

Abstract
α-Synuclein (αS) and tau have a lot in common. Dyshomeostasis and aggregation of both proteins is central in the pathogenesis of neurodegenerative diseases: Parkinson’s disease, dementia with Lewy bodies, multi-system atrophy and other’synucleinopathies’ in the case of αS; Alzheimer’s disease, frontotemporal dementia, progressive supranuclear palsy and other’tauopathies’ in the case of tau. The aggregated states of αS and tau are found to be (hyper)phosphorylated, but the relevance of the phosphorylation in health or disease is not well understood. Both tau and αS are typically characterized as ‘intrinsically disordered’ proteins, while both engage in transient interactions with cellular components, there by undergoing structural changes and context-specific folding. αS transiently binds to (synaptic) vesicles forming a membrane-induced amphipathic helix; tau transiently interacts with MTs forming an ‘extended structure’. The regulation and exact nature of the interactions are not fully understood. Here we review recent and previous insights into the dynamic, transient nature of αS and tau with regard to the mode of interaction with their targets, the dwell-time while bound, and the cis and trans factors underlying the frequent switching between bound and unbound states. These aspects are intimately linked to hypotheses on how subtle changes in their transient behaviors may trigger the earliest steps in the pathogenesis of the respective brain diseases. Based on a deeper understanding of transient αS and tau conformations in the cellular context, new therapeutic strategies may emerge, and it may become clearer why existing approaches have failed or how they could be optimized.
1. Introduction
In a sense, the study of protein folding is a quandary: conventional NMR, cryo-EM, or X-ray crystallography can solve the structure of a purified protein but does so by taking the protein out of its biological context. On the other hand, various cell biological and biochemical techniques can provide insight into a protein’s behavior with in a cellular context but the information on its structure at atomic-level resolution is limited. That does not seem to be a problem for many proteins because their in vitro structures are a good approximation of those in the cell. However, it becomes a greater challenge when studying ‘intrinsically disordered’ proteins. Such proteins (or at least significant portions of them) adopt a more or less random-coil (‘unfolded’) structure in solution in vitro, but this does not rule out that context-specific folding/assembly states can arise inside cells. On the contrary, for many ‘intrinsically disordered’ proteins, transient folding has been proposed, but the dynamic nature of such states can render the proteins elusive to precise characterization. The stakes arguably become higher when the proteins under investigation are implicated in disease. Indeed, two key proteins in neurodegenerative diseases, α-synuclein (αS) in Parkinson’s disease (PD), Dementia with Lewy bodies (DLB) and other ‘synucleinopathies’ and tau in Alzheimer’s disease (AD), frontotemporal dementia (FTD) and other ‘tauopathies’, are still not yet well understood regarding their dynamic conformational behavior in cells. αS and tau, which both form insoluble aggregates in the brain under disease conditions, have been described to be largely soluble in their native states while transiently interacting with cellular targets: αS with (synaptic) vesicles, tau with MTs. This indicates that a contextual understanding of the transient and dynamic behavior of both proteins at the chemical, molecular, and cellular level may be required to understand synucleinopathies and tauopathies and develop therapeutic strategies through new perspectives. Here, we discuss recent advances in understanding cellular αS and tau dynamics in the context of the previous literature. Our special focus will be molecular’cis’ and ‘trans’ determinants undergirding αS and tau transient behavior. We conclude with some remarks on possible therapeutic strategies in light of our analysis.
2. αS
2A. αS in health and disease
Since the first discoveries of αS aggregation in Lewy bodies1 (see below), hundreds of studies have probed the precise structure and characteristics of physiological αS. There are many fine reviews on structure/function2–6, genetic interactions7,8, and neurotoxicity9,4,10–13 of αS, but what is significant to highlight here is that despite years of research, the precise function, subcellular distribution, and context-specific behavior of αS are still under investigation. It is likely that these properties are all intimately linked.
In mature neurons, including dopaminergic neurons14,15, αS predominantly resides in presynaptic nerve terminals, which has prompted the hypothesis that αS may be involved in regulating dopamine release.16–18 Considerable evidence suggests that αS helps regulate (synaptic) vesicle trafficking.19–25 This role of αS is consistent with its well-documented propensity to bind to small vesicles in vitro, which is mediated by its formation of transient amphipathic helices at highly curved membranes (see below).26–29 PD and DLB cytopathology and neurodegeneration are characterized by αS accumulation into Lewy bodies, large intracellular aggregates whose major componentis αS. However, the exact relationship of these lesions with dopaminergic neuronal death in the substantia nigra pars compacta and eventual PD symptoms, such as bradykinesia and muscular tremors30, is not fully understood. Similarly, we lack understanding of the exact pathogenic significance of cortical Lewy bodies in DLB. How and under what conditions αS aggregates have been areas of much inquiry and debate.
2B. αS transiently binds to synaptic vesicles
Early biochemical characterizations of purified bacterially-expressed αS identified the protein to be soluble14–31, which confirmed previous immunogold-EM data that detected αS throughout the cytoplasmic matrices in axon terminals.3 Subsequent biophysical and biochemical studies, however, reported that αS can bind to small unilamellar and multilamellar vesicles33,34 and detergent micelles35. Chemically crosslinked αS in SH-SY5Y cell homogenates was also identified in vesicle fractions by flotation centrifugation,36 but this observation did not seem to directly contradict the generally soluble nature of αS, because fractionated brain extracts revealed only a weak association of αS with synaptic vesicles37,38. Similarly, photobleaching microscopy indicated that αS interacts only weakly with membranous elements of the nerve terminal, and each molecule appears to switch rapidly between the aqueous cytosol and the membrane.38,39 While methods to interrogate the function of αS vis-à-vis synaptic vesicles40–42,25,43,15 became a natural implication of such studies, this function is still not well understood (for other proposed functions, see review by Bendor et al.2). Nevertheless, what has emerged is a nuanced picture of αS – a aqueously soluble protein that can transiently interact with vesicular membranes.
Recent studies have supplemented established biophysical and microscopy techniques by using new technologies to quantify the transient nature of αS. Single-molecule microscopy combined with photo-bleaching identified an average of seventy αS molecules bound to each vesicle and situated 10 nm apart from each other.46 Moreover, the use of phospholipid bilayer nanodiscs (which had been previously used to study αS-Ca+2 interactions)47 and bacterially purified αS allowed for αS-membrane association and dissociation kinetics to be calculated in vitro (e.g., an off-rate of 0.015 ± 0.006 s−1).48 However, data generated by in vivo multiphoton fluorescence recovery after bleaching (FRAP) coupled with murine cranial window surgery were consistent with the existence of at least two pools of αS in terminals with lower levels of mobility than measured previously. The observed t1/2 for axonal terminal photobleaching recovery (~2 min) in the mice was much slower than that measured in a photobleaching study of GFP-tagged αS at presynaptic terminals in an acute dissociated hippocampal cell culture system (<10sec)39, or YFP-tagged αS in C. elegans body wall muscle (<10sec)49. In addition to the presynaptic terminal, the turnover and mobility of αS in the somatic compartment of cortical neurons was measured by Unni et al. 51,52 In contrast to the discrepancy between their presynaptic terminal results and those reported in the literature, the rapid mobility of αS-GFP measured within the soma (<5 sec) was in good agreement with these previous studies of αS mobility. This suggested that the cellular context of αS may influence transient binding affinity and kinetics.50,51
This state of equilibrium between soluble and membrane-associated forms of αS is expected to be finely balanced and tightly regulated. Familial PD mutations as well as engineered variants have been observed to shift the balance towards misfolding, insolubility, inclusion formation and cell toxicity52–55 (discussed in further detail below). Consequently, such adverse changes in neuronal cells pose functional problems in vesicle trafficking, recycling, and neurotransmitter release56–59 (even overexpressed wildtype αS has been shown to undergo accelerated aggregation60,61 and display many of the aforementioned phenotypes62,18,63–65). In short, these recent observations highlight the transient nature of wildtype αS and the importance for the molecule to remain dynamic to keep cell toxicity at bay.
2C. αS transiently exists in a variety of monomeric and multimeric forms
Soluble αS had long been believed to exist solely as an unfolded monomer, both in vitro and in vivo. Less than a decade ago, emerging studies on the conformation of αS began to posit that the protein can exist in part physiologically as a soluble, higher-ordered multimer, apparently primarily a tetramer.66–71 Several biochemical as well as in-cell NMR studies responded by defending the classical view of αS as a natively unfolded monomer in cells72–75. A review on αS homeostasis76 highlighted the debate and suggested specific reasons for why certain studies reported the successful purification of tetrameric/multimeric αS whereas others did not: 1) multimer stability might be tissue-specific,67,74,77,71 2) αS purification in vitro under non-denaturing conditions could stabilize multimers, but the exact parameters for successful multimer purification are not fully understood 67,78,3; and 3) under certain conditions an undiscovered multimer-stabilizing molecule (perhaps one or more lipid molecules) may be lost during purification.71 Because of the difficulty of αS multimer purification and stabilization, intact-cell methods such as the application of cell-penetrant cross-1 inkers to cells and brain tissue68,79–81,54,82 as well as fluorescent-protein complementation79,83–86 became increasingly important for documenting multimeric αS arrangements. Intact-cell NMR, an elegant method that was used to propose the in vivo relevance of unfolded soluble αS75, is ‘blind’ to multimeric or membrane-associated αS molecules87. A crosslinking study that compared the outcome of crosslinking intact cells vs. lysates demonstrated that αS and βS putative tetramers (60 kDa) were prominent in intact cells but largely disassembled in crude cell lysates; only in highly concentrated (‘crowded’) cell lysates could some multimeric αS be trapped by crosslinking.88 These findings were consistent with a model in which αS multimerization is highly dependent on the intact cellular environment. Relevant factors that promote αS multimerization inside cells could be general ‘molecular crowding’ or the transient but constant interaction with specific cellular factors such as membrane lipids, organelles or other biomolecules. Obviously, dilution by cell lysis reduces crowding and disrupts any weak transient interactions that may exist. Recently, an elegant in vitro study demonstrated that transient αS-membrane interactions may indeed be the first step towards native αS-αS assembly.89 The authors investigated the interplay of αS with 13:0 phosphatidyl choline small unilamellar vesicles. By modulating αS binding through phase transitions of the vesicle lipids, soluble helical αS species could be reconstituted that behaved as multimers. While folding of certain ‘intrinsically disordered proteins’ upon binding to ligands is well established, no previous report had described folding assisted by intermittent contact with a cofactor (in this case: membrane lipids). This remarkable study is in agreement with several reports on soluble αS multimers67,66,90,88,91,77,78,71,70,79,80,92,54,82,93. The relevance of multimeric αS at membranes has also been highlighted in other studies (e.g., in the context of mediating SNARE complex assembly94,95 and vesicle clustering23). Fig. 1A illustrates a model of dynamic cellular αS behavior in health, centered around the idea of (vesicle) membrane-assisted, transient αS folding and assembly89. In this model, αS monomer and multimers levels are highly transient, dynamic, and sensitive to environmental factors. Fig. 1B highlights potential mechanisms of synucleinopathy initiation based on this model; the illustration takes into account reports on excess soluble αS monomers as the starting point of cytosolic fibrillar αS aggregation95 (top left), fibrillar αS aggregation that is mediated by excess membrane binding via ‘nucleatin’96 (bottom right), as well as membranous non-fibrillar αS aggregation as an emerging concept82,97,98 (bottom left).
Figure 1. Transient cellular behavior of αS.
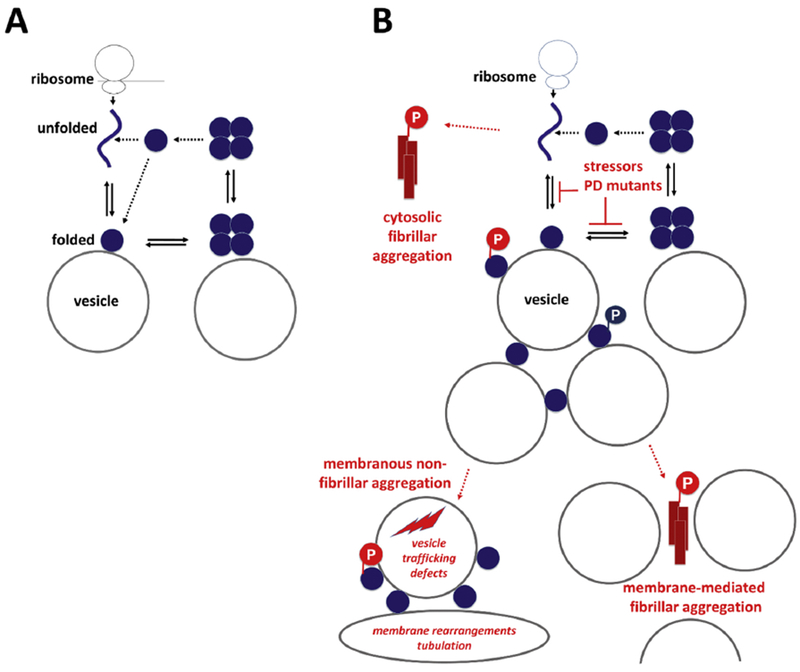
A, Physiological situation: Coming off the ribosome, αS is soluble, unfolded and monomeric. Upon binding to vesicular membranes, it adopts helical fold. Folded monomers transiently assemble to form metastable multimers/tetramers on membranes. Multimers/tetramers are only weakly membrane-associated and likely in an equilibrium with cytosolic multimers/tetramers. Cytosolic tetramers/multimers may have an intrinsic propensity to disassemble - and eventually unfold, initiating a new cycle. B, Pathological situation (pathological states are in red): perturbed cellular αS homeostasis increases i) the levels of aggregation-prone unfolded monomers in the cytosol or ii) the level of membrane-associated monomeric αS. Excess soluble αS monomers may be the starting point of cytosolic fibrillar αS aggregation (top left), while excess membrane association may either cause fibrillar αS aggregation via ‘nucleatin’96 (bottom right) or membranous non-fibrillar αS aggregation (bottom left). The role of phosphorylation atSerl29 is unclear. While primarily considered a pathological event before or in response to fibrillar aggregation (red), there may also be physiological aspects of this post-translational modification (blue).
2D. Molecular determinants of the transient nature of αS
Across the 140 amino acids that span αS, the protein is often subdivided into three regions: the N-terminal region (amino acids 1-60), the ‘NAC’ (non-amyloid component) domain (amino acids 61-95), and the C-terminal region (amino acids 96-140).3 The N-terminal region has been of immense interest to better understand the molecular determinants for αS transient membrane-binding behavior, because early studies identified this region to contain amino acid repeat motifs that resemble lipid-binding domains often observed in apolipoproteins14. Biochemical, cell biological and biophysical studies have shed much light on how αS ‘cis’ determinants99–101,54 and ‘trans’ factors such as vesicle membrane composition102–104,89,105 may govern αS transient binding to artificial and biological membranes.
