

Abstract
Treatment and prognosis of Fanconi anaemia (FA) and acquired aplastic anaemia (AA) differ. However, delayed and inappropriate treatments are administered in FA due to its similarities to AA in presentation. The objective of the current study was to elucidate differences between the molecular mechanisms underlying FA and AA as well as to identify biomarkers and pathways associated with FA via bioinformatics analyses. Proteomic data were obtained from bone marrow samples of patients with FA and AA. Gene ontology analysis was performed using a Database for Annotation, Visualization and Integrated Discovery. KEGG pathway enrichment analyses were conducted using the ClueGO plug-in in Cytoscape. A DEP-associated protein-protein interaction (PPI) network was constructed using STRING and visualized in Cytoscape. A total of 114 DEPs, including 71 upregulated proteins and 43 downregulated proteins, were present in the FA samples, compared with those in the AA samples. Upregulated proteins were enriched in the nucleosome assembly, canonical glycolysis, glycolytic process, and the glycolysis/gluconeogenesis pathway, whereas downregulated proteins were enriched in relation to immune response, negative regulation of apoptosis, proteolysis and CoA biosynthesis. Eight hub proteins with a high degree of connectivity were obtained as follows: alpha-enolase (ENO1), HSP90AA1, phosphoglycerate kinase 1 (PGK1), HSP90AB1, ACTC1, ACTBL2, EEF1A1 and CFL1. Upregulation of ENO1 and CFL1 in patients with FA was confirmed through a WB experiment, and substantiated by the results of data analyses. Bioinformatics analyses are useful for identification of biomarkers and pathways associated with FA and AA. Some crucial DEPs, such as ENO1, PGK1, ACTC1, ACTBL2, EEF1A1 and CFL1, may play an important role in FA and show potential as serological markers for its early diagnosis.
Keywords: Fanconi anemia, aplastic anemia, proteomics, bioinformatics analysis, biomarker
Introduction
Fanconi anaemia (FA) is a rare autosomal recessive genetic disease. Most patients are characterized by bone marrow failure as well as congenital malformations. However, some patients present with complex and diverse clinical manifestations, indicating the difficulties involved in differentiating between FA and AA [1-4]. Aplastic anaemia comprises a group of pathogenies leading to the bone marrow failure syndrome, bone marrow hematopoietic hypoplasia, and peripheral blood pancytopenia. It is characterized by clinical anaemia, bleeding, and infection [5,6]. Valid methods for identifying biomarkers useful for the screening and diagnosis of FA as opposed to AA are very important. Proteomics focuses on the composition, content and signalling pathways of proteins in cells, tissues or organisms. Networks, pathways and protein-protein interactions (PPIs) involved in FA and AA can be elucidated using this new technology [7].
In this analysis, we performed a proteomic analysis of the marrow of patients with FA and AA to detect differentially expressed proteins (DEPs). We examined biological processes (BP), molecular functions (MF), and cellular components (CC) using the Database for Annotation, Visualization and Integrated Discovery (DAVID) [8,9] and the Kyoto Encyclopaedia of Genes and Genomes (KEGG) pathways of DEPs via ClueGO [10]. We identified 5 key modules, established the PPI network of DEPs and selected 8 hub proteins with a high degree of connectivity as follows; alpha enolase (ENO1), HSP90AA1, phosphoglycerate kinase 1 (PGK1), HSP90AB1, ACTC1, ACTBL2, EEF1A1 and CFL1. These proteins may play an important role in FA, serve as new biomarkers for its diagnosis and guide its combination therapy.
Materials and methods
Patient samples
The study group was composed of a patient with FA and a patient with AA. The findings of chromosomal fracture experiments of FA patients examined at the Children’s Hospital of Soochow University were negative. Information was obtained from primary pathological reports. All human bone marrow tissue samples used in this study were approved by the Hospital Research Ethics Committee.
Protein preparation and TMT labelling
Cells in the marrow and blood were harvested at 12,000 g for 10 min at 4°C. Collected sediment was washed thrice with PBS buffer (pH 7.4). The samples were ground into powder in liquid nitrogen, and proteins extracted using lysis buffer. The suspension was sonicated at 200 W for 15 min and centrifuged at 12,000 g for 15 min at 4°C. The supernatant was mixed well with 5 volumes of chilled acetone containing 10% (v/v) TCA, incubated overnight at 20°C, centrifuged at 12,000 g for 15 min at 4°C, and discarded. The precipitate was washed thrice with chilled acetone. The pellet was air dried and dissolved in lysis buffer. Next, in-gel digestion was performed.
Proteins were digested with trypsin at a protein:trypsin ratio of 30:1 at 37°C for 16 h. Peptides were dried via vacuum centrifugation following digestion by trypsin. The peptides were reconstituted and processed with a TMT label in accordance with the manufacturer’s protocols. The labelled peptides were used for mass spectrometry and identification.
LC-MS/MS analysis using an Orbitrap elite hybrid mass spectrometer
Each fraction was resuspended using buffer A (5% CAN and 0.1% FA), and centrifuged at 12,000 g for 10 min. The final peptide concentration was approximately 0.5 μg/μL. Approximately 10 μL of the peptides were injected into nanoHPLC using an auto sampler on a 2 cm C18 trap column.
Identification of proteins through mass spectrometry
The largest dataset selected was Swiss-Prot (Human), which included 20,316 protein sequences. The results were exported to Microsoft Excel for further analysis. Quantitative protein ratios were weighted and normalized. Ratios with fold changes >1.50 were considered significant.
