

Summary
Contact-dependent growth inhibition (CDI) is a form of interbacterial competition mediated by CdiB-CdiA two-partner secretion systems. CdiA effector proteins carry polymorphic C-terminal toxin domains (CdiA-CT), which are neutralized by specific CdiI immunity proteins to prevent self-inhibition. Here, we present the crystal structures of CdiA-CT•CdiI complexes from Klebsiella pneumoniae 342 and Escherichia coli 3006. The toxins adopt related folds that resemble the ribonuclease domain of colicin D, and both are isoacceptor specific tRNases that cleave the acceptor stem of deacylated tRNAGAUIle. Though the toxins are similar in structure and substrate specificity, CdiA-CTKp342 activity requires translation factors EF-Tu and EF-Ts, whereas CdiA-CTEC3006 is intrinsically active. Further, the corresponding immunity proteins are unrelated in sequence and structure. CdiIKp342 forms a dimeric β-sandwich, whereas CdiIEC3006 is an α-solenoid monomer. Given that toxin-immunity genes co-evolve as linked pairs, these observations suggest that the similarities in toxin structure and activity reflect functional convergence.
Keywords: bacterial competition, toxin-antitoxin systems, two-partner secretion, type V secretion system
Graphical Abstract
eTOC
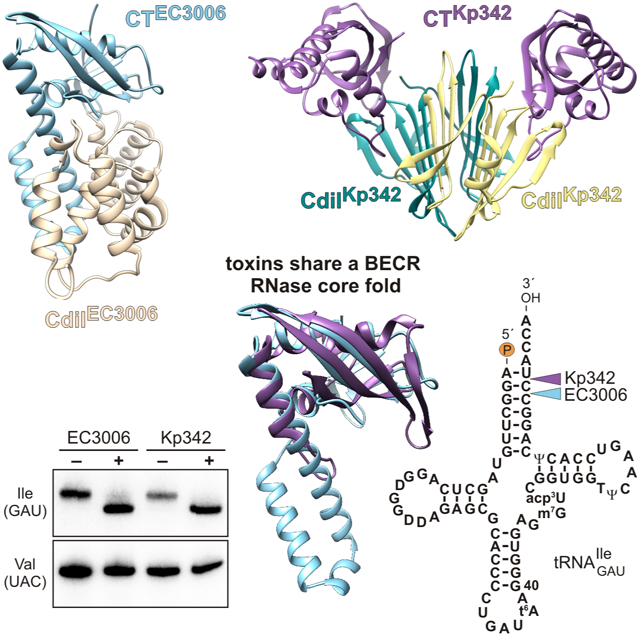
Gucinski et al. present the structures of two CdiA toxin domains in complex with their cognate immunity proteins. These toxins adopt the same fold and exhibit similar tRNase activities. The immunity proteins are completely unrelated in sequence and structure, suggesting that similarities shared by the toxins arose through convergent evolution.
Introduction
Contact-dependent toxin delivery has emerged as an important general mechanism by which bacteria compete with neighboring cells. Gram-negative bacteria use type I (Garcia-Bayona et al., 2017), type IV (Souza et al., 2015), type V (Aoki et al., 2005) and type VI (Hood et al., 2010; MacIntyre et al., 2010) secretion systems to transfer toxic effector proteins directly into target cells. Similarly, Gram-positive species use the general secretory pathway and type VII secretion systems to compete with neighboring bacteria (Cao et al., 2016; Koskiniemi et al., 2013; Whitney et al., 2017). Direct cell-to-cell toxin delivery was first discovered in Escherichia coli isolate EC93, which uses a specialized type V secretion system – comprised of the CdiB and CdiA proteins – to inhibit the growth of other E. coli strains in a process termed contact-dependent growth inhibition or CDI (Aoki et al., 2005). CdiB is an Omp85 β-barrel transport protein that exports the CdiA effector across the outer membrane. During its biogenesis, CdiA undergoes a programmed secretion arrest that sequesters the C-terminal half of the effector in the periplasm (Ruhe et al., 2018). The N-terminal half of CdiA forms an extracellular filament that projects ~33 nm from the cell surface (Ruhe et al., 2018). The distal tip of the filament contains an adhesin domain that recognizes a specific receptor on target E. coli cells. Once receptor is engaged, CdiA export resumes, and the C-terminal toxin region (CdiA-CT) is transferred into the target bacterium. The CdiA-CT toxin from E. coli EC93 forms membrane pores, which dissipate the proton gradient and interfere with ATP synthesis in target bacteria (Aoki et al., 2009). The cdi locus also encodes a small CdiI immunity protein that neutralizes CdiA-CT activity to protect the cell from auto-intoxication (Aoki et al., 2005). Thus, CDI allows E. coli EC93 cells to inhibit non-isogenic competitors, while simultaneously providing immunity to the toxins delivered by siblings.
Genome surveys reveal that cdi loci are wide-spread throughout Gram-negative species and are particularly common in pathogenic proteobacteria (Aoki et al., 2010; Willett et al., 2015b; Zhang et al., 2012). A hallmark of CDI systems is the striking polymorphism of CdiA-CT and CdiI sequences across bacteria. Different isolates of a given species often deploy distinct toxin domains, which are encoded together with equally unique immunity proteins (Anderson et al., 2012; Aoki et al., 2010; Nikolakakis et al., 2012). The variable CdiA-CT region is usually demarcated by a conserved peptide motif, exemplified by the VENN sequence in enterobacteria. We currently recognize 130 CdiA-CT sequence types and note that toxin-immunity pairs within the same family can exhibit significant sequence variation. These latter polymorphisms are concentrated at the CdiA-CT•CdiI binding interface, and therefore immunity proteins generally do not provide cross-protection against "near-cognate" toxins (Michalska et al., 2018; Morse et al., 2015; Poole et al., 2011). CDI system evolution is also heavily influenced by horizontal gene transfer. E. coli cdi loci are invariably located within genomic islands or on plasmids, and the 3′-regions are frequently littered with fragmented cdiA-CT/cdiI sequences and interspersed integrase and transposase genes (Aoki et al., 2010; Poole et al., 2011; Ruhe et al., 2016). These observations suggest that bacteria acquire new cdiA-CT/cdiI sequences via horizontal gene transfer and integrate the modules into the cdi locus to change the toxin they deploy (Arenas et al., 2013; Poole et al., 2011). This hypothesis is supported by experimental work showing that cdiA-CT/cdiI modules from various species can be fused at the VENN coding sequence of cdiA from E. coli EC93 to produce functional chimeras (Aoki et al., 2010; Beck et al., 2014; Webb et al., 2013; Willett et al., 2015a). Together, these observations suggest that novel toxins confer a selective advantage, which in turn drives the rapid diversification and exchange of toxin-immunity gene pairs. In this manner, CDI toxin-immunity protein diversity contributes to self-nonself recognition, allowing bacteria to establish kin groups that ultimately shape the structure of microbial communities (Anderson et al., 2012, 2014).
Though there are more than a hundred CDI toxin sequence types, only 42 have predicted biochemical activities and/or functional annotations (Table S1). Some CdiA-CT sequences are clearly related to the C-terminal nuclease domains of colicins, which are diffusible protein toxins released by many strains of E. coli (Cascales et al., 2007). CdiA proteins from Erwinia chrysanthemi EC16 (NCBI reference: AAN38708.1), Bordetella petrii BAA-461 (CAP40933.1), Burkholderia pseudomallei K96243 (WP_011205723.1) and Moraxella catarrhalis McGHS1 (ABQ43332.1) carry toxins that are homologous to the nuclease domains of colicin E3, colicin D, colicin E5 and colicin E2, respectively (Table S1) (Aoki et al., 2010; Walker et al., 2004). Biochemical analyses have confirmed that CdiA-CTEC16 cleaves 16S rRNA at the same site as colicin E3, and that CdiA-CTK96243 cleaves the anticodon loops of queuosine-containing tRNAs in the same manner as colicin E5 (Beck et al., 2014; Nikolakakis et al., 2012). Aravind and coworkers have identified numerous novel toxin (Ntox) families, many of which are associated with CDI systems and are predicted to have RNase activities (Table S1) (Zhang et al., 2012; Zhang et al., 2011). Of these, the Ntox21 (Pfam: PF15526), Ntox28 (PF15605) and EndoU (PF14436) families have been confirmed experimentally. The Ntox21 domain has the same fold and 16S rRNA nuclease activity as colicin E3 (Beck et al., 2014; Zhang et al., 2012). The Ntox28 domain forms a small α-helical bundle that cleaves the anticodon loops of several tRNAs (Aoki et al., 2010; Diner et al., 2012; Johnson et al., 2016a). The EndoU domain resembles eukaryotic and viral RNA processing enzymes (Holberger et al., 2012; Jamet et al., 2015; Michalska et al., 2018; Zhang et al., 2012). Most of the remaining annotated CDI toxin types are predicted to be DNases and nucleobase deaminases (Table S1) (Zhang et al., 2012), though these activities have yet to be verified experimentally.
Given that most CdiA-CT domains lack functional annotations, we have used a combination of crystallography and biochemical approaches to identify new toxin activities and provide insight into the diversity of toxin-immunity protein interactions. This work has revealed that many CDI toxins adopt known nuclease folds. CdiA-CT domains from B. pseudomallei strains 1026b and 1655 share the core α/β-fold of PD(D/E)-XK superfamily phosphodiesterases (Johnson et al., 2016b; Morse et al., 2012; Nikolakakis et al., 2012). This fold forms the catalytic core of restriction endonucleases, but the Burkholderia CDI toxins are specific for tRNA. CdiA-CTYkris from Yersinia kristensenii ATCC 33638 is the first RNase A superfamily member to be found outside of vertebrates, though the toxin shares no detectable sequence similarity with RNase A orthologs and lacks the superfamily's characteristic disulfide bonds (Batot et al., 2017). Finally, CdiA-CTNC101 from E. coli NC101 is a member of the Barnase/EndoU/colicin E5-D/RelE (BECR) RNase family. This latter toxin is remarkable because it requires the essential translation factors EF-Tu and EF-Ts to cleave tRNA substrates (Jones et al., 2017; Michalska et al., 2017). In each instance, computational approaches did not predict that the toxin domains were related to known nuclease families, underscoring the inherent difficulty in predicting structure and function solely from protein sequence. Based on these examples, it seems likely that many uncharacterized CDI toxins belong to known enzyme families.
Here, we extend our survey to CdiA-CT•CdiI complexes from E. coli 3006 (EC3006) and Klebsiella pneumoniae 342 (Kp342). Position-specific iterated (PSI) BLAST analysis suggests that these toxin domains are related, though they only share ~13% sequence identity. Crystal structures of the complexes support this relationship, revealing that the C-terminal subdomain of each toxin adopts the BECR RNase fold. However, the corresponding immunity proteins are completely unrelated in sequence and structure. The toxins are structurally similar to the tRNase domain of colicin D, which cleaves the anticodon loops of tRNAArg isoacceptors (Tomita et al., 2000). Though CdiA-CTKp342 and CdiA-CTEC3006 are also isoacceptor specific tRNases, they cleave the aminoacyl acceptor stem of tRNAGAUIle. Further, purified CdiA-CTEC3006 alone is sufficient to cleave tRNAGAUIle in vitro, whereas CdiA-CTKp342 requires translation factors EF-Tu and EF-Ts for its activity. The dramatically different structures of CdiI immunity proteins, together with the differential requirement for EF-Tu and EF-Ts, strongly suggest that these BECR RNase domains are related through convergent evolution.