Different regions of the protein have been shown to have different binding affinities for charged lipid membranes. The first 25 residues in the N-terminus have been suggested to ‘anchor’ into the bilayer106, with certain key residues (M1, V3, F4, and L8) stabilizing αS at membranes via Van der Waals interactions, while other residues (K6, K10, and K12) interact electrostatically with negatively-charged lipid head groups.107,108 Due to the repeated structure of αS (6-7 imperfect repeats of 11 aa with the consensus sequence of the repeat motifs being KTKEGV), this mechanism can be extended to the first ~95 aa of the protein. The positively-charged Lys residues have been suggested to interact with negatively-charged lipid head groups when the N-terminus of αS forms an α-helix (3.67 amino acids per turn)109, which allows the Lys residues to lie perpendicular to the helical axis on a membrane.110 In contrast, nonpolar amino acid residues can partially dip into the membrane bilayer (~1-5 Å below lipid head groups)111–114, especially where lipid packing defects exist, and interact with the lipid carbon chains.115,116 However, in the αS membrane-induced 3-11 helix, some non-polar residues are exposed to the aqueous phase, and some polar residues are embedded into the nonpolar lipid bilayer, most prominently threonine residues (see Fig. 2), leading to imperfect amphipathic helix formation. Consequently, the specific arrangement of charged residues, polar lipid head groups, and nonpolar carbon chains seem to manifest the transient nature of αS-membrane binding5,54 (see further discussion in Dettmer et al. 201797).
Figure 2. Molecular determinants of the transient nature of αS.
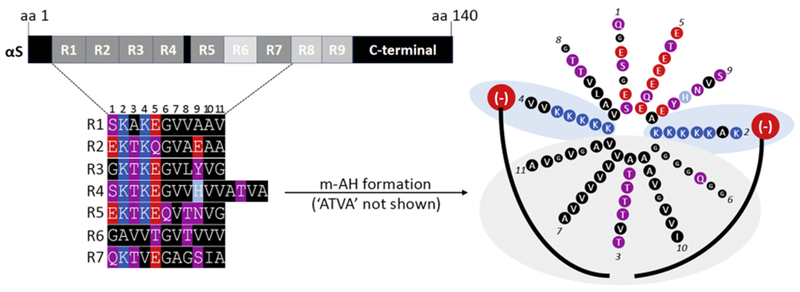
Top left: the 140-aa sequence of human wildtype αS contains 9 semi-conserved 11-aa repeats with the core consensus motif ‘KTKEGV’. Highly conserved repeats are indicated in dark grey, poorly conserved repeats in light grey. Bottom left: color-coded schematic of repeats 1-7 of human αS by aligning the aa sequence via the KTKEGV motifs. Blue indicates basic (light blue: histidine), red: acidic, purple: polar uncharged, and black: non-polar residues. In addition to KTKEGV, the polar character of positions 1 and 9 as well as the non-polar, hydrophobic character of positions 8, 10, and 11 are relatively well conserved. Repeats 1–9 are interrupted only once: by ’ATVA’ between repeat 4 and 5. Right: Color-coded schematic of αS repeats 1-7 (omitting ’ATVA’ between repeats 4 and 5) in an 11/3 helical wheel, embedded in the outer leaflet of a curved vesicle membrane (negatively charged lipid head-groups in red, fatty acid ‘tails’ in black). The helix is stabilized by hydrophobic interactions (gray area) and electrostatic interactions (blue area). Certain structural aspects such as the presence of polar threonines as well as the small and ‘helix-breaking’ glycine prevent the formation of a highly stable helix.
This principle can be confirmed by observing how rationally engineered mutants within the canonical repeat motif, KTKEGV, that’correct’the imperfect hydrophobicity within the hydrophobic half of the αS membrane-induced amphipathic helix can there by abolish the transient nature of αS.80,54,117,118 Six or 7-repeats of a KLKEGV or KTKEIV or KTKKGV mutant can destabilize native tetramers/multimers in neurons and presumably lead to the accumulation of helical monomeric αS at membranes and perhaps some unfolded cytosolic αS, as these mutants remained monomeric when tested by DSG crosslinking and were highly enriched in PBS-insoluble fractions that required Triton-X 100 to solubilize membranes.80 Beyond such artificially engineered αS, an amplification of the E46K fPD-linked mutant (i.e., KTKKGV in repeat 4) in the two adjacent repeat motifs strongly reduces tetramers/multimers, as suggested by intact cell-crosslinking and YFP-fluorescent-protein complementation assays, and it also accumulates in PBS-insobluble fractions.79,97 In addition, the mutant H50Qwas reported to decrease αS solubility 10-fold.119 However, also the membrane-binding deficient, cytosol-enriched fPD variants A30P and G51D cause PD, and it was suggested that they expose their hydrophobic core in solution, thus enabling other αS molecules to bind and aggregate.120 A study based on intact-cell crosslinking and YFP complementation concluded that all known fPD-linked αS variants lead to a relative decrease of αS multimers and a gain in aggregation-prone monomers (either in the cytosol or at membranes, depending on the specific mutants121). In short, certain regions of αS as well as specific residues have been shown to play a role in either increasing or decreasing binding affinities to lipid bilayers through electrostatic or Van der Waals interactions; increasing αS dwell-time either in the cytosol or at membranes both seem to decrease multimerization, with negative consequences on cell health, as evidenced by the αS fPD-causing variants.
Lastly, post-translational modifications add nuance to αS proteostasis. αS phosphorylation is of particular interest because 90% of aggregated αS present in Lewy bodies were reported to be phosphorylated at serine 129 (pS129),122 implying that this specific phosphorylation event is connected to pathology (Fig. 1B). In addition to the largely insoluble Lewy bodies, however, soluble pS129 αS has been found in CSF samples from PD patients and even controls (e.g., 123). Growing evidence also indicates that Lewy bodies can contain large amounts of pS129-positive αS without being fibrillary (e.g., a very recent study98). The interplay between αS tetramerization/multimerization has not yet been studied. Moreover, it has been proposed that pS129 may actually be a neuroprotective mechanism to accelerate the clearance of aggregated αS.124,125 This may be achieved at least with the aid of PLK-2126. Still other groups have concluded that rather than neurotoxic or neuroprotective roles for pS129, it serves a normal regulatory function in αS turnover127 and even in controlling gene expression.128 In short, whether pS129 unequivocally indicates pathology under all circumstances is unclear. The trigger for pS129, whether certain kinases phosphorylate only when αS interacts with membranes, or whether a “master kinase” exists may all be important questions, the answers to which might further reveal whether pS129 plays multiple roles in contributing to synucleinopathy.
3. Tau
3A. Tau in health and disease
One of the hallmarks of AD, frontotemporal dementia (FTD), progressive supranuclear palsy (PSP), Pick’s disease and other neurodegenerative diseases collectively called tauopathies are the so-called neurofibrillary tangles (NFTs), which consist mainly of aggregated tau protein129–132. There are numerous efforts to target tau with the hope of slowing disease progression and subsequent cognitive decline. In the case of AD this is done in addition to targeting amyloid beta, the other, more upstream neuropathological hallmark of AD133. Interestingly, the level of dementia in AD patients and AD animal models may correlate well with tau lesions134–136 –one of several reasons to thoroughly examine the therapeutic potential of tau. Since the discovery of tau from porcine brain MT fractions in 1975137 and its identification as the main NFT component129–132, major advances have been made in understanding the biology of tau especially focusing on its aggregation and hyper-phosphorylation in tauopathies137,138. Tau is a MT-associated protein (MAP) that may be involved in regulating MT stability (tau function and structure-function relationships are discussed below in sections 3B-D). The tau protein exists in six isoforms ranging in size from 352-441 amino acids as a result of alternative splicing of exons139. The isoforms are marked by the presence or absence of exons 2, 3 and 10 and are regulated based on the stage of development of the brain, though their specific function is not known 140. The isoforms are named based on how many MT binding repeats (R) are expressed, with the 3R isoform having three MT binding repeats and the 4R isoform four binding repeats138. The 3R isoform is expressed mainly during the early development stage of the brain and the 4R isoform is expressed later in adulthood 139 140. Most of what we know about tau originates from its function in neuronal cells. Besides its neuronal expression, tau was found to be expressed in non-neuronal tissues such as liver, muscle, kidney and even cancer cell lines141,142. Tau’s characterization as an ‘intrinsically disordered’ protein in solution, while having numerous (proposed) functions within the cell, challenged the thought that proteins have to be pre-folded to exhibit specific functions in a cell.143,144
3B. Tau transiently binds to MTs
As one of the three major component of the cytoskeleton, microtubules (MTs) control the formation of mitotic spindles, cilia and flagella and play an important role in cell motility and polarity145,146. MTs are built from α- and β-tubulin monomers into a long cylindrical hollow shaped protofilaments. MTs undergo dynamic changes between growth and shortening known as dynamic instability147. Individual MT polymers elongate by the addition of GTP-tubulin dimers to the end of the MT and shrink due to the competitive activity of end-binding proteins preventing new tubulin dimers to bind 148,149. A group of proteins known as MT associated proteins (MAPs) binds to and promotes the assembly and stabilization of MTs. Tau is a MAP that transiently interacts with MTs to regulate their dynamics and spatial organization. In addition, tau is believed to stabilize MTs in axons which serve as ‘roads’ for transport within the cell.150 There are many proposed mechanisms for the MT-associated function of tau.151–153 Several lines of investigation substantiate that tau bridges linear rows of tubulin dimers, by binding to several protofilaments via its MT binding repeats.154,155 Others have shown that tau stabilizes MTs by bridging the interfaces of tubulin heterodimers.156 Recent findings, however, suggest that tau may not stabilize axonal MTs at all, but rather modifies the biology of MTs by enabling them to have long labile domains.157,158 Various techniques such as protein biochemistry and NMR have been used to delineate the physical and structural basis of the transient tau-MT interaction leading to different hypotheses/models about the interplay between these two binding partners.
It has been suggested that a dynamic equilibrium between free tau and tau bound to MTs is dictated by tau phosphorylation and de-phosphorylation cycles159–161. Phosphorylation sites such as Ser262, Ser365, Ser205 and Thr231 are typically considered to reduce the strength of the interaction between tau and MTs when phosphorylated.159Tau phosphorylation was proposed to alter its conformation and affinity to MTs, leading to a detachment from MTs162,163 In line with this idea, dysregulation of certain kinases has been proposed to perturb or even eliminate tau’s MT binding in models of tauopathies164–166. While tau hyper-phosphorylation is certainly a hallmark of tauopathies, recent studies challenge the view that phosphorylation cycles are the key mechanism underlying transient (instead of stable) tau-MT interaction. In various binding and decay assays, it was shown that tau may be bound tau on each MT filament for only ~40 ms.167 This dwell-time is ~100 times shorter than previously reported168 and too short for regulation by kinases: the average catalytic ability of a kinase is approximately 10 interactions per second.169 Hence, Janning et al. proposed a ‘kiss-and-hop mechanism’ of regulating tau-MT interaction.167 Rather than a phosphorylation/de-phosphorylation mechanism of initiating the transient MT-tau interaction, the authors suggest that tau may ‘hop’ between MTs in an undirected manner. Despite its high mobility, it was postulated that tau can shifts the tubulin equilibrium toward polymerization: the short interaction time between tau and MTs was proposed to suppress the natural dissociation of tubulin dimers, allowing the addition of extra GTP cap size to initiate an interaction. Janning et al. further speculate that the rapid kiss-and-hop interaction that they observe may explain why tau, although binding to MTs, does not interfere with axonal transport. 167 It is worth noting that the ‘kiss and hop’ model was developed in PC12 cells which do not express axonal components. Therefore, the distance between two MTs is ~70 nm instead of the average ~20 nm for MT-MT interactions170. It is conceivable that details of the protein conformation and the effect of phosphorylation amongst others are model-dependent.171 While probably not regulating the binding/unbinding cycles per se, its exact phosphorylation state is still expected to contribute to tau’s overall probability MT binding affinity (see 3B). Reduced MT binding may in turn cause accumulation in the cytsosol and aggregation. Figure 3A summarizes several aspects discussed in this paragraph and emphasizes the proposed kiss-and-hop mechanism of the tau-MT interaction167. Figure 3B illustrates potential patho-mechanisms of AD, FTD and other tauopathies in the context of impaired tau-MT interaction.
Figure 3. Transient cellular behavior of tau.
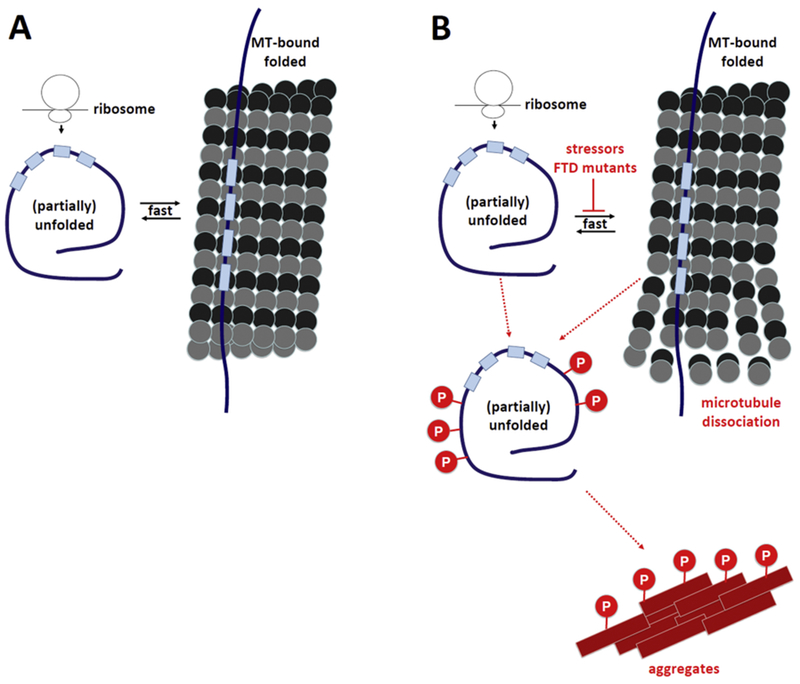
A, Physiological situation: Coming off the ribosome, tau is soluble, unfolded and monomeric. Upon binding to MTs, tau adopts a different fold, but almost immediately becomes soluble again. Tau phosphorylation may contribute to regulating the transient tau-MT interaction. B, Pathological situation (pathological states are in red): perturbed cellular tau homeostasis is characterized by tau hyper-phosphorylation, lack of tau-MT interaction, disintegration of MTs and the formation of proteinaceous tau aggregates.
