

Abstract
Dystonia and levodopa-induced dyskinesia (LID) are both hyperkinetic movement disorders. Dystonia arises most often spontaneously, although it may be seen after stroke, injury, or as a result of genetic causes. LID is associated with Parkinson disease (PD), emerging as a consequence of chronic therapy with levodopa, and may be either dystonic or choreiform. LID and dystonia share important phenomenological properties and mechanisms. Both LID and dystonia are generated by an integrated circuit involving the cortex, basal ganglia, thalamus and cerebellum. They also share dysregulation of striatal cholinergic signaling and abnormalities of striatal synaptic plasticity. The long duration nature of both LID and dystonia suggests that there may be underlying epigenetic dysregulation as a proximate cause. While both may improve after interventions such as deep brain stimulation (DBS), neither currently has a satisfactory medical therapy, and many people are disabled by the symptoms of dystonia and LID. Further study of the fundamental mechanisms connecting these two disorders may lead to novel approaches to treatment or prevention.
Keywords: Dystonia, dyskinesia, levodopa, basal ganglia, cerebellum, dopamine, acetylcholine, plasticity
Introduction
Dystonia and levodopa-induced dyskinesia (LID) are both involuntary hyperkinetic movement disorders. Although generally considered to be distinct conditions, dystonia and LID share certain phenomenological aspects and dysfunction of common neuronal circuits (Calabresi et al., 2016). Dystonia arises most often spontaneously, although it may be seen after stroke, injury, or as a result of genetic causes. LID is associated with Parkinson disease (PD), emerging as a consequence of chronic therapy with levodopa, and may be either dystonic or choreiform. Recent work, however, has revealed that these two conditions have more in common than has been appreciated. They share not only clinical features at times, but also underlying physiological and neurochemical alterations. At present, there is no satisfactory medical treatment for either dystonia or dyskinesia, but by exploring the common links it may be possible to find novel approaches to these problems.
Commonalities in clinical aspects
Levodopa-induced dyskinesia (LID)
Levodopa or L-dopa is the gold-standard pharmacological therapy used to control motor symptoms of Parkinson disease (PD). A major limitation of this therapy, however, are the motor complications, such as wearing off and LID, which commonly occur after a few years of levodopa treatment (Calabresi et al., 2010). LIDs are represented by a spectrum of movement disorders including chorea, dystonia, ballism, and myoclonus. The clinical manifestations can be divided into three main categories based on movement patterns and the temporal correlation between the occurrence of LID and levodopa intake: peak-dose dyskinesias (occurring with high blood levodopa concentrations), biphasic dyskinesias (occurring during the rising and falling phase of blood levodopa), and off dyskinesias (at the trough of blood levodopa concentration) (Calabresi et al., 2010; Espay et al., 2018). The risk of developing LID correlates with age at onset (with earlier onset more prone to dyskinesia), gender (more common in women), disease duration and severity. The risk of LID is also modified by levodopa dose and body weight (Eusebi et al., 2018). In addition, there is some evidence for genetic factors which may contribute to the risk of dyskinesia (Lee et al., 2011), but these studies have been relatively limited in scope so far and the full extent of genetic contributions to LID remains to be explored.
Dystonia
Dystonia is characterized by sustained muscle contraction associated with twisting, repetitive, and patterned movements and abnormal posture (Breakefield et al., 2008). Dystonia may be isolated, where dystonia is the only feature (except perhaps tremor) or combined, with features such as parkinsonism or myoclonus (Albanese et al., 2013). The most common forms of dystonia seen in clinical practice are isolated and focal, affecting only one body region. Isolated dystonia may also affect larger regions of the body, becoming segmental, multifocal, or generalized. In focal isolated dystonia, clinical investigation most commonly does not reveal a specific etiology, and the cause is considered idiopathic. Some focal isolated dystonias, and a larger proportion of generalized and early-onset dystonias, may be determined genetically; more than 20 distinct genes can trigger a dystonic syndrome of this kind (Klein, 2014). Dystonia combined with parkinsonism can be seen in a number of neurodegenerative, genetic, toxic, and metabolic disorders (Jankovic and Tintner, 2001; LeWitt et al., 1986; Tolosa and Compta, 2006).
Dystonia and LID in Parkinson disease
Parkinson disease is a clinical syndrome characterized by resting tremor, bradykinesia, rigidity, and postural instability. Patients with PD often exhibit a therapeutic response to treatment with levodopa, and observation of this response supports the diagnosis of PD. Recently, it has become clear that there are additional clinical features associated with PD. Anosmia, constipation, and sleep disorders (REM behavior disorder) may appear in the prodromal phase, and later depression and cognitive impairment may become prominent. Pathologically, the typical findings are depletion of dopaminergic neurons from the substantia nigra pars compacta, along with abnormal accumulation of the protein alpha-synuclein, as Lewy bodies and fibrillar structures. There is currently a great deal of debate as to whether all forms of PD share a common etiology, or whether there are in fact multiple physiological pathways to a similar endpoint (Espay et al., 2017), but in either case the clinical syndrome is usually distinct enough to be readily recognizable.
Dystonia is often a feature of PD in the absence of any treatment. A seminal clinical description of dystonia in PD reported this symptom as an initial feature in patients with both early- and late-onset PD and described action dystonia of the limbs and cranial dystonia (Poewe et al., 1988). The authors speculated that that the coexistence of PD and dystonia might indicate a common pathophysiology. In modern clinical practice, it is common for patients to report dystonia of the feet early in the morning, as part of morning akinesia, or when dopaminergic medications are temporarily withdrawn. Dystonia as a feature of levodopa-induced dystonia was reported as an off- period, biphasic, or peak-dose phenomenon. Interestingly, these different subtypes of dystonia had differential pattern of localization suggesting distinct receptor and biochemical correlates of basal ganglia somatotopy (Poewe et al., 1988). The phenotypes of dystonia seen in PD are discussed in detail in the recent review by Shetty et al. (Shetty et al., 2019).
It worth noting that PD has been commonly associated with the degeneration of midbrain dopamine neurons (Jellinger, 1999) while various forms of parkinsonism and symptomatic dystonia occur after focal lesions localized in some structures of the basal ganglia, and in particular of the striatum and globus pallidus ((Bhatia and Marsden, 1994; Kuoppamaki, 2005; Munchau, 2000; Tambasco et al., 2018); reviewed in (Standaert, 2011)). Also, experimental studies have produced evidence that alterations of distinct anatomical areas of the basal ganglia might contribute to the pathophysiology of parkinsonism and dystonia (Kumbhare et al., 2017). Although the mechanisms underlying the co-existence of parkinsonism, LID, and dystonia have not been fully elucidated, clinical features of dystonia in PD and LID have been well characterized. It has been widely reported that dystonia occurs as an off symptom or as a peak-dose effect of levodopa. Moreover, dystonia is reduced by levodopa treatment when it is observed in PD, while in atypical parkinsonism levodopa has detrimental effects on dystonic symptoms (Yoon, 2018).
An important commonality among PD, dystonia, and LID is related to the therapeutic response to the deep brain stimulation (DBS) of the globus pallidus internus (GPi) in all these conditions. It is now clear that DBS of the GPi ameliorates both hypokinetic (PD) and hyperkinetic disorders (dystonia and LID) (Wichmann and DeLong, 2016). This observation is similar to the clinical experience with pallidotomy for these disorders. Experimental and clinical findings support the hypothesis that improvements following the lesion or DBS of specific basal ganglia nuclei, such as the GPi, are not directly related to specific neurochemical changes of the target nucleus, but rather to the change of the activity of upstream and downstream areas of the brain (Walker et al., 2012; Wichmann and DeLong, 2016). These areas, in fact, might be released from pathologic basal ganglia activity since DBS or ablation might lead to a new equilibrium that is responsible for the amelioration of both hypokinetic and hyperkinetic clinical conditions. In this context, it is interesting that the effects of DBS on LID are often rapid, whereas the effects on dystonia are often delayed by weeks or months, suggesting different underlying adaptive mechanisms (Quartarone and Hallett, 2013).