Results
Crystallization and structure determination
We cloned the cdiA-CT and cdiI coding sequences from K. pneumoniae 342 and E. coli 3006 into T7 RNA polymerase expression plasmids. Each CdiA-CT construct includes the VENN peptide motif, which begins at Val2905 in CdiAKp342 and Val2921 in CdiAEC3006 (Figure S1). To facilitate comparisons of homologous toxins carried by different CdiA effectors, CdiA-CT residues are typically numbered from Val1 of the conserved VENN motif. The CdiA-CT•CdiIKp342 complex has the wild-type sequence, but CdiA-CTEC3006 lacks residue Asn332, for which the codon was inadvertently deleted during PCR amplification. Protein complexes were labeled with selenomethionine and purified by Ni2+-affinity chromatography using His6 epitope tags appended to the N-terminus of CdiA-CTKp342 and the C-terminus of CdiIEC3006. Prior to crystallization, both complexes were subjected to limited proteolysis, which removed the N-terminal "cytoplasm-entry" domain from each CdiA-CT construct (Figure S1). Cytoplasm-entry domains have no toxin activity, but are required to translocate the toxin across the cytoplasmic membrane of target bacteria (Willett et al., 2015a). The CdiA-CT•CdiIKp342 complex crystallized in space group P21212 with two heterotetrameric complexes in the asymmetric unit (Table 1). CdiA-CT•CdiIEC3006 crystallized in space group P21 with two heterodimeric complexes in the asymmetric unit (Table 1). Structures were determined at 2.55 Å for CdiA-CT•CdiIKp342 and 2.20 Å for CdiA-CT•CdiIEC3006 by SAD phasing using anomalous signals from selenium atoms.
Table 1.
Data processing and refinement statistics.
| Data processing | ||
|---|---|---|
| Protein | CdiA-CT•CdiIEC3006 | CdiA-CT•CdiIKp342 |
| Wavelength (Å) | 0.9792 | 0.9792 |
| Resolution range (Å)a | 30.0 - 2.20 (2.24 - 2.20) | 30.0 - 2.55 (2.59 - 2.55) |
| Space group | P21 | P21212 |
| Unit cell parameters (Å, °) | 50.71 106.53 72.65 101.0 | 102.64 145.45 84.28 |
| Unique reflections | 38,085 (1,879) | 41,852 (2,053) |
| Multiplicity | 4.2 (3.8) | 25.6 (22.7) |
| Completeness (%) | 99.9 (100) | 99.9 (100) |
| <Ι >/< σΙ> | 11.28 (1.96) | 34.5 (1.8) |
| Rmergeb | 0.127 (0.749) | 0.144 (2.27) |
| CC1/2c | 0.664 | 0.722 |
| CC*c | 0.893 | 0.916 |
| Refinement | ||
| Resolution (Å) | 30.0 - 2.20 | 30.0 - 2.55 |
| Reflections work/test set | 36,865/1,189 | 39,752/2,035 |
| Rwork/ Rfreed | 0.176/0.218 | 0.181/0.232 |
| Average B factor (Å2) (No of atoms) | ||
| macromolecule | 40.6 (5,010) | 88.0 (7,377) |
| ligands | 51.6 (27) | |
| solvent | 40.6 (174) | 77.6 (23) |
| Rmsd bond lengths (Å) | 0.011 | 0.008 |
| Rmsd bond angles (°) | 1.06 | 0.965 |
| Ramachandran favorede (%) | 98.1 | 99.2 |
| Ramachandran outliers | 0 | 0 |
| Clashscoree | 3.51 | 5.8 |
| PDB ID | 6CP8 | 6CP9 |
Values in parentheses correspond to the highest resolution shell.
Rmerge = ΣhΣj|Ihj−<Ih>|/ΣhΣjIhj, where Ihj is the intensity of observation j of reflection h.
As defined by (Karplus and Diederichs, 2012)
R = Σh|Fo|−|Fc|/Σh|Fo| for all reflections, where Fo and Fc are observed and calculated structure factors, respectively. Rfree is calculated analogously for the test reflections, randomly selected and excluded from the refinement.
As defined by Molprobity (Davis et al., 2004)
Structure of the CdiA-CT•CdiIKp342 complex
CdiA-CTKp342 and CdiIKp342 form a heterotetrameric complex in the crystal with two toxin domains bound to each CdiIKp342 homodimer (Figure 1A). The crystallized toxin domain (chains A, C, E and G) likely spans residues Val139 – Lys264, with the most complete chain containing residues Thr143 – Gly261 (chain E). The CdiA-CTKp342 toxin domain consists of four N-terminal α-helices and a four-stranded antiparallel β-sheet at the C-terminus (Figure 1A). The β-sheet is twisted and partially wraps around helix α4. All CdiIKp342 immunity proteins (chains B, D, F and H) are nearly complete. Each heterotetramer is covalently linked to its crystallographic symmetry mate through a disulfide bond between CdiIKp342 residue Cys38. This linkage is likely an artifact of crystallization, because the CdiA-CT•CdiIKp342 complex migrates as a tetramer, not an octamer, during size-exclusion chromatography (data not shown). CdiIKp342 forms a dimeric β-sandwich with each protomer contributing two antiparallel β-sheets (Figure 1A). The N-terminal (β1′-β5′) and C-terminal (β6′-β9′) sheets from each protomer interact in parallel via β5′ and β9′ to form a 9-stranded mixed β-sheet (Figure 1B). In addition to the β-complementation H-bond network, the backbone amides of Lys93 make direct H-bond contacts with the side-chains of Glu94, and the amide side-chains of Asn97 also interact with one another across the dimer interface (Figure 1B).
Figure 1. Structure of the CdiA-CT•CdiIKp342 complex.

A) Secondary structure elements of the CdiA-CT•CdiIKp342 complex. B) CdiIKp342 forms a dimeric -sandwich. C) Direct H-bond and ion-pair contacts. See also Figures S1 and S2.
The CdiA-CT•CdiIKp342 binding interface buries ~1,800 Å2 (on average) of the solvent-accessible surface area. In general, peripheral elements of CdiIKp342 interact with the toxin domain. N-terminal residues Glu4, Lys6 and Glu9 of the immunity protein interact with Lys213, Tyr215 and Tyr219 (respectively) within the β1-β2 hairpin of the toxin domain (Figure 1C). CdiIKp342 residues Arg25 and Asp26 form salt-bridges with Glu244 and Arg252/Lys157 (respectively) in the toxin domain (Figure 1C). The N-terminal β-sheet of CdiIKp342 interacts extensively with the long loop connecting helices α2 and α3 of the toxin. CdiIKp342 Asp70 forms a salt-bridge with toxin residue Arg174, and CdiIKp342 residues Ser22 and Ser29 both bind to His170 in CdiA-CTKp342 (Figure 1C). There are additional hydrophobic contacts between CdiIKp342 residues Phe2, Phe23, Phe30, Val48 and Val49 and CdiA-CTKp342 residues Tyr215, Tyr219 and Phe222 within the β1-β2 hairpin.
Structure of the CdiA-CT•CdiIEC3006 complex
The CdiA-CT•CdiIEC3006 asymmetric unit contains two heterodimeric complexes. The modeled CdiA-CTEC3006 toxin domains (chains A & B) contain residues Asn175 – Lys336 (numbered from Val1 of the VENN peptide motif), and residues Val4 – Asn161 of CdiIEC3006 are resolved (chains C & D). The CdiA-CTEC3006 toxin domain is composed of a globular α/β-head subdomain, from which extends an α-helical protrusion. The head subdomain is composed of two three-stranded antiparallel β-sheets (β1-β3 and β4-β6) that wrap around helix α5 (Figure 2A). The N-terminal sheet (β1-β3) interacts with the C-terminal sheet (β4-β6) in parallel via β3 and β6. The short helix α1 follows the N-terminal sheet and leads into the long helical extension formed by α2, α3 and α4 (Figure 2A). The CdiIEC3006 immunity protein adopts an α-solenoid structure with α1′-α2′, α3′-α4, α5′-α6′ and α7′-α8′ forming α-hairpins, of which the latter two are linked by short 310 helices, G1′ and G2′ (Figure 2A).
Figure 2. Structure of the CdiA-CT•CdiIEC3006 complex.

A) Secondary structure elements of the CdiA-CT•CdiIEC3006 complex. B) Direct H-bond and ion-pair contacts. See also Figures S1 and S3.
CdiIEC3006 binds between the two subdomains of CdiA-CTEC3006 through an extended network of direct H-bonds and salt-bridges. The primary anchoring interactions link the internal helical layer (α2′, α4′ and α6′) of CdiIEC3006 to the α-helical protrusion of the toxin. Many of these contacts involve toxin helix α4, with residues Glu249 and Glu252 both interacting with Lys103 of CdiIEC3006 (Figure 2B). Toxin residue Arg260 forms a salt bridge with Asp140 of CdiIEC3006 (Figure 2B). In addition, the loop connecting α1′ to α2′ of CdiIEC3006 inserts into the cleft between the β-sheets of the toxin α/β-head subdomain (Figure 2B). The main-chain carbonyl groups of Ala25, Asn26 and Phe28 in this loop interact with the amide backbone of Ala185 and the side-chain of Lys204, respectively (Figure 2B). The side-chains of CdiIEC3006 residues Asn26, Asn62 and Asp140 also form direct H-bonds with toxin residues Arg327, Arg299 and Thr334, respectively (Figure 2B). Hydrophobic contacts further stabilize the complex, with immunity protein residues Phe67, Phe143 and Tyr144 stacking against the toxin in the vicinity of Lys253, His256 and Arg260. Altogether, the CdiA-CT•CdiIEC3006 interface buries ~4,300 Å2 of the solvent-accessible surface area of the monomeric units.
CdiA-CTKp342 and CdiA-CTEC3006 are structurally similar
Although CdiA-CTKp342 and CdiA-CTEC3006 share only ~13% sequence identity, each is captured by iterative PSI-BLAST search when the other domain is submitted as the query (Table S2). In accordance with this sequence similarity, the CdiA-CTKp342 structure superimposes onto the α/β-head subdomain of CdiA-CTEC3006 with rmsd of 2.7 Å (Figure 3A). The toxins share the C-terminal β-sheet, though this element consists of four strands in CdiA-CTKp342. CdiA-CTKp342 helices α2 and α4 correspond to α1 and α5 in CdiA-CTEC3006 (Figures 3B & 3C). The N-terminal β-sheet of CdiA-CTEC3006 is replaced by helix α1 in CdiA-CTKp342, and the α-helical protuberance of CdiA-CTEC3006 is significantly reduced to a long loop and short helix α3 in CdiA-CTKp342. DALI searches for structural homologs of CdiA-CTKp342 and CdiA-CTEC3006 recover a number of ribonucleases that share the barnase/EndoU/colicin D-E5/RelE (BECR) fold (Holm and Rosenstrom, 2010; Zhang et al., 2012). The closest structural homologs of CdiA-CTKp342 include the C-terminal domain of colicin D and the BrnT toxin from Brucella abortus (Table 2). Intriguingly, we also recovered the CDI toxin from E. coli strain NC101 (Table 2), which requires translation factors EF-Tu and EF-Ts to cleave tRNA substrates in vitro (Michalska et al., 2017). Hits from the CdiA-CTEC3006 search were dominated by proteins that superimpose onto the α-helical extension subdomain, and only four of the top several hundred hits match the α/β-head subdomain. These include type II ParE toxins from Mesorhizobium opportunistum, E. coli O157:H7 str. SS52 and Caulobacter vibrioides (Table 2). Thus, the CdiA-CTKp342 and CdiA-CtEC3006 toxin domains are heretofore unrecognized members of the BECR ribonuclease superfamily.
Figure 3. CdiA-CTKp342 and CdiA-CTEC3006 are structurally similar.

A) Superimposition of the CdiA-CTKp342 and CdiA-CTEC3006 toxin domains. See also Table S2. B) Binding position of the CdiIKp342 immunity protein. C) Binding position of the CdiIEC3006 immunity protein.
Table 2.
