]
- 98.Gadducci A, Biglia N, Tana R, Cosio S, Gallo M. Metformin use and gynecological cancers: A novel treatment option emerging from drug repositioning. Crit Rev Oncol Hematol. 2016;105:73–83. doi: 10.1016/j.critrevonc.2016.06.006. [DOI] [PubMed] [Google Scholar]
- 99.Jove M, Gatius S, Yeramian A, Portero-Otin M, Eritja N, Santacana M, Colas E, Ruiz M, Pamplona R, Matias-Guiu X. Metabotyping human endometrioid endometrial adenocarcinoma reveals an implication of endocannabinoid metabolism. Oncotarget. 2016;7:52364–52374. doi: 10.18632/oncotarget.10564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Altadill T, Dowdy TM, Gill K, Reques A, Menon SS, Moiola CP, Lopez-Gil C, Coll E, Matias-Guiu X, Cabrera S, Garcia A, Reventos J, Byers SW, Gil-Moreno A, Cheema AK, Colas E. Metabolomic and lipidomic profiling identifies the role of the RNA editing pathway in endometrial carcinogenesis. Sci Rep. 2017;7:8803. doi: 10.1038/s41598-017-09169-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Makowski L, Noland RC, Koves TR, Xing W, Ilkayeva OR, Muehlbauer MJ, Stevens RD, Muoio DM. Metabolic profiling of PPARalpha-/- mice reveals defects in carnitine and amino acid homeostasis that are partially reversed by oral carnitine supplementation. FASEB J. 2009;23:586–604. doi: 10.1096/fj.08-119420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Sampey BP, Freemerman AJ, Zhang J, Kuan PF, Galanko JA, O’Connell TM, Ilkayeva OR, Muehlbauer MJ, Stevens RD, Newgard CB, Brauer HA, Troester MA, Makowski L. Metabolomic profiling reveals mitochondrial-derived lipid biomarkers that drive obesity-associated inflammation. PLoS One. 2012;7:e38812. doi: 10.1371/journal.pone.0038812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Gomes AS, Ramos H, Soares J, Saraiva L. p53 and glucose metabolism: an orchestra to be directed in cancer therapy. Pharmacol Res. 2018;131:75–86. doi: 10.1016/j.phrs.2018.03.015. [DOI] [PubMed] [Google Scholar]
- 104.Stine JE, Bae-Jump V. Metformin and gynecologic cancers. Obstet Gynecol Surv. 2014;69:477–489. doi: 10.1097/OGX.0000000000000092. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.