Circuits in LID and dystonia: cortico-basal ganglia system
The classical model of the basal ganglia, described as the direct/indirect pathway model, hypothesizes a dual interaction between glutamatergic and dopaminergic neurotransmission in the striatum (Albin et al., 1989; DeLong, 1990). In this model it has been hypothesized that cortical activation produces a release of glutamate that activates striatal spiny projecting neurons (SPNs) reaching the substantia nigra pars reticulata (SNpr) and the globus pallidus pars interna (GPi). The GABAergic SPNs originating in the striatum and projecting to the SNpr and GPi are considered as the “direct pathway” and they exert an inhibitory action on the target structures. Since both these structures are also GABAergic, this inhibition leads to a disinhibitory effect on the thalamic glutamatergic neurons projecting to the cortex, which finally results in cortical activation and motor activity. Conversely, activation of SPNs which indirectly project to the SNpr via the globus pallidus pars externa (GPe) and the subthalamic nucleus (STN) (indirect pathway), inhibits the GABAergic neurons of the GPe. This latter event causes a disinhibition of the glutamatergic neurons of the STN. The increased discharge of these excitatory STN neurons in turn activates the GPi and SNpr GABAergic neurons projecting to the thalamus, resulting in thalamic inhibition, reduced thalamocortical activation and motor inactivation. SPNs of the direct and indirect pathway have distinct expression of dopamine (DA) receptors: excitatory D1 DA receptors are expressed by direct pathway SPNs, while inhibitory D2 receptors are localized on SPNs of the indirect pathway. Through this differential action, the net effect of dopamine on both pathways is to promote movement.
Recently, the rigid distinction proposed by the direct/indirect pathway model and its functional relevance has been matter of debate. It has been demonstrated that optogenetic activation of striatal direct and indirect pathway SPNs does in fact induce distinct cellular responses in SNr neurons, supporting the dichotomous function of these pathways (Kravitz et al., 2013). Alternatively, it has been suggested that coordinated activation of both pathways may be required during action selection, and this coordinated activity of the direct and indirect pathways is critical for the appropriate timing and synchrony of BG circuits during movement (Cui et al., 2013). Another critical aspect regarding the model of motor control is how activity in cortical networks regulates direct and indirect pathways. It has been shown that cortical information about motor planning is directed to both direct and indirect pathway SPNs (Kress et al., 2013), suggesting that the two pathways act in conjunction to initiate movements as also postulated by Cui and colleagues (Cui et al., 2013).
It is important to stress that in both LID, as well as in dystonia, the direct and indirect pathways should not be seen as separate systems, as hypothesized in the classical interpretation of the model. In fact the two pathways in both physiological and in hyperkinetic conditions, such as LID and dystonia, are structurally and functionally intertwined at least at two distinct levels: in the striatum, where the direct and indirect pathways communicate via striatal cholinergic interneurons (ChIs), as well as other types of interneurons, and axon collaterals of SPN’s; and outside of the striatum, where striato-GPe collaterals may bridge the two pathways, potentially allowing the direct pathway to modulate the indirect pathway (Calabresi et al., 2015; Cazorla et al., 2014).
Disease states may also modulate the neurochemical architecture of the basal ganglia. In models of striatal dopamine deficiency, levodopa treatment leads to upregulation of striatal dopamine D3 receptors, which are otherwise sparse (Bordet et al., 1997). This is important because these D3 receptors are expressed primarily in SPN’s of the direct pathway, where they can interact and synergize with D1 receptors (Lanza et al, 2018). D3 receptors have a potent effect, such that overexpression alone in normal animals is sufficient to create susceptibility to LID (Cote et al, 2014). Much less is known about disease-specific alterations in striatal dopamine receptor systems in dystonia, although imaging studies in human isolated focal dystonia have produced some evidence for selective reductions in striatal D3 ligand binding (Karimi et al, 2011).
Circuits in LID and dystonia: thalamus and cerebellar system
While much of the work on LID and dystonia has focused on the basal ganglia, it is clear that other brain structures are involved in the pathogenesis of these movements, particularly the cerebellum and thalamus. Indeed, rather than trying to identify the “locus” of hyperkinetic disorders, it may be more fruitful to approach these conditions as network disorders, involving abnormalities of communication between the cerebral cortex, the basal ganglia, thalamus, cerebellum, and ultimately the descending motor pathways and spinal motor neurons.
Thalamus and cerebellar system in dystonia
The cerebellum clearly has a role in many, if not most, forms of dystonia, a topic recently reviewed in detail by Shakkottai et al. (Shakkottai et al., 2017). Some of the earliest evidence for the importance of the cerebellum comes from work in the spontaneously dystonic (DT) rat. In these animals, surgical removal of the cerebellum is curative, preventing or abolishing dystonia (LeDoux et al., 1993). Although the genetic abnormality in the DT rat is not known to have a human correlate, another genetic form of dystonia that does arise in patients, rapid onset dystonia parkinsonism (RDP, or DYT13) does appear to be linked to cerebellar dysfunction. RDP is caused by a mutation of the α3 subunit of electrogenic sodium pump (de Carvalho Aguiar et al., 2004). Animals with localized cerebellar inhibition of this pump, using either genetic knockdown of the α3 subunit or the inhibitor oubain, develop burst firing of Purkinje cells and behavioral dystonia (Calderon et al., 2011; Fremont et al., 2015).
In humans, the evidence for the involvement of the cerebellum in dystonia is more indirect. Dystonia arising as a symptom of stroke in the cerebellum is well described (LeDoux and Brady, 2003; Suri et al., 2018). Imaging studies support the importance of the cerebellum, with evidence of increased glucose metabolism in myoclonus-dystonia (Carbon et al., 2013) and alterations in fractional anisotropy suggesting microstructural changes (Carbon et al., 2008). Functional abnormalities of the cerebellum in human dystonia can also be detected using transcranial magnetic stimulation (Hubsch et al., 2013).
The cerebellum, of course, does not induce dystonia in isolation, but rather through its network connections through the thalamus to basal ganglia and other regions. The dentate nucleus of the cerebellum communicates through the thalamus to the striatum (Bostan and Strick, 2010). In two genetic forms of isolated dystonia, DYT1 and DYT6, studies in manifesting and non-manifesting carriers have shown that they have reduced connectivity in cerebellothalamic tracts. Interestingly, in the non-manifesting carriers, the lack of dystonic phenotype was associated with an additional abnormality in cortico-thalamic connectivity, suggesting that this second abnormality offsets the effect of the cerebellothalamic abnormality (Argyelan et al., 2009). Interestingly, genetic knockdown of TorsinA (the protein responsible for DYT1 dystonia) in the cerebellum but not the basal ganglia of mice leads to dystonic movements (Fremont et al., 2017).
Two recent studies of cerebellar and thalamic connectivity with basal ganglia in animal models provide some insight into how the reciprocal interactions between these systems may contribute to dystonia. Sciamanna et al. (Sciamanna et al., 2012b) examined the influence of projection from the thalamus on striatal cholinergic neurons. In normal mice, electrical activation of these projections causes a pause in the spontaneous firing of cholinergic interneurons. In a mouse model with knockin of the DYT1 mutation, stimulation of the thalamostriatal axons causes a shortened pause and abnormal spiking in interneurons. This abnormality is driven by altered cholinergic tone in the striatum, and suggest that there is impaired integration between thalamic efferents and striatal signaling in DYT1 and possibly other forms of dystonia. Chen et al. (Chen et al., 2014) subsequently tested the effect of cerebellar dentate activation, using electric stimulation or optogenetics, in awake freely moving mice and found that the dentate, acting through the intralaminar thalamus, could directly activate striatal neurons. Further, they show that optogenetic blockade or lesioning of the intralaminar thalamus can block the appearance of cerebellar-induced dystonia.
Thalamus and cerebellar system in LID
The cerebellum has also been implicated in the pathogenesis of LID (Kishore and Popa, 2014). The anatomical substrates for cerebellar interactions with the basal ganglia in LID are similar to those thought to be involved in dystonia, consisting of bidirectional multi-synaptic pathways linking the cerebellum, thalamus, striatum and basal ganglia (reviewed in (Caligiore et al., 2017)). A recent study using resting state MRI has documented an important effect of levodopa on connectivity between the brainstem and cerebellum in PD, demonstrating the importance of dopaminergic effects on cerebellar function (Mueller et al., 2018). In patients with LID, both repetitive transcranial magnetic stimulation (rTMS) (Koch et al., 2009) and transcranial direct current stimulation (tDCS) of the cerebellum (Ferrucci et al., 2016) have been shown to attenuate dyskinesias. As is dystonia, these observations illustrate the important functional connections between basal ganglia and cerebellar circuits, and may offer some novel approaches to the therapy of these hyperkinetic disorders.