Structural homologs of CdiA-CT•CdiIKp342 and CdiA-CT•CdiIEC3006, See also Figure S5.
| Query protein |
Homolog (organism) | PDB IDa |
Z- score |
rmsdb (Å) |
lali/nresc | % identity |
|---|---|---|---|---|---|---|
| CdiIKp342 (B) | β-galactosidase (Escherichia coli) | 5TTG (A) | 5.6 | 3.6 | 85/1015 | 9 |
| galactose mutarotase (Lactococcus lactis) | 1NSS (B) | 5.2 | 4.3 | 89/346 | 9 | |
| lysyl oxidase (Komagataella pastoris) | 1N9E (A) | 5.2 | 4.4 | 71/735 | 8 | |
| copper amine oxidase (Arthrobacter globiformis) | 5ZPE (B) | 5.1 | 4.3 | 80/620 | 6 | |
| CdiA-CTKp342 (A) | colicin D (E. coli) | 1V74 (A) | 5.5 | 3.5 | 81/107 | 9 |
| colicin D (E. coli) | 1TFO (A) | 5.5 | 3.4 | 79/103 | 9 | |
| BrnT (B. abortus) | 3U97 (A) | 5.1 | 2.5 | 65/77 | 9 | |
| HigB2 (V. cholerae) | 5JA8 (C) | 5.0 | 3.4 | 73/108 | 10 | |
| colicin D (E. coli) | 1TFK (A) | 4.5 | 3.5 | 69/94 | 7 | |
| CdiA-CTNC101 (E. coli) | 5I4Q (A) | 4.5 | 3.1 | 70/88 | 7 | |
| CdiIEC3006 (C) | AP-1 complex subunit β1 (Homo sapiens) | 4P6Z (B) | 9.1 | 3.4 | 133/570 | 8 |
| Puf5p (Saccharomyces cerevisiae) | 5BZV (A) | 8.9 | 3.5 | 133/364 | 9 | |
| importin β1 (Saccharomyces cerevisiae) | 2BKU (B) | 8.7 | 3.5 | 143/857 | 9 | |
| glomulin (Homo sapiens) | 4F52 (F) | 8.6 | 3.5 | 143/512 | 8 | |
| importin-4 (Homo sapiens) | 5×BK (D) | 8.6 | 3.7 | 137/369 | 8 | |
| transportin-1 (Homo sapiens) | 4JLQ (A) | 8.6 | 3.5 | 132/840 | 8 | |
| CdiA-CtEC3006 (A) | ParE (Mesorhizobium opportunistum) | 5CEG (B) | 4.6 | 3.9 | 74/103 | 5 |
| ParE2 (E. coli str. SS52) | 5CW7 (P) | 4.4 | 3.3 | 67/95 | 4 | |
| ParE (Caulobacter vibrioides CB15) | 3KXE (B) | 4.1 | 3.5 | 71/94 | 6 |
Homologous chains are given in parentheses.
root-mean-square deviation.
length of alignment (lali)/total number of residues (nres)
In contrast to the toxin domains, the immunity proteins are unrelated in sequence and structure (Figures 3B & 3C). The β-sandwich module of CdiIKp342 is present in several bacterial enzymes, including β-galactosidase and amine oxidases (Table 2). Though this β-motif is common in various proteins, CdiIKp342 appears to be the first example of an immunity protein with this fold. The DALI search for CdiIEC3006 homologs recovered several eukaryotic proteins with armadillo-like repeats (Figure 2A). These latter structural homologs include a subunit of the clathrin associated AP-1 complex, human importin and transportin proteins that function in nuclear import, and the yeast RNA-scaffolding protein Puf5p (Table 2). Although the CdiI proteins are structurally distinct, both bind to their cognate toxins at the same relative position on the α/β core (Figures 3B & 3C), suggesting that they neutralize their cognate toxins using similar mechanisms.
CdiA-CTEC3006 and CdiA-CTKp342 cleave tRNAGAUIle
We previously showed that tRNAGAUIle is cleaved in response to CdiA-CTEC3006 intoxication (Willett et al., 2015a). Given the structural homology between CdiA-CTEC3006 and CdiA-CTKp342, we tested whether the toxins also exhibit similar tRNase activities. We co-expressed each toxin with cognate CdiI that carries a C-terminal ssrA(DAS) degron (McGinness et al., 2006). Degron-tagged immunity proteins are degraded by the ClpXP protease, thereby liberating the toxins to act on cellular substrates. We note that wild-type CdiA-CTEC3006 (containing Asn332) was used for this and all following experiments. Northern blot analysis confirmed that CdiA-CTEC3006 activation leads to tRNAGAUIle cleavage (Figure 4A, compare lanes 1, 2 & 3). This activity is specific, because no cleavage of tRNAUACVal, tRNAGUATyr or tRNAUAALeu was detected (Figure 4A). Analysis of RNA from CdiA-CTKp342 intoxicated cells revealed similar RNase activity and substrate specificity (Figure 4A, compare lanes 1, 4 & 5), suggesting that both toxins are isoacceptor specific tRNases.
Figure 4. CdiA-CTEC3006 and CdiA-CTKp342 cleave tRNAGAUIle.

A) Toxin activation inside E. coli cells leads to tRNAGAUIle cleavage. Toxin expression was induced L-arabinose where indicated and RNA isolated for Northern blot hybridization using probes to the indicated tRNAs. Aminoacyl acceptor stem sequences and toxin cleavage sites are indicated on the right. B) tRNAGAUIle sequence showing the hybridized S1 probe and oligonucleotide standards used to map toxin cleavage sites. C) S1 protection analysis of tRNAGAUIle cleaved by CdiA-CTKp342. RNA samples were isolated from i) cells intoxicated by intracellular CdiA-CTKp342 expression, ii) competition co-cultures and iii) in vitro nuclease reactions. Where indicated, the neutralizing effect of CdiIKp342 was examined. Samples were hybridized with 3′-radiolabeled S1 probe and treated with S1 nuclease as described in Methods. A portion of the S1 probe-tRNAGAUIle heteroduplex sequence is shown to the right of the autoradiogram. D) S1 protection analysis of tRNAGAUIle cleaved by CdiA-CTEC3006. Samples were analyzed as described for panel C.
Though CdiA-CTKp342 and CdiA-CTEC3006 have similar activities, Northern blot analysis lacks the resolution to determine whether the toxins cleave substrate at the same positions. Because other CDI toxins degrade the 3′-ends of tRNA (Jones et al., 2017; Michalska et al., 2017; Nikolakakis et al., 2012), we used S1 nuclease protection analysis to map cleavage sites in the 3′-arm of the aminoacyl acceptor stem (Figure 4B). This analysis revealed that tRNAGAUIle is truncated after nucleotide C72 in CdiA-CTKp342 intoxicated cells (Figures 4C, lanes 3 & 4). CdiA-CTEC3006 also acts on the acceptor stem, but cleaves after nucleotide C71 (Figures 4D, lanes 3 & 4). These cleaved tRNAs cannot be repaired by the CCA-adding nucleotidyl transferase, because the unpaired discriminator nucleotide is removed (Betat et al., 2010). Thus, both toxins inactivate tRNAGAUIle by releasing short 3′-oligonucleotide fragments.
tsf mutants are resistant to CdiA-CTKp342, but not CdiA-CTEC3006 intoxication
As noted above, CdiA-CTKp342 shares structural homology with the CdiA-CTNC101 toxin domain from E. coli NC101. CdiA-CTNC101 requires translation factors EF-Tu and EF-Ts to support its tRNase activity in vitro; and tsf mutations that alter the coiled-coil domain of EF-Ts protect target bacteria from inhibition by this toxin (Michalska et al., 2017). Therefore, we tested tsf mutants for resistance to CdiA-CTKp342 and CdiA-CTEC3006 to determine whether the toxins are translation factor-dependent. We first fused CdiA-CTKp342 and CdiA-CTEC3006 sequences to the VENN motif of CdiAEC93 and confirmed that the resulting chimeras are active in competition co-cultures. Cells expressing the chimeric effectors outcompete wild-type target bacteria ~1,000-fold after 1 h of co-culture in shaking broth (Figure 5A). This growth advantage is due to CdiA-CT toxin activity, because competitive fitness is restored to target cells that express cognate cdiI, whereas non-cognate immunity genes are not protective (Figure 5A). We then examined tsf(A202E) and tsf(Δcoil) target cells, which encode EF-Ts with an Ala202Glu substitution at the tip of the coiled-coil domain and a deletion of the entire coiled-coil domain, respectively. Both tsf mutants are completely resistant to inhibition by CdiA-CTKp342, but remain as susceptible to CdiA-CTEC3006 intoxication as tsf+ target bacteria (Figure 5A). We also used Northern blotting to monitor toxin activity in the co-cultures. Cleaved tRNAGAUIle was readily detected in RNA isolated from co-cultures with non-immune tsf+ target bacteria (Figure 5B, lanes 1 & 6). Because these co-cultures were seeded with inhibitor and target cells at a 1:1 ratio, only half of the tRNAGAUIle should be cleaved because inhibitor bacteria are immune to their own toxin. Accordingly, the expression of cognate cdiI in target cells blocked this nuclease activity (Figure 5B, lanes 4 & 10). Together, these observations indicate that the toxins degrade most, if not all, tRNA substrate in target cells. S1 protection analysis confirmed that tRNAGAUIle is cleaved at the same sites identified from intra-cytoplasmic expression (Figures 4C & 4D, lanes 5 & 6). Analysis of RNA isolated from co-cultures with tsf target bacteria revealed that CdiA-CTEC3006 retains full tRNase activity (Figure 5B, lanes 2 & 3), whereas the CdiA-CTKp342 toxin appears to be inactive in these target cells (Figure 5B, lanes 7 & 8). Thus, wild-type EF-Ts is required for CdiA-CTKp342 mediated nuclease activity and cell killing, but has no influence on intoxication by CdiA-CTEC3006.
Figure 5. tsf mutants are resistant to CdiA-CTKp342, but not CdiA-CTEC3006 intoxication.

A) Competition co-cultures. Target bacteria were co-cultured at a 1:1 ratio with inhibitor cells that deliver either CdiA-CTEC3006 or CdiA-CTKp342. Competitive index is the ratio of viable target to inhibitor cells at 1 h divided by the initial ratio. Data are from three independent experiments together with the average ± SEM. B) Toxin activity in co-cultures. Inhibitor strains were co-cultured with the indicated target cells for 30 min, then RNA was isolated for Northern blot analysis of tRNAGAUIle.
CdiA-CTKp342 and CdiA-CTC3006 are specific for deacylated tRNAIle
To confirm that CdiA-CTKp342 and CdiA-CTEC3006 directly catalyze tRNA cleavage, we tested the nuclease activities of purified toxin domains in vitro. Initial experiments with CdiA-CTEC3006 showed that only a fraction of the tRNAGAUIle substrate in total RNA preparations is cleaved (Figure 6A). We attempted to optimize the reactions using different buffers and Mg2+ supplementation, but still found that about half of the substrate could be converted (Figure 6A). Given that CdiA-CTEC3006 cleaves near the 3′-end of tRNAGAUIle, we reasoned that aminoacylation may affect substrate selection. Using acid-urea gel electrophoresis to resolve aminoacylated from deacylated tRNAs, we found that about half of the tRNAGAUIle is charged in total RNA isolated from E. coli (Figure 6B, lane 1). Analysis of in vitro nuclease reactions on acid-urea gels revealed that CdiA-CTEC3006 preferentially cleaves the deacylated fraction of tRNAGAUIle (Figure 6B, compare lanes 1 & 2). This activity is not due to contaminating RNases, because it can be blocked with purified CdiIEC3006 (Figure 6B, lane 3). Based on these results, we repeated the in vitro nuclease assays with deacylated tRNA and found that this substrate was cleaved efficiently by CdiA-CTEC3006 (Figure 6C, lanes 1 & 2). Though CdiA-CTEC3006 is specific for tRNAGAUIle in vivo, we detected significant activity against tRNAUACVal and tRNAUAALeu in vitro (Figure 6C). In contrast to CdiA-CTEC3006, purified CdiA-CTKp342 shows weak tRNase activity in vitro (Figure 6C, lanes 4 & 5). Because tsf mutants are resistant to CdiA-CTKp342, we reasoned that this toxin probably requires EF-Tu and EF-Ts for full nuclease activity. Indeed, supplementation with purified EF-Tu and EF-Ts greatly stimulates CdiA-CTKp342 activity in vitro (Figure 6C, compare lanes 4 & 6). Both translation factors are required for nuclease activity, because CdiA-CTKp342 does not cleave substrate in reactions supplemented with either EF-Ts or EF-Tu individually (Figure 6D, lanes 3 & 4). The activity is also GTP-dependent (Figure 6D, compare lanes 5 & 6), suggesting that CdiA-CTKp342 recognizes its substrate in the context of tRNA•EF-Tu•GTP ternary complexes. Further, we found that translation factor-activated CdiA-CTKp342 only cleaves deacylated tRNAGAUIle in vitro (Figure 6B, lane 4).