Bioinformatics analysis
For each gene list, pathway and process enrichment analyses were carried out using the following ontology sources: KEGG Pathway, GO Biological Processes, Reactome Gene Sets, Canonical Pathways, and CORUM. All genes in the genome were used as the enrichment background. Terms with a p-value <0.01, a minimum count of 3, and an enrichment factor (ratio of observed counts to expected counts due to chance) >1.5 were collected and grouped into clusters based on their membership similarities. The most statistically significant term within a cluster was chosen to represent that cluster. PPI enrichment analysis reflected the interactions of DEPs, wherein the interacting protein complex functionally affected the physiological process. The MCODE algorithm was used to identify densely connected network components. GO analysis was performed to detect molecular functions, biological processes and cellular components with DAVID (https://david.ncifcrf.gov/). ClueGo and MCODE plug-ins in Cytoscape were used to perform pathway and process enrichment analyses and PPI enrichment analysis.
Statistical analysis
Differences between groups were analysed using Student’s t-test or the one-way ANOVA test. Statistical significance was set at P<0.05. Statistical analyses and the drawing of plots were performed using GraphPad Prism 6.01 software (La Jolla, CA, USA).
Results
Sample information and gene test results of patients with FA
Experimental samples were obtained from the bone marrow of one patient with FA and one patient with AA. The patient diagnosed with FA exhibited defects as indicated by the FA genetic test, and carried 2 heterozygous mutations in BRCA2: c.T943A (exon10) and c.T7469C (exon15) (Table 1), inherited from the father and the mother, respectively.
Table 1.
General information of children with AA and FA
| Number | Samples | Disease type | Chromosomal break test | Gene mutation sites | Parents carry genetic conditions |
|---|---|---|---|---|---|
| 1 | Marrow | FA | Negative | BRCA2 gene with two heterozygous mutations: c.T943A (exon10), c.T7469C (exon15) | Each of the parents carry a mutant gene |
| 2 | Marrow | AA | Negative | Negative |
Statistical analysis of FA and AA proteomics
The largest dataset selected was Swiss-Prot (Human). A total of 20,316 unique protein sequences were obtained and matched with 564 proteins in the bone marrow. The workflow of the proteomic analysis is shown in Figure 1A. Identification of the physicochemical properties of proteins generally reflect protein derivation in peptides and proteome sample identification, and therefore require technical and biological evaluation. In our study, the sequence coverage of most identified proteins was satisfactory (Figure 1B). In the sequence coverage of 350 proteins, 62% proteins were distributed within the 95% confidence interval (CI), and 17.9% proteins (101 proteins) were within the 80% CI. Therefore, a mass distribution was constructed (Figure 1C); the molecular weights of 90% of the proteins were between 10-90 kDa, whereas those of 5 proteins were less than 10 kDa, and those of 65 proteins were higher than 90 kDa. The number of PSMs that matched the proteins is shown in Figure 1D. The number of PSMs may be used to quantify map counts. Hence, this index provides an approximate indication of the distribution of proteins with different levels of abundance. The distribution of isoelectric points of identified proteins is displayed in Figure 1E. Sequence coverage and the number of PSMs matching the proteins showed that protein identification was credible. The distribution of molecular weights and isoelectric points of the identified proteins were reasonable, as well. A total of 114 DEPs, including 71 upregulated proteins and 43 downregulated proteins, were identified in the FA samples compared with those in the AA samples.
Figure 1.

Statistical analysis of proteomic data of FA and AA. A. Sample processing and data analysis procedure used for proteomic analysis of FA and AA. B. Sequence coverage distribution of identified proteins. Results showed that protein coverage distribution was reasonable. C. The protein mass distribution of identified proteins. D. Number of PSMs matching the identified proteins. E. The distribution of protein isoelectric points of identified proteins.
GO function and KEGG pathway enrichment analysis of DEPs
GO analysis of BP, MF, and CC of the bone marrow samples were performed using the DAVID website. The results of DAVID revealed that upregulated and downregulated DEPs had a total of 117 GO terms. GO analysis indicated that upregulated DEPs were associated with nucleosome assembly, canonical glycolysis, respiratory burst, cell or subcellular component movement and the glycolytic process, whereas downregulated genes were mainly involved in cellular protein metabolic processes, immune response, negative regulation of apoptosis, proteolysis and retinal homeostasis. Details of the top 5 GO terms of BP, MF, and CC are shown in Table 2.
Table 2.