Striatal cholinergic signaling
Striatal cholinergic signaling in LID
The striatum has the highest level of acetylcholine in brain, and although ChIs are few in number, they have widespread axonal arborizations which can modulate striatal activity via muscarinic and nicotinic receptors controlling synaptic transmission, plasticity and motor outputs (Calabresi et al., 2000). Increased cholinergic signaling seems to play a role in LID induction. It has been reported that chronic levodopa administration in parkinsonian mice induces LID with enhanced response to dopamine in terms of cholinergic interneurons (ChIs) excitability and extracellular signal-regulated kinase (ERK) activation in ChIs of denervated striatum (Ding et al., 2010). Moreover, the expression of LID was partially attenuated by the administration of a muscarinic receptor antagonist. Ablation of ChIs via Cre-dependent viral expression of the diphtheria toxin A subunit (DT-A) in hemiparkinsonian transgenic mice significantly reduced LID without altering the beneficial efficacy of levodopa, providing good evidence that ChIs play a key role in LID development (Won et al., 2014). It has been also reported that a positive allosteric modulator (PAM) acting on M4 muscarinic receptors (M4Rs) attenuated dyskinetic behavior through the blockade of aberrant plasticity in SPNs, (Shen et al., 2015). Interestingly, this M4R modulation was also effective in reducing LIDs in a primate PD model (Shen et al., 2015).
Although clinical evidence suggesting a pathophysiological link between the nicotinic cholinergic system and LIDs is limited (Brumberg et al., 2017), several preclinical studies showed that nicotine and nicotinic acetylcholine receptor (nAChR) drugs reduce LIDs by up to 60% in several parkinsonian animal models (Bordia et al., 2010; Huang et al., 2011; Zhang et al., 2014; Zhang et al., 2015). Indeed, nAChR agonists reduce LID in partially DA denervated animals (Bordia et al., 2010; Quik et al., 2013). Probably this effect occurs through activation of presynaptic nicotinic receptors on DA terminals to promote DA release (Quik et al., 2013) and reduction of the release of glutamate from terminals originating from cortex and thalamus (Ding et al., 2008; Parikh et al., 2010).
Striatal cholinergic signaling in dystonia
Striatal acetylcholine also appears to play a critical role in the pathophysiology of dystonia. An early observation was that anticholinergic treatments, usually non-selective antagonists of muscarinic receptors, were effective in reducing motor symptoms in some cases of dystonia (Burke et al., 1986; Fahn, 1983). Cholinergic agonists can also induce dystonia in humans (Shafrir et al., 1986).
Enhanced action of striatal acetylcholine has been demonstrated in animal model systems of dystonia (Eskow Jaunarajs et al., 2015). In mouse models of DYT1, there is increased striatal extracellular acetylcholine that can be measured directly with microdialysis (Scarduzio et al., 2017). This hypercholinergic state has a number of functional consequences, leading to alterations in cholinergic neuron function, with activation, rather than inhibition by dopamine D2 receptor agonists (a “paradoxical response,” (Pisani et al., 2006)) as well as altered synaptic plasticity (Martella et al., 2009b) and downstream changes in GABA transmission (Sciamanna et al., 2009). The molecular basis for the “paradoxical response” appears to involve competition among G protein coupled receptors for downstream signaling pathways, resulting in activation of beta-arrestin signaling in the dystonia model (Scarduzio et al., 2017).
The mutation in DYT1 dystonia is believed to cause loss of torsinA function (Breakefield et al., 2008), and the consequences of deletion of torsinA from cholinergic neurons have been explored. Pappas et al (Pappas et al., 2015) found that forebrain deletion of Dyt1, the mouse homolog of the human TOR1A gene, caused both dystonic behavior and loss of cholinergic neurons from the striatum. Further, selective deletion of Dyt1 from ChI’s also led to loss of cholinergic neurons and dystonic posturing (Pappas et al., 2018). These studies stand in contrast to earlier work in which deletion of Dyt1 from ChI’s led to an electrophysiological phenotype, but not loss of ChI’s or dystonic behavior (Sciamanna et al., 2012a). Although these differing phenotypes with similar manipulations of Dyt1 in ChI’s have not been fully explained, the main difference from an experimental perspective is that the studies from Pappas et al. used a loxP mouse designed to achieve deletion of exons 3-5 of Dyt1, coding for much of the torsinA protein including the ATPase domain (Liang et al., 2014), while the earlier studies used a different loxP mouse designed to produce a deletion of only exons 3 and 4 (Yokoi et al., 2008), perhaps creating a protein with some residual function.
The efficacy of anticholinergic therapy in at least some forms of dystonia, and the corresponding failure of most other pharmacological approaches which have been studied, has generated enthusiasm for examining cholinergic signaling in greater detail. Most of the current therapies, such as trihexyphenidyl, are non-selective cholinergic antagonists and lead to adverse effects including sedation and memory impairment. A more selective approach to modulating cholinergic function in dystonia, and perhaps targeting the nicotinic receptor system, might be an important therapeutic advance (Eskow Jaunarajs et al., 2015; Zimmerman et al., 2017).
Synaptic plasticity in LID and dystonia
Striatal synaptic plasticity alterations in LID
The development of LID is closely related to dysfunctional synaptic plasticity at corticostriatal synapses and, specifically, to the loss of synaptic depotentiation (Picconi et al., 2003). In particular, dopamine denervation induced by nigral 6-hydroxy-dopamine (6-OHDA) injection in rats causes loss of corticostriatal LTP in SPNs. Chronic treatment with levodopa is able to restore this form of synaptic plasticity. However, in about 50% of the animals the long-term treatment with this drug causes LID. Interestingly, in dyskinetic animals low frequency stimulation of corticostriatal projections is unable to reverse LTP in SPNs (loss of depotentiation) (Picconi et al., 2003). Conversely, this homeostatic form of synaptic plasticity was preserved in parkinsonian rodents that did not develop LID. It has been proposed that loss of synaptic depotentiation might destabilize neuronal circuits in the basal ganglia, resulting in dyskinesias. Abnormalities of synaptic plasticity in dyskinetic animals are related to changes in the D1 - protein kinase A (PKA)–DARPP-32 signaling pathway that inhibit the activity of protein phosphatase (PP) PP-1, a molecule involved in depotentiation at corticostriatal synapses. In particular, the activation of D1 DA receptors triggers the PKA-induced phosphorylation on Thr34 of DARPP-32, which in turn inhibits PP-1. Notably, only the 6-OHDA-lesioned animals that develop LID show increased phosphorylation of Thr34-DARPP-32. The Ras-ERK pathway regulates both activity-dependent striatal LTP and synaptic depotentiation and possibly plays a critical role in LID induction (Cerovic et al., 2015).
In dyskinetic rats, long-term depression (LTD), the opposite form of plasticity at corticostriatal synapses, is significantly reduced (Picconi et al., 2010). LTD disruption has been related to abnormal modulation of intracellular cGMP levels in SPNs. Accordingly, the pharmacological modulation of striatal phosphodiesterases through inhibitors (zaprinast and UK-343664) was able to restore LTD to control levels. Moreover, a complex serotonergic regulation of striatal bidirectional synaptic plasticity may be of critical relevance in mediating the reported antidyskinetic effect exerted by serotonergic agonists (Ghiglieri et al., 2016).
Another study suggests that bidirectional plasticity of both striatonigral and striatopallidal outputs is a necessary condition to ensure correct motor control, while dyskinetic movements are caused by an abnormal segregation of plasticity between the two pathways (Thiele et al., 2014). In particular, these authors explored spike-timing-dependent plasticity (STDP) at corticostriatal synapses and found that in dopamine-denervated mice, direct and indirect pathway neurons exhibit only unidirectional plasticity, regardless of the stimulation paradigm (Thiele et al., 2014). A recent study suggests that STDP in PD and in LID is cell-type specific. In fact, the intrinsic excitability and corticostriatal synaptic connectivity of SPNs in the indirect pathway are lower in parkinsonian animals than in control rodents. Conversely, these properties of SPNs in the direct pathway are enhanced in tissues from experimental PD and suppressed in LID models (Fieblinger et al., 2014). Moreover, it has been recently reported that with SPN type-specific chemogenetic stimulation the role of the two striatal pathways in LID can be distinguished: stimulation of direct pathway SPNs exacerbated dyskinetic responses to levodopa, while stimulation of indirect pathway SPNs inhibited these responses (Alcacer et al., 2017).