3C. Lack of strong evidence for physiological multimeric forms of tau
The tau literature does not provide ample evidence for the existence of native tau higher-order assemblies. One study172 employed a surface force apparatus to determine the force profile for each of six tau isoforms physisorbed through self-assembly from solution to negatively charged mica surfaces. The authors interpreted their data as incompatible with tau acting on MTs as a monomer and speculated that two tau molecules may associate in an antiparallel configuration. This structure was suggested to be held together by an electrostatic ‘zipper’ of complementary salt bridges composed of the N-terminal and central regions of each tau monomer, with the C-terminal MT-binding regions extending outward from each end of the dimeric backbone. This tau dimer would then determine the length and strength of the linker holding two MTs together and to be the fundamental structural unit of tau, underlying both its normal and pathological action. Independent confirmation of such a mechanism is lacking.
3D. Molecular determinants of the transient nature of tau
Tau is typically characterized as consisting of an N-terminal-, a proline-rich, a MT-binding domain (MBD) and a C-terminal domain (Fig. 4). As it is evident from their names, the MT-binding domains are important for MT interactions139,173. Each isoform of tau has three to four repeats (‘R-repeats’) located in the C-terminus.174 Using synthetic tau fragments, it has been shown that these repeats form MT binding units.175,176 Positively charged repeat sequences of the MBD were proposed to interact with negatively charged residues in tubulin and hence to facilitate the tau-tubulin interaction155,162. Recent studies based on NMR177 and cryo-EM178 provide detailed insight into a complex mode of interaction. Both studies reiterate the previous notion that MT-bound tau does not become helical - in contrast to vesicle-bound αS that forms membrane-induced amphipathic 3-11 helices. Instead, tau adopts an ‘extended’ secondary structure, a state that is both different from unfolded and from helically or beta-sheet folded. In the extended structure, each of tau’s R-repeats was observed to span both intra- and inter-dimer interfaces, centered on α-tubulin and connecting three tubulin monomers (Fig. 4). In each individual interaction site, key tau residues engage in specific interactions with key MT residues. Examples from the cryo-EM study178 are: Ser258 and Ser262 in tau form hydrogen bonds with α-tubulin Glu434. Tau’s conserved Lys259 interacts with an acidic α-tubulin patch formed by Asp424, Glu420, and Glu423. Ile260 is buried within a hydrophobic pocket formed by α-tubulin’s Ile265, Val435, and Tyr262 at the interdimer interface. Lys267 may be positioned to interact with the acidic α-tubulin C-terminal tail. The universally conserved Ser262, was proposed to be critically involved in tight contacts with tubulin near a polymerization interface. The new structure thus now offers a detailed explanation for how Ser262 phosphorylation may disrupt tau-tubulin interactions.
Figure 4. Molecular determinants of the transient nature of tau.
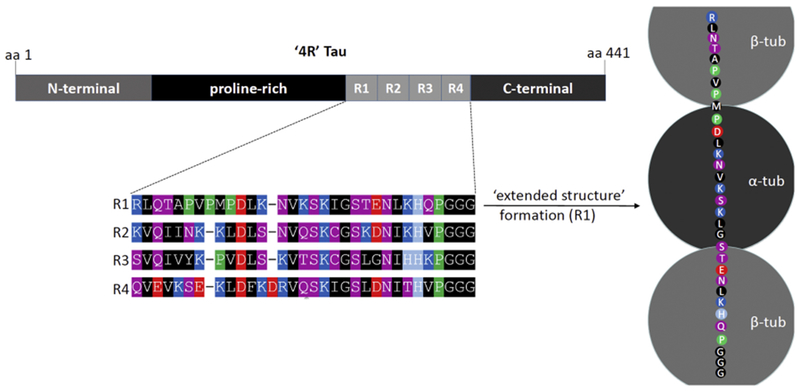
Top left: the 441-aa sequence of human wildtype ‘4R’ tau contains 4 semi-conserved 31 or 32-aa ‘R repeats’, plus proline-rich, N-terminal and C-terminal domains. Bottom left: color-coded schematic of the 4 R repeats. Blue indicates basic (light blue: histidine), red: acidic, purple: polar uncharged, black: non-polar, green: proline residues. Right: Color-coded schematic of R repeat 1 bound to MTs via 3 tubulin subunits (2β, 1α).The proposed structure is neither unfolded nor helically or β-sheet folded. Instead, it is characterized as ‘extended’. The exact molecular basis fora highly transient, instead of a strong binding to MTs will require further research.
What these new studies leave open, however, is the question why tau binding to MTs is actually dynamic and tau’s dwell-time on MTs is so short. On the contrary, the studies should not be misinterpreted as evidence for the presence of stable MT-tau assemblies within cells. The authors do not point to specific amino acids that are ‘sub-optimal’ for achieving energetic minima (such as the central threonine in the hydrophobic half of αS membrane-associated helices; see Fig. 2). The rapid switch of tau between soluble and MT-bound tau suggests that the energetic minimum of the soluble unfolded state is similar to that of MT-bound state(s). Though it may contribute to it, Ser262 phosphorylation is not expected to be the sole reason for tau’s dynamic behavior, for reasons discussed in section 3B.
While the 2015 and 2018 structural studies mainly focused on the R-repeats, the proline-rich regions in tau are also believed to influence the tau-MT interaction179,180. The proline regions line the repeat domain of tau and may initiate the formation of the tau-MT interaction by binding to the MT-binding repeats of tubulin, a ‘green light’ to trigger tau-MT interactions152,181. Notably, there are many phosphorylation sites on the proline-rich region which may affect interactions with other proteins as well as tau-MT interactions182,183. Mutagenesis studies on human tau isoforms revealed that loss of the proline-rich domains reduces MT binding180. Tau mutants with a deletion of the repeat domain, but with an intact proline-rich domain, were found to still bind to MTs (see below)181,184. Combining the proline-rich regions with the adjacent repeat regions increased the binding affinity of tau 10-fold compared to mutants lacking the proline-rich region185. Taken together, the data show that the proline-rich domain and the repeat regions in tau synergistically promote its MT binding. Accordingly, Mandelkow and colleagues proposed the ‘jaws’ model of MT-tau interaction to explain these observations184,186. Mutants of tau with deletions or multiplications of different domains were generated and tested for tau-MT interactions in cells. The authors found evidence for the binding of tau with MTs in the absence of the repeat regions. However, these mutants were unable to stabilize MTs. Increasing the number of repeats in the repeat region enhanced the interaction and effect of tau on MTs. On the other hand, mutants with deletion of the proline-rich regions flanking the repeat regions showed only low affinity to MTs and did not induce reorganization of the MTs. The authors concluded that the proline-rich regions flanking the binding repeats serve as targeting domains to position tau on the MTs and that the repeat domain act as the primary interacting domains, hence the ‘jaw’ model of tau-MT interaction184. According to the authors, stabilization of MTs is only achieved if both domains of tau act together.
4. Therapeutic intervention via correcting transient behaviors of αS and tau?
The transient behavior and context-specific folding of αS and tau is not a contradiction to their frequent characterization as’intrinsically disordered proteins’ because this term can simply be interpreted as lack of a sole and fixed three-dimensional structure. However, all current major protein classes considered ‘druggable’ are stably folded in their native states. Proteins like αS and tau on the contrary seem to exist as heterogeneous conformational ensembles, which renders them unsuitable for standard rational drug design approaches.187 Nonetheless, several different small molecule strategies are currently under investigation, including: (1) stabilizing the proteins in their natively disordered states, (2) inhibiting interactions with binding partners, and (3) inducing allosteric inhibition.187
In agreement with scenario (1), Collier et al.188 proposed that the tricyclic antidepressant compound nortriptyline can inhibit the aggregation of αS by directly binding to the soluble, monomeric form. The authors further proposed that nortriptyline-mediated ‘reconfiguration of the monomer’ can inhibit the formation of toxic conformations of the protein. Similarly, a recent study reported that stabilizing αS monomers with the polyphenol Oleuropein aglycone (OleA) reduced the likelihood for αS to aggregate into toxic molecules189 (see Singh et al.’s review for further comment on phenol as therapeutic targets190). More in line with scenario (2), Perni et al.191 demonstrated that squalamine (a natural product with proposed anticancer and antiviral activity that can be extracted from shark liver) can prevent αS aggregation by displacing αS from the surfaces of vesicles. This was suggested to block a first, membrane-assisted, step in the αS aggregation process. The same group then reported that the related compound trodusquemine not only inhibits αS-membrane interactions, but also blocks the fibril-dependent secondary pathways in the aggregation reaction, thereby effectively suppressing the toxicity of αS oligomers in neuronal cells.192 However, other groups had suggested that pharmacological stabilization of monomeric αS in its helical form at membranes may inhibit pathogenic misfolding and aggregation.193 And yet, both approaches could eventually turn out to be problematic since αS monomers both in membrane-associated96 and soluble conformation194,195 have been reported to be aggregation-prone. Thus, another attractive option of preventing αS aggregation would be stabilizing native multimeric/tetrameric states of αS that have been reported to be aggregation-resistant.92,196 An example of the direct stabilization of a tetrameric protein via a small molecule, thereby preventing amyloidosis, is the drug tafamidis that stabilizes transthyretin.197 While the design of direct stabilizers of αS tetramers will likely depend on the successful generation of αS tetramer/multimer high-resolution structural data, it was recently shown that stabilizing native αS self-assembly can be achieved in an indirect fashion: the inhibition of stearoyl-CoA-desaturase (SCD) can increase αS multimer: monomer ratios in the multimerization-deficient familial-PD-linked mutant E46K.198 The exact mechanism is not clear, but it seems likely that blocking the SCD-catalyzed production of oleic acid (18:1) causes a reduced occurrence of this fatty acid in membrane lipids, leading to higher levels of saturated fatty acids. A higher degree of saturation has been reported to interfere with αS-membrane interaction because ‘membrane defects’ that favor αS binding are less likely to occur.199 Thus, the correction of excess αS E46K membrane binding and the correction of E46K’s lack of multimerization seem to go hand in hand. The target SCD was confirmed in a different study that was based on αS (yeast) toxicity.200
In the case of tau, a recent study investigated the structural basis of small molecule druggability of native tau monomers.201 As an example, the authors showed that methylene blue binds to monomeric full-length tau selectively with high affinity (Kd = 86.6 nM). The authors interpret their study as evidence that tau can be a viable drug target for small molecules that bind to monomeric tau and influence the way in which the protein interacts among itself and with other proteins.201 Clinical trials based on methylene blue have thus far turned out to be rather disappointing202–204. Authors’ claims of some efficacy after a post-hoc subgroup secondary analysis have been controversial. 205 A 2013 review206 lists several classes of potential inhibitors/modulators of tau aggregation such as polyphenols, rhodanines, phenyl-thiazolylhydrazide, N-phenylamines, benzothiazoles and aminothienopyridazines. Only few of these structures are expected to actually bind to and stabilize monomeric soluble tau; most inhibitors/modulators likely redirect the self-assembly toward ‘off-pathway’ oligomeric forms.206 In light of the topic of this review, such compounds are not considered modulators of normal dynamic behavior of tau. Moreover, many putative aggregation inhibitors have demonstrated to be potent in in vitro assays, but evidence of inhibiting tau aggregation in vivo and, even more importantly, cognitive improvement is still lacking.205 It is also worth mentioning that the specificity of tau aggregation inhibitors, including methylene blue, is limited and pleiotropic effects can be expected.205 Harnessing the hyper-phosphorylation of aggregated tau207 for therapy has been considered a viable approach for a long time. However, tau phosphorylation cannot solely be considered a pathological event because the protein’s transient binding to MTs is likely to be regulated at least in part via phosphorylation as well (see above).208 The kinases and phosphatases that have been demonstrated to be involved in tau phosphorylation comprise GSK3β, Cdk5 and p25209,210. Interfering with tau phosphorylation promises to affect protein homeostasis upstream of proteinaceous aggregation and remains a valuable strategy. However, the exact regulation as well as physiological and/or pathological significance of tau phosphorylation at all its different phosphorylation sites remains a question to be answered.
Lastly, while modifying the composition of the vesicles that αS binds to (e.g., via SCD inhibition) has emerged only recently as a new potential strategy for treating synucleinopathies198,200, tubulin-stabilizing compound have already been tested in clinical trials211,212, albeit unsuccessfully in phase III212. MT imbalance is typically considered a downstream effect of tau dyshomeostasis. The interplay between αS and fatty acid/lipid/vesicles, however, may be more complicated. For example, a previous study demonstrated that expressing αS alters fatty acid composition of dopaminergic neurons in a way that reflects alterations in human brains with synucleinopathies.213 Genetic evidence, on the other hand, suggests that impaired glucocerebrosidase activity may be upstream of αS dyshomeostasis in PD pathogenesis214,215. Mazzuli et al. proposed a ‘bidirectional pathogenic loop’ between glucocerebrosidase and αS.216 Other lipid-related genes that emerged from GWAS as PD risk factors comprise a diacylglycerol kinase, DGKQ, which controls cellular diglyceride content217–221, and fatty acid elongase 7, a determinant of acyl-chain length and hence lipid composition/membrane fluidity222. Both indicate that changes in lipid metabolism can be upstream of αS dyshomeostasis (which does not exclude additional effects on lipids downstream of αS). In turn, modifying cellular lipid composition could indeed help keep cellular αS folding homeostasis intact or correct it back to normal. A similar approach for correcting tau homeostasis via modification of MT biology may be less evident from genetics or the literature in general.