Top five GO functions from the enrichment analyses of upregulated and downregulated DEPs
| Expression | Category | Term | Count | % | p value |
|---|---|---|---|---|---|
| Upregulated | GOTERM_BP_DIRECT | Nucleosome assembly | 8 | 12.3 | 2.70E-07 |
| GOTERM_BP_DIRECT | Canonical glycolysis | 5 | 7.7 | 2.40E-06 | |
| GOTERM_BP_DIRECT | Respiratory burst | 4 | 6.2 | 1.30E-05 | |
| GOTERM_BP_DIRECT | Movement of cell or subcellular component | 6 | 9.2 | 1.60E-05 | |
| GOTERM_BP_DIRECT | Glycolytic process | 4 | 6.2 | 2.60E-04 | |
| GOTERM_CC_DIRECT | Extracellular exosome | 51 | 78.5 | 8.50E-29 | |
| GOTERM_CC_DIRECT | Focal adhesion | 17 | 26.2 | 2.80E-13 | |
| GOTERM_CC_DIRECT | Membrane | 27 | 41.5 | 6.90E-09 | |
| GOTERM_CC_DIRECT | Cytosol | 32 | 49.2 | 3.20E-08 | |
| GOTERM_CC_DIRECT | Vesicle | 8 | 12.3 | 3.20E-07 | |
| GOTERM_MF_DIRECT | Poly(A) RNA binding | 18 | 27.7 | 6.50E-07 | |
| GOTERM_MF_DIRECT | Protein binding | 52 | 80 | 4.20E-06 | |
| GOTERM_MF_DIRECT | Nucleosomal DNA binding | 5 | 7.7 | 2.70E-05 | |
| GOTERM_MF_DIRECT | Structural constituent of cytoskeleton | 6 | 9.2 | 6.00E-05 | |
| GOTERM_MF_DIRECT | Superoxide-generating NADPH oxidase activity | 3 | 4.6 | 7.60E-04 | |
| Downregulated | GOTERM_BP_DIRECT | Cellular protein metabolic process | 5 | 14.3 | 8.10E-05 |
| GOTERM_BP_DIRECT | Immune response | 6 | 17.1 | 1.30E-03 | |
| GOTERM_BP_DIRECT | Negative regulation of apoptotic process | 6 | 17.1 | 1.80E-03 | |
| GOTERM_BP_DIRECT | Proteolysis | 6 | 17.1 | 2.70E-03 | |
| GOTERM_BP_DIRECT | Retina homeostasis | 3 | 8.6 | 2.80E-03 | |
| GOTERM_CC_DIRECT | Extracellular exosome | 28 | 80 | 1.80E-16 | |
| GOTERM_CC_DIRECT | Extracellular space | 18 | 51.4 | 3.70E-11 | |
| GOTERM_CC_DIRECT | Lysosome | 7 | 20 | 3.40E-06 | |
| GOTERM_CC_DIRECT | Melanosome | 5 | 14.3 | 3.60E-05 | |
| GOTERM_CC_DIRECT | Cell surface | 7 | 20 | 4.50E-04 | |
| GOTERM_MF_DIRECT | Serine-type endopeptidase activity | 7 | 20 | 8.80E-06 | |
| GOTERM_MF_DIRECT | Protein binding | 25 | 71.4 | 1.30E-02 | |
| GOTERM_MF_DIRECT | Histone binding | 3 | 8.6 | 2.40E-02 | |
| GOTERM_MF_DIRECT | Heparin binding | 3 | 8.6 | 3.90E-02 | |
| GOTERM_MF_DIRECT | Lysophospholipase activity | 2 | 5.7 | 4.80E-02 |
KEGG pathway enrichment analysis was performed using ClueGO in Cytoscape. KEGG results indicated that upregulated DEPs were mainly associated with glycolysis/gluconeogenesis, leukocyte trans-endothelial migration, the estrogen signalling pathway, the pentose phosphate pathway, the glucagon signalling pathway, propanoate metabolism and pyruvate metabolism. Downregulated DEPs were associated with lysosomes, autophagy, phagosomes, pantothenate, CoA biosynthesis and the neurotrophin signalling pathway (Figure 2A, 2B and Table 3).
Figure 2.
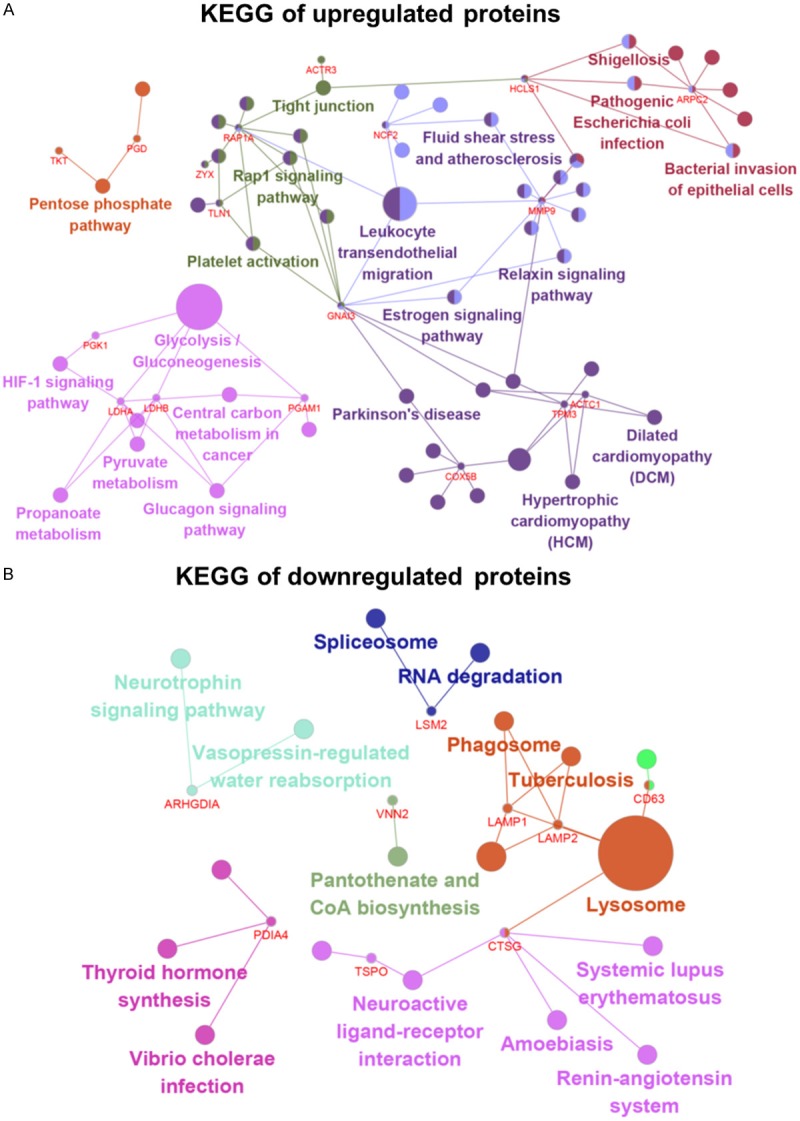
Signalling pathway enrichment analysis of DEP function in AA and FA. A. KEGG pathways of upregulated proteins. Upregulated DEPs were mainly associated with glycolysis/gluconeogenesis, leukocyte transendothelial migration, the estrogen signalling pathway and the pentose phosphate pathway. B. KEGG pathways of downregulated proteins. Downregulated DEPs were associated with lysosomes, autophagy, phagosome, pantothenate and CoA biosynthesis and the neurotrophin signalling pathway. DEPs functional and signalling pathway enrichment was conducted using Cytoscape. DEPs, differentially expressed proteins; KEGG, Kyoto Encyclopaedia of Genes and Genomes.