NMDA glutamate receptors are key modulators of striatal plasticity. Molecular studies have confirmed a rearrangement of NMDA receptor subunits expressed by SPNs in PD dyskinetic models (Hallett and Standaert, 2004; Mellone and Gardoni, 2018; Mellone et al., 2015b; Sgambato-Faure and Cenci, 2012). In particular there is an altered ratio of synaptic GluN2A/GluN2B-containing NMDA receptors in the striatum of dyskinetic rats and monkeys, as well as in post-mortem tissue from dyskinetic PD patients (Dunah et al., 2004; Dunah and Standaert, 2001; Dunah et al., 2000; Hallett et al., 2005; Mellone et al., 2015a). Accordingly, the modulation of synaptic NMDA receptor composition by a cell-permeable peptide interfering with GluN2A subunit interaction with the scaffolding protein postsynaptic density protein 95 (PSD-95) leads to a reduction in the dyskinetic motor behavior in two animal models of LID. Recent work has also revealed that the GluN2D subunit, which in normal animals is found only in interneurons in the striatum (Standaert et al., 1996), is upregulated and expressed in SPN’s in dyskinetic animals (Mellone et al., 2019). These findings suggest that synaptic NMDA receptor subunit composition could represent a target for therapeutic approaches aimed at ameliorating levodopa motor side effects (Mellone et al., 2015b).
Striatal synaptic plasticity alterations in dystonia
The study of dystonia in experimental models is limited by the fact that most of the rodent genetic models do not exhibit overt dystonia. One exception is the mouse model of dopamine-responsive dystonia produced by deletion of GTP cyclohydrolase, which has dystonic features and profound dopamine deficiency (Rose et al., 2015). Most other genetically authentic models of human dystonia show only subtle motor abnormalities and yet have peculiar neurochemical and neurophysiological alterations. In transgenic mice overexpressing mutant torsinA, modeling DYT1 dystonia, no corticostriatal LTD can be elicited, whereas LTP is greater than in control animals (Martella et al., 2009a). Moreover, while low frequency stimulation reverts potentiated synapses to resting levels (depotentiation) in control mice, this phenomenon is absent in mutant mice (Martella et al., 2009a). Interestingly, these alterations of synaptic plasticity are similarly observed across species and distinct mouse lines (Martella et al., 2014). All together, these observations suggest that the loss of downscaling is a distinctive feature in multiple models of DYT1 dystonia.
The aberrant control of synaptic plasticity in the DYT1 model seems to be related to a dysfunctional ACh transmission in the striatum (Breakefield et al., 2008; Eskow Jaunarajs et al., 2015; Pisani et al., 2007). In particular, it has been reported that antimuscarinic drugs, in particular antagonists of M1 muscarinic ACh receptors, rescue bidirectional synaptic plasticity at corticostriatal synapses (Maltese et al., 2014), suggesting that exaggerated acetylcholine release is a final step for plasticity disruption.
Dopamine D2 receptors also play a role. In fact, quinpirole activation of D2 receptors on cholinergic interneurons triggers a paradoxical excitation of cholinergic firing, rather than inhibition as observed in WT mice (Pisani et al., 2007; Scarduzio et al., 2017; Sciamanna et al., 2011), which could exacerbate the hypercholinergic tone. Moreover, antagonism of adenosine-2A (A2A) receptors recovers bidirectional plasticity in mutant mice, suggesting an interaction between D2/A2A receptors located on indirect SPNs in balancing synaptic plasticity induction (Napolitano et al., 2010).
A recent elegant work demonstrated that early loss of functional and structural synaptic homeostasis represents a unique endophenotypic trait during striatal maturation, promoting the appearance of clinical manifestations in mutation carriers (Maltese et al., 2018). Indeed, although it is well-known that the onset of abnormal movements in DYT1 dystonia is between childhood and adolescence, the pathophysiological mechanisms underlying their appearance in this temporal window are still unclear. Pisani’s group has investigated the SPNs synaptic plasticity in this critical developmental window, discovering that in the Tor1a+/Δgag DYT1 dystonia mouse model, LTP in SPNs appeared prematurely while LTD was never recorded (Maltese et al., 2018). Moreover, analysis of dendritic spines showed an increase of both spine width and mature mushroom spines in neurons from mutant mice, paralleled by an enhanced AMPA receptors accumulation and increased BDNF levels. Consistently, antagonism of BDNF rescued synaptic plasticity deficits and AMPA currents in this genetic DYT1 model.
Epigenetic regulation in LID and dystonia
Much of the work on LID and dystonia has emphasized short-term alterations such synaptic potentiation and neurochemical changes, but a core feature of both LID and dystonia is persistence of the symptoms over very long periods of time. In patients with PD, once LID appears they retain a persistent sensitivity to levodopa, with re-emergence of the dyskinesias with re-exposure, even after long intervals. The only reliable way to prevent the recurrence of dyskinesia is surgical interventions such as DBS. Similarly, the clinical features of most forms of dystonia are very long-lasting; usually once dystonia develops in a patient, it is unlikely to remit spontaneously, and usually persists throughout life unless there is intervention such as DBS or botulinum toxin, or in the rare cases where they may be treatable by neurotransmitter replacement.
Long duration symptoms point towards long-term mechanisms of neural plasticity, and among the most long-lasting of these are epigenetic changes, alterations in cellular DNA and associated histones. This is best documented in LID models, where chronic treatment with levodopa can lead to alterations in both histones (Nicholas et al., 2008) and DNA methylation in striatal neurons (Figge et al., 2016). Much less is known about potential epigenetic mechanisms in dystonia, although one recent study has looked at imprinting, a mechanism dependent on DNA methylation in iPSC derived neurons in genetic myoclonus-dystonia (Grutz et al., 2017). Further study of these mechanisms is important because they may lead to ways to modify the long-term alterations which lead to the lifelong persistence of LID and dystonia.
Conclusions
LID and dystonia share important phenomenological properties and mechanisms. Both are hyperkinetic disorders. Dystonic symptoms may appear as part of LID, and when present may be identical in clinical appearance to dystonia of other causes. In contrast, patients with dystonia may exhibit some choreiform movements but they are generally modest in amplitude and would never be mistaken for the dramatic chorea that can occur in LID. Both LID and dystonia are generated by an integrated circuit involving the cortex, basal ganglia, thalamus and cerebellum. They also share dysregulation of striatal cholinergic signaling, and abnormalities of striatal synaptic plasticity. The long duration nature of both LID and dystonia suggests that there may be underlying epigenetic dysregulation as a proximate cause, but the studies of epigenetic change in these disorders is at an early state. On the whole, we believe it is reasonable to view both dystonia and LID as two different manifestations of aberrant striatal (and perhaps cerebellar) synaptic plasticity.
LID and dystonia share another important property, which is that neither can be adequately treated with currently available medications. These are disabling disorders, and there is an urgent need to develop better approaches to manage these difficult symptoms.
Fig. 1. Possible common synaptic and molecular mechanisms underlying L-DOPA-induced and dystonia.
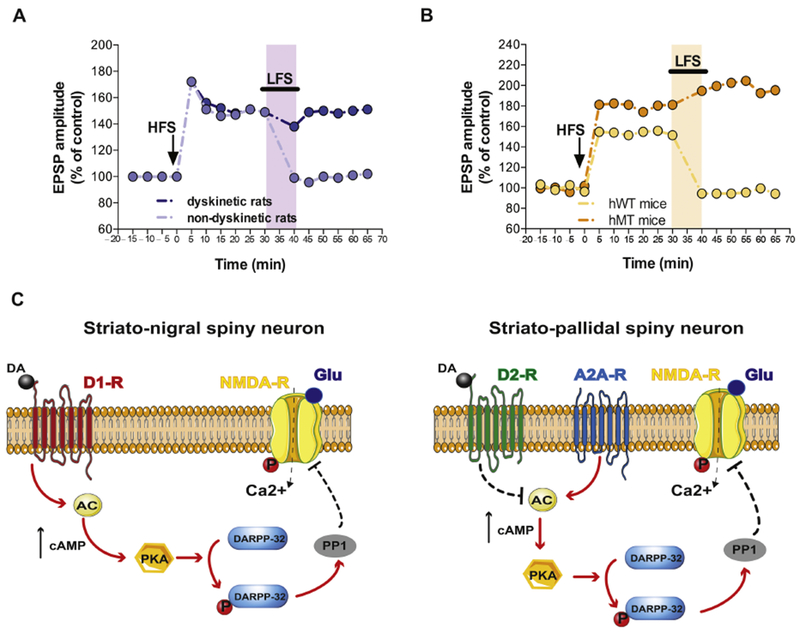
A) Time course of striatal synaptic plasticity (long term potentiation, LTP, and depotentiation) following HFS and LFS protocols in dyskinetic and non-dyskinetic 6-OHDA-lesioned rats. Animals showing therapeutic effects of l-DOPA (light violet) display a full recovery of bidirectional synaptic plasticity (modified from Picconi et al., Nat Neurosci 2003). B) Time course of LTP and synaptic depotentiation in transgenic mice overexpressing human mutant (hMT, orange) and wild type torsinA controls (hWT, yellow) a dystonia model (modified from Martella et al., Brain 2009). Shaded areas in light colors represent LFS protocol application. C) Schematic representation of the signaling pathways underlying the synaptic deficits of striato-nigral (D1-positive, left) and striato-pallidal (D2-positive, right) spiny neurons in levodopa-induced dyskinesia hyperkinetic rats and dystonic mice. In particular, the left panel shows that activation of D1 dopamine receptors leads to an increase of cAMP which in turn stimulates PKA favouring DARPP-32 phosphorylation. This latter event activates PP1 and influence NMDA receptor function. The right panel shows a similar downstream pathway that is initiated by the activation of A2 adenosine receptors rather than by D1 receptors. Note the inhibitory effect on this pathway exerted by D2 dopamine receptors.