It is also important to note that in both tauopathies and synucleinopathies the pathway toward fibrillary assembly may be quite different from one disease to another. As far as the endpoints are concerned, there is growing evidence for disease-specific aggregate formation. For example, the single protofilaments with an elongated and loosely arranged structure found in Pick’s Disease are markedly different from the compact bundles of C-shaped paired helical tau filaments found in AD.223,224 Similarly, PD, DLB and MSA have been reported to be characterized by distinct αS aggregate ‘strains’ that exhibit different ‘seeding’ characteristics225–227, and the notion of non-fibrillar, membranous αS aggregation98 adds further complexity. These differences in advanced pathology may point at differences in disease initiation. As a consequence, drugs targeting certain transient forms within tau and αS dynamic equilibria could be disease-specific and inefficient toward another assembly pathway.
tau and α-synuclein are central in the pathogenesis of a variety of neurologic diseases
both engage in transient interactions with cellular components
both constantly undergo structural changes and context-specific folding in the cell
new insights on regulation and exact nature of this dynamic biology are emerging
a deeper understanding of transient conformations may lead to new therapies
Acknowledgments
We thank Dennis Selkoe, Nagendran Ramalingam, Thibaut Imberdis, Saranna Fanning, Elizabeth Terry-Kantor, Luis Fonseca-Ornelas, and Arati Tripathi (BWH/Harvard Medical School) as well as Tim Bartels and Tong Guo (University College London) for discussions and critical revision of the manuscript. UD’s research is funded by NIH grant NS099328.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Literature
- 1.Spillantini MG, Schmidt ML, Lee VM-Y, Trojanowski JQ, Jakes R, Goedert M, α-Synuclein in Lewy bodies, Nature. 388 (1997) 839–840. doi: 10.1038/42166. [DOI] [PubMed] [Google Scholar]
- 2.Bendor JT, Logan TP, Edwards RH, The function of α-synuclein, Neuron. 79(2013) 1044–1066. doi: 10.1016/j.neuron.2013.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mor DE, Ugras SE, Daniels MJ, Ischiropoulos H, Dynamic structural flexibility of α-synuclein, Neurobiol. Dis 88(2016) 66–74. doi: 10.1016/j.nbd.2015.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Villar-Piqué A, Lopes da Fonseca T, Outeiro TF, Structure, function and toxicity of alpha-synuclein: the Bermuda triangle in synucleinopathies, J. Neurochem 139 Suppl 1 (2016) 240–255. doi: 10.1111/jnc.13249. [DOI] [PubMed] [Google Scholar]
- 5.Wang C, Zhao C, Li D, Tian Z, Lai Y, Diao J, Liu C, Versatile Structures of α-Synuclein, Front. Mol. Neurosci 9(2016) 48. doi: 10.3389/fnmol.2016.00048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cremades N, Chen SW, Dobson CM, Structural Characteristics of α-Synuclein Oligomers, Int. Rev. Cell Mol. Biol 329 (2017) 79–143. doi: 10.1016/bs.ircmb.2016.08.010. [DOI] [PubMed] [Google Scholar]
- 7.Schneider SA, Alcalay RN, Neuropathology of genetic synucleinopathies with parkinsonism: Review of the literature, Mov. Disord. Off. J. Mov. Disord. Soc 32 (2017) 1504–1523. doi: 10.1002/mds.27193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nussbaum RL, Genetics of Synucleinopathies, Cold Spring Harb. Perspect. Med 8 (2018). doi: 10.1101/cshperspect.a024109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lashuel HA, Overk CR, Oueslati A, Masliah E,The many faces of α-synuclein: from structure and toxicity to therapeutic target, Nat. Rev. Neurosci 14(2013) 38–48. doi: 10.1038/nrn3406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Berrocal R, Vasquez V, Rao Krs S, Gadad BS, Rao KS, α-Synuclein Misfolding Versus Aggregation Relevance to Parkinson’s Disease: Critical Assessment and Modeling, Mol. Neurobiol 51(2015) 1417–1431. doi: 10.1007/s12035-014-8818-2. [DOI] [PubMed] [Google Scholar]
- 11.Ghosh D, Singh PK, Sahay S, Jha NN, Jacob RS, Sen S, Kumar A, Riek R, Maji SK, Structure based aggregation studies reveal the presence of helix-rich intermediate during α-Synuclein aggregation, Sci. Rep 5 (2015) 9228. doi: 10.1038/srep09228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wong YC, Krainc D, α-synuclein toxicity in neurodegeneration: mechanism and therapeutic strategies, Nat. Med 23 (2017) 1–13. doi: 10.1038/nm.4269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Roberts HL, Brown DR, Seeking a mechanism for the toxicity of oligomeric α-synuclein, Biomolecules. 5(2015) 282–305. doi: 10.3390/biom5020282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.George JM, Jin H, Woods WS, Clayton DF, Characterization of a novel protein regulated during the critical period for song learning in the zebra finch, Neuron. 15 (1995) 361–372. doi: 10.1016/0896-6273(95)90040-3. [DOI] [PubMed] [Google Scholar]
- 15.Iwai A, Masliah E, Yoshimoto M, Ge N, Flanagan L, de Silva HA, Kittel A, Saitoh T, The precursor protein of non-A beta component of Alzheimer’s disease amyloid is a presynaptic protein of the central nervous system, Neuron. 14(1995) 467–475. [DOI] [PubMed] [Google Scholar]
- 16.Murphy DD, Rueter SM, Trojanowski JQ, Lee VM-Y, Synucleins are developmentally expressed, and α-synuclein regulates the size of the presynaptic vesicular pool in primary hippocampal neurons, J. Neurosci 20(2000) 3214–3220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gedalya TB, Loeb V, Israeli E, Altschuler Y, Selkoe DJ, Sharon R, α-Synuclein and Polyunsaturated Fatty Acids Promote Clathrin-Mediated Endocytosis and Synaptic Vesicle Recycling, Traffic. 10(2009) 218–234. doi: 10.1111/j.1600-0854.2008.00853.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nemani VM, Lu W, Berge V, Nakamura K, Onoa B, Lee MK, Chaudhry FA, Nicoll RA, Edwards RH, Increased Expression of α-Synuclein Reduces Neurotransmitter Release by Inhibiting Synaptic Vesicle Reclustering after Endocytosis, Neuron. 65 (2010) 66–79. doi: 10.1016/j.neuron.2009.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Abeliovich A, Schmitz Y, Fariñas I, Choi-Lundberg D, Ho WH, Castillo PE, Shinsky N, Verdugo JM, Armanini M, Ryan A, Hynes M, Phillips H, Sulzer D, Rosenthal A, Mice lacking alpha-synuclein display functional deficits in the nigrostriatal dopamine system, Neuron. 25(2000) 239–252. [DOI] [PubMed] [Google Scholar]
- 20.Chandra S, Fornai F, Kwon H-B, Yazdani U, Atasoy D, Liu X, Hammer RE, Battaglia G, German DC, Castillo PE, Südhof TC, Double-knockout mice for alpha- and beta-synucleins: effect on synaptic functions, Proc. Natl. Acad. Sci. U. S. A 101 (2004) 14966–14971. doi: 10.1073/pnas.0406283101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nemani VM, Lu W, Berge V, Nakamura K, Onoa B, Lee MK, Chaudhry FA, Nicoll RA, Edwards RH, Increased expression of alpha-synuclein reduces neurotransmitter release by inhibiting synaptic vesicle reclustering after endocytosis, Neuron. 65(2010) 66–79. doi: 10.1016/j.neuron.2009.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Scott D, Roy S, α-Synuclein inhibits intersynaptic vesicle mobility and maintains recycling-pool homeostasis, J. Neurosci. Off. J. Soc. Neurosci 32(2012) 10129–10135. doi: 10.1523/JNEUROSCI.0535-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang L, Das U, Scott DA, Tang Y, McLean PJ, Roy S, α-synuclein multimers cluster synaptic vesicles and attenuate recycling, Curr. Biol. CB 24 (2014) 2319–2326. doi: 10.1016/j.cub.2014.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vargas KJ, Makani S, Davis T, Westphal CH, Castillo PE, Chandra SS, Synucleins regulate the kinetics of synaptic vesicle endocytosis, J. Neurosci. Off. J. Soc. Neurosci 34(2014) 9364–9376. doi: 10.1523/JNEUROSCI.4787-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Logan T, Bendor J, Toupin C, Thorn K, Edwards RH, α-Synuclein promotes dilation of the exocytotic fusion pore, Nat. Neurosci 20(2017) 681–689. doi: 10.1038/nn.4529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Davidson WS, Jonas A, Clayton DF, George JM, Stabilization of alpha-synuclein secondary structure upon binding to synthetic membranes, J. Biol. Chem 273 (1998) 9443–9449. [DOI] [PubMed] [Google Scholar]
- 27.Chandra S, Chen X, Rizo J, Jahn R, Südhof TC, A broken alpha-helix in folded alpha-Synuclein, J. Biol. Chem 278 (2003) 15313–15318. doi: 10.1074/jbc.M213128200. [DOI] [PubMed] [Google Scholar]
- 28.Jao CC, Der-Sarkissian A, Chen J, Langen R, Structure of membrane-bound alpha-synuclein studied by site-directed spin labeling, Proc. Natl. Acad. Sci. U. S. A 101 (2004) 8331–8336. doi: 10.1073/pnas.0400553101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jao CC, Hegde BG, Chen J, Haworth IS, Langen R, Structure of membrane-bound alpha-synuclein from site-directed spin labeling and computational refinement, Proc. Natl. Acad. Sci. U. S. A 105 (2008) 19666–19671. doi: 10.1073/pnas.0807826105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kalia LV, Lang AE, Parkinson’s disease, The Lancet. 386 (2015) 896–912. doi: 10.1016/S0140-6736(14)61393-3. [DOI] [PubMed] [Google Scholar]
- 31.Irizarry MC, Kim TW, McNamara M, Tanzi RE, George JM, Clayton DF, Hyman BT, Characterization of the precursor protein of the non-A beta component of senile plaques (NACP) in the human central nervous system, J. Neuropathol. Exp. Neurol 55(1996) 889–895. [DOI] [PubMed] [Google Scholar]
- 32.Nakajo S, Shioda S, Nakai Y, Nakaya K, Localization of phosphoneuroprotein 14(PNP 14) and its mRNA expression in rat brain determined by immunocytochemistry and in situ hybridization, Brain Res. Mol. Brain Res 27 (1994) 81–86. [DOI] [PubMed] [Google Scholar]
- 33.Davidson WS, Jonas A, Clayton DF, George JM, Stabilization of alpha-synuclein secondary structure upon binding to synthetic membranes, J. Biol. Chem 273 (1998) 9443–9449. [DOI] [PubMed] [Google Scholar]
- 34.Jo E, McLaurin J, Yip CM, George-Hyslop PS, Fraser PE, α-Synuclein Membrane Interactions and Lipid Specificity, J. Biol. Chem 275 (2000) 34328–34334. doi: 10.1074/jbc.M004345200. [DOI] [PubMed] [Google Scholar]
- 35.Eliezer D, Kutluay E, Bussell R, Browne G, Conformational properties of alpha-synuclein in its free and lipid-associated states, J. Mol. Biol 307 (2001) 1061–1073. doi: 10.1006/jmbi.2001.4538. [DOI] [PubMed] [Google Scholar]
- 36.Kim YS, Laurine E, Woods W, Lee S-J, A novel mechanism of interaction between alpha-synuclein and biological membranes, J. Mol. Biol 360 (2006) 386–397. doi: 10.1016/j.jmb.2006.05.004. [DOI] [PubMed] [Google Scholar]
- 37.Kahle PJ, Neumann M, Ozmen L, Müller V, Odoy S, Okamoto N, Jacobsen H, Iwatsubo T, Trojanowski JQ, Takahashi H, Wakabayashi K, Bogdanovic N, Riederer P, Kretzschmar HA, Haass C, Selective Insolubility of α-Synuclein in Human Lewy Body Diseases Is Recapitulated in a Transgenic Mouse Model, Am. J. Pathol 159 (2001) 2215–2225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fortin DL, Troyer MD, Nakamura K, Kubo S, Anthony MD, Edwards RH, Lipid rafts mediate the synaptic localization of alpha-synuclein, J. Neurosci. Off. J. Soc. Neurosci 24 (2004) 6715–6723. doi: 10.1523/JNEUROSCI.1594-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fortin DL, Nemani VM, Voglmaier SM, Anthony MD, Ryan TA, Edwards RH, Neural activity controls the synaptic accumulation of alpha-synuclein, J. Neurosci. Off. J. Soc. Neurosci 25 (2005) 10913–10921. doi: 10.1523/JNEUROSCI.2922-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bendor JT, Logan TP, Edwards RH, The function of α-synuclein, Neuron. 79(2013) 1044–1066. doi: 10.1016/j.neuron.2013.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Burré J, The Synaptic Function of α-Synuclein, J. Park. Dis 5 (2015) 699–713. doi: 10.3233/JPD-150642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lou X, Kim J, Hawk BJ, Shin Y-K, α-Synuclein may cross-bridge v-SNARE and acidic phospholipids to facilitate SNARE-dependent vesicle docking, Biochem. J 474 (2017) 2039–2049. doi: 10.1042/BCJ20170200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Almandoz-Gil L, Persson E, Lindström V, Ingelsson M, Erlandsson A, Bergström J, In Situ Proximity Ligation Assay Reveals Co-Localization of Alpha-Synuclein and SNARE Proteins in Murine Primary Neurons, Front. Neurol 9(2018) 180. doi: 10.3389/fneur.2018.00180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liang B, Tamm LK, Solution NMR of SNAREs, complexin and α-synuclein in association with membrane-mimetics, Prog. Nucl. Magn. Reson. Spectrosc 105 (2018) 41–53. doi: 10.1016/j.pnmrs.2018.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhang Z, Jiang X, Xu D, Zheng W, Liu M, Li C, Calcium accelerates SNARE-mediated lipid mixing through modulating α-synuclein membrane interaction, Biochim. Biophys. Acta (2018). doi: 10.1016/j.bbamem.2018.03.025. [DOI] [PubMed] [Google Scholar]
- 46.Fakhree MAA, Zijlstra N, Raiss CC, Siero CJ, Grabmayr H, Bausch AR, Blum C, Claessens MMAE, The numberof α-synuclein proteins per vesicle gives insights into its physiological function, Sci. Rep 6 (2016) 30658. doi: 10.1038/srep30658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang Z, Dai C, Bai J, Xu G, Liu M, Li C, Ca2+ modulating α-synuclein membrane transient interactions revealed by solution NMR spectroscopy, Biochim. Biophys. Acta BBA - Biomembr 1838 (2014) 853–858. doi: 10.1016/j.bbamem.2013.11.016. [DOI] [PubMed] [Google Scholar]