Table 3.
KEGG pathway analysis of DEPs associated with FA
| Expression | Terms | Description | p Value | Number of proteins |
|---|---|---|---|---|
| Upregulated | KEGG:00010 | Glycolysis/Gluconeogenesis | 2.78435E-05 | 4 |
| KEGG:04670 | Leukocyte trans-endothelial migration | 0.000197977 | 4 | |
| KEGG:04260 | Cardiac muscle contraction | 0.000943429 | 3 | |
| KEGG:00030 | Pentose phosphate pathway | 0.002019859 | 2 | |
| KEGG:04922 | Glucagon signalling pathway | 0.002117107 | 3 | |
| KEGG:00640 | Propanoate metabolism | 0.00229856 | 2 | |
| KEGG:00620 | Pyruvate metabolism | 0.00341019 | 2 | |
| KEGG:04611 | Platelet activation | 0.003521256 | 3 | |
| Downregulated | KEGG:04142 | Lysosome | 4.65034E-06 | 4 |
| KEGG:04140 | Autophagy | 0.005296114 | 2 | |
| KEGG:04145 | Phagosome | 0.007417878 | 2 | |
| KEGG:05152 | Tuberculosis | 0.010204301 | 2 | |
| KEGG:00770 | Pantothenate and CoA biosynthesis | 0.011122788 | 1 | |
| KEGG:04614 | Renin-angiotensin system | 0.014211982 | 1 | |
| KEGG:04080 | Neuroactive ligand-receptor interaction | 0.023839615 | 2 | |
| KEGG:04962 | Vasopressin-regulated water reabsorption | 0.027181831 | 1 | |
| KEGG:05110 | Vibrio cholerae infection | 0.030885554 | 1 | |
| KEGG:04918 | Thyroid hormone synthesis | 0.045688094 | 1 |
PPI network of DEPs
PPI relationships of the 114 DEPs were obtained using STRING and visualized using Cytoscape. In STRING, active interaction sources included textming, experiments, databases, co-expression, neighbourhood, gene fusion, and co-occurrence. PPIs with an interaction score >0.4 (medium confidence) were selected, and a total of 415 PPI relationships were generated. The total PPI network of DEPs is shown in Figure 3. Based on the PPI network, the top 8 hub genes showing a high degree of connectivity were selected as the hub proteins (Table 4).
Figure 3.
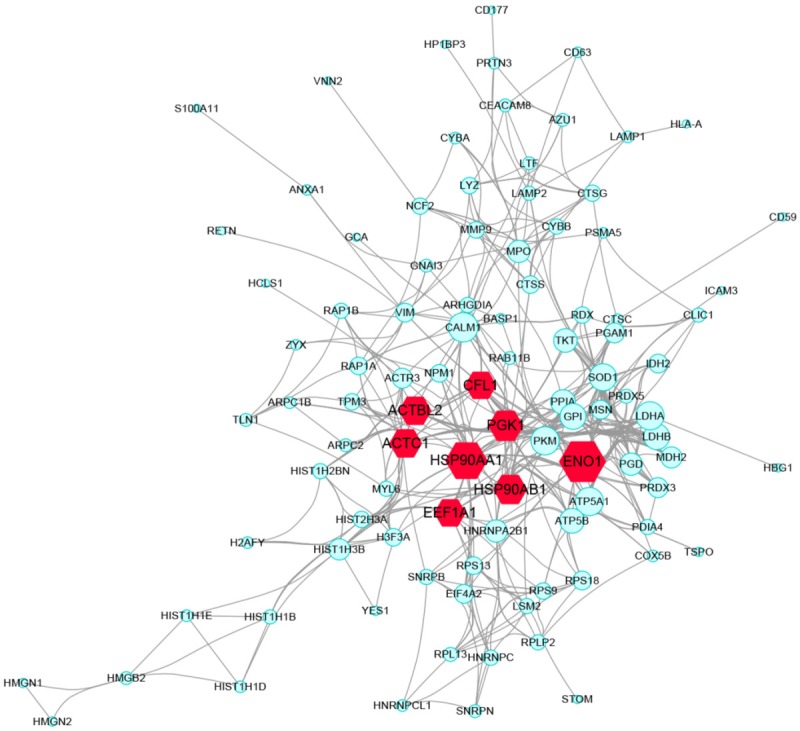
Protein-protein interaction between DEPs, (PPI) network complex and modular analysis. Using the STRING online database, total of 114 DEPs (including 71 upregulated proteins and 43 downregulated proteins) were filtered into the DEGs PPI network complex. Red labels represent hub genes. The size of the edge was determined on the basis of the degree score of proteins.