Abbreviation: AC, adenylate cyclase; DARPP-32, dopamine- and cAMP-regulated phosphoprotein 32 kDa; P, phosphorylation; PP1, Protein Phosphatase 1; PKA, Protein Kinase A; solid arrows, proposed increasing molecular action; dotted arrows, possible blocking molecular action; black arrows, increased levels.
Highlights.
Levodopa-induced dyskinesia (LID) and dystonia are hyperkinetic movement disorders that share important phenomenological properties and mechanisms.
Both LID and dystonia are generated by an integrated circuit involving the cortex, basal ganglia, thalamus and cerebellum.
LID and dystonia share dysregulation of striatal cholinergic signaling, and abnormalities of striatal synaptic plasticity.
The long duration nature of both LID and dystonia suggests that there may be underlying epigenetic dysregulation as a proximate cause, but the studies of epigenetic change in these disorders is at an early state.
Acknowledgements
This work was supported in part by Marlene and Paolo Fresco Institute for Parkinson’s and Movement Disorders to PC and National Institutes of Health grants P01NS087997 and P50NS108675 to DGS. The authors thanks Drs. Karen Eskow-Jaunarajs and Mariangela Scarduzio for critical reading of the manuscript and helpful comments.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Paolo Calabresi, Neurological Clinic, Department of Medicine, “Santa Maria della Misericordia” Hospital, University of Perugia, Perugia 06132, Italy and IRCCS Fondazione Santa Lucia, Rome, Italy.
David G. Standaert, Department of Neurology, University of Alabama at Birmingham, Birmingham, AL, USA 35294.
References
- Albanese A, Bhatia K, Bressman SB, Delong MR, Fahn S, Fung VS, Hallett M, Jankovic J, Jinnah HA, Klein C, Lang AE, Mink JW, Teller JK, 2013. Phenomenology and classification of dystonia: a consensus update. Mov Disord. 28,863–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albin RL, Young AB, Penney JB, 1989. The functional anatomy of basal ganglia disorders. Trends in Neurosciences. 12, 366–375. [DOI] [PubMed] [Google Scholar]
- Alcacer C, Andreoli L, Sebastianutto I, Jakobsson J, Fieblinger T, Cenci MA, 2017. Chemogenetic stimulation of striatal projection neurons modulates responses to Parkinson,Äôs disease therapy. Journal of Clinical Investigation. 127,720–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Argyelan M, Carbon M, Niethammer M, Ulug AM, Voss HU, Bressman SB, Dhawan V, Eidelberg D, 2009. Cerebellothalamocortical connectivity regulates penetrance in dystonia. J Neurosci. 29,9740–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhatia KP, Marsden CD, 1994. The behavioural and motor consequences of focal lesions of the basal ganglia in man. Brain. 117,859–876. [DOI] [PubMed] [Google Scholar]
- Bordet R, Ridray S, Carboni S, Diaz J, Sokoloff P, Schwartz JC, 1997. Induction of dopamine D3 receptor expression as a mechanism of behavioral sensitization to levodopa. Proc Natl Acad Sci U S A. 94, 3363–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bordia T, Campos C, McIntosh JM, Quik M, 2010. Nicotinic Receptor-Mediated Reduction in L-DOPA-Induced Dyskinesias May Occur via Desensitization. Journal of Pharmacology and Experimental Therapeutics. 333, 929–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bostan AC, Strick PL, 2010. The cerebellum and basal ganglia are interconnected. Neuropsychol Rev. 20,261–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breakefield XO, Blood AJ, Li Y, Hallett M, Hanson PI, Standaert DG, 2008. The pathophysiological basis of dystonias. Nature Reviews Neuroscience. 9, 222–234. [DOI] [PubMed] [Google Scholar]
- Brumberg J, Küsters S, Al-Momani E, Marotta G, Cosgrove KP, van Dyck CH, Herrmann K, Homola G. r. A., Pezzoli G, Buck AK, Volkmann J, Samnick S, Isaias IU, 2017. Cholinergic activity and levodopa-induced dyskinesia: a multitracer molecular imaging study. Annals of Clinical and Translational Neurology. 4,632–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burke RE, Fahn S, Marsden CD, 1986. Torsion dystonia: a double-blind, prospective trial of high-dosage trihexyphenidyl. Neurology. 36, 160–4. [DOI] [PubMed] [Google Scholar]
- Calabresi P, Centonze D, Gubellini P, Pisani A, Bernardi G, 2000. Acetylcholine-mediated modulation of striatal function. Trends in Neurosciences. 23,120–126. [DOI] [PubMed] [Google Scholar]
- Calabresi P, Filippo MD, Ghiglieri V, Tambasco N, Picconi B, 2010. Levodopa-induced dyskinesias in patients with Parkinson’s disease: filling the bench-to-bedside gap. The Lancet Neurology. 9,1106–1117. [DOI] [PubMed] [Google Scholar]
- Calabresi P, Ghiglieri V, Mazzocchetti P, Corbelli I, Picconi B, 2015. Levodopa-induced plasticity: a double-edged sword in Parkinson’s disease? Philosophical Transactions of the Royal Society B: Biological Sciences. 370,20140184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calabresi P, Pisani A, Rothwell J, Ghiglieri V, Obeso JA, Picconi B, 2016. Hyperkinetic disorders and loss of synaptic downscaling. Nature Neuroscience. 19, 868–875. [DOI] [PubMed] [Google Scholar]
- Calderon DP, Fremont R, Kraenzlin F, Khodakhah K, 2011. The neural substrates of rapid-onset Dystonia-Parkinsonism. Nat Neurosci. 14,357–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caligiore D, Pezzulo G, Baldassarre G, Bostan AC, Strick PL, Doya K, Helmich RC, Dirkx M, Houk J, Jorntell H, Lago-Rodriguez A, Galea JM, Miall RC, Popa T, Kishore A, Verschure PF, Zucca R, Herreros I, 2017. Consensus Paper: Towards a Systems-Level View of Cerebellar Function: the Interplay Between Cerebellum, Basal Ganglia, and Cortex. Cerebellum. 16,203–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carbon M, Kingsley PB, Tang C, Bressman S, Eidelberg D, 2008. Microstmctural white matter changes in primary torsion dystonia. Mov Disord. 23,234–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carbon M, Raymond D, Ozelius L, Saunders-Pullman R, Frucht S, Dhawan V, Bressman S, Eidelberg D, 2013. Metabolic changes in DYT11 myoclonus-dystonia. Neurology. 80, 385–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cazorla M, de Carvalho FD, Chohan MO, Shegda M, Chuhma N, Rayport S, Ahmari SE, Moore H, Kellendonk C, 2014. Dopamine D2 Receptors Regulate the Anatomical and Functional Balance of Basal Ganglia Circuitry. Neuron. 81, 153–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerovic M, Bagetta V, Pendolino V, Ghiglieri V, Fasano S, Morella I, Hardingham N, Heuer A, Papale A, Marchisella F, Giampà C, Calabresi P, Picconi B, Brambilla R, 2015. Derangement of Ras-Guanine Nucleotide-Releasing Factor 1 (Ras-GRF1) and Extracellular Signal-Regulated Kinase (ERK) Dependent Striatal Plasticity in L-DOPA-Induced Dyskinesia. Biological Psychiatry. 77,106–115. [DOI] [PubMed] [Google Scholar]
- Chen CH, Fremont R, Arteaga-Bracho EE, Khodakhah K, 2014. Short latency cerebellar modulation of the basal ganglia. Nat Neurosci. 17,1767–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cote SR, Chitravanshi VC, Bleickardt C, Sapru HN, Kuzhikandathil EV, 2014. Overexpression of the dopamine D3 receptor in the rat dorsal striatum induces dyskinetic behaviors. Behav Brain Res. 263,46–50. [DOI] [PubMed] [Google Scholar]