- 48.Viennet T, Wördehoff MM, Uluca B, Poojari C, Shaykhalishahi H, Willbold D, Strodel B, Heise H, Buell AK, Hoyer W, Etzkorn M, Structural insights from lipid-bilayer nanodiscs link α-Synuclein membrane-binding modes to amyloid fibril formation, Commun. Biol 1(2018) 44. doi: 10.1038/s42003-018-0049-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.van Ham TJ, Thijssen KL, Breitling R, Hofstra RMW, Plasterk RHA, Nollen EAA, C. elegans model identifies genetic modifiers of alpha-synuclein inclusion formation during aging, PLoS Genet. 4(2008) e1000027. doi: 10.1371/journal.pgen.1000027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Unni VK, Weissman TA, Rockenstein E, Masliah E, McLean PJ, Hyman BT, In vivo imaging of alpha-synuclein in mouse cortex demonstrates stable expression and differential subcellular compartment mobility, Plos One. 5(2010) e10589. doi: 10.1371/journal.pone.0010589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Spinelli KJ, Taylor JK, Osterberg VR, Churchill MJ, Pollock E, Moore C, Meshul CK, Unni VK, Presynaptic Alpha-Synuclein Aggregation in a Mouse Model of Parkinson’s Disease, J. Neurosci 34 (2014) 2037–2050. doi: 10.1523/JNEUROSCI.2581-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Brucale M, Sandal M, Di Maio S, Rampioni A, Tessari I, Tosatto L, Bisaglia M, Bubacco L, Samori B, Pathogenic mutations shift the equilibria of alpha-synuclein single molecules towards structured conformers, Chembiochem Eur. J. Chem. Biol 10(2009) 176–183. doi: 10.1002/cbic.200800581. [DOI] [PubMed] [Google Scholar]
- 53.Lázaro DF, Rodrigues EF, Langohr R, Shahpasandzadeh FI, Ribeiro T, Guerreiro P, Gerhardt E, Kröhnert K, Klucken J, Pereira MD, Popova B, Kruse N, Mollenhauer B, Rizzoli SO, Braus GH, Danzer KM, Outeiro TF, Systematic Comparison of the Effects of Alpha-synuclein Mutations on Its Oligomerization and Aggregation, PLoS Genet. 10 (2014). doi: 10.1371/journal.pgen.1004741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dettmer U, Ramalingam N, von Saucken VE, Kim T-E, Newman AJ, Terry-Kantor E, Nuber S, Ericsson M, Fanning S, Bartels T, Lindquist S, Levy OA, Selkoe D, Loss of native α-synuclein multimerization by strategically mutating its amphipathic helix causes abnormal vesicle interactions in neuronal cells, Hum. Mol. Genet 26 (2017) 3466–3481. doi: 10.1093/hmg/ddx227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Íñigo-Marco I, Valencia M, Larrea L, Bugallo R, Martínez-Goikoetxea M, Zuriguel I, Arrasate M, E46K α-synuclein pathological mutation causes cell-autonomous toxicity without altering protein turnoveror aggregation, Proc. Natl. Acad. Sci 114 (2017) E8274–E8283. doi: 10.1073/pnas.1703420114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Xu J, Kao S-Y, Lee FJS, Song W, Jin L-W, Yankner BA, Dopamine-dependent neurotoxicity of alpha-synuclein: a mechanism for selective neurodegeneration in Parkinson disease, Nat. Med 8 (2002) 600–606. doi: 10.1038/nm0602-600. [DOI] [PubMed] [Google Scholar]
- 57.Lotharius J, Barg S, Wiekop P, Lundberg C, Raymon HK, Brundin P, Effect of Mutant α-Synuclein on Dopamine Homeostasis in a New Human Mesencephalic Cell Line, J. Biol. Chem 277 (2002) 38884–38894. doi: 10.1074/jbc.M205518200. [DOI] [PubMed] [Google Scholar]
- 58.Xu J, Wu X-S, Sheng J, Zhang Z, Yue H-Y, Sun L, Sgobio C, Lin X, Peng S, Jin Y, Gan L, Cai H, Wu L-G, α-Synuclein Mutation Inhibits Endocytosis at Mammalian Central Nerve Terminals, J. Neurosci 36 (2016) 4408–4414. doi: 10.1523/JNEUROSCI.3627-15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Busch DJ, Oliphint PA, Walsh RB, Banks SML, Woods WS, George JM, Morgan JR, Acute increase of α-synuclein inhibits synaptic vesicle recycling evoked during intense stimulation, Mol. Biol. Cell 25 (2014) 3926–3941. doi: 10.1091/mbc.E14-02-0708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lee H-J, Choi C, Lee S-J, Membrane-bound α-Synuclein Has a High Aggregation Propensity and the Ability to Seed the Aggregation of the Cytosolic Form, J. Biol. Chem 277 (2002) 671–678. doi: 10.1074/jbc.M107045200. [DOI] [PubMed] [Google Scholar]
- 61.Raiss CC, Braun TS, Konings IBM, Grabmayr H, Hassink GC, Sidhu A, le Feber J, Bausch AR, Jansen C, Subramaniam V, Claessens MMAE, Functionally different α-synuclein inclusions yield insight into Parkinson’s disease pathology, Sci. Rep 6(2016) 23116. doi: 10.1038/srep23116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Larsen KE, Schmitz Y, Troyer MD, Mosharov E, Dietrich P, Quazi AZ, Savalle M, Nemani V, Chaudhry FA, Edwards RH, Stefanis L, Sulzer D, α-Synuclein Overexpression in PC12 and Chromaffin Cells Impairs Catecholamine Release by Interfering with a Late Step in Exocytosis, J. Neurosci 26(2006) 11915–11922. doi: 10.1523/JNEUROSCI.3821-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Soper JH, Roy S, Stieber A, Lee E, Wilson RB, Trojanowski JQ, Burd CG, Lee VM-Y, α-Synuclein–induced Aggregation of Cytoplasmic Vesicles in Saccharomyces cerevisiae, Mol. Biol. Cell 19 (2008) 1093–1103. doi: 10.1091/mbc.E07-08-0827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lundblad M, Decressac M, Mattsson B, Björklund A, Impaired neurotransmission caused by overexpression of α-synuclein in nigral dopamine neurons, Proc. Natl. Acad. Sci 109 (2012) 3213–3219. doi: 10.1073/pnas.1200575109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lautenschläger J, Stephens AD, Fusco G, Ströhl F, Curry N, Zacharopoulou M, Michel CH, Laine R, Nespovitaya N, Fantham M, Pinotsi D, Zago W, Fraser P, Tandon A, George-Hyslop PS, Rees E, Phillips JJ, Simone AD, Kaminski CF, Schierle GSK, C-terminal calcium binding of α-synuclein modulates synaptic vesicle interaction, Nat. Commun 9 (2018) 712. doi: 10.1038/s41467-018-03111-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wang W, Perovic I, Chittuluru J, Kaganovich A, Nguyen LTT, Liao J, Auclair JR, Johnson D, Landeru A, Simorellis AK, Ju S, Cookson MR, Asturias FJ, Agar JN, Webb BN, Kang C, Ringe D, Petsko GA, Pochapsky TC, Hoang QQ, A soluble α-synuclein construct forms a dynamic tetramer, Proc. Natl. Acad. Sci 108 (2011) 17797–17802. doi: 10.1073/pnas.H13260108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bartels T Choi JG, Selkoe DJ, α-Synuclein occurs physiologically as a helically folded tetramer that resists aggregation, Nature. 477 (2011) 107–110. doi: 10.1038/nature10324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Dettmer U, Newman AJ, Luth ES, Bartels T, Selkoe D, In Vivo Cross-linking Reveals Principally Oligomeric Forms of α-Synuclein and β-Synuclein in Neurons and Non-neural Cells, J. Biol. Chem 288 (2013) 6371–6385. doi: 10.1074/jbc.M112.403311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gurry T, Ullman O, Fisher CK, Perovic I, Pochapsky T, Stultz CM, The Dynamic Structure of α-Synuclein Multimers, J. Am. Chem. Soc 135 (2013) 3865–3872. doi: 10.1021/ja310518p. [DOI] [PubMed] [Google Scholar]
- 70.Killinger BA, Moszczynska A, Characterization of α-Synuclein Multimer Stoichiometry in Complex Biological Samples by Electrophoresis, Anal. Chem 88(2016) 4071–4084. doi: 10.1021/acs.analchem.6b00419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Luth ES, Bartels T, Dettmer U, Kim NC, Selkoe DJ, Purification of α-Synuclein from Human Brain Reveals an Instability of Endogenous Multimers as the Protein Approaches Purity, Biochemistry(Mosc.).54(2015) 279–292. doi: 10.1021/bi501188a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Binolfi A, Theillet F-X, Selenko P, Bacterial in-cell NMR of human α-synuclein: a disordered monomer by nature?, Biochem. Soc. Trans 40 (2012) 950–954. doi: 10.1042/BST20120096. [DOI] [PubMed] [Google Scholar]
- 73.Fauvet B, Mbefo MK, Fares M-B, Desobry C, Michael S, Ardah MT, Tsika E, Coune P, Prudent M, Lion N, Eliezer D, Moore DJ, Schneider B, Aebischer P, El-Agnaf OM, Masliah E, Lashuel HA, α-Synuclein in central nervous system and from erythrocytes, mammalian cells, and Escherichia coli exists predominantly as disordered monomer, J. Biol. Chem 287 (2012) 15345–15364. doi: 10.1074/jbc.M111.318949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Burré J, Vivona S, Diao J, Sharma M, Brunger AT, Südhof TC, Properties of native brain α-synuclein, Nature. 498(2013) E4–E6. doi: 10.1038/nature12125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Theillet F-X, Binolfi A, Bekei B, Martorana A, Rose HM, Stuiver M, Verzini S, Lorenz D, van Rossum M, Goldfarb D, Selenko P, Structural disorder of monomeric α-synuclein persists in mammalian cells, Nature. 530(2016) 45–50. doi: 10.1038/nature16531. [DOI] [PubMed] [Google Scholar]
- 76.Dettmer U, Selkoe D, Bartels T, New insights into cellular α-synuclein homeostasis in health and disease, Curr. Opin. Neurobiol 36(2016) 15–22. doi: 10.1016/j.conb.2015.07.007. [DOI] [PubMed] [Google Scholar]
- 77.Westphal CH, Chandra SS, Monomeric Synucleins Generate Membrane Curvature, J. Biol. Chem 288 ( 2013) 1829–1840. doi: 10.1074/jbc.M112.418871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Gould N, Mor DE, Lightfoot R, Malkus K, Giasson B, Ischiropoulos H, Evidence of Native α-Synuclein Conformers in the Human Brain, J. Biol. Chem 289 (2014) 7929–7934. doi: 10.1074/jbc.C113.538249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Dettmer U, Newman AJ, Soldner F, Luth ES, Kim NC, von Saucken VE, Sanderson JB, Jaenisch R, Bartels T, Selkoe D, Parkinson-causing α-synuclein missense mutations shift native tetramers to monomers as a mechanism for disease initiation, Nat. Commun 6(2015). doi: 10.1038/ncomms8314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Dettmer U, Newman AJ, von Saucken VE, Bartels T, Selkoe D, KTKEGV repeat motifs are key mediators of normal α-synuclein tetramerization: Their mutation causes excess monomers and neurotoxicity, Proc. Natl. Acad. Sci. U. S. A 112 (2015) 9596–9601. doi: 10.1073/pnas.1505953112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kim T, Mehta SL, Kaimal B, Lyons K, Dempsey RJ, Vemuganti R, Poststroke Induction of α-Synuclein Mediates Ischemic Brain Damage, J. Neurosci 36(2016) 7055–7065. doi: 10.1523/JNEUROSCI.1241-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Nuber S, Rajsombath M, Minakaki G, Winkler J, Müller CP, Ericsson M, Caldarone B, Dettmer U, Selkoe DJ, Abrogating Native α-Synuclein Tetramers in Mice Causes a L-DOPA-Responsive Motor Syndrome Closely Resembling Parkinson’s Disease, Neuron. 100(2018) 75–90.e5. doi: 10.1016/j.neuron.2018.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kothawala A, Kilpatrick K, Novoa JA, Segatori L, Quantitative Analysis of α-Synuclein Solubility in Living Cells Using Split GFP Complementation, PLOS ONE. 7 (2012) e43505. doi: 10.1371/journal.pone.0043505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Dimant H, Kalia SK, Kalia LV, Zhu LN, Kibuuka L, Ebrahimi-Fakhari D, McFarland NR, Fan Z, Hyman BT, McLean PJ, Direct detection of alpha synuclein oligomers in vivo, Acta Neuropathol. Commun 1(2013) 6. doi: 10.1186/2051-5960-1-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Moussaud S, Malany S, Mehta A, Vasile S, Smith LH, McLean PJ, Targeting alpha-synuclein oligomers by protein-fragment complementation for drug discovery in synucleinopathies, Expert Opin. Ther. Targets 19 (2015) 589–603. doi: 10.1517/14728222.2015.1009448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Cai W, Feng D, Schwarzschild MA, McLean PJ, Chen X, Bimolecular Fluorescence Complementation of Alpha-synuclein Demonstrates its Oligomerization with Dopaminergic Phenotype in Mice, EBioMedicine. 29(2018) 13–22. doi: 10.1016/j.ebiom.2018.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Alderson TR, Bax A, Parkinson’s disease: Disorder in the court, Nature. (2016). doi: 10.1038/nature16871. [DOI] [PubMed] [Google Scholar]
- 88.Dettmer U, Newman AJ, Luth ES, Bartels T, Selkoe D, In vivo cross-linking reveals principally oligomeric forms of α-synuclein and β-synuclein in neurons and non-neural cells, J. Biol. Chem 288 (2013) 6371–6385. doi: 10.1074/jbc.M112.403311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Rovere M, Sanderson JB, Fonseca-Ornelas L, Patel DS, Bartels T, Refolding of helical soluble α-synuclein through transient interaction with lipid interfaces, FEBS Lett. 592 (2018) 1464–1472. doi: 10.1002/1873-3468.13047. [DOI] [PubMed] [Google Scholar]
- 90.Trexler AJ, Rhoades E, N-Terminal acetylation is critical for forming α-helical oligomer of α-synuclein, Protein Sci. Publ. Protein Soc 21(2012) 601–605. doi: 10.1002/pro.2056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Gurry T, Ullman O, Fisher CK, Perovic I, Pochapsky T, Stultz CM, The dynamic structure of α-synuclein multimers, J. Am. Chem. Soc 135 (2013) 3865–3872. doi: 10.1021/ja310518p. [DOI] [PubMed] [Google Scholar]