Figure 6. CdiA-CTKp342 and CdiA-CTC3006 are specific for deacylated tRNAGAUIle.

A) In vitro nuclease reactions. Total RNA isolated from E. coli was incubated with purified CdiA-CTEC3006 and analyzed by Northern blotting as described in Methods. Where indicated, reactions were supplemented with CdiIEC3006 or 5 mM MgCl2. B) Acid-urea gel analysis of nuclease reactions. C) In vitro nuclease assays using deacylated tRNA. Where indicated, purified EF-Tu and EF-Ts were included in the reactions. D) CdiA-CTKp342 requires EF-Tu, EF-Ts and GTP to support tRNase activity. Deacylated tRNA was treated with the indicated proteins and tRNAGAUIle analyzed by Northern blotting. E) Acid-urea gel analysis of tRNAGAUIle isolated from mupirocin-treated cells. F) Northern blot analysis of tRNAGAUIle isolated from competition co-cultures in the presence of mupirocin.
The toxins' specificity for uncharged tRNA is unusual and perhaps unprecedented. This finding is particularly surprising for CdiA-CTKp342, because this toxin presumably acts on tRNA•EF-Tu•GTP, and EF-Tu has relatively low affinity for uncharged tRNA (Janiak et al., 1990). Therefore, we asked whether deacylated tRNA is a relevant substrate inside target bacteria. We first treated E. coli target bacteria with the antibiotic mupirocin – which inhibits isoleucyl-tRNA synthetase (Yanagisawa et al., 1994) – to convert all cellular tRNAGAUIle into the deacylated form (Figure 6E). We then introduced mupirocin-resistant inhibitor cells that deploy CdiA-CTEC3006 or CdiA-CTKp342 and incubated the mixed populations for 30 min in the presence of mupirocin. Northern blot analysis revealed tRNase activity in the co-cultures with mupirocin-treated target cells that lack immunity (Figure 6F). Moreover, cleaved tRNAGAUIle levels were roughly equivalent to those observed in co-cultures without mupirocin treatment (compare Figure 6F, lanes 1 & 3 with Figure 5B, lanes 1 & 6), indicating that the antibiotic has no discernable effect on toxin activity in vivo. Taken together, these results demonstrate that both toxins are specific for deacylated tRNAGAUIle.
The nuclease active sites of CdiA-CTKp342 and CdiA-CTEC3006
Finally, we probed the active site of each toxin domain to gain insight into catalysis. BECR RNases are diverse and use a variety of catalytic residues to cleave substrate (Zhang et al., 2014). We chose colicin D as a model to guide mutagenesis because its C-terminal nuclease domain is the closest structural homolog of CdiA-CTKp342 (Table 2), and its active site has been examined in detail (Graille et al., 2004). Structure superimposition shows that CdiA-CTKp342 residues Lys157, Tyr160 and Thr255 are arranged in the same relative positions as catalytic residues Lys608, His611 and Ser677 in colicin D (Figure 7A). The CdiA-CTEC3006 toxin appears to have a similarly configured active site with residues Lys204, Tyr208 and Thr330 (Figure 7B). In addition, Arg174 and Arg252 of CdiA-CTKp342 superimpose with CdiA-CTEC3006 Arg260 and Arg327 (respectively) (Figure 7B), suggesting that they may contribute to substrate binding. The putative catalytic triad of CdiA-CTKp342 is completely conserved between close homologs (Figure S2), whereas homologs of CdiA-CTEC3006 sometimes contain Phe or Trp residues in place of Tyr208 (Figure S3). CdiA-CTKp342 homologs also contain an invariant His170 residue directed toward the putative active site, and this residue may be equivalent to His256 in CdiA-CTEC3006 (Figures S2 & 7B). We first mutated these residues in the context of full-length CdiA chimeras to examine the effects on growth inhibition activity in competition co-cultures. Ala substitutions of CdiA-CTKp342 residues Lys157, Tyr160 and Arg252 completely abrogate inhibition activity (Figure 7C). Mutation of His170 and Thr255A significantly attenuated growth inhibition activity, whereas the Arg174Ala mutation had no discernable effect (Figure 7C). Similar results were obtained with the corresponding mutations made in the CdiA-CTEC3006 domain, though the Arg260Ala mutation abrogates CdiA-CTEC3006 inhibition activity (Figure 7D). We note that each CdiA variant is produced at the same level as wild-type (Figure S4), indicating that functional defects are not the result of low expression or protein destabilization. Northern blot analyses of RNA from these co-cultures showed that inhibition activities are closely correlated with tRNAGAUIle cleavage in target bacteria (Figures 7E & 7F). To further test whether the mutations directly influence nuclease activity, we purified the CdiA-CTEC3006 variant toxin domains and examined their activities on deacylated tRNA in vitro. Overall, CdiA-CTEC3006 in vitro activity correlates well with the competitive fitness of inhibitor bacteria. For example, the Glu236Ala substitution has no apparent effect on competitive fitness (Figure 7D) and the corresponding purified CdiA-CT has the same nuclease activity as wild-type in vitro (Figure 7G). Taken together, these results indicate that the mutations directly affect tRNase activity, either by interfering with substrate binding or catalysis.
Figure 7. The nuclease active sites of CdiA-CTKp342 and CdiA-CTEC3006.

A) Superimposition of CdiA-CTKp342 onto the nuclease domain of colicin D. B) Putative active sites of CdiA-CTKp342 and CdiA-CTEC3006 nuclease domains. C & D) Competition co-cultures. Target bacteria were co-cultured at a 1:1 ratio with inhibitor cells that deliver the indicated CdiA-CTKp342 (panel C) or CdiA-CTEC3006 (panel D) variants. Competitive indices are calculated as the ratio of viable target to inhibitor cells at 1 h divided by the initial ratio. Data are from three independent experiments together with the average ± SEM. See also Figure S4. E & F) Toxin activities in co-cultures. Inhibitor strains were co-cultured with the indicated target cells for 30 min, then total RNA was isolated for Northern blot analysis. G) In vitro nuclease activity of CdiA-CTEC3006 variants. The indicated CdiA-CTEC3006 domains were purified and incubated with deacylated tRNA substrate.
Discussion
The C-terminal toxin domains of CdiAKp342 and CdiAEC3006 share structural homology, and both are specific tRNases that cleave the acceptor stem of tRNAGAUIle. In contrast, the CdiIKp342 and CdiIEC3006 immunity proteins differ radically in both sequence and structure, suggesting that the similarities between their cognate toxins arose through convergent evolution. Because immunity protein function is essential to protect toxin producing cells from self-inhibition, toxin-immunity gene pairs are thought to evolve through iterative mutation cycles in which substitutions that perturb complex formation are compensated by reciprocal changes in the cognate partner that reestablish binding affinity (Morse et al., 2015; Riley, 1993a, b). This phenomenon is apparent in alignments of CdiA-CT•CdiIKp342 and CdiA-CT•CdiIEC3006 with homologous toxin-immunity protein pairs from other bacterial species (Figures S2 & S3). For example, toxin-interacting residues within CdiIEC3006 are not conserved between close homologs (Figure S3B). The predicted immunity proteins for CdiA-CTKp342 homologs exhibit even greater variability, with no significant identity shared by CdiIKp342 and the Pseudomonas immunity proteins in Figure S2B. This latter observation shows that immunity protein sequences can diverge radically during evolution. Nevertheless, each immunity protein in Figure S2B has the same predicted secondary structure as CdiIKp342, suggesting that all fold into dimeric β-sandwiches. The fact that CdiIEC3006 does not share this predominately β fold strongly suggests that it, and its cognate toxin domain, evolved from a different primordial ancestor than the CdiA-CT•CdiIKp342 complex.
CdiA-CTKp342 and CdiA-CTEC3006 also differ in their requirements for extrinsic activation. Purified CdiA-CTEC3006 is competent to cleave substrate in vitro, but CdiA-CTKp342 activity is dependent on translation factors EF-Tu and EF-Ts. Perhaps the unique α-helical subdomain in CdiA-CTEC3006 contributes to substrate binding, and this function is fulfilled by EF-Tu/EF-Ts for CdiA-CTKp342. CDI toxins from E. coli EC869 and E. coli NC101 also require EF-Tu and EF-Ts for activity (Jones et al., 2017; Michalska et al., 2017). These latter toxins also cleave the 3′-ends of specific tRNAs and are thought to use EF-Tu as a scaffold to position substrate in their active sites. Structures of the EF-Tu•CdiA-CT•CdiINC101 complex show that the toxin binds to domain 2 of EF-Tu at a position that partially overlaps with the aminoacylated-tRNA binding site (Michalska et al., 2017). Though CdiA-CTKp342 and CdiA-CTNC101 do not share significant sequence homology, they have similar tertiary structures (see Table 2), raising the possibility that they bind EF-Tu in the same manner. However, the CdiA-CTKp342 toxin clashes with several elements of EF-Tu when modeled in place of CdiA-CTNC101 in the EF-Tu•CdiA-CT•CdiINC101 crystal structure. Helix α4 of CdiA-CTKp342 interferes with EF-Tu strand e2, and there are clashes between the a2-b2 hairpin of the translation factor and α5, β2 and β3 of the toxin (Figure S5). Therefore, CdiA-CTKp342 and CdiA-CTNC101 presumably interact with EF-Tu using distinct contacts, consistent with differences in their cleavage sites on tRNA. CdiA-CTKp342 cleaves within the double-stranded acceptor stem, whereas CdiA-CTNC101 removes the single-stranded 3′ tail from substrate tRNA (Michalska et al., 2017). CdiA-CTKp342 and CdiA-CTNC101 also differ in their responses to aminoacylated substrate. CdiA-CTNC101 appears to act on both aminoacylated and deacylated tRNAs, whereas CdiA-CTKp342 activity is blocked by aminoacylation. The latter result is surprising because EF-Tu-dependent toxins are presumed to recognize substrate in the context of tRNA•EF-Tu•GTP ternary complexes, and EF-Tu binds deacylated tRNA with ~103-fold lower affinity than aminoacyl-tRNA (Janiak et al., 1990). Nonetheless, we find that a substantial proportion of tRNAGAUIle is deacylated in E. coli cells (see Figure 6A), and this substrate is rapidly degraded upon intoxication with CdiA-CTKp342. These observations suggest that EF-Tu binds deacylated tRNA in vivo. In principle, this interaction could be driven by mass action, because the intracellular concentration of EF-Tu (~50 μM) is about 20-fold higher than the dissociation constant (~2.5 μM) for binding to deacylated tRNA. Alternatively, EF-Tu may promote nuclease activity independent of its tRNA-binding properties, perhaps functioning as a chaperone to stabilize the toxin. We note that translation factors are not intrinsically required for acceptor-stem tRNase activity, because CdiA-CTEC3006 catalyzes a similar reaction in the absence of EF-Tu and EF-Ts.