- Cui G, Jun SB, Jin X, Pham MD, Vogel SS, Lovinger DM, Costa RM, 2013. Concurrent activation of striatal direct and indirect pathways during action initiation. Nature. 494,238–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Carvalho Aguiar P, Sweadner KJ, Penniston JT, Zaremba J, Liu L, Caton M, Linazasoro G, Borg M, Tijssen MA, Bressman SB, Dobyns WB, Brashear A, Ozelius LJ, 2004. Mutations in the Na+/K+ -ATPase alpha3 gene ATP1A3 are associated with rapid-onset dystonia parkinsonism. Neuron. 43, 169–75. [DOI] [PubMed] [Google Scholar]
- DeLong MR, 1990. Primate models of movement disorders of basal ganglia origin. Trends in Neurosciences. 13,281–285. [DOI] [PubMed] [Google Scholar]
- Ding J, Peterson JD, Surmeier DJ, 2008. Corticostriatal and Thalamostriatal Synapses Have Distinctive Properties. Journal of Neuroscience. 28,6483–6492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding Y, Won L, Britt JP, Lim SAO, McGehee DS, Kang UJ, 2010. Enhanced striatal cholinergic neuronal activity mediates L-DOPA-induced dyskinesia in parkinsonian mice. Proceedings of the National Academy of Sciences. 108,840–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunah AW, Sirianni AC, Fienberg AA, Bastia E, Schwarzschild MA, Standaert DG, 2004. Dopamine D1-dependent trafficking of striatal N-methyl-D-aspartate glutamate receptors requires Fyn protein tyrosine kinase but not DARPP-32. Mol Pharmacol. 65, 121–9. [DOI] [PubMed] [Google Scholar]
- Dunah AW, Standaert DG, 2001. Dopamine D1 receptor-dependent trafficking of striatal NMDA glutamate receptors to the postsynaptic membrane. J Neurosci. 21, 5546–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunah AW, Wang Y, Yasuda RP, Kameyama K, Huganir RL, Wolfe BB, Standaert DG, 2000. Alterations in subunit expression, composition, and phosphorylation of striatal N-methyl-D-aspartate glutamate receptors in a rat 6-hydroxydopamine model of Parkinson’s disease. Mol Pharmacol. 57, 342–52. [PubMed] [Google Scholar]
- Eskow Jaunarajs KL, Bonsi P, Chesselet MF, Standaert DG, Pisani A, 2015. Striatal cholinergic dysfunction as a unifying theme in the pathophysiology of dystonia. Prog Neurobiol. 127–128,91–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espay AJ, Morgante F, Merola A, Fasano A, Marsili L, Fox SH, Bezard E, Picconi B, Calabresi P, Lang AE, 2018. Levodopa-induced dyskinesia in Parkinson disease: Current and evolving concepts. Annals of Neurology. 84,797–811. [DOI] [PubMed] [Google Scholar]
- Espay AJ, Schwarzschild MA, Tanner CM, Fernandez HFL, Simon DK, Leverenz JB, Merola A, Chen-Plotkin A, Brundin P, Kauffman MA, Erro R, Kieburtz K, Woo D, Macklin EA, Standaert DG, Lang AE, 2017. Biomarker-driven phenotyping in Parkinson’s disease: A translational missing link in disease-modifying clinical trials. Mov Disord. 32,319–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eusebi P, Romoli M, Paoletti FP, Tambasco N, Calabresi P, Pametti L, 2018. Risk factors of levodopa-induced dyskinesia in Parkinson,Aos disease: results from the PPMI cohort, npj Parkinson’s Disease. 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fahn S, 1983. High dosage anticholinergic therapy in dystonia. Neurology. 33, 1255–61. [DOI] [PubMed] [Google Scholar]
- Ferrucci R, Cortese F, Bianchi M, Pittera D, Turrone R, Bocci T, Borroni B, Vergari M, Cogiamanian F, Ardolino G, Di Fonzo A, Padovani A, Priori A, 2016. Cerebellar and Motor Cortical Transcranial Stimulation Decrease Levodopa-Induced Dyskinesias in Parkinson’s Disease. Cerebellum. 15,43–47. [DOI] [PubMed] [Google Scholar]
- Fieblinger T, Graves SM, Sebel LE, Alcacer C, Plotkin JL, Gertler TS, Chan CS, Heiman M, Greengard P, Cenci MA, Surmeier DJ, 2014. Cell type-specific plasticity of striatal projection neurons in parkinsonism and L-DOPA-induced dyskinesia. Nature Communications. 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Figge DA, Eskow Jaunarajs KL, Standaert DG, 2016. Dynamic DNA Methylation Regulates Levodopa-Induced Dyskinesia. J Neurosci. 36,6514–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fremont R, Tewari A, Angueyra C, Khodakhah K, 2017. A role for cerebellum in the hereditary dystonia DYT1. Elife. 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fremont R, Tewari A, Khodakhah K, 2015. Aberrant Purkinje cell activity is the cause of dystonia in a shRNA-based mouse model of Rapid Onset Dystonia-Parkinsonism. Neurobiol Dis. 82,200–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghiglieri V, Mineo D, Vannelli A, Cacace F, Mancini M, Pendolino V, Napolitano F, di Maio A, Mellone M, Stanic J, Tronci E, Fidalgo C, Stancampiano R, Carta M, Calabresi P, Gardoni F, Usiello A, Picconi B, 2016. Modulation of serotonergic transmission by eltoprazine in L-DOPA-induced dyskinesia: Behavioral, molecular, and synaptic mechanisms. Neurobiology of Disease. 86, 140–153. [DOI] [PubMed] [Google Scholar]
- Grutz K, Seibler P, Weissbach A, Lohmann K, Carlisle FA, Blake DJ, Westenberger A, Klein C, Grunewald A, 2017. Faithful SGCE imprinting in iPSC-derived cortical neurons: an endogenous cellular model of myoclonus-dystonia. Sci Rep. 7, 41156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallett PJ, Dunah AW, Ravenscroft P, Zhou S, Bezard E, Crossman AR, Brotchie JM, Standaert DG, 2005. Alterations of striatal NMDA receptor subunits associated with the development of dyskinesia in the MPTP-lesioned primate model of Parkinson’s disease. Neuropharmacology. 48,503–16. [DOI] [PubMed] [Google Scholar]
- Hallett PJ, Standaert DG, 2004. Rationale for and use of NMDA receptor antagonists in Parkinson’s disease. Pharmacol Ther. 102, 155–74. [DOI] [PubMed] [Google Scholar]
- Huang LZ, Campos C, Ly J, Ivy Carroll F, Quik M, 2011. Nicotinic receptor agonists decrease L-dopa-induced dyskinesias most effectively in partially lesioned parkinsonian rats. Neuropharmacology. 60, 861–868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubsch C, Roze E, Popa T, Russo M, Balachandran A, Pradeep S, Mueller F, Brochard V, Quartarone A, Degos B, Vidailhet M, Kishore A, Meunier S, 2013. Defective cerebellar control of cortical plasticity in writer’s cramp. Brain. 136,2050–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jankovic J, Tintner R, 2001. Dystonia and parkinsonism. Parkinsonism & Related Disorders. 8, 109–121. [DOI] [PubMed] [Google Scholar]
- Jellinger KA, Post mortem studies in Parkinson,Äôs disease ,Äî is it possible to detect brain areas for specific symptoms? , Journal of Neural Transmission. Supplementa. Springer Vienna, 1999, pp. 1–29. [DOI] [PubMed] [Google Scholar]
- Karimi M, Moerlein SM, Videen TO, Luedtke RR, Taylor M, Mach RH, Perlmutter JS, 2011. Decreased striatal dopamine receptor binding in primary focal dystonia: a D2 or D3 defect? Mov Disord. 26, 100–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishore A, Popa T, 2014. Cerebellum in levodopa-induced dyskinesias: the unusual suspect in the motor network. Front Neurol. 5, 157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein C, 2014. Genetics in dystonia. Parkinsonism Relat Disord. 20 Suppl 1, S137–42. [DOI] [PubMed] [Google Scholar]
- Koch G, Brusa L, Carrillo F, Lo Gerfo E, Torriero S, Oliveri M, Mir P, Caltagirone C, Stanzione P, 2009. Cerebellar magnetic stimulation decreases levodopa-induced dyskinesias in Parkinson disease. Neurology. 73,113–9. [DOI] [PubMed] [Google Scholar]