- 92.Iljina M, Tosatto L, Choi ML, Sang JC, Ye Y, Hughes CD, Bryant CE, Gandhi S, Klenerman D, Arachidonic acid mediates the formation of abundant alpha-helical multimers of alpha-synuclein, Sci. Rep 6 (2016) 33928. doi: 10.1038/srep33928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kim S, Yun SP, Lee S, Umanah GE, Bandaru VVR, Yin X, Rhee P, Karuppagounder SS, Kwon S-H, Lee H, Mao X, Kim D, Pandey A, Lee G, Dawson VL, Dawson TM, Ko HS, GBA1 deficiency negatively affects physiological α-synuclein tetramers and related multimers, Proc. Natl. Acad. Sci. U. S. A 115 (2018) 798–803. doi: 10.1073/pnas.1700465115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Burré J, Sharma M, Südhof TC, α-Synuclein assembles into higher-order multimers upon membrane binding to promote SNARE complex formation, Proc. Natl. Acad. Sci 111 (2014) E4274–E4283. doi: 10.1073/pnas.1416598111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Burré J, Sharma M, Südhof TC, Definition of a molecular pathway mediating α-synuclein neurotoxicity, J. Neurosci. Off. J. Soc. Neurosci 35(2015) 5221–5232. doi: 10.1523/JNEUROSCI.4650-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Galvagnion C, Buell AK, Meisl G, Michaels TCT, Vendruscolo M, Knowles TPJ, Dobson CM, Lipid vesicles trigger α-synuclein aggregation by stimulating primary nucleation, Nat. Chem. Biol 11 (2015) 229–234. doi: 10.1038/nchembio.1750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Dettmer U, Ramalingam N, von Saucken VE, Kim T-E, Newman AJ, Terry-Kantor E, Nuber S, Ericsson M, Fanning S, Bartels T, Lindquist S, Levy OA, Selkoe D, Loss of native α-synuclein multimerization by strategically mutating its amphipathic helix causes abnormal vesicle interactions in neuronal cells, Hum. Mol. Genet 26 (2017) 3466–3481. doi: 10.1093/hmg/ddx227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Shahmoradian SH, Lewis AJ, Genoud C, Hench J, Moors TE, Navarro PP, Castaño-Díez D, Schweighauser G, Graff-Meyer A, Goldie KN, Sütterlin R, Huisman E, Ingrassia A, de Gier Y, Rozemuller AJM, Wang J, Paepe AD, Erny J, Staempfli A, Hoernschemeyer J, Großerüschkamp F, Niedieker D, El-Mashtoly SF, Quadri M, Van IJcken WFJ, Bonifati V, Gerwert K, Bohrmann B, Frank S, Britschgi M, Stahlberg H, Van de Berg WDJ, Lauer ME, Lewy pathology in Parkinson’s disease consists of crowded organelles and lipid membranes, Nat. Neurosci 22(2019) 1099–1109. doi: 10.1038/S41593-019-0423-2. [DOI] [PubMed] [Google Scholar]
- 99.Fusco G, Simone AD, Gopinath T, Vostrikov V, Vendruscolo M, Dobson CM, Veglia G, Direct observation of the three regions in α-synuclein that determine its membrane-bound behaviour, Nat. Commun 5 (2014) 3827. doi: 10.1038/ncomms4827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Lokappa SB, Suk J-E, Balasubramanian A, Samanta S, Situ AJ, Ulmer TS, Sequence and membrane determinants of the random coil-helix transition of α-synuclein, J. Mol. Biol 426 (2014) 2130–2144. doi: 10.1016/j.jmb.2014.02.024. [DOI] [PubMed] [Google Scholar]
- 101.Lee JH, Ying J, Bax A, Nuclear Magnetic Resonance Observation of α-Synuclein Membrane Interaction by Monitoring the Acetylation Reactivity of Its Lysine Side Chains, Biochemistry (Mosc.).55 (2016) 4949–4959. doi: 10.1021/acs.biochem.6b00637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Pfefferkorn CM, Jiang Z, Lee JC, Biophysics of α-Synuclein Membrane Interactions, Biochim. Biophys. Acta 1818 (2012) 162–171. doi: 10.1016/j.bbamem.2011.07.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Zhang Z, Dai C, Bai J, Xu G, Liu M, Li C, Ca(2+) modulating α-synuclein membrane transient interactions revealed by solution NMR spectroscopy, Biochim. Biophys. Acta 1838 (2014) 853–858. doi: 10.1016/j.bbamem.2013.11.016. [DOI] [PubMed] [Google Scholar]
- 104.Galvagnion C, The Role of Lipids Interacting with α-Synuclein in the Pathogenesis of Parkinson’s Disease, J. Park. Dis 7 (2017) 433–450. doi: 10.3233/JPD-171103. [DOI] [PubMed] [Google Scholar]
- 105.Fecchio C, Palazzi L, Polverino de Laureto P, Fecchio C, Palazzi L, Polverino de Laureto P, α-Synuclein and Polyunsaturated Fatty Acids: Molecular Basis of the Interaction and Implication in Neurodegeneration, Molecules. 23(2018) 1531. doi: 10.3390/molecules23071531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Fusco G, Sanz-Hernandez M, De Simone A, Order and disorder in the physiological membrane binding of α-synuclein, Curr. Opin. Struct. Biol 48(2018) 49–57. doi: 10.1016/j.sbi.2017.09.004. [DOI] [PubMed] [Google Scholar]
- 107.Pirc K, Ulrih NP, α-Synuclein interactions with phospholipid model membranes: Key roles for electrostatic interactions and lipid-bilayer structure, Biochim. Biophys. Acta BBA - Biomembr 1848 (2015) 2002–2012. doi: 10.1016/j.bbamem.2015.06.021. [DOI] [PubMed] [Google Scholar]
- 108.Fusco G, De Simone A, Arosio P, Vendruscolo M, Veglia G, Dobson CM, Structural Ensembles of Membrane-bound α-Synuclein Reveal the Molecular Determinants of Synaptic Vesicle Affinity, Sci. Rep 6 (2016). doi: 10.1038/srep27125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Alderson TR, Markley JL, Biophysical characterization of α-synuclein and its controversial structure, Intrinsically Disord. Proteins. 1(2013) e26255. doi: 10.4161/idp.26255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Jao CC, Hegde BG, Chen J, Haworth IS, Langen R, Structure of membrane-bound alpha-synuclein from site-directed spin labeling and computational refinement, Proc. Natl. Acad. Sci. U. S. A 105 (2008) 19666–19671. doi: 10.1073/pnas.0807826105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Wietek J, Haralampiev I, Amoussouvi A, Herrmann A, Stöckl M, Membrane bound α-synuclein is fully embedded in the lipid bilayer while segments with higher flexibility remain, FEBS Lett. 587 (2013) 2572–2577. doi: 10.1016/j.febslet.2013.06.034. [DOI] [PubMed] [Google Scholar]
- 112.Tsigelny IF, Sharikov Y, Wrasidlo W, Gonzalez T, Desplats PA, Crews L, Spencer B, Masliah E, Role of α-synuclein penetration into the membrane in the mechanisms of oligomer pore formation, FEBS J. 279 (2012) 1000–1013. doi: 10.1111/j.1742-4658.2012.08489.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.West A, Brummel BE, Braun AR, Rhoades E, Sachs JN, Membrane remodeling and mechanics: Experiments and simulations of α-Synuclein, Biochim. Biophys. Acta BBA - Biomembr 1858 (2016) 1594–1609. doi: 10.1016/j.bbamem.2016.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Cheng C-Y, Varkey J, Ambroso MR, Langen R, Han S, Hydration dynamics as an intrinsic ruler for refining protein structure at lipid membrane interfaces, Proc. Natl. Acad. Sci. U. S. A 110 (2013) 16838–16843. doi: 10.1073/pnas.1307678110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Nuscher B, Kamp F, Mehnert T, Odoy S, Haass C, Kahle PJ, Beyer K, Alpha-synuclein has a high affinity for packing defects in a bilayer membrane: a thermodynamics study, J. Biol. Chem 279 (2004) 21966–21975. doi: 10.1074/jbc.M401076200. [DOI] [PubMed] [Google Scholar]
- 116.Ouberai MM, Wang J, Swann MJ, Galvagnion C, Guilliams T, Dobson CM, Welland ME, α-Synuclein senses lipid packing defects and induces lateral expansion of lipids leading to membrane remodeling, J. Biol. Chem 288 (2013) 20883–20895. doi: 10.1074/jbc.M113.478297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Pranke IM, Morello V, Bigay J, Gibson K, Verbavatz J-M, Antonny B, Jackson CL, α-Synuclein and ALPS motifs are membrane curvature sensors whose contrasting chemistry mediates selective vesicle binding, J. Cell Biol 194 (2011) 89–103. doi: 10.1083/jcb.201011118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Harada R, Kobayashi N, Kim J, Nakamura C, Han S-W, Ikebukuro K, Sode K, The effect of amino acid substitution in the imperfect repeat sequences of α-synuclein on fibrillation, Biochim. Biophys. Acta BBA - Mol. Basis Dis 1792 (2009) 998–1003. doi: 10.1016/j.bbadis.2009.06.010. [DOI] [PubMed] [Google Scholar]
- 119.Porcari R, Proukakis C, Waudby CA, Bolognesi B, Mangione PP, Paton JFS, Mullin S, Cabrita LD, Penco A, Relini A, Verona G, Vendruscolo M, Stoppini M, Tartaglia GG, Camilloni C, Christodoulou J, Schapira AHV, Bellotti V, The H50Q Mutation Induces a 10-fold Decrease in the Solubility of α-Synuclein, J. Biol. Chem 290 (2015) 2395–2404. doi: 10.1074/jbc.M114.610527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Ysselstein D, Joshi M, Mishra V, Griggs AM, Asiago JM, McCabe GP, Stanciu LA, Post CB, Rochet JC, Effects of impaired membrane interactions on α-synuclein aggregation and neurotoxicity, Neurobiol. Dis 79(2015) 150–163. doi: 10.1016/j.nbd.2015.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Dettmer U, Rationally Designed Variants of α-Synuclein Illuminate Its in vivo Structural Properties in Health and Disease, Front. Neurosci 12(2018) 623. doi: 10.3389/fnins.2018.00623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Fujiwara H, Hasegawa M, Dohmae N, Kawashima A, Masliah E, Goldberg MS, Shen J, Takio K, Iwatsubo T, α-synuclein is phosphorylated in synucleinopathy lesions, Nat. Cell Biol (2002). doi: 10.1038/ncb748. [DOI] [PubMed] [Google Scholar]
- 123.Wang Y, Shi M, Chung KA, Zabetian CP, Leverenz JB, Berg D, Srulijes K, Trojanowski JQ, Lee VM-Y, Siderowf AD, Hurtig H, Litvan I, Schiess MC, Peskind ER, Masuda M, Hasegawa M, Lin X, Pan C, Galasko D, Goldstein DS, Jensen PH, Yang H, Cain KC, Zhang J, Phosphorylated α-synuclein in Parkinson’s disease, Sci. Transl. Med 4 (2012) 121ra20. doi: 10.1126/scitranslmed.3002566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Machiya Y, Hara S, Arawaka S, Fukushima S, Sato H, Sakamoto M, Koyama S, Kato T, Phosphorylated α-Synuclein at Ser-129 is targeted to the proteasome pathway in a ubiquitin-independent manner, J. Biol. Chem 285 (2010) 40732–40744. doi: 10.1074/jbc.M110.141952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Oueslati A, Implication of Alpha-Synuclein Phosphorylation at S129 in Synucleinopathies: What Have We Learned in the Last Decade?, J. Park. Dis 6 (2016) 39–51. doi: 10.3233/JPD-160779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Oueslati A, Schneider BL, Aebischer P, Lashuel HA, Polo-like kinase 2 regulates selective autophagic-synuclein clearance and suppresses its toxicity in vivo, Proc. Natl. Acad. Sci 110 (2013) E3945–54. doi: 10.1073/pnas.1309991110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Tenreiro S, Reimão-Pinto MM, Antas P, Rino J, Wawrzycka D, Macedo D, Rosado-Ramos R, Amen T, Waiss M, Magalhães F, Gomes A, Santos CN, Kaganovich D, Outeiro TF, Phosphorylation Modulates Clearance of Alpha-Synuclein Inclusions in a Yeast Model of Parkinson’s Disease, PLoS Genet. 10(2014). doi: 10.1371/journal.pgen.1004302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Pinho R, Paiva I, Jerčić KG, Fonseca-Ornelas L, Gerhardt E, Fahlbusch C, Garcia-Esparcia P, Kerimoglu C, Pavlou MAS, Villar-Piqué A, Szego É, Lopes Da Fonseca T, Odoardi F, Soeroes S, Rego AC, Fischle W, Schwamborn JC, Meyer T, Kügler S, Ferrer I, Attems J, Fischer A, Becker S, Zweckstetter M, Borovecki F, Outeiro TF, Nuclear localization and phosphorylation modulate pathological effects of alpha-synuclein, Hum. Mol. Genet (2019). doi: 10.1093/hmg/ddy326. [DOI] [PubMed] [Google Scholar]
- 129.Grundke-Iqbal I, Iqbal K, Quinlan M, Tung YC, Zaidi MS, Wisniewski HM, Microtubule-associated protein tau. A component of Alzheimer paired helical filaments, J Biol Chem. 261(1986) 6084–9. [PubMed] [Google Scholar]
- 130.Kosik KS, Joachim CL, Selkoe DJ, Microtubule-associated protein tau (tau) is a major antigenic component of paired helical filaments in Alzheimer disease, Proc. Natl. Acad. Sci. U. S. A 83 (1986) 4044–4048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Wood JG, Mirra SS, Pollock NJ, Binder LI, Neurofibrillary tangles of Alzheimer disease share antigenic determinants with the axonal microtubule-associated protein tau (tau), Proc. Natl. Acad. Sci. U. S. A 83 (1986) 4040–4043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Wolozin BL, Pruchnicki A, Dickson DW, Davies P, A neuronal antigen in the brains of Alzheimer patients, Science. 232 (1986) 648–650. [DOI] [PubMed] [Google Scholar]
- 133.Hardy J, Selkoe DJ, The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics, Science. 297(2002) 353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- 134.Braak H, Braak E, Evolution of the neuropathology of Alzheimer’s disease, Acta Neurol Scand Suppl. 165 (1996) 3–12. [DOI] [PubMed] [Google Scholar]
- 135.Nelson PT, Alafuzoff I, Bigio EH, Bouras C, Braak H, Cairns NJ, Castellani RJ, Crain BJ, Davies P, Del Tredici K, Duyckaerts C, Frosch MP, Haroutunian V, Hof PR, Hulette CM, Hyman BT, Iwatsubo T, Jellinger KA, Jicha GA, Kovari E, Kukull WA, Leverenz JB, Love S, Mackenzie IR, Mann DM, Masliah E, McKee AC, Montine TJ, Morris JC, Schneider JA, Sonnen JA, Thal DR, Trojanowski JQ, Troncoso JC, Wisniewski T, Woltjer RL, Beach TG, Correlation of Alzheimer disease neuropathologic changes with cognitive status: a review of the literature, J Neuropathol Exp Neurol. 71 (2012) 362–81. doi: 10.1097/NEN.0b013e31825018f7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Golde TE, Open questions for Alzheimer’s disease immunotherapy, Alzheimers Res Ther. 6(2014) 3. doi: 10.1186/alzrt233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Weingarten MD, Lockwood AH, Hwo SY, Kirschner MW, A protein factor essential for microtubule assembly, Proc Natl Acad Sci U A. 72 (1975) 1858–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Lee VM, Goedert M, Trojanowski JQ, Neurodegenerative tauopathies, Annu Rev Neurosci. 24 (2001) 1121–59. doi: 10.1146/annurev.neuro.24.1.1121. [DOI] [PubMed] [Google Scholar]