To our knowledge, CdiA-CTKp342 and CdiA-CTEC3006 are the first RNases reported to act specifically on tRNAGAUIle. Though this substrate specificity is novel, other isoacceptor specific tRNases have been identified over the past 30 years and several of these nucleases mediate interbacterial competition. Colicins E5 and D are diffusible protein toxins released by certain strains of E. coli to kill non-isogenic competitors (Cascales et al., 2007). Colicin E5 carries a C-terminal tRNase domain that cleaves the anticodon loops of tRNAAsn, tRNAAsp, tRNAHis and tRNATyr (Ogawa et al., 1999). Similarly, colicin D carries an anticodon nuclease domain that is specific for tRNAArg isoacceptors (Tomita et al., 2000). We note that the colicin D nuclease domain is the closest known structural homolog of CdiA-CTKp342, demonstrating that the BECR fold can adapt to recognize different tRNA subdomains. Other CdiA effectors also deliver toxic tRNase domains. CdiABp1026b from B. pseudomallei 1026b carries a C-terminal PD-(D/E)XK phosphodiesterase domain that preferentially cleaves the acceptor stem of tRNAAla (Koskiniemi et al., 2013; Nikolakakis et al., 2012), and the EndoU RNase domain in CdiASTECO31 from E. coli STEC_O31 is a tRNAGlu specific anticodon nuclease (Michalska et al., 2018). Specific tRNase toxins are also deployed for inter-strain competition between Gram-positive bacteria. The large cell wall-associated proteins (WapA) of Bacillus subtilis are functionally analogous to CdiA effectors and inhibit neighboring bacteria with their C-terminal toxin domains. The WapA toxin domain from B. subtilis subsp. 'natto' cleaves tRNAGlu in a similar manner as the CdiA-CTSTECO31 toxin, and the toxin from B. subtilis subsp. spizizenii T-UB-10 cleaves the tRNASer anticodon (Koskiniemi et al., 2013). Isoacceptor specific tRNases also play roles beyond competition. E. coli PrrC was the first isoacceptor specific anticodon nuclease to be discovered, and this intriguing enzyme functions as a type of innate immunity to viral infection (Levitz et al., 1990). PrrC is a latent anticodon nuclease that is activated in response to bacteriophage T4 infection to cleave tRNALys. This activity blocks protein synthesis, thereby inhibiting phage production to protect neighboring sibling cells that have yet to become infected. More recently, type II proteic toxin-antitoxin (TA) loci have been discovered to encode specific anticodon nucleases. The biological functions of type II TA systems remain enigmatic despite considerable research over the past two decades, but prevailing models postulate that these toxins regulate bacterial growth in response to environmental cues (Gerdes and Maisonneuve, 2012; Van Melderen, 2010). VapC toxins from Salmonella and Shigella inactivate initiator tRNAiMet to arrest protein synthesis (Winther and Gerdes, 2011), and the numerous VapC paralogs of Mycobacterium tuberculosis exhibit specificities for tRNAAla, tRNACys, tRNALeu, tRNAGln, tRNASer and tRNATrp (Cruz et al., 2015; Winther et al., 2016). Finally, the MazF-mt9 toxin from M. tuberculosis was recently shown to have tRNALys specific anticodon nuclease activity (Schifano et al., 2016). The growing number of isoacceptor specific nucleases underscores the critical role of tRNA in the control of bacterial growth and viability.
CdiA proteins are characterized by their variable C-terminal toxin domains, which can be exchanged between effectors to produce functional chimeras (Aoki et al., 2010; Nikolakakis et al., 2012). This modularity allows the cell to periodically change the toxin it deploys through genetic recombination. Metagenomic analyses strongly suggest that new cdiA-CT/cdiI gene pairs are acquired through horizontal gene transfer and fused to cdiA via homologous recombination or site-specific integrase activity (Arenas et al., 2013; Poole et al., 2011). Thus, closely related toxin domains are often found distributed across phylogenetically diverse species. This is illustrated in database searches for CdiA-CTKp342 and CdiA-CTEC3006 homologs, which return CdiA proteins from several different species. For example, CdiA-CTKp342 homologs are found in predicted CdiA effectors from Yersinia, Dickeya, Achromobacter, Pseudomonas and Burkholderia species (Table S2 & Figure S2). CdiA-CTKp342 and CdiA-CTEC3006 homologs are also commonly found at the C-terminus of Rhs/YD-peptide repeat proteins (Table S2), which constitute another important group of anti-bacterial effectors. In Gram-negative bacteria, Rhs proteins are exported through type VI secretion systems (T6SS) by virtue of their non-covalent interactions with VgrG (Hachani et al., 2014; Koskiniemi et al., 2013; Shneider et al., 2013; Whitney et al., 2014). The Rhs effectors identified in Table S2 are presumably also deployed in a T6SS-dependent manner, because they contain N-terminal PAAR domains, which interact with the C-terminal β-spike of trimeric VgrG (Shneider et al., 2013). The YD-repeat containing WapA protein of B. subtilis carries an N-terminal signal peptide for export through the SecYEG translocon (Koskiniemi et al., 2013). In contrast, the Streptomyces YD-peptide repeat proteins identified in Table S2 lack recognizable signal peptides, raising the possibility that they are deployed through a specialized secretion system. Lastly, we identified two PrsW metalloproteases from Gram-positive Paenibacillus species that carry C-terminal domains that share 32-36% sequence identity with CdiA-CTEC3006 (Table S2). PrsW-mediated toxin delivery has yet to be demonstrated experimentally, but their linkage to several polymorphic toxin domain families strongly suggests that these integral membrane proteins function in intercellular competition as first suggested by Aravind and colleagues (Zhang et al., 2012; Zhang et al., 2011).
Finally, this study highlights the limitations of homology modeling to predict protein structure and function. CdiA-CTKp342 and CdiA-CTEC3006 are both BECR-fold RNases, yet the domains are not annotated as such in current databases. The BECR superfamily was first defined by Aravind and coworkers, who recognized that the αββββ core of barnase is present in other microbial RNase families (Zhang et al., 2012). In addition to known RNases, Zhang et al. also identified 10 uncharacterized toxin subgroups that are predicted to adopt the BECR core fold. These latter novel toxin (Ntox) subgroups are differentiated by distinct sequence motifs that presumably contribute to substrate binding and catalysis. To date, only the Ntox21 group (Pfam: PF15526) has been examined experimentally, and that study confirmed predictions that the domain adopts the BECR fold and cleaves 16S rRNA in the same manner as colicin E3 (Beck et al., 2014; Zhang et al., 2012). Most databases recognize the Ntox35 (PF15534), Ntox47 (PF15540) and Ntox50 (PF15542) BECR groups, but the Ntox7, Ntox19, Ntox36, Ntox41 and Ntox48 families are not listed in the current Pfam 32.0 database. The Ntox48 group may have been misclassified initially, because the CdiA-CT domain from Dickeya zeae Ech1591 (NCBI: ACT06855.1) that was originally cited as the family paragon is now annotated as an EHHH/Endo VII nuclease (PF15657). It is less clear why the remaining five BECR groups are no longer extant. CdiA effectors commonly carry C-terminal Ntox7, Ntox19, Ntox41 and Ntox49 domains, and the CdiA-CT Ntox7 domain from Y. pestis Pestoides A inhibits target bacteria when fused to an E. coli CdiA protein (Willett et al., 2015a). We note that the diverse active sites of BECR nucleases pose a significant challenge for homology-based predictions. Some BECR nucleases have unusual active sites that lack the canonical His and Glu catalytic residues typically found in RNases. For example, E. coli RelE contains no His residues and instead appears to use Lys54 as general base to abstract a proton from the 2′-hydroxyl of its substrate, and Arg81 as an acid to protonate the 5′ leaving group (Dunican et al., 2015; Griffin et al., 2013). Similarly, colicin E5 lacks a catalytic His residue and likely uses Lys or Gln residues to initiate cleavage (Yajima et al., 2006). RelE and colicin E5 also carry unique C-terminal extensions that are critical for substrate recognition (Neubauer et al., 2009; Yajima et al., 2006). BECR active-site plasticity is further compounded by the tendency for catalytic residues to migrate between secondary structure elements (Zhang et al., 2014). The catalytic triad of barnase is contained within the last two strands of the core, but the Lys-His and Lys-Tyr motifs of colicin D and CdiA-CTKp342 emanate from N-terminal α-helices that are not part of the BECR core. Similar structural plasticity is well documented for the PD-(D/E)-XK superfamily of phosphodiesterases, which are notoriously difficult to identify though sequence analysis (Steczkiewicz et al., 2012). We anticipate that many more BECR core variations will be discovered as more superfamily members are identified and characterized.
STAR Methods
Lead contact and materials availability
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Christopher Hayes (chayes@lifesci.ucsb.edu).
Experimental model and subject details
Bacterial growth conditions.
Bacterial strains are presented in the Key Resources Table. Bacteria were cultured in M9 minimal medium, lysogeny broth (LB) or on LB agar at 37 °C. Unless indicated otherwise, media were supplemented with antibiotics at the following concentrations: 150 μg/mL ampicillin (Amp), 100 μg/mL chloramphenicol (Cm), 50 μg/mL kanamycin (Kan), 200 μg/mL spectinomycin (Spc), and 25 μg/mL tetracycline (Tet).