- Kravitz AV, Owen SF, Kreitzer AC, 2013. Optogenetic identification of striatal projection neuron subtypes during in vivo recordings. Brain Research. 1511,21–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kress GJ, Yamawaki N, Wokosin DL, Wickersham IR, Shepherd GMG, Surmeier DJ, 2013. Convergent cortical innervation of striatal projection neurons. Nature Neuroscience. 16, 665–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumbhare D, Holloway KL, Baron MS, 2017. Parkinsonism and dystonia are differentially induced by modulation of different territories in the basal ganglia. Neuroscience. 353,42–57. [DOI] [PubMed] [Google Scholar]
- Kuoppamaki M, 2005. Parkinsonism following bilateral lesions of the globus pallidus: performance on a variety of motor tasks shows similarities with Parkinson’s disease. Journal of Neurology, Neurosurgery & Psychiatry. 76, 482–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanza K, Meadows SM, Chambers NE, Nuss E, Deak MM, Ferre S, Bishop C, 2018. Behavioral and cellular dopamine D1 and D3 receptor-mediated synergy: Implications for L-DOPA-induced dyskinesia. Neuropharmacology. 138,304–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeDoux MS, Brady KA, 2003. Secondary cervical dystonia associated with structural lesions of the central nervous system. Mov Disord. 18,60–9. [DOI] [PubMed] [Google Scholar]
- LeDoux MS, Lorden JF, Ervin JM, 1993. Cerebellectomy eliminates the motor syndrome of the genetically dystonic rat. Exp Neurol. 120,302–10. [DOI] [PubMed] [Google Scholar]
- Lee JY, Cho J, Lee EK, Park SS, Jeon BS, 2011. Differential genetic susceptibility in diphasic and peak-dose dyskinesias in Parkinson’s disease. Mov Disord. 26, 73–9. [DOI] [PubMed] [Google Scholar]
- LeWitt PA, Bums RS, Newman RP, 1986. Dystonia in Untreated Parkinsonism. Clinical Neuropharmacology. 9, 293–297. [DOI] [PubMed] [Google Scholar]
- Liang CC, Tanabe LM, Jou S, Chi F, Dauer WT, 2014. TorsinA hypofunction causes abnormal twisting movements and sensorimotor circuit neurodegeneration. J Clin Invest. 124,3080–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maltese M, Stanic J, Tassone A, Sciamanna G, Ponterio G, Vanni V, Martella G, Imbriani P, Bonsi P, Mercuri NB, Gardoni F, Pisani A, 2018. Early stmctural and functional plasticity alterations in a susceptibility period of DYT1 dystonia mouse striatum. eLife. 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martella G, Maltese M, Nisticò R, Schirinzi T, Madeo G, Sciamanna G, Ponterio G, Tassone A, Mandolesi G, Vanni V, Pignatelli M, Bonsi P, Pisani A, 2014. Regional specificity of synaptic plasticity deficits in a knock-in mouse model of DYT1 dystonia. Neurobiology of Disease. 65, 124–132. [DOI] [PubMed] [Google Scholar]
- Martella G, Tassone A, Sciamanna G, Platania P, Cuomo D, Viscomi ΜT, Bonsi P, Cacci E, Biagioni S, Usiello A, Bemardi G, Sharma N, Standaert DG, Pisani A, 2009a. Impairment of bidirectional synaptic plasticity in the striatum of a mouse model of DYT1 dystonia: role of endogenous acetylcholine. Brain. 132,2336–2349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martella G, Tassone A, Sciamanna G, Platania P, Cuomo D, Viscomi ΜT, Bonsi P, Cacci E, Biagioni S, Usiello A, Bemardi G, Sharma N, Standaert DG, Pisani A, 2009b. Impairment of bidirectional synaptic plasticity in the striatum of a mouse model of DYT1 dystonia: role of endogenous acetylcholine. Brain. 132,2336–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellone M, Gardoni F, 2018. Glutamatergic mechanisms in L-DOPA-induced dyskinesia and therapeutic implications. J Neural Transm (Vienna). 125, 1225–1236. [DOI] [PubMed] [Google Scholar]
- Mellone M, Stanic J, Hernandez LF, Iglesias E, Zianni E, Longhi A, Prigent A, Picconi B, Calabresi P, Hirsch EC, Obeso JA, Di Luca M, Gardoni F, 2015a. NMDA receptor GluN2A/GluN2B subunit ratio as synaptic trait of levodopa-induced dyskinesias: from experimental models to patients. Front Cell Neurosci. 9,245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellone M, Stanic J, Hernandez LF, Iglesias E, Zianni E, Longhi A, Prigent A, Picconi B, Calabresi P, Hirsch EC, Obeso JA, Di Luca M, Gardoni F, 2015b. NMDA receptor GluN2A/GluN2B subunit ratio as synaptic trait of levodopa-induced dyskinesias: from experimental models to patients. Frontiers in Cellular Neuroscience. 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellone M, Zianni E, Stanic J, Campanelli F, Marino G, Ghiglieri V, Longhi A, Thiolat ML, Li Q, Calabresi P, Bezard E, Picconi B, Di Luca M, Gardoni F, 2019. NMDA receptor GluN2D subunit participates to levodopa-induced dyskinesia pathophysiology. Neurobiol Dis. 121,338–349. [DOI] [PubMed] [Google Scholar]
- Mueller K, Jech R, Ballarini T, Holiga S, Ruzicka F, Piecha FA, Moller HE, Vymazal J, Ruzicka E, Schroeter ML, 2018. Modulatory Effects of Levodopa on Cerebellar Connectivity in Parkinson’s Disease. Cerebellum. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munchau A, 2000. Unilateral lesions of the globus pallidus: report of four patients presenting with focal or segmental dystonia. Journal of Neurology, Neurosurgery & Psychiatry. 69, 494–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Napolitano F, Pasqualetti M, Usiello A, Santini E, Pacini G, Sciamanna G, Errico F, Tassone A, Di Dato V, Martella G, Cuomo D, Fisone G, Bemardi G, Mandolesi G, Mercuri NB, Standaert DG, Pisani A, 2010. Dopamine D2 receptor dysfunction is rescued by adenosine A2A receptor antagonism in a model of DYT1 dystonia. Neurobiol Dis. 38,434–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholas AP, Lubin FD, Hallett PJ, Vattem P, Ravenscroft P, Bezard E, Zhou S, Fox SH, Brotchie JM, Sweatt JD, Standaert DG, 2008. Striatal histone modifications in models of levodopa-induced dyskinesia. JNeurochem. 106,486–94. [DOI] [PubMed] [Google Scholar]
- Pappas SS, Darr K, Holley SM, Cepeda C, Mabrouk OS, Wong JM, LeWitt TM, Paudel R, Houlden H, Kennedy RT, Levine MS, Dauer WT, 2015. Forebrain deletion of the dystonia protein torsinA causes dystonic-like movements and loss of striatal cholinergic neurons. Elife. 4, e08352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pappas SS, Li J, LeWitt TM, Kim JK, Monani UR, Dauer WT, 2018. A cell autonomous torsinA requirement for cholinergic neuron survival and motor control. Elife. 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parikh V, Ji J, Decker MW, Sarter M, 2010. Prefrontal β2 Subunit-Containing and α7 Nicotinic Acetylcholine Receptors Differentially Control Glutamatergic and Cholinergic Signaling. Journal of Neuroscience. 30, 3518–3530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picconi B, Bagetta V, Ghiglieri V, Paillè V, Di Filippo M, Pendolino V, Tozzi A, Giampà C, Fusco FR, Sgobio C, Calabresi P, 2010. Inhibition of phosphodiesterases rescues striatal long-term depression and reduces levodopa-induced dyskinesia. Brain. 134, 375–387. [DOI] [PubMed] [Google Scholar]
- Picconi B, Centonze D, Håkansson K, Bernardi G, Greengard P, Fisone G, Cenci MA, Calabresi P, 2003. Loss of bidirectional striatal synaptic plasticity in L-DOPA-induced dyskinesia. Nature Neuroscience. 6,501–506. [DOI] [PubMed] [Google Scholar]
- Pisani A, Bemardi G, Ding J, Surmeier DJ, 2007. Re-emergence of striatal cholinergic interneurons in movement disorders. Trends in Neurosciences. 30, 545–553. [DOI] [PubMed] [Google Scholar]