- 139.Goedert M, Spillantini MG, Jakes R, Rutherford D, Crowther RA, Multiple isoforms of human microtubule-associated proteintau: sequences and localization in neurofibrillary tangles of Alzheimer’s disease, Neuron. 3(1989) 519–26. [DOI] [PubMed] [Google Scholar]
- 140.Kosik KS, Orecchio LD, Bakalis S, Neve RL, Developmentally regulated expression of specific tau sequences, Neuron. 2(1989) 1389–97. [DOI] [PubMed] [Google Scholar]
- 141.Rouzier R, Rajan R, Wagner P, Hess KR, Gold DL, Stec J, Ayers M, Ross JS, Zhang P, Buchholz TA, Kuerer H, Green M, Arun B, Hortobagyi GN, Symmans WF, Pusztai L, Microtubule-associated protein tau: a marker of paclitaxel sensitivity in breast cancer, Proc Natl Acad Sci U A. 102 (2005) 8315–20. doi: 10.1073/pnas.0408974102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Kenner L, el-Shabrawi Y, Hutter H, Forstner M, Zatloukal K, Hoefler G, Preisegger KH, Kurzbauer R, Denk H, Expression of three- and four-repeat tau isoforms in mouse liver, Hepatology. 20 (1994) 1086–9. [DOI] [PubMed] [Google Scholar]
- 143.Goedert M, Spillantini MG, Crowther RA, Tau proteins and neurofibrillary degeneration, Brain Pathol. Zurich Switz 1(1991) 279–286. [DOI] [PubMed] [Google Scholar]
- 144.Lee VM, Trojanowski JQ, The disordered neuronal cytoskeleton in Alzheimer’s disease, Curr. Opin. Neurobiol 2(1992) 653–656. [DOI] [PubMed] [Google Scholar]
- 145.Viswanadha R, Sale WS, Porter ME, Ciliary Motility: Regulation of Axonemal Dynein Motors, Cold Spring Harb Perspect Biol. 9 (2017). doi: 10.1101/cshperspect.a018325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Barlan K, Gelfand VI, Microtubule-Based Transport and the Distribution, Tethering, and Organization of Organelles, Cold Spring Harb Perspect Biol. 9(2017). doi: 10.1101/cshperspect.a025817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Mitchison T, Kirschner M, Dynamic instability of microtubule growth, Nature. 312 (1984) 237–42. [DOI] [PubMed] [Google Scholar]
- 148.Akhmanova A, Steinmetz MO, Control of microtubule organization and dynamics: two ends in the limelight, Nat Rev Mol Cell Biol. 16 (2015) 711–26. doi: 10.1038/nrm4084. [DOI] [PubMed] [Google Scholar]
- 149.Akhmanova A, Steinmetz MO, Tracking the ends: a dynamic protein network controls the fate of microtubule tips, Nat Rev Mol Cell Biol. 9(2008) 309–22. doi: 10.1038/nrm2369. [DOI] [PubMed] [Google Scholar]
- 150.Hirokawa N, Takemura R, Molecular motors and mechanisms of directional transport in neurons, Nat. Rev. Neurosci 6 (2005) 201–214. doi: 10.1038/nrnl624. [DOI] [PubMed] [Google Scholar]
- 151.Choi MC, Raviv U, Miller HP, Gaylord MR, Kiris E, Ventimiglia D, Needleman DJ, Kim MW, Wilson L, Feinstein SC, Safinya CR, Human microtubule-associated-protein tau regulates the number of protofilaments in microtubules: a synchrotron x-ray scattering study, Biophys J. 97 (2009) 519–27. doi: 10.1016/j.bpj.2009.04.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Butner KA, Kirschner MW, Tau protein binds to microtubules through a flexible array of distributed weak sites, J Cell Biol. 115 (1991) 717–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Santarella RA, Skiniotis G, Goldie KN, Tittmann P, Gross H, Mandelkow EM, Mandelkow E, Hoenger A, Surface-decoration of microtubules by human tau, J Mol Biol. 339 (2004) 539–53. doi: 10.1016/j.jmb.2004.04.008. [DOI] [PubMed] [Google Scholar]
- 154.Al-Bassam J, Ozer RS, Safer D, Halpain S, Milligan RA, MAP2 and tau bind longitudinally along the outer ridges of microtubule protofilaments, J Cell Biol. 157 (2002) 1187–96. doi: 10.1083/jcb.200201048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Kar S, Fan J, Smith MJ, Goedert M, Amos LA, Repeat motifs of tau bind to the insides of microtubules in the absence of taxol, EMBO J. 22 (2003) 70–7. doi: 10.1093/emboj/cdg001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Kadavath H, Hofele RV, Biernat J, Kumar S, Tepper K, Urlaub H, Mandelkow E, Zweckstetter M, Tau stabilizes microtubules by binding at the interface between tubulin heterodimers, Proc Natl Acad Sci U A. 112 (2015) 7501–6. doi: 10.1073/pnas.1504081112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Qiang L, Sun X, Austin TO, Muralidharan H, Jean DC, Liu M, Yu W, Baas PW, Tau Does Not Stabilize Axonal Microtubules but Rather Enables Them to Have Long Labile Domains, Curr. Biol. CB 28(2018) 2181–2189.e4. doi: 10.1016/j.cub.2018.05.045. [DOI] [PubMed] [Google Scholar]
- 158.Baas PW, Qiang L, Tau: It’s Not What You Think, Trends Cell Biol. (2019). doi: 10.1016/j.tcb.2019.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Ballatore C, Lee VM, Trojanowski JQ, Tau-mediated neurodegeneration in Alzheimer’s disease and related disorders, Nat Rev Neurosci. 8(2007) 663–72. doi: 10.1038/nrn2194. [DOI] [PubMed] [Google Scholar]
- 160.Biernat J, Gustke N, Drewes G, Mandelkow EM, Mandelkow E, Phosphorylation of Ser262 strongly reduces binding of tau to microtubules: distinction between PHF-like immunoreactivity and microtubule binding, Neuron. 11 (1993) 153–63. [DOI] [PubMed] [Google Scholar]
- 161.Scott CW, Spreen RC, Herman JL, Chow FP, Davison MD, Young J, Caputo CB, Phosphorylation of recombinant tau by cAMP-dependent protein kinase. Identification of phosphorylation sites and effect on microtubule assembly, J. Biol. Chem 268(1993) 1166–1173. [PubMed] [Google Scholar]
- 162.Jho YS, Zhulina EB, Kim MW, Pincus PA, Monte carlo simulations of tau proteins: effect of phosphorylation, Biophys J. 99 (2010) 2387–97. doi: 10.1016/j.bpj.2010.06.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Fischer D, Mukrasch MD, Biernat J, Bibow S, Blackledge M, Griesinger C, Mandelkow E, Zweckstetter M, Conformational changes specific for pseudophosphorylation at serine 262 selectively impair binding of tau to microtubules, Biochemistry (Mosc.). 48 (2009) 10047–55. doi: 10.1021/bi901090m. [DOI] [PubMed] [Google Scholar]
- 164.Trinczek B, Biernat J, Baumann K, Mandelkow EM, Mandelkow E, Domains of tau protein, differential phosphorylation, and dynamic instability of microtubules, Mol Biol Cell. 6(1995) 1887–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Sun Q, Gamblin TC, Pseudohyperphosphorylation causing AD-like changes in tau has significant effects on its polymerization, Biochemistry (Mosc.). 48(2009) 6002–11. doi: 10.1021/bi900602h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Leger J, Kempf M, Lee G, Brandt R, Conversion of serine to aspartate imitates phosphorylation-induced changes in the structure and function of microtubule-associated protein tau, J Biol Chem. 272 (1997) 8441–6. [DOI] [PubMed] [Google Scholar]
- 167.Janning D, Igaev M, Sundermann F, Bruhmann J, Beutel O, Heinisch JJ, Bakota L, Piehler J, Junge W, Brandt R, Single-molecule tracking of tau reveals fast kiss-and-hop interaction with microtubules in living neurons, Mol Biol Cell. 25(2014) 3541–51. doi: 10.1091/mbc.E14-06-1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Konzack S, Thies E, Marx A, Mandelkow EM, Mandelkow E, Swimming against the tide: mobility of the microtubule-associated protein tau in neurons, J Neurosci. 27 (2007) 9916–27. doi: 10.1523/JNEUROSCI.0927-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Newton AC, Protein kinase C: structure, function, and regulation, J Biol Chem. 270 (1995) 28495–8. [DOI] [PubMed] [Google Scholar]
- 170.Jacobs JR, Stevens JK, Changes in the organization of the neuritic cytoskeleton during nerve growth factor-activated differentiation of PC12 cells: a serial electron microscopic study of the development and control of neurite shape, J Cell Biol. 103 (1986) 895–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Narayanan RL, Durr UH, Bibow S, Biernat J, Mandelkow E, Zweckstetter M, Automatic assignment of the intrinsically disordered protein Tau with 441-residues, J Am Chem Soc. 132 (2010) 11906–7. doi: 10.1021/ja105657f. [DOI] [PubMed] [Google Scholar]
- 172.Rosenberg KJ, Ross JL, Feinstein HE, Feinstein SC, Israelachvili J. Complementary dimerization of microtubule-associated tau protein: Implications for microtubule bundling and tau-mediated pathogenesis, Proc. Natl. Acad. Sci. U. S. A 105 ( 2008) 7445–7450. doi: 10.1073/pnas.0802036105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Mandelkow EM, Schweers O, Drewes G, Biernat J, Gustke N, Trinczek B, Mandelkow E, Structure, microtubule interactions, and phosphorylation of tau protein, Ann N Acad Sci. 777 (1996) 96–106. [DOI] [PubMed] [Google Scholar]
- 174.Goedert M, Jakes R, Expression of separate isoforms of human tau protein: correlation with the tau pattern in brain and effects on tubulin polymerization, EMBO J. 9(1990) 4225–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Himmler A, Drechsel D, Kirschner MW, Martin DW, Tau consists of a set of proteins with repeated C-terminal microtubule-binding domains and variable N-terminal domains, Mol. Cell. Biol 9 (1989) 1381–1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Lee G, Neve RL, Kosik KS, The microtubule binding domain of tau protein, Neuron. 2(1989) 1615–24. [DOI] [PubMed] [Google Scholar]
- 177.Kadavath H, Jaremko M, Jaremko t., Biernat J, Mandelkow E, Zweckstetter M, Folding of the Tau Protein on Microtubules, Angew. Chem. Int. Ed Engl 54 (2015) 10347–10351. doi: 10.1002/anie.201501714. [DOI] [PubMed] [Google Scholar]
- 178.Kellogg EH, Hejab NMA, Poepsel S, Downing KH, DiMaio F, Nogales E, Near-atomic model of microtubule-tau interactions, Science. 360 (2018) 1242–1246. doi: 10.1126/science.aat1780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Lewis SA, Ivanov IE, Lee GH, Cowan NJ, Organization of microtubules in dendrites and axons is determined by a short hydrophobic zipper in microtubule-associated proteins MAP2 and tau, Nature. 342 (1989) 498–505. doi: 10.1038/342498a0. [DOI] [PubMed] [Google Scholar]
- 180.Kanai Y, Chen J, Hirokawa N, Microtubule bundling by tau proteins in vivo: analysis of functional domains, EMBO J. 11 (1992) 3953–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Gustke N, Trinczek B, Biernat J, Mandelkow EM, Mandelkow E, Domains of tau protein and interactions with microtubules, Biochemistry (Mosc.). 33(1994) 9511–22. [DOI] [PubMed] [Google Scholar]
- 182.Steinhilb ML, Dias-Santagata D, Fulga TA, Felch DL, Feany MB, Tau phosphorylation sites work in concert to promote neurotoxicity in vivo, Mol Biol Cell. 18(2007) 5060–8. doi: 10.1091/mbc.e07-04-0327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Gong CX, Liu F, Grundke-Iqbal I, Iqbal K, Post-translational modifications of tau protein in Alzheimer’s disease, J Neural Transm Vienna. 112(2005) 813–38. doi: 10.1007/s00702-004-0221-0. [DOI] [PubMed] [Google Scholar]
- 184.Preuss U, Biernat J, Mandelkow EM, Mandelkow E, The “jaws” model of tau-microtubule interaction examined in CHO cells, J Cell Sci. 110 ( Pt 6) (1997) 789–800. [DOI] [PubMed] [Google Scholar]
- 185.Goode BL, Denis PE, Panda D, Radeke MJ, Miller HP, Wilson L, Feinstein SC, Functional interactions between the proline-rich and repeat regions of tau enhance microtubule binding and assembly, Mol Biol Cell. 8(1997) 353–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Mukrasch MD, von Bergen M, Biernat J, Fischer D, Griesinger C, Mandelkow E, Zweckstetter M, The “jaws” of the tau-microtubule interaction, J. Biol. Chem 282(2007) 12230–12239. doi: 10.1074/jbe.M607159200. [DOI] [PubMed] [Google Scholar]
- 187.Joshi P, Vendruscolo M, Druggability of Intrinsically Disordered Proteins, Adv. Exp. Med. Biol 870 (2015) 383–400. doi: 10.1007/978-3-319-20164-1_13. [DOI] [PubMed] [Google Scholar]
- 188.Collier TJ, Srivastava KR, Justman C, Grammatopoulous T, Hutter-Paier B, Prokesch M, Havas D, Rochet J-C, Liu F, Jock K, de Oliveira P, Stirtz GL, Dettmer U, Sortwell CE, Feany MB, Lansbury P, Lapidus L, Paumier KL, Nortriptyline inhibits aggregation and neurotoxicity of alpha-synuclein by enhancing reconfiguration of the monomeric form, Neurobiol. Dis 106(2017) 191–204. doi: 10.1016/j.nbd.2017.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Palazzi L, Bruzzone E, Bisello G, Leri M, Stefani M, Bucciantini M, de Laureto PP, Oleuropein aglycone stabilizes the monomeric α-synuclein and favours the growth of non-toxic aggregates, Sci. Rep 8 (2018) 8337. doi: 10.1038/s41598-018-26645-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Singh SK, Dutta A, Modi G, α-Synuclein aggregation modulation: an emerging approach for the treatment of Parkinson’s disease, Future Med. Chem 9(2017) 1039–1053. doi: 10.4155/fmc-2017-0016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Perni M, Galvagnion C, Maltsev A, Meisl G, Müller MBD, Challa PK, Kirkegaard JB, Flagmeier P, Cohen SIA, Cascella R, Chen SW, Limboker R, Sormanni P, Heller GT, Aprile FA, Cremades N, Cecchi C, Chiti F, Nollen EAA, Knowles TPJ, Vendruscolo M, Bax A, Zasloff M, Dobson CM, A natural product inhibits the initiation of α-synuclein aggregation and suppresses its toxicity, Proc. Natl. Acad. Sci. U. S. A 114 (2017) E1009–E1017. doi: 10.1073/pnas.1610586114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Perni M, Flagmeier P, Limbocker R, Cascella R, Aprile FA, Galvagnion C, Heller GT, Meisl G, Chen SW, Kumita JR, Challa PK, Kirkegaard JB, Cohen SIA, Mannini B, Barbut D, Nollen EAA, Cecchi C, Cremades N, Knowles TPJ, Chiti F, Zasloff M, Vendruscolo M, Dobson CM, Multistep Inhibition of α-Synuclein Aggregation and Toxicity in Vitro and in Vivo by Trodusquemine, ACS Chem. Biol 13 (2018) 2308–2319. doi: 10.1021/acschembio.8b00466. [DOI] [PubMed] [Google Scholar]