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| rabbit polyclonal antisera to the CdiAEC393 TPS transport domain (residues Val33 – Gly285) | (Ruhe et al., 2015) | N/A |
| IRDye® 800CW goat anti-rabbit IgG | LI-COR | Cat# P/N 925-32211 |
| Bacterial and Virus Strains | ||
| E. coli B sub-strain BL21 (DE3) | Agilent | Cat# 200131 |
| E. coli K-12 sub-strain MG1655 | E. coli Genetic Stock Center | Strain #7740 |
| E. coli K-12 sub-strain EPI100 | Epicentre/Lucigen | Cat# EC10010 |
| E. coli K-12 sub-strain X90 | N/A | |
| E. coli: CH2016: X90 (DE3) Δrna ΔslyD::kan | (Garza-Sanchez et al., 2011) | N/A |
| E. coli: CH7157: X90 ΔclpX ΔclpA::kan | (Nikolakakis et al., 2012) | N/A |
| E. coli: CH8251: MC4100 rifR | (Willett et al., 2015) | N/A |
| E. coli: CH12738: MG1655 ara::spc | (Jones et al., 2017) | N/A |
| E. coli: CH12739: MG1655 ara::spc tsf(A202E) | (Jones et al., 2017) | N/A |
| E. coli: CH12740: MG1655 ara::spc tsf(Δcoil) | (Jones et al., 2017) | N/A |
| E. coli: CH15087: EPI100 mupR | This paper | N/A |
| E. coliI: DY378: W3110 lcI857 Δ(cro-bioA) | (Thomason et al., 2007) | N/A |
| Biological Samples | ||
| Chemicals, Peptides, and Recombinant Proteins | ||
| Critical Commercial Assays | ||
| The Protein Complex Suite | Qiagen | |
| Deposited Data | ||
| Structure data and refinement statistics for CdiA-CT•CdiIKp342 complex | This paper | PDB: 6CP9 |
| Structure data and refinement statistics for CdiA-CT•CdiIEC3006 complex | This paper | PDB: 6CP8 |
| Experimental Models: Cell Lines | ||
| Experimental Models: Organisms/Strains | ||
| Oligonucleotides | ||
| Complete list of oligonucleotides and PCR primers | This paper | see Table S3 |
| Recombinant DNA | ||
| pET21b | Novagen | Cat# 69741-3 |
| pTrc99A | Amersham | N/A |
| pCH978 | This paper | N/A |
| pCH6283 | Genscript | N/A |
| pCH6289 | Genscript | N/A |
| pCH7171 | (Johnson et al., 2016) | N/A |
| pCH8001 | (Beck et al., 2014) | N/A |
| pCH10163 | (Morse et al., 2012) | N/A |
| pCH11483 | (Willett et al., 2015) | N/A |
| pCH11526 | (Willett et al., 2015) | N/A |
| pCH11948 | This paper | N/A |
| pCH12158 | This paper | N/A |
| pCH12602 | (Jones et al., 2017) | N/A |
| pCH12603 | (Jones et al., 2017) | N/A |
| pCH12802 | This paper | N/A |
| pCH12861 | This paper | N/A |
| pCH12865 | This paper | N/A |
| pCH12898 | This paper | N/A |
| pCH13677 | (Willett et al., 2015) | N/A |
| pCH13813 | This paper | N/A |
| pCH13887 | This paper | N/A |
| pCH13888 | This paper | N/A |
| pCH13890 | This paper | N/A |
| pCH13892 | This paper | N/A |
| pCH13893 | This paper | N/A |
| pCH13894 | This paper | N/A |
| pCH13895 | This paper | N/A |
| pCH13896 | This paper | N/A |
| pCH14200 | This paper | N/A |
| pCH14201 | This paper | N/A |
| pCH14278 | This paper | N/A |
| pCH14279 | This paper | N/A |
| pCH14280 | This paper | N/A |
| pCH14281 | This paper | N/A |
| pCH14282 | This paper | N/A |
| pCH14301 | This paper | N/A |
| pCH14912 | This paper | N/A |
| pCH14913 | This paper | N/A |
| pCH14982 | This paper | N/A |
| pMCSG63 | (Eschenfeldt et al., 2009) | N/A |
| pMCSG63-APC111476 | This paper | N/A |
| pMCSG63-APC200209 | This paper | N/A |
| pMCSG63-APC200215 | This paper | N/A |
| Software and Algorithms | ||
| Chimera | (Pettersen et al., 2004) | https://www.cgl.ucsf.edu/chimera/ |
| PHENIX | (Adams et al., 2010) | https://www.phenix-online.org/ |
| Coot | (Emsley and Cowtan, 2004) | https://www2.mrc-lmb.cam.ac.uk/personal/pemsley/coot/ |
| HKL-3000 | (Minor et al., 2006) | http://www.hkl-xray.com/hkl-3000 |
| Other | ||
Method details
Plasmid constructions.
Plasmids are presented in the Key Resources Table and oligonucleotide primers are listed in Table S3.
Protein over-production plasmids
The coding sequences for CdiA-CT/CdiIKp342 and CdiA-CT/CdiIEC3006 were synthesized by Genscript (Piscataway, NJ) and supplied in the pUC57 vector (pCH6289 and pCH6283). The cdiIEC3006 gene was amplified using primers 209CdiIF52/209CdiIR45, and the product inserted into the SspI ligation independent cloning (LIC) site of pMCSG63 (Eschenfeldt et al., 2009; Eschenfeldt et al., 2013) to generate pMCSG63-APC111476, which produces CdiIEC3006 carrying an N-terminal His6 tag. The cdiA-CTEC006 coding sequence was amplified with primers 209CdiAF43/209CdiAR46, and the product inserted into the SmaI LIC site of plasmid pMCSG63-APC111476. Sequencing revealed that most clones acquired frame-shift mutations that inactivate the toxin gene. One clone (pMCSG63-APC200209) was identified with no frame-shifts, though it contains an in-frame deletion of the codon for Asn332. The cdiA-CT-cdiIKp342 module was amplified with primers 215CdiAF41/215CdiIR57, and the product inserted into pMCSG63 as described above to produce plasmid pMCSG63-APC200215, which produces CdiA-CTKp342 with an N-terminal His6 tag and untagged CdiIKp342.
For biochemical analyses, the cdiA-CT/cdiIKp342 and cdiA-CT/cdiIEC3006 modules were amplified with primers CH3962/CH3685 and CH4304/CH3425 (respectively), and ligated to pCH8001 (Beck et al., 2014) via KpnI/SpeI restriction sites to generate plasmids pCH12861 and pCH978 (respectively). Plasmid sequences were confirmed by DNA sequencing, and therefore wild-type toxins were used for all functional assays. For in vivo toxin activation experiments, the cdiA-CT/cdiI modules were subcloned into pCH7171 using NcoI/SpeI restriction sites to append ssrA(DAS) degrons onto CdiIKp342 (pCH12158) and CdiIEC3006 (pCH13677). The cdiIKp342 and cdiIEC3006 genes were amplified with primers CH3965/CH3685 and CH3244/CH3425 (respectively) and ligated via KpnI/SpeI to pCH8001 to generate plasmids pCH12898 and pCH12802 for the purification of CdiIKp342-His6 and CdiIEC3006-His6, respectively. The immunity genes were also amplified with CH3965/CH3570 and CH3244/CH3245 (respectively) and ligated to pTrc99aKX via KpnI/XhoI restriction site to generate plasmids pCH12865 and pCH11526, which were used to express native immunity genes in target bacteria during competition co-culture experiments.
Site-directed mutagenesis
Site-directed mutagenesis of cdiA-CTEC3006 was performed by overlap-extension (OE-PCR) or megaprimer PCR (Aiyar et al., 1996) using primer pair CH4304/CH3425 in conjunction with: CH4276 for Lys204Ala (pCH14301); CH4699/CH4700 for Tyr208Ala (pCH14982); CH4252/CH4253 for His256Ala (pCH13887); CH4250/CH4251 for Glu259Ala (pCH14200); CH4254/CH4255 for Arg260Ala (pCH13888); CH4256/CH4257 for Arg327Ala (pCH14201); and CH4277 for Thr330Ala (pCH13890). All final products were digested with KpnI/SpeI and ligated to pCH8001 to generate protein overexpression constructs. The same general procedure was used introduce site-directed mutations into cdiA-CTKp342. Primer pair CH3962/CH3685 was used in conjunction with CH4438/CH4439 for Lys157Ala; CH4697/CH4698 for Tyr160Ala; CH4440/CH4441 for His170Ala; CH4442/CH4443 for Arg174Ala; CH4444/CH4445 for Arg252Ala; and CH4446/CH4447 for Thr255Ala.
Chimeric CdiA constructions
Chimeric cdiAEC93-CTKp342 expression constructs were generated by allelic exchange of the counter-selectable pheS* marker from plasmid pCH10163 (Morse et al., 2012). All cdiA-CT/cdiIKp342 alleles were first amplified using primers CH3517/CH3518. The resulting products were fused to DNA fragments amplified from regions upstream and downstream of the cdiAEC93 gene. The upstream homology fragment was amplified using primers CH4100/CH4101, and the downstream fragment with primers CH4102/CH4103. The upstream and downstream homology fragments were then fused to cdiA-CT/cdiIKp342 modules using OE-PCR with primers CH4100/CH4103. The final DNA products (100 ng) were electroporated together with plasmid pCH10163 (300 ng) into E. coli DY378 cells (Thomason et al., 2007). Recombinant clones encoding wild-type (pCH11948), Lys157Ala (pCH14278), Tyr160Ala (pCH14912), His170Ala (pCH14279), Arg174Ala (pCH14280), Arg252Ala (pCH14281), Thr255Ala (pCH14282) alleles of CdiA-CTKp342 were isolated on yeast extract glucose-agar supplemented with 33 μg/mL chloramphenicol and 10 mm D/L-p-chlorophenylalanine. The chimeric cdiAEC93-CTEC3006 expression plasmid pCH11483 has been described (Willett et al., 2015a), and derivative constructs encoding Lys204Ala (pCH13892), Tyr208Ala (pCH14913), His256Ala (pCH13893), Glu259Ala (pCH13813), Arg260Ala (pCH13894), Arg327Ala (pCH13895) and Thr330Ala (pCH13896) variants of CdiA-CTEC3006 were isolated as described above.
Protein expression, purification and crystallization.
A single colony was inoculated into 2 mL of LB medium supplemented with 100 μg/mL ampicillin and incubated at 37 °C with shaking for 6 h. LB cultures were then diluted 1:100 into 50 mL of M9 medium supplemented with 0.5% glycerol, 100 μg/mL of ampicillin, trace minerals and vitamins. Cells continued to grow overnight at 37 °C for preparation of large scale growth. Overnight culture was diluted 1:100 into 1 L M9 medium containing 0.5% glycerol, 100 μg/mL ampicillin and trace minerals, cells were grown to an OD600 ~ 0.8 and cooled to 18 °C. Selenomethionine (SeMet) was added at a final concentration of 60 μg/mL together with L-isoleucine, L-leucine, L-lysine, L-phenylalanine L-threonine and L-valine to a final concentration of 100 μg/mL and cells were induced with 0.5 mM isopropyl-D-thiogalactopyranoside (IPTG). Cultures were grown overnight at 18°C. The cells were harvested, cell pellets washed and resuspended in 50 mM Tris-HCl (pH 8.0), 500 mM NaCl, 10 mM 2-mercaptoethanol (2-ME), 10% glycerol. Cells were broken in Fast Break reagent (Promega Madison, Wisconsin) supplemented with 10 μg/mL lysozyme and Complete Protease Inhibitor Cocktail (Roche Mannheim, Germany). Cell lysates were centrifuged and the supernatants filtered. A Nickel (II) Sepharose HisTrap column (GE Healthcare Uppsala Sweden) was used for purification of the proteins. Fractions were loaded onto a Hiload 26/60 Superdex200 size exclusion column equilibrated with 20 mM Tris-HCl (pH 7.5) 150 mM NaCl, 2 mM dithiothreitol. Fractions were pooled and concentrated using an Amicon Ultracel 10K centrifugal concentrator. The CdiA-CT•CdiIEC3006 and CdiA-CT•CdiIKp342 complexes were concentrated to 12 mg/mL or 10 mg/mL and subsequently incubated overnight on ice with chymotrypsin (20 ng/μL) or trypsin (40 ng/μL). This treatment produced a ~19 kDa fragment of CdiA-CTEC3006, whereas CdiA-CTKp342 was converted into two species of ~14 and ~15 kDa. The digested samples were crystallized directly using the Protein Complex Suite (Qiagen) crystallization screen at 4 °C. CdiA-CT•CdiI EC3006 crystallized from 0.1 M HEPES (pH 7.0), 18% PEG-12,000. CdiA-CT•CdiIKp342 crystal grew in the presence of 0.1 M sodium citrate (pH 5.0), 8% PEG-8000.
Data collection, structure solution and refinement.
Crystals were cryo-protected in reservoir solution supplemented with 17% (CdiA-CT•CdiIEC3006) or 27% (CdiA-CT•CdiIKp342) glycerol and flash-cooled in liquid nitrogen. Diffraction images were recorded on the ADSC Q315r detector at Structural Biology Center 19-ID beamline at the Advanced Photon Source, Argonne National Laboratory. Single-wavelength anomalous diffraction (SAD) datasets were collected at 100K near the selenium K-absorption edge to utilize selenium anomalous signal for phasing. Complex crystals diffracted to 2.20 and 2.55 Å for CdiA-CT•CdiIEC3006 and CdiA-CT•CdiIKp342, respectively. The images were processed with the HKL3000 suite (Minor et al., 2006). Intensities were converted to structure factor amplitudes in the Ctruncate program (French and Wilson, 1978; Padilla and Yeates, 2003) from the CCP4 package (Winn et al., 2011). The data collection and processing statistics are given in Table 1.