Table 4.
Top hub proteins in the PPI network based on degree
| Name | Degree | Eccentricity | Betweenness | Stress | Bridging | Centroid |
|---|---|---|---|---|---|---|
| ENO1 | 33 | 0.2 | 1496.064246 | 11498 | 15.15034088 | 0 |
| HSP90AA1 | 29 | 0.25 | 1487.316057 | 14224 | 17.33456018 | 0 |
| PGK1 | 23 | 0.2 | 244.8566235 | 3848 | 6.987651367 | -26 |
| HSP90AB1 | 22 | 0.25 | 696.3301408 | 9648 | 16.4548778 | -12 |
| ACTC1 | 21 | 0.25 | 546.361113 | 8242 | 12.90466075 | -21 |
| ACTBL2 | 21 | 0.25 | 546.361113 | 8242 | 12.90466075 | -21 |
| EEF1A1 | 20 | 0.25 | 512.4611003 | 6102 | 15.37382408 | -25 |
| CFL1 | 20 | 0.2 | 387.9301232 | 3856 | 11.66773024 | -25 |
The hub proteins, which play an important role in FA progression, were alpha-enolase (ENO1, degree: 33), isoform 2 of heat shock protein HSP 90-alpha (HSP90AA1, degree: 29), phosphoglycerate kinase 1 (PGK1; degree: 23), heat shock protein HSP 90 beta (HSP90AB1, degree: 22), actin (degree: 21), beta-actin-like protein 2 (ACTBL2, degree: 21), elongation factor 1-alpha 1 (EEF1A1, degree: 20) and cofilin-1 (CFL1, degree: 20).
Module and hub protein analysis
A PPI enrichment analysis was conducted, and the 5 most significant sub-modules of DEPs were extracted from the PPI network [11]. Module 1 (Molecular Complex Detection [MCODE] score =7.263) was constructed with 20 nodes and 69 edges (Figure 4A); module 2 (MCODE score =6.5000) was constructed with 9 nodes and 26 edges (Figure 4B); module 3 (MCODE score =5.000) was constructed with 5 nodes and 10 edges (Figure 4C); module 4 (MCODE score =4.000) was constructed with 4 nodes and 6 edges (Figure 4D); and module 5 (MCODE score =4.000) was constructed with 4 nodes and 6 edges (Figure 4E). Further GO analysis of the MCODE 1 component mainly focused on translational initiation, SRP-dependent co-translational protein targeting the membrane, nuclear-transcribed mRNA catabolic process, nonsense-mediated decay, rRNA processing, translation and canonical glycolysis (Table 5). KEGG pathway enrichment analyses were associated with carbon metabolism, antibiotic biosynthesis, ribosomes, glycolysis/gluconeogenesis, amino acid biosynthesis, glucagon signalling pathway, pentose phosphate pathway, and the estrogen signalling pathway (Table 6). Furthermore, to verify results of the data analysis, we collected samples from patients with AA and FA, and investigated changes in key proteins using western blot analysis. The results showed that compared with AA, ENO1 and CFL1 were upregulated in FA, while WB results were consistent with those of the data analysis (Figure 4F).
Figure 4.
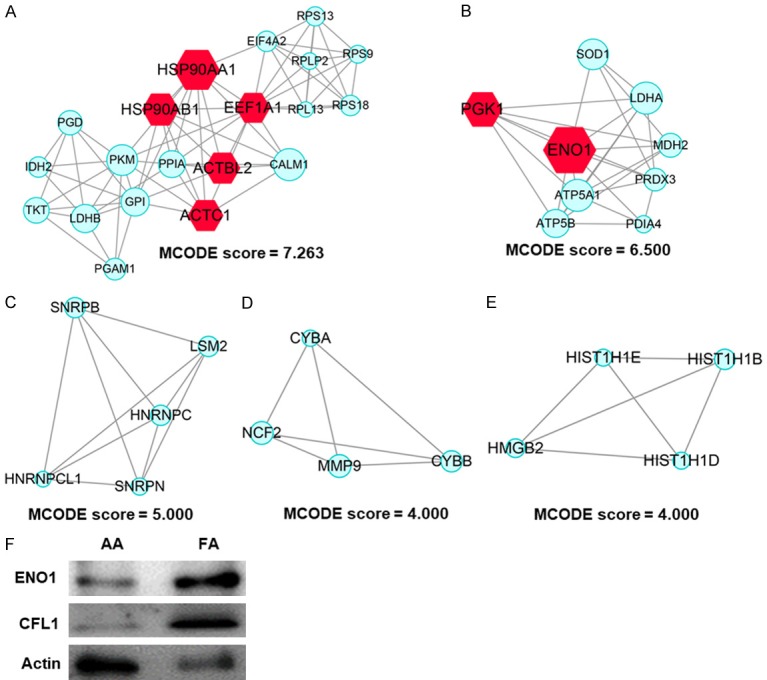
Significant modules in the PPI networks. A. Module 1 consists of 20 nodes and 69 edges, and MCODE score was 7.263; B. Module 2 consists of 9 nodes and 26 edges; C. Module 3 consists of 5 nodes and 10 edges; D. Module 4 consists of 4 nodes and 6 edges; E. Module 5 consists of 4 nodes and 6 edges; F. Western blot results of ENO1 and CFL1 in AA and FA patients.