- 193.Fonseca-Ornelas L, Eisbach SE, Paulat M, Giller K, Fernández CO, Outeiro TF, Becker S, Zweckstetter M, Small molecule-mediated stabilization of vesicle-associated helical α-synuclein inhibits pathogenic misfolding and aggregation, Nat. Commun 5(2014) 5857. doi: 10.1038/ncomms6857. [DOI] [PubMed] [Google Scholar]
- 194.Conway KA, Harper JD, Lansbury PT, Accelerated in vitro fibril formation by a mutant alpha-synuclein linked to early-onset Parkinson disease, Nat. Med 4 (1998) 1318–1320. doi: 10.1038/3311. [DOI] [PubMed] [Google Scholar]
- 195.Conway KA, Harper JD, Lansbury PT, Fibrils formed in vitro from alpha-synuclein and two mutant forms linked to Parkinson’s disease are typical amyloid, Biochemistry (Mosc.). 39(2000) 2552–2563. [DOI] [PubMed] [Google Scholar]
- 196.Bartels T, Choi JG, Selkoe DJ, α-Synuclein occurs physiologically as a helically folded tetramer that resists aggregation, Nature. 477 (2011) 107–110. doi: 10.1038/nature10324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Johnson SM, Connelly S, Fearns C, Powers ET, Kelly JW, The transthyretin amyloidoses: from delineating the molecular mechanism of aggregation linked to pathology to a regulatory-agency-approved drug, J. Mol. Biol 421 (2012) 185–203. doi: 10.1016/j.jmb.2011.12.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Fanning S, Haque A, Imberdis T, Baru V, Barrasa MI, Nuber S, Termine D, Ramalingam N, Flo GPH, Noble T, Sandoe J, Lou Y, Landgraf D, Freyzon Y, Newby G, Soldner F, Terry-Kantor E, Kim T-E, Hofbauer HF, Becuwe M, Jaenisch R, Pincus D, Clish CB, Walther TC, Farese RV, Srinivasan S, Welte MA, Kohlwein SD, Dettmer U, Lindquist S, Selkoe D, Lipidomic Analysis of α-Synuclein Neurotoxicity Identifies Stearoyl CoA Desaturase as a Target for Parkinson Treatment, Mol. Cell (2018). doi: 10.1016/j.molcel.2018.11.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Nuscher B, Kamp F, Mehnert T, Odoy S, Haass C, Kahle PJ, Beyer K, Alpha-synuclein has a high affinity for packing defects in a bilayer membrane: a thermodynamics study, J Biol Chem. 279 (2004) 21966–75. doi: 10.1074/jbc.M401076200. [DOI] [PubMed] [Google Scholar]
- 200.Vincent BM, Tardiff DF, Piotrowski JS, Aron R, Lucas MC, Chung CY, Bacherman H, Chen Y, Pires M, Subramaniam R, Doshi DB, Sadlish H, Raja WK, Solís EJ, Khurana V, Le Bourdonnec B, Scannevin RH, Rhodes KJ, Inhibiting Stearoyl-CoA Desaturase Ameliorates α-Synuclein Cytotoxicity, Cell Rep. 25(2018) 2742–2754.e31. doi: 10.1016/j.celrep.2018.11.028. [DOI] [PubMed] [Google Scholar]
- 201.Kiss R, Csizmadia G, Solti K, Keresztes A, Zhu M, Pickhardt M, Mandelkow E, Tóth G, Structural Basis of Small Molecule Targetability of Monomeric Tau Protein, ACS Chem. Neurosci (2018). doi: 10.1021/acschemneuro.8b00182. [DOI] [PubMed] [Google Scholar]
- 202.Wischik CM, Staff RT, Wischik DJ, Bentham P, Murray AD, Storey JMD, Kook KA, Harrington CR, Tau aggregation inhibitor therapy: an exploratory phase 2 study in mild or moderate Alzheimer’s disease, J. Alzheimers Dis. JAD 44 (2015) 705–720. doi: 10.3233/JAD-142874. [DOI] [PubMed] [Google Scholar]
- 203.Gauthier S, Feldman HH, Schneider LS, Wilcock GK, Frisoni GB, Hardlund JH, Moebius HJ, Bentham P, Kook KA, Wischik DJ, Schelter BO, Davis CS, Staff RT, Bracoud L, Shamsi K, Storey JMD, Harrington CR, Wischik CM, Efficacy and safety of tau-aggregation inhibitor therapy in patients with mild or moderate Alzheimer’s disease: a randomised, controlled, double-blind, parallel-arm, phase 3 trial, Lancet Lond. Engl 388 ( 2016) 2873–2884. doi: 10.1016/S0140-6736(16)31275-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Wilcock GK, Gauthier S, Frisoni GB, Jia J, Hardlund JH, Moebius HJ, Bentham P, Kook KA, Schelter BO, Wischik DJ, Davis CS, Staff RT, Vuksanovic V, Ahearn T, Bracoud L, Shamsi K, Marek K, Seibyl J, Riedel G, Storey JMD, Harrington CR, Wischik CM, Potential of Low Dose Leuco-Methylthioninium Bis(Hydromethanesulphonate) (LMTM) Mono therapy for Treatment of Mild Alzheimer’s Disease: Cohort Analysis as Modified Primary Outcome in a Phase III Clinical Trial, J. Alzheimers Dis. JAD 61(2018) 435–457. doi: 10.3233/JAD-170560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Medina M, An Overview on the Clinical Development of Tau-Based Therapeutics, Int. J. Mol. Sci 19 (2018). doi: 10.3390/ijms19041160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Bulic B, Pickhardt M, Mandelkow E, Progress and developments in tau aggregation inhibitors for Alzheimer disease, J. Med. Chem 56 (2013) 4135–4155. doi: 10.1021/jm3017317. [DOI] [PubMed] [Google Scholar]
- 207.Grundke-Iqbal I, Iqbal K, Tung YC, Quinlan M, Wisniewski HM, Binder LI, Abnormal phosphorylation of the microtubule-associated protein tau (tau) in Alzheimer cytoskeletal pathology, Proc Natl Acad Sci U A. 83 (1986) 4913–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Lindwall G, Cole RD, Phosphorylation affects the ability of tau protein to promote microtubule assembly, J Biol Chem. 259 (1984) 5301–5. [PubMed] [Google Scholar]
- 209.Tell V, Hilgeroth A, Recent developments of protein kinase inhibitors as potential AD therapeutics, Front Cell Neurosci. 7 (2013) 189. doi: 10.3389/fncel.2013.00189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Shukla V, Skuntz S, Pant HC, Deregulated Cdk5 activity is involved in inducing Alzheimer’s disease, Arch Med Res. 43 (2012) 655–62. doi: 10.1016/j.arcmed.2012.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Morimoto BH, Schmechel D, Hirman J, Blackwell A, Keith J, Gold M, AL-108-211 Study, A double-blind, placebo-controlled, ascending-dose, randomized study to evaluate the safety, tolerability and effects on cognition of AL-108 after 12 weeks of intranasal administration in subjects with mild cognitive impairment, Dement. Geriatr. Cogn. Disord 35(2013) 325–336. doi: 10.1159/000348347. [DOI] [PubMed] [Google Scholar]
- 212.Boxer AL, Lang AE, Grossman M, Knopman DS, Miller BL, Schneider LS, Doody RS, Lees A, Golbe LI, Williams DR, Corvol J-C, Ludolph A, Burn D, Lorenzl S, Litvan I, Roberson ED, Höglinger GU, Koestler M, Jack CR, Van Deerlin V, Randolph C, Lobach IV, Heuer HW, Gozes I, Parker L, Whitaker S, Hirman J, Stewart AJ, Gold M, Morimoto BH, AL-108-231 Investigators, Davunetide in patients with progressive supra nuclear palsy: a randomised, double-blind, placebo-controlled phase 2/3 trial, Lancet Neurol. 13(2014) 676–685. doi: 10.1016/S1474-4422(14)70088-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Sharon R, Bar-Joseph I, Mirick GE, Serhan CN, Selkoe DJ, Altered fatty acid composition of dopaminergic neurons expressing alpha-synuclein and human brains with alpha-synucleinopathies, J. Biol. Chem 278 (2003) 49874–49881. doi: 10.1074/jbc.M309127200. [DOI] [PubMed] [Google Scholar]
- 214.Aharon-Peretz J, Rosenbaum H, Gershoni-Baruch R, Mutations in the glucocerebrosidase gene and Parkinson’s disease in Ashkenazi Jews, N. Engl. J. Med 351 (2004) 1972–1977. doi: 10.1056/NEJMoa033277. [DOI] [PubMed] [Google Scholar]
- 215.Goker-Alpan O, L Giasson B, Eblan MJ, Nguyen J, Hurtig HI, Lee VM-Y, Trojanowski JQ, Sidransky E, Glucocerebrosidase mutations are an important risk factor for Lewy body disorders, Neurology. 67(2006) 908–910. doi: 10.1212/01.wnl.0000230215.41296.18. [DOI] [PubMed] [Google Scholar]
- 216.Mazzulli JR, Xu Y-H, Sun Y, Knight AL, McLean PJ, Caldwell GA, Sidransky E, Grabowski GA, Krainc D, Gaucher disease glucocerebrosidase and α-synuclein form a bidirectional pathogenic loop in synucleinopathies, Cell. 146(2011) 37–52. doi: 10.1016/j.cell.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Simon-Sanchez J, Schulte C, Bras JM, Sharma M, Gibbs JR, Berg D, Paisan-Ruiz C, Lichtner P, Scholz SW, Hernandez DG, Kruger R, Federoff M, Klein C, Goate A, Perlmutter J, Bonin M, Nalls MA, Illig T, Gieger C, Houlden H, Steffens M, Okun MS, Racette BA, Cookson MR, Foote KD, Fernandez HH, Tray nor BJ, Schreiber S, Arepalli S, Zonozi R, Gwinn K, van der Brug M, Lopez G, Chanock SJ, Schatzkin A, Park Y, Hollenbeck A, Gao J, Huang X, Wood NW, Lorenz D, Deuschl G, Chen H, Riess O, Hardy JA, Singleton AB, Gasser T, Genome-wide association study reveals genetic risk underlying Parkinson’s disease, Nat Genet. 41 (2009) 1308–12. doi: 10.1038/ng.487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Simon-Sanchez J, van Hilten JJ, van de Warren burg B, Post B, Berendse HW, Arepalli S, Hernandez DG, de Bie RM, Velseboer D, Scheffer H, Bloem B, van Dijk KD., Rivadeneira F, Hofman A, Uitterlinden AG, Rizzu P, Bochdanovits Z, Singleton AB, Heutink P, Genome-wide association study confirms extant PD risk loci among the Dutch, Eur J Hum Genet. 19 (2011) 655–61. doi: 10.1038/ejhg.2010.254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Chen YP, Song W, Huang R, Chen K, Zhao B, Li J, Yang Y, Shang HF, GAK rs1564282 and DGKQ rs11248060 increase the risk for Parkinson’s disease in a Chinese population, J Clin Neurosci. 20 (2013) 880–3. doi: 10.1016/j.jocn.2012.07.011 [DOI] [PubMed] [Google Scholar]
- 220.Nalls MA, Pankratz N, Lill CM, Do CB, Hernandez DG, Saad M, DeStefano AL, Kara E, Bras J, Sharma M, Schulte C, Keller MF, Arepalli S, Letson C, Edsall C, Stefansson H, Liu X, Pliner H, Lee JH, Cheng R, C. International Parkinson’s Disease Genomics, Gen.l. Parkinson’s Study Group Parkinson’s Research: The Organized, andMe, GenePd, C. NeuroGenetics Research, G. Hussman Institute of Human, I. Ashkenazi Jewish Dataset, H. Cohorts for, E. Aging Research in Genetic, C. North American Brain Expression, C. United Kingdom Brain Expression, C. Greek Parkinson’s Disease,G. Alzheimer Genetic Analysis, Ikram MA, Ioannidis JP, Hadjigeorgiou GM, Bis JC, Martinez M, Perlmutter JS, Goate A, Marder K, Fiske B, Sutherland M, Xiromerisiou G, Myers RH, Clark LN, Stefansson K, Hardy JA, Heutink P, Chen H, Wood NW, Houlden H, Payami H, Brice A, Scott WK, Gasser T, Bertram L., Eriksson N, Foroud T, Singleton AB, Large-scale meta-analysis of genome-wide association data identifies six new risk loci for Parkinson’s disease, Nat Genet. 46 (2014) 989–93. doi: 10.1038/ng.3043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Zhu XC, Cao L, Tan MS, Jiang T, Wang HF, Lu H, Tan CC, Zhang W, Tan L, Yu JT, Association of Parkinson’s Disease GWAS-Linked Loci with Alzheimer’s Disease in Han Chinese, Mol Neurobiol. 54 ( 2017) 308–318. doi: 10.1007/s12035-015-9649-5. [DOI] [PubMed] [Google Scholar]
- 222.Chang D, Nalls MA, Hallgrímsdóttir IB, Hunkapiller J, van der Brug M, Cai F, International Parkinson’s Disease Genomics Consortium, 23andMe Research Team, Kerchner GA, Ayalon G, Bingol B, Sheng M, Hinds D, Behrens TW, Singleton AB, Bhangale TR, Graham RR, A meta-analysis of genome-wide association studies identifies 17 new Parkinson’s disease risk loci, Nat. Genet 49 (2017) 1511–1516. doi: 10.1038/ng.3955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Goedert M, Eisenberg DS, Crowther RA, Propagation of Tau Aggregates and Neurodegeneration, Annu. Rev. Neurosci 40(2017) 189–210. doi: 10.1146/annurev-neuro-072116-031153. [DOI] [PubMed] [Google Scholar]
- 224.Fitzpatrick AWP, Falcon B, He S, Murzin AG, Murshudov G, Garringer HJ, Crowther RA, Ghetti B, Goedert M, Scheres SHW, Cryo-EM structures of tau filaments from Alzheimer’s disease, Nature. 547 (2017) 185–190. doi: 10.1038/nature23002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Peelaerts W, Bousset L, Van der Perren A, Moskalyuk A, Pulizzi R, Giugliano M, Van den Haute C, Melki R, Baekelandt V, α-Synuclein strains cause distinct synucleinopathies after local and systemic administration, Nature. 522(2015) 340–344. doi: 10.1038/nature14547. [DOI] [PubMed] [Google Scholar]
- 226.Peelaerts W, Bousset L, Baekelandt V, Melki R, α-Synuclein strains and seeding in Parkinson’s disease, incidental Lewy body disease, dementia with Lewy bodies and multiple system atrophy: similarities and differences, Cell Tissue Res. 373(2018) 195–212. doi: 10.1007/s00441-018-2839-5. [DOI] [PubMed] [Google Scholar]
- 227.Yamasaki TR, Holmes BB, Furman JL, Dhavale DD, Su BW, Song E-S, Cairns NJ, Kotzbauer PT, Diamond MI, Parkinson’s disease and multiple system atrophy have distinct α-synuclein seed characteristics, J. Biol. Chem 294 (2019) 1045–1058. doi: 10.1074/jbe.RA118.004471. [DOI] [PMC free article] [PubMed] [Google Scholar]