The structures were solved by the SAD phasing with selenium peak data in the HKL-3000 software pipeline (Minor et al., 2006), utilizing SHELXD, SHELXE (Sheldrick, 2008), MLPHARE (Otwinowski, 1991) and DM (Cowtan, 1994) for heavy atom search and phasing. The initial protein models were built by the HKL builder utilizing Buccaneer (Cowtan, 2006). The final models were obtained through alternating manual rebuilding in Coot (Emsley and Cowtan, 2004) and crystallographic refinement in Refmac (Murshudov et al., 1997; Winn et al., 2011) and Phenix (Adams et al., 2010). In both cases, refinement included optimization of TLS parameters with 37 groups for the CdiA-CT•CdiIEC3006 model and 62 groups for CdiA-CT•CdiIKp342. The CdiA-CT•CdiIEC3006 structure contains two copies of the complex, with the following residues most likely present in the crystallized material but not modeled due to the lack of interpretable electron density: Asn174 (chains A and B), Met1 – Asn3 (chain C) and Met1 – Val4 (chain D). The CdiA-CT•CdiIKp342 model contains four copies of the complex, with the following residues missing from the final model: Val139 – Thr143 and Val262 – Lys264 (chain A), Arg116 (chain B), Val139 – 144 and Val262 – Lys264 (chain C), Val139 – Ile142 and Val262 – Lys264 (chain E), Val139 – Asn150 and Asn260 – Arg264 (chain G), and Ile115 – Arg116 (chain H). Refinement statistics are presented in Table 1. The atomic coordinates and structure factors have been deposited in the Protein Data Bank under accession codes 6CP8 (CdiA-CT•CdiIEC3006) and 6CP9 (CdiA-CT•CdiIKp342). All structure figures were prepared using UCSF Chimera software (Pettersen et al., 2004).
Protein purification for biochemical analyses.
E. coli CH2016 cells harboring expression plasmids were cultured at 37 °C with shaking in LB media suppleme nted with 150 μg/mL ampicillin. Cultures were adjusted to 1 mM IPTG at mid-log phase (OD600 ~ 0.7) and incubated for an additional 2 h. Cells were harvested by centrifugation and frozen at −80 °C. C ell pellets were re-suspended in lysis buffer [50 mM Tris-HCl (pH 7.5), 150 mM NaCl, 10 mM 2-ME, 0.05% Triton X-100, 20 mM imidazole] and broken by two passages through a French press at 20,000 psi. Cell debris was removed by two rounds of centrifugation at 16,000 ×g at 4 °C. His6-TrxA-EF-Tu (from pCH12603), His6-TrxA-EF-Ts (from pCH12602) and CdiI-His6 proteins were purified by Ni2+-affinity chromatography in lysis buffer and eluted with 20 mM Tris-HCl (pH 7.5), 250 mM imidazole. Imidazole was removed by dialysis against 20 mM sodium phosphate (pH 7.8), 150 mM NaCl, 10 mM 2-ME. Native EF-Tu and EF-Ts were cleaved from the fusion proteins with TEV protease, followed by passage over a Ni2+-NTA agarose column to remove the N-terminal His6-TrxA fragments and TEV protease. Toxins were purified by first isolating the CdiA-CT•CdiIKp342-His6 and CdiA-CT•CdiIEC3006-His6 complexes on Ni2+-NTA agarose as described above. Toxins were eluted with denaturing buffer, 6 M guanidine-HCl in 20 mM Tris-HCl (pH 7.5). The isolated toxins were then refolded by dialysis against 20 mM sodium phosphate (pH 7.8), 150 mM NaCl, 10 mM 2-ME. Purified proteins were quantified by absorbance at 280 nm using the following extinction coefficients: CdiA-CTKp342, 13,075 cm−1 M−1; CdiIKp342-His6, 11,460 cm−1 M−1; CdiA-CTEC3006, 23,505 cm−1 M−1, CdiIEC3006-His6, 21,890 cm−1 M−1; EF-Tu, 20,400 cm−1 M−1 and EF-Ts, 4,470 cm−1 M−1
In vivo toxin activity and competition co-cultures.
E. coli X90 cells carrying plasmids pCH12158 (CdiA-CT•CdiIKp342-DAS) and pCH12599 (CdiA-CT•CdiIEC3006-DAS) were grown in tetracycline-supplemented LB media for 1 h (OD600 ~ 0.1), then expression was induced with 0.4% L-arabinose. Induced cultures were incubated at 37 °C with shaking for 3 h, harve sted into an equal volume of ice-cold methanol and cell pellets frozen at −80 °C prior to RNA extracti on.
E. coli EPI100 inhibitor strains that deliver CdiA-CTKp342 and CdiA-CTEC3006 derivatives were mixed with at a 1:1 ratio with rifampicin-resistant E. coli MC4100 target cells that harbor empty vector plasmid pTrc99A (cdiI−) pCH12865 (cdiIKp342) or pCH11526 (cdiIEC3006) in shaking LB media without antibiotics. Viable inhibitor and target bacteria were enumerated (as colony forming units (cfu) per mL on selective media) upon initial mixing and after 1 h of co-culture. Target cell fitness is expressed as the competitive index, which is calculated as the final ratio of target to inhibitor cells divided by the initial ratio at t = 0. Competitive indices from three independent experiments are reported together with the average ± standard error of the mean. For the analysis of toxic tRNase activity, samples were harvested into an equal volume of ice-cold methanol after 30 min of co-culture. Cells were collected by centrifugation and frozen at −80 °C for subsequent RNA extraction.
SDS-PAGE and immunoblot analysis.
E. coli EPI100 cells carrying chimeric CDI expression plasmids were diluted to OD600 ~ 0.05 in LB medium supplemented with Amp and cultured with shaking at 37 °C. Once in mid-log phase, cultures were treated with Spc for 20 min to block protein synthesis. Cells were harvested by centrifugation and frozen at −80 °C. F rozen cell pellets were re-suspended in urea-lysis buffer [50% urea, 150 mM NaCl, 20 mM Tris-HCl (pH 8.0)] and subjected a freeze-thaw cycle to extract proteins for SDS-PAGE and immunoblotting. Urea-soluble protein samples (5 μL) were analyzed by SDS-PAGE on Tris-tricine 6% polyacrylamide gels run at 100 V (constant) for 3 h. Gels were soaked for 15 min in 25 mM Tris, 192 mM glycine (pH 8.6), 10% methanol, then electroblotted to low-fluorescence PVDF membranes using a semi-dry transfer apparatus at 17 V (constant) for 1 h. Membranes were blocked with 4% non-fat milk in PBS for 1 h at room temperature, and incubated with primary antibodies in 0.1% non-fat milk, PBS overnight at 4 °C. Rabbit polyclonal antisera to the N-terminal TPS domain was used at a 1:10,000 dilution (Ruhe et al., 2015). Blots were incubated with 800CW-conjugated goat anti-rabbit IgG (1:40,000 dilution, LICOR) in 0.1% non-fat milk in PBS. Immunoblots were visualized with a LI-COR Odyssey infrared imager.
RNA isolation and analyses.
Frozen cell pellets were resuspended in guanidinium isothiocyanate (GITC)-phenol and total RNA extracted as described (Garza-Sánchez et al., 2006). RNAs (5 μg) were run on 50% urea/7.5% polyacrylamide gels buffered with 0.5× Tris-borate EDTA and electroblotted to positively charged nylon membranes for Northern blot analysis. Charged and deacylated tRNAs were resolved on 50% urea/10% polyacrylamide gels buffered with 100 mM sodium acetate (pH 5.5) as described (Janssen and Hayes, 2009). Blots were hybridized with [32P]-labeled oligonucleotide probes specific for E. coli tRNAGAUIle (CH577), tRNAUACVal (CH1248), tRNAUUCGlu (CH1417), tRNAUAALeu (CH2036) and tRNAGUATyr (CH798) (Table S3) and visualized by phosphorimaging using Bio-Rad Quantity One software. S1 nuclease protection analysis was performed as described (Hayes and Sauer, 2003) to map cleavage sites at the 3′-end of tRNAGAUIle. The S1 probe (CH3931) and marker oligonucleotides (CH3932, CH3933 and CH3934) were first radiolabeled at their 3′-termini with [α-32P]-cordycepin triphosphate (Perkin Elmer) and terminal transferase. Labeled oligonucleotides were then passed over a G-25 spin column and treated with unlabeled ATP and T4 polynucleotide kinase to phosphorylate the 5′-termini. The radiolabeled probe was hybridized with RNA samples for 4 h at 50 °C. Hybridization reactions were digested with S1 n uclease at 37 °C for 30 min, then quenched with sodium acetate (pH 5.0) and precipitated with 90% ethanol. The S1 nuclease reactions were run on 50% urea/10% polyacrylamide gels buffered with 0.5× Tris-borate-EDTA and visualized on a Bio-Rad phosphorimager using Quantity One software.
In vitro nuclease assays.
In vitro nuclease assays were performed in reaction buffer [20 mM Tris-HCl (pH 7.5), 100 mM NaCl, 5 mM MgCl2, 10 mM 2-ME and 100 μg/μL bovine serum albumin] with toxins used at 1 μM final concentration. Where indicated, purified EF-Tu (1 μM) and EF-Ts (1 μM) were included, and these reactions were supplemented with 1 mM GTP. Immunity proteins were used at 3 μM final concentration. Protein mixtures were equilibrated for 30 min at room temperature. Reactions were then initiated by addition of E. coli total RNA to final concentration of 800 ng/μL, followed by incubation for 1 h at 37 °C. Substrate tRNAs were d eacylated at pH 8.9 for 1 h at 37 °C. Reactions were quenched with an equal volume of 2× SDS-urea gel loading buffer and run on 50% urea, 7.5% polyacrylamide gels buffered with 0.5× Tris-borate-EDTA. Gels were electroblotted to nylon membranes for hybridization with radiolabeled oligonucleotide probes as described above.
Quantification and statistical analysis
All competition co-cultures were performed as three independent experiments on separate days. Competitive indices are reported as the average ± SEM as outlined in the figure legends and Method Details.
Data and software availability
Structure datasets generated during this study are available in the Protein Data Bank under accession numbers 6CP8 (http://www.rcsb.org/structure/6CP8) and 6CP9 (http://www.rcsb.org/structure/6CP9).
Supplementary Material
Figure S1. Domain architectures of CdiAKp342 and CdiAEC3006, Related to Figures 1 & 2. The N-terminal signal sequence (ss) and TPS transport domain are required for secretion of CdiA across the cytoplasmic and outer membranes (respectively) of inhibitor cells. The filamentous hemagglutinin-1 (FHA-1) domain is composed of peptide repeats (Pfam: PF05594) that form a β-helix; and the FHA-2 repeat (PF13332) domain is predicted to translocate CdiA-CT toxins across the target-cell outer membrane. The receptor-binding domain (RBD) resides between the FHA repeat domains. The CdiAEC3006 RBD recognizes BamA on target cells (Ruhe et al., 2017), and the binding specificity of the RBD from CdiAKp342 has not been determined. The Tyr/Pro-enriched (YP) domain is required for stable presentation of CdiA on the surface of inhibitor cells (Ruhe et al., 2018). The pre-toxin (PT) domain is thought to act as an auto-protease, cleaving the C-terminal (CdiA-CT) region near the conserved VENN peptide motif to enable toxin delivery into target bacteria. The polymorphic CdiA-CT region is composed of two domains. The extreme C-terminal domains is typically a toxic nuclease, whereas the N-terminal "cytoplasm-entry" domain mediates toxin translocation into the target-cell cytosol. The entry domain from CdiAEC3006 hijacks the D-glucose transporter PtsG to translocate into target cells, whereas the entry domain of CdiAKp342 is uncharacterized. NCBI Refseq identifiers for each effector are given in parentheses.