Table 5.
Top 10 GO functional enrichment analyses of the most significant DEP modules
| Category | Term | Count | % | p Value |
|---|---|---|---|---|
| GOTERM_BP_DIRECT | Translational initiation | 6 | 30 | 2.60E-07 |
| GOTERM_BP_DIRECT | SRP-dependent cotranslational protein targeting to membrane | 5 | 25 | 2.70E-06 |
| GOTERM_BP_DIRECT | Viral transcription | 5 | 25 | 5.30E-06 |
| GOTERM_BP_DIRECT | Nuclear-transcribed mRNA catabolic process, nonsense-mediated decay | 5 | 25 | 6.80E-06 |
| GOTERM_BP_DIRECT | rRNA processing | 5 | 25 | 6.80E-05 |
| GOTERM_BP_DIRECT | Translation | 5 | 25 | 1.30E-04 |
| GOTERM_BP_DIRECT | Canonical glycolysis | 3 | 15 | 3.50E-04 |
| GOTERM_BP_DIRECT | Response to salt stress | 2 | 10 | 9.60E-03 |
| GOTERM_BP_DIRECT | Positive regulation of cell size | 2 | 10 | 1.10E-02 |
| GOTERM_BP_DIRECT | Positive regulation of protein import into nucleus, translocation | 2 | 10 | 1.10E-02 |
Table 6.
Top 10 KEGG pathway enrichment analyses in the most significant DEP module
| Category | Term | Description | Count | % | p Value |
|---|---|---|---|---|---|
| KEGG_PATHWAY | hsa01200 | Carbon metabolism | 6 | 30 | 5.70E-06 |
| KEGG_PATHWAY | hsa01130 | Biosynthesis of antibiotics | 7 | 35 | 7.20E-06 |
| KEGG_PATHWAY | hsa03010 | Ribosome | 5 | 25 | 2.80E-04 |
| KEGG_PATHWAY | hsa00010 | Glycolysis/Gluconeogenesis | 4 | 20 | 5.40E-04 |
| KEGG_PATHWAY | hsa01230 | Biosynthesis of amino acids | 4 | 20 | 7.20E-04 |
| KEGG_PATHWAY | hsa04922 | Glucagon signalling pathway | 4 | 20 | 1.70E-03 |
| KEGG_PATHWAY | hsa00030 | Pentose phosphate pathway | 3 | 15 | 2.20E-03 |
| KEGG_PATHWAY | hsa04915 | Estrogen signalling pathway | 3 | 15 | 2.40E-02 |
Discussion
FA is an autosomal recessive disorder characterized by progressive bone marrow hematopoietic failure, multiple congenital anomalies, and predisposition to neoplastic disease [12]. Some studies have shown that clinical manifestations of FA may not be typical, especially in those patients who present as chimeras, and easily lead to misdiagnoses or missed diagnoses [13]. Currently, only chromosomal aberrations induced by mitomycin C or diepoxybutane can be distinguished from other diseases, such as AA [14,15]. To date, systematic studies comparing FA and AA, or comparative proteomic analyses of FA and AA have been rarely performed. As a part of this study, proteomics was conducted to examine the differences in the functions, signalling pathways and PPI in bone marrow samples of FA and AA patients. This study could be used for the identification of appropriate proteomic protocols and biomarkers as well as for the determination of FA and AA. A total of 114 DEPs, including 71 upregulated proteins and 43 downregulated proteins, were identified. GO function and KEGG pathway analyses of DEPs were performed to enhance the understanding of these DEPs. GO analysis indicated that the upregulated proteins were mainly associated with the following functions: nucleosome assembly, canonical glycolysis, respiratory burst, cell or subcellular component movement, and glycolytic process, whereas downregulated proteins were mainly associated with cellular protein metabolic processes, immune response, negative regulation of apoptosis, proteolysis, and retinal homeostasis. The hub proteins were analysed and selected through degree calculation. Our findings indicate that ENO1, HSP90AA1, PGK1, HSP90AB1, ACTC1, ACTBL2, EEF1A1 and CFL1 may play key roles in FA and also function as diagnostic markers.
ENO1 was the most significant hub protein. It is a multifunctional enzyme that participates in glycolysis and other processes, such as hypoxia tolerance, growth control and allergic responses [16-19]. It can activate plasminogen on cell surfaces and also functions in the intravascular and pericellular fibrinolytic system. It stimulates immunoglobulin production and participates in the immune system. ENO1 is upregulated in FA. Thus, its functional features may partially explain changes in the immune function of FA. PGK1 acts as a polymerase alpha cofactor protein and glycolytic enzyme [20-23]. It is involved in the synthesis of pyruvate from D-glyceraldehyde-3-phosphate and is thereby related to energy metabolism. Molecular function analysis indicated that PGK1 is involved in ATP-binding as well as in phosphoglycerate kinase activities and protein-disulfide reductase activities. Biological process analysis revealed that PGK1 is associated with canonical glycolysis, cellular response to hypoxia, epithelial cell differentiation and gluconeogenesis.