- Pisani A, Martella G, Tscherter A, Bonsi P, Sharma N, Bemardi G, Standaert DG, 2006. Altered responses to dopaminergic D2 receptor activation and N-type calcium currents in striatal cholinergic interneurons in a mouse model of DYT1 dystonia. Neurobiol Dis. 24, 318–25. [DOI] [PubMed] [Google Scholar]
- Poewe WH, Lees AJ, Stem GM, 1988. Dystonia in parkinsoris disease: Clinical and pharmacological features. Annals of Neurology. 23, 73–78. [DOI] [PubMed] [Google Scholar]
- Quartarone A, Hallett M, 2013. Emerging concepts in the physiological basis of dystonia. Mov Disord. 28, 958–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quik M, Campos C, Grady SR, 2013. Multiple CNS nicotinic receptors mediate l-dopa-induced dyskinesias: Studies with parkinsonian nicotinic receptor knockout mice. Biochemical Pharmacology. 86, 1153–1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose SJ, Yu XY, Heinzer AK, Harrast P, Fan X, Raike RS, Thompson VB, Pare JF, Weinshenker D, Smith Y, Jinnah HA, Hess EJ, 2015. A new knock-in mouse model of l-DOPA-responsive dystonia. Brain. 138,2987–3002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scarduzio M, Zimmerman CN, Jaunarajs KL, Wang Q, Standaert DG, McMahon LL, 2017. Strength of cholinergic tone dictates the polarity of dopamine D2 receptor modulation of striatal cholinergic interneuron excitability in DYT1 dystonia. Exp Neurol. 295,162–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sciamanna G, Bonsi P, Tassone A, Cuomo D, Tscherter A, Viscomi ΜT, Martella G, Sharma N, Bemardi G, Standaert DG, Pisani A, 2009. Impaired striatal D2 receptor function leads to enhanced GABA transmission in a mouse model of DYT1 dystonia. Neurobiol Dis. 34, 133–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sciamanna G, Hollis R, Ball C, Martella G, Tassone A, Marshall A, Parsons D, Li X, Yokoi F, Zhang L, Li Y, Pisani A, Standaert DG, 2012a. Cholinergic dysregulation produced by selective inactivation of the dystonia-associated protein torsinA. Neurobiol Dis. 47,416–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sciamanna G, Tassone A, Mandolesi G, Puglisi F, Ponterio G, Martella G, Madeo G, Bemardi G, Standaert DG, Bonsi P, Pisani A, 2012b. Cholinergic Dysfunction Alters Synaptic Integration between Thalamostriatal and Corticostriatal Inputs in DYT1 Dystonia. Journal of Neuroscience. 32, 11991–12004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sciamanna G, Tassone A, Martella G, Mandolesi G, Puglisi F, Cuomo D, Madeo G, Ponterio G, Standaert DG, Bonsi P, Pisani A, 2011. Developmental Profile of the Aberrant Dopamine D2 Receptor Response in Striatal Cholinergic Interneurons in DYT1 Dystonia. PLoS ONE. 6, e24261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sgambato-Faure V, Cenci MA, 2012. Glutamatergic mechanisms in the dyskinesias induced by pharmacological dopamine replacement and deep brain stimulation for the treatment of Parkinson’s disease. Prog Neurobiol. 96, 69–86. [DOI] [PubMed] [Google Scholar]
- Shafrir Y, Levy Y, Beharab A, Nitzam M, Steinherz R, 1986. Acute dystonic reaction to bethanechol—a direct acetylcholine receptor agonist. Dev Med Child Neurol. 28,646–8. [DOI] [PubMed] [Google Scholar]
- Shakkottai VG, Batla A, Bhatia K, Dauer WT, Dresel C, Niethammer M, Eidelberg D, Raike RS, Smith Y, Jinnah HA, Hess EJ, Meunier S, Hallett M, Fremont R, Khodakhah K, LeDoux MS, Popa T, Gallea C, Lehericy S, Bostan AC, Strick PL, 2017. Current Opinions and Areas of Consensus on the Role of the Cerebellum in Dystonia. Cerebellum. 16, 577–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen W, Plotkin JL, Francardo V, Ko WK, Kin D, Xie Z, Li Q, Fieblinger T, Wess J, Neubig RR, Lindsley CW, Conn PJ, Greengard P, Bezard E, Cenci MA, Surmeier DJ, 2015. M4 Muscarinic Receptor Signaling Ameliorates Striatal Plasticity Deficits in Models of L-DOPA-Induced Dyskinesia. Neuron. 88, 762–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shetty AS, Bhatia KP, Lang AE, 2019. Dystonia and Parkinson’s disease: What is the relationship? Neurobiol Dis. [DOI] [PubMed] [Google Scholar]
- Standaert DG, 2011. Update on the pathology of dystonia. Neurobiology of Disease. 42, 148–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Standaert DG, Landwehrmeyer GB, Kemer JA, Penney JB Jr., Young AB, 1996. Expression of NMDAR2D glutamate receptor subunit mRNA in neurochemically identified interneurons in the rat neostriatum, neocortex and hippocampus. Brain Res Mol Brain Res. 42,89–102. [DOI] [PubMed] [Google Scholar]
- Suri R, Rodriguez-Porcel F, Donohue K, Jesse E, Lovera L, Dwivedi AK, Espay AJ, 2018. Post-stroke Movement Disorders: The Clinical, Neuroanatomic, and Demographic Portrait of 284 Published Cases. J Stroke Cerebrovasc Dis 27,2388–2397. [DOI] [PubMed] [Google Scholar]
- Tambasco N, Romoli M, Calabresi P, 2018. Selective basal ganglia vulnerability to energy deprivation: Experimental and clinical evidences. Progress in Neurobiology. 169, 55–75. [DOI] [PubMed] [Google Scholar]
- Thiele SL, Chen B, Lo C, Gertier TS, Warre R, Surmeier JD, Brotchie JM, Nash JE, 2014. Selective loss of bi-directional synaptic plasticity in the direct and indirect striatal output pathways accompanies generation of parkinsonism and l-DOPA induced dyskinesia in mouse models. Neurobiology of Disease. 71,334–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tolosa E, Compta Y, 2006. Dystonia in Parkinson,Aos disease. Journal of Neurology. 253, vfi7–vfil3. [DOI] [PubMed] [Google Scholar]
- Walker HC, Huang H, Gonzalez CL, Bryant JE, Killen J, Cutter GR, Knowlton RC, Montgomery EB, Guthrie BL, Watts RL, 2012. Short latency activation of cortex during clinically effective subthalamic deep brain stimulation for Parkinson’s disease. Mov Disord. 27,864–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wichmann T, DeLong MR, 2016. Deep Brain Stimulation for Movement Disorders of Basal Ganglia Origin: Restoring Function or Functionality? Neurotherapeutics. 13,264–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Won L, Ding Y, Singh P, Kang UJ, 2014. Striatal Cholinergic Cell Ablation Attenuates L-DOPA Induced Dyskinesia in Parkinsonian Mice. Journal of Neuroscience. 34,3090–3094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokoi F, Dang MT, Mitsui S, Li J, Li Y, 2008. Motor deficits and hyperactivity in cerebral cortex-specific Dyt1 conditional knockout mice. JBiochem. 143,39–47. [DOI] [PubMed] [Google Scholar]
- Yoon WT, 2018. Comparison of dystonia between Parkinson’s disease and atypical parkinsonism: The clinical usefulness of dystonia distribution and characteristics in the differential diagnosis of parkinsonism. Neurologia i Neurochirurgia Polska. 52,48–53. [DOI] [PubMed] [Google Scholar]
- Zhang D, Bordia T, McGregor M, McIntosh JM, Decker MW, Quik M, 2014. ABT-089 and ABT-894 reduce levodopa-induced dyskinesias in a monkey model of Parkinson’s disease. Movement Disorders. 29,508–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang D, McGregor M, Bordia T, Perez XA, McIntosh JM, Decker MW, Quik M, 2015. (Œ±7 nicotinic receptor agonists reduce levodopa-induced dyskinesias with severe nigrostriatal damage. Movement Disorders. 30,1901–1911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmerman CN, Eskow Jaunarajs KL, Meringolo M, Rizzo FR, Santoro M, Standaert DG, Pisani A, 2017. Evaluation of AZD1446 as a Therapeutic in DYT1 Dystonia. Front Syst Neurosci. 11, 43. [DOI] [PMC free article] [PubMed] [Google Scholar]