GO and KEGG pathway analyses were performed to enhance our understanding of DEP interactions. Biological process analysis indicated that nucleosome assembly was mainly associated with upregulated proteins. Nucleosome subunits of eukaryotic chromatin were formed by spontaneous reactions between histones and DNA. The nucleosome assembly pathway has been studied in disease and disorder research, and was found to be associated with FA.
In marrow proteomics, differentially expressed networks and pathways of FA and AA were mainly involved in metabolic processes, monosaccharide bio-syntheses and gluconeogenesis, thereby reflecting energy metabolism differences between FA and AA. In addition, protein translation and replication differed between the two diseases. Moreover, the activation of DNA fragmentation factor, DNA fragmentation induced by apoptosis, formation of senescence-associated heterochromatin foci and the regulation of cell morphogenesis were enriched by DEP analysis. These pathways and functions, which are associated with leukaemia, provide useful information related to the treatment of FA.
FA related research has undergone a shift from chromosomal aberrations to DNA repair and oncology studies, which, via monitoring of pathogenesis and mediation of DNA hinge injury repair pathways, have made remarkable breakthroughs. High-tumour susceptibility studies have also made considerable progress. Proteomic enrichment has also revealed certain tumour-related functions and pathways. KEGG pathways for upregulated DEPs were mainly associated with glycolysis/gluconeogenesis, leukocyte trans-endothelial migration, the pentose phosphate pathway and the glucagon signalling pathway, indicating that energy metabolism of FA patients was stronger than that of AA patients. Downregulated DEPs were associated with lysosome, autophagy, phagosome, tuberculosis, pantothenate, and CoA biosynthesis. Cell function may be further reduced in FA patients compared to that of AA patients. Enrichment of these functions and pathways may provide insight into the conversion of FA to leukaemia [24].
To the best of our knowledge, this is the first study to use bioinformatics to compare differences between FA and AA. Our findings may also provide information related to the identification and differential diagnosis of FA and AA for follow-up studies.
Conclusions
The present study examined the DEPs of FA and AA through a systematic bioinformatics analysis and revealed that immunological processes and functions of FA patients exhibiting cellular macromolecule localization ability were reduced compared with that of AA. ENO1, PGK1, ACTC1, ACTBL2, EEF1A1 and CFL1 may be used as serologic markers for the early diagnosis of FA. These findings may remarkably improve our understanding of the differences between molecular mechanisms underlying FA and AA. Candidate hub proteins and pathways may be used as therapeutic targets.
Acknowledgements
The work was supported by the Natural Science Foundation of China (NO. 81770193), Jiangsu Province Projects (NO. BE2017659, BRA2017541, and CXTDA2017014), and Suzhou projects (SZS201615, SZZX201504, SS201809, and SYS201643).
Disclosure of conflict of interest
None.
References
- 1.Ameziane N, May P, Haitjema A, van de Vrugt HJ, van Rossum-Fikkert SE, Ristic D, Williams GJ, Balk J, Rockx D, Li H, Rooimans MA, Oostra AB, Velleuer E, Dietrich R, Bleijerveld OB, Maarten Altelaar AF, Meijers-Heijboer H, Joenje H, Glusman G, Roach J, Hood L, Galas D, Wyman C, Balling R, den Dunnen J, de Winter JP, Kanaar R, Gelinas R, Dorsman JC. A novel Fanconi anaemia subtype associated with a dominant-negative mutation in RAD51. Nat Commun. 2015;6:8829. doi: 10.1038/ncomms9829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bijangi-Vishehsaraei K, Saadatzadeh MR, Werne A, McKenzie KA, Kapur R, Ichijo H, Haneline LS. Enhanced TNF-alpha-induced apoptosis in Fanconi anemia type C-deficient cells is dependent on apoptosis signal-regulating kinase 1. Blood. 2005;106:4124–4130. doi: 10.1182/blood-2005-05-2096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Guardiola P, Socie G, Li X, Ribaud P, Devergie A, Esperou H, Richard P, Traineau R, Janin A, Gluckman E. Acute graft-versus-host disease in patients with Fanconi anemia or acquired aplastic anemia undergoing bone marrow transplantation from HLA-identical sibling donors: risk factors and influence on outcome. Blood. 2004;103:73–77. doi: 10.1182/blood-2003-06-2146. [DOI] [PubMed] [Google Scholar]
- 4.Auerbach AD, Rogatko A, Schroeder-Kurth TM. International Fanconi anemia registry: relation of clinical symptoms to diepoxybutane sensitivity. Blood. 1989;73:391–396. [PubMed] [Google Scholar]
- 5.Zhang J, Wu Q, Zheng Y. Persistent elevated bone marrow plasma levels of thrombopoietin in patients with aplastic anemia. Cytokine. 2016;85:11–13. doi: 10.1016/j.cyto.2016.05.020. [DOI] [PubMed] [Google Scholar]
- 6.Bar C, Povedano JM, Serrano R, Benitez-Buelga C, Popkes M, Formentini I, Bobadilla M, Bosch F, Blasco MA. Telomerase gene therapy rescues telomere length, bone marrow aplasia, and survival in mice with aplastic anemia. Blood. 2016;127:1770–1779. doi: 10.1182/blood-2015-08-667485. [DOI] [PubMed] [Google Scholar]
- 7.Bachi A, Bonaldi T. Quantitative proteomics as a new piece of the systems biology puzzle. J Proteomics. 2008;71:357–367. doi: 10.1016/j.jprot.2008.07.001. [DOI] [PubMed] [Google Scholar]
- 8.Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 9.Huang DW, Sherman BT, Tan Q, Kir J, Liu D, Bryant D, Guo Y, Stephens R, Baseler MW, Lane HC, Lempicki RA. DAVID bioinformatics resources: expanded annotation database and novel algorithms to better extract biology from large gene lists. Nucleic Acids Res. 2007;35:W169–175. doi: 10.1093/nar/gkm415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bindea G, Mlecnik B, Hackl H, Charoentong P, Tosolini M, Kirilovsky A, Fridman WH, Pages F, Trajanoski Z, Galon J. ClueGO: a cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics. 2009;25:1091–1093. doi: 10.1093/bioinformatics/btp101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Micale G, Continella A, Ferro A, Giugno R, Pulvirenti A. GASOLINE: a cytoscape app for multiple local alignment of PPI networks. Version 2. F1000Res. 2014;3:140. doi: 10.12688/f1000research.4537.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Przychodzen B, Makishima H, Sekeres MA, Balasubramanian SK, Thota S, Patel BJ, Clemente M, Hirsch C, Dienes B, Maciejewski JP. Fanconi anemia germline variants as susceptibility factors in aplastic anemia, MDS and AML. Oncotarget. 2018;9:2050–2057. doi: 10.18632/oncotarget.23328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Milner RD, Khallouf KA, Gibson R, Hajianpour A, Mathew CG. A new autosomal recessive anomaly mimicking Fanconi’s anaemia phenotype. Arch Dis Child. 1993;68:101–103. doi: 10.1136/adc.68.1.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Martinez A, Hinz JM, Gomez L, Molina B, Acuna H, Jones IM, Frias S, Coleman MA. Differential expression of TP53 associated genes in Fanconi anemia cells after mitomycin C and hydroxyurea treatment. Mutat Res. 2008;656:1–7. doi: 10.1016/j.mrgentox.2008.06.012. [DOI] [PubMed] [Google Scholar]
- 15.Pagano G, Zatterale A, Korkina LG. Mitomycin C-induced DNA damage in Fanconi anemia: cross-linking or redox-mediated effects? Blood. 1999;93:1116–1118. [PubMed] [Google Scholar]
- 16.Okudela K, Mitsui H, Matsumura M, Arai H, Shino K, Sekine A, Woo T, Suzuki T, Ishikawa Y, Umeda S, Tajiri M, Masuda M, Ohashi K. The potential significance of alpha-enolase (ENO1) in lung adenocarcinomas - a utility of the immunohistochemical expression in pathologic diagnosis. Pathol Int. 2017;67:602–609. doi: 10.1111/pin.12607. [DOI] [PubMed] [Google Scholar]
- 17.Principe M, Borgoni S, Cascione M, Chattaragada MS, Ferri-Borgogno S, Capello M, Bulfamante S, Chapelle J, Di Modugno F, Defilippi P, Nistico P, Cappello P, Riganti C, Leporatti S, Novelli F. Alpha-enolase (ENO1) controls alpha v/beta 3 integrin expression and regulates pancreatic cancer adhesion, invasion, and metastasis. J Hematol Oncol. 2017;10:16. doi: 10.1186/s13045-016-0385-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cappello P, Principe M, Bulfamante S, Novelli F. Alpha-Enolase (ENO1), a potential target in novel immunotherapies. Front Biosci (Landmark Ed) 2017;22:944–959. doi: 10.2741/4526. [DOI] [PubMed] [Google Scholar]
- 19.Liu YQ, Huang ZG, Li GN, Du JL, Ou YP, Zhang XN, Chen TT, Liang QL. Effects of alpha-enolase (ENO1) over-expression on malignant biological behaviors of AGS cells. Int J Clin Exp Med. 2015;8:231–239. [PMC free article] [PubMed] [Google Scholar]
- 20.Prasanth KR, Chuang C, Nagy PD. Co-opting ATP-generating glycolytic enzyme PGK1 phosphoglycerate kinase facilitates the assembly of viral replicase complexes. PLoS Pathog. 2017;13:e1006689. doi: 10.1371/journal.ppat.1006689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Falkenberg VR, Whistler T, Murray JR, Unger ER, Rajeevan MS. Identification of Phosphoglycerate Kinase 1 (PGK1) as a reference gene for quantitative gene expression measurements in human blood RNA. BMC Res Notes. 2011;4:324. doi: 10.1186/1756-0500-4-324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Riley DE, Goldman MA, Gartler SM. Nucleotide sequence of the 3’ nuclease-sensitive region of the human phosphoglycerate kinase 1 (PGK1) gene. Genomics. 1991;11:212–214. doi: 10.1016/0888-7543(91)90121-t. [DOI] [PubMed] [Google Scholar]
- 23.Mazzoni C, Torella M, Petrera A, Palermo V, Falcone C. PGK1, the gene encoding the glycolitic enzyme phosphoglycerate kinase, acts as a multicopy suppressor of apoptotic phenotypes in S. cerevisiae. Yeast. 2009;26:31–37. doi: 10.1002/yea.1647. [DOI] [PubMed] [Google Scholar]
- 24.Yeo CJ, Gilman AL. Interleukin-2-induced graft-versus-leukemia for the treatment of AML in a BRCA2 Fanconi anemia patient. J Pediatr Hematol Oncol. 2014;36:e78–80. doi: 10.1097/MPH.0b013e31828e5c56. [DOI] [PubMed] [Google Scholar]