Figure S2. Homologs of CdiA-CTKp342 and CdiIKp342, Related to Figure 1C. A) Homologs of CdiA-CTKp342 were aligned using Clustal omega and rendered using the ESPript 3.0 server. Secondary structure elements for CdiA-CTKp342 are shown above the alignment. Residues that make direct contacts with CdiIKp342 are indicated by purple circles. Sequences are from the following bacteria with NCBI protein identifiers given in parentheses: K. pneumoniae 342 (WP_012542573.1), Dickeya zeae Ech586 (WP_012884592.1), Yersinia pseudotuberculosis IP 31758 (WP_012104437.1), Snodgrassella alvi wkB237 (WP_100141475.1), Burkholderia latens RF32-BP12 (WP_059545330.1), Achromobacter sp. 2789STDY5608608 (CUJ27995.1), Pseudomonas sp. BS3767 (SDI04727.1), Pseudomonas syringae Riq4 (WP_080370524.1), Pseudomonas sp. URIL14HWK12:I9 (WP_097084422.1). B) CdiIKp342 homologs were aligned as in panel A. Residues that make direct contacts with CdiA-CTKp342 are indicated by yellow circles. Sequences are from the following bacteria with NCBI protein identifiers given in parentheses: K. pneumoniae 342 (WP_041165391.1), D. zeae Ech586 (WP_012884593.1), Y. pseudotuberculosis IP 31758 (WP_011193122.1), S. alvi wkB237 (WP_100141474.1), B. latens RF32-BP12 (unannotated), Achromobacter sp. 2789STDY5608608 (unannotated), Pseudomonas sp. BS3767 (SDI04696.1), P. syringae Riq4 (WP_052966432.1), Pseudomonas sp. URIL14HWK12:I9 (WP_097084423.1).
Figure S3. Homologs of CdiA-CTEC3006 and CdiIEC3006, Related to Figure 2B. A) Homologs of CdiA-CTEC3006 were aligned using Clustal omega and rendered using the ESPript 3.0 server. Secondary structure elements for CdiA-CTEC3006 are shown above the alignment. Residues that make direct contacts with CdiIEC3006 are indicated by light blue circles. Sequences are from the following bacteria with NCBI protein identifiers given in parentheses: E. coli 3006 (EKI34460.1), Pectobacterium carotovorum F157 (PPE62299.1), Dickeya zeae MS2 (WP_102802545.1), Shigella sp. FC130 (WP_069368300.1), Paenibacillus sp. UNC451MF (WP_051620561.1), Kosakonia radicincitans YD4 (WP_052502032.1), Paenibacillaceae bacterium GAS479 (SDS42377.1), Cronobacter sakazakii cro795W1 (WP_105572611.1), Klebsiella michiganensis RC10 (WP_052698991.1). B) CdiIEC3006 homologs were aligned as in panel A. Residues that make direct contacts with CdiA-CTEC3006 are indicated by yellow circles. Sequences are from the following bacteria with NCBI protein identifiers given in parentheses: E. coli 3006 (EKI34459.1), P. carotovorum F157 (PPE62298.1), D. zeae MS2 (WP_102802544.1), Shigella sp. FC130 (WP_069368301.1), Paenibacillus sp. UNC451MF (WP_028549579.1), K. radicincitans YD4 (WP_043953995.1), Paenibacillaceae bacterium GAS479 (SDS42341.1), C. sakazakii cro795W1 (WP_087600961.1), K. michiganensis RC10 (WP_045783059.1).
Figure S4. Immunoblot analysis of chimeric CdiA proteins, Related to Figure 7. Chimeric CdiAEC93 proteins that carry the indicated CdiA-CTKp342 (panel A) or CdiA-CTEC3006 (panel B) sequences were analyzed by immunoblotting using polyclonal antibodies to the N-terminal TPS domain of CdiAEC93. Molecular mass standards (in kDa) appear in the red fluorescent channel.
Figure S5. Predicted inter-molecular clashes between EF-Tu and CdiA-CTKp342, Related to Table 2. CdiA-CTKp342 was superimposed onto the CdiA-CTNC101 toxin domain from PDB:5I4Q. EF-Tu secondary structure elements are labeled according to (Nissen et al., 1999).
Table S1. CDI toxin sequence types, Related to Figure 3A.
Table S2. PSI-BLAST search results, Related to Figure 3A.
Table S3. Oligonucleotides, Related to STAR Methods.
Highlights.
The crystal structures of two CDI toxin•immunity protein complexes are presented
Both toxins are isoacceptor specific ribonucleases that cleave deacylated tRNAGAUIle
The immunity proteins do not share any sequence or structural homology
Similarities between these tRNase toxins likely arose through convergent evolution
Acknowledgments
This work was supported by National Institutes of Health grants GM117373 (C.W.G & C.S.H.), GM102318 (C.W.G., C.S.H. & subcontract to A.J.), GM094585 (A.J.), GM115586 (A.J.) and the U. S. Department of Energy, Office of Biological and Environmental Research, under contract DE-AC02-06CH11357 (A.J.).
Footnotes
Declaration of Interests
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, et al. (2010). PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr 66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aiyar A, Xiang Y, and Leis J (1996). Site-directed mutagenesis using overlap extension PCR. Methods Mol Biol 57, 177–191. [DOI] [PubMed] [Google Scholar]
- Anderson MS, Garcia EC, and Cotter PA (2012). The Burkholderia bcpAIOB genes define unique classes of two-partner secretion and contact dependent growth inhibition systems. PLoS Genet 8, e1002877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson MS, Garcia EC, and Cotter PA (2014). Kind discrimination and competitive exclusion mediated by contact-dependent growth inhibition systems shape biofilm community structure. PLoS Pathog 10, e1004076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoki SK, Diner EJ, de Roodenbeke CT, Burgess BR, Poole SJ, Braaten BA, Jones AM, Webb JS, Hayes CS, Cotter PA, et al. (2010). A widespread family of polymorphic contact-dependent toxin delivery systems in bacteria. Nature 468, 439–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoki SK, Pamma R, Hernday AD, Bickham JE, Braaten BA, and Low DA (2005). Contact-dependent inhibition of growth in Escherichia coli. Science 309, 1245–1248. [DOI] [PubMed] [Google Scholar]
- Aoki SK, Webb JS, Braaten BA, and Low DA (2009). Contact-dependent growth inhibition causes reversible metabolic downregulation in Escherichia coli. Journal of bacteriology 191, 1777–1786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arenas J, Schipper K, van Ulsen P, van der Ende A, and Tommassen J (2013). Domain exchange at the 3′ end of the gene encoding the fratricide meningococcal two-partner secretion protein A. BMC Genomics 14, 622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batot G, Michalska K, Ekberg G, Irimpan EM, Joachimiak G, Jedrzejczak R, Babnigg G, Hayes CS, Joachimiak A, and Goulding CW (2017). The CDI toxin of Yersinia kristensenii is a novel bacterial member of the RNase A superfamily. Nucleic Acids Res 45, 5013–5025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck CM, Morse RP, Cunningham DA, Iniguez A, Low DA, Goulding CW, and Hayes CS (2014). CdiA from Enterobacter cloacae delivers a toxic ribosomal RNase into target bacteria. Structure 22, 707–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betat H, Rammelt C, and Morl M (2010). tRNA nucleotidyltransferases: ancient catalysts with an unusual mechanism of polymerization. Cell Mol Life Sci 67, 1447–1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao Z, Casabona MG, Kneuper H, Chalmers JD, and Palmer T (2016). The type VII secretion system of Staphylococcus aureus secretes a nuclease toxin that targets competitor bacteria. Nat Microbiol 2, 16183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cascales E, Buchanan SK, Duche D, Kleanthous C, Lloubes R, Postle K, Riley M, Slatin S, and Cavard D (2007). Colicin biology. Microbiol. Mol. Biol. Rev 71, 158–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowtan K (1994). DM: an automated procedure for phase improvement by density modification. Joint CCP4 and ESF-EACBM newsletter on protein crystallography 31, 34–38. [Google Scholar]
- Cowtan K (2006). The Buccaneer software for automated model building. 1. Tracing protein chains. Acta Crystallogr D Biol Crystallogr 62, 1002–1011. [DOI] [PubMed] [Google Scholar]
- Cruz JW, Sharp JD, Hoffer ED, Maehigashi T, Vvedenskaya IO, Konkimalla A, Husson RN, Nickels BE, Dunham CM, and Woychik NA (2015). Growth-regulating Mycobacterium tuberculosis VapC-mt4 toxin is an isoacceptor-specific tRNase. Nat Commun 6, 7480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis IW, Murray LW, Richardson JS, and Richardson DC (2004). MOLPROBITY: structure validation and all-atom contact analysis for nucleic acids and their complexes. Nucleic Acids Res 32, W615–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diner EJ, Beck CM, Webb JS, Low DA, and Hayes CS (2012). Identification of a target cell permissive factor required for contact-dependent growth inhibition (CDI). Genes Dev 26, 515–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunican BF, Hiller DA, and Strobel SA (2015). Transition State Charge Stabilization and Acid-Base Catalysis of mRNA Cleavage by the Endoribonuclease RelE. Biochemistry 54, 7048–7057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, and Cowtan K (2004). Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 60, 2126–2132. [DOI] [PubMed] [Google Scholar]
- Eschenfeldt WH, Lucy S, Millard CS, Joachimiak A, and Mark ID (2009). A family of LIC vectors for high-throughput cloning and purification of proteins. Methods Mol Biol 498, 105–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eschenfeldt WH, Makowska-Grzyska M, Stols L, Donnelly MI, Jedrzejczak R, and Joachimiak A (2013). New LIC vectors for production of proteins from genes containing rare codons. J Struct Funct Genomics 14, 135–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- French S, and Wilson K (1978). On the treatment of negative intensity observations. Acta Crystallogr A A34, 517–525. [Google Scholar]
- Garcia-Bayona L, Guo MS, and Laub MT (2017). Contact-dependent killing by Caulobacter crescentus via cell surface-associated, glycine zipper proteins. Elife 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garza-Sánchez F, Janssen BD, and Hayes CS (2006). Prolyl-tRNA(Pro) in the A-site of SecM-arrested ribosomes inhibits the recruitment of transfer-messenger RNA. J Biol Chem 281, 34258–34268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerdes K, and Maisonneuve E (2012). Bacterial persistence and toxin-antitoxin loci. Annu Rev Microbiol 66, 103–123. [DOI] [PubMed] [Google Scholar]
- Graille M, Mora L, Buckingham RH, van Tilbeurgh H, and de Zamaroczy M (2004). Structural inhibition of the colicin D tRNase by the tRNA-mimicking immunity protein. EMBO J 23, 1474–1482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffin MA, Davis JH, and Strobel SA (2013). Bacterial toxin RelE: a highly efficient ribonuclease with exquisite substrate specificity using atypical catalytic residues. Biochemistry 52, 8633–8642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hachani A, Allsopp LP, Oduko Y, and Filloux A (2014). The VgrG proteins are "a la carte" delivery systems for bacterial type VI effectors. J Biol Chem 289, 17872–17884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayes CS, and Sauer RT (2003). Cleavage of the A site mRNA codon during ribosome pausing provides a mechanism for translational quality control. Mol. Cell 12, 903–911. [DOI] [PubMed] [Google Scholar]
- Holberger LE, Garza-Sanchez F, Lamoureux J, Low DA, and Hayes CS (2012). A novel family of toxin/antitoxin proteins in Bacillus species. FEBS Lett 586, 132–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holm L, and Rosenstrom P (2010). Dali server: conservation mapping in 3D. Nucleic Acids Res 38, W545–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hood RD, Singh P, Hsu F, Guvener T, Carl MA, Trinidad RR, Silverman JM, Ohlson BB, Hicks KG, Plemel RL, et al. (2010). A type VI secretion system of Pseudomonas aeruginosa targets a toxin to bacteria. Cell Host Microbe 7, 25–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jamet A, Jousset AB, Euphrasie D, Mukorako P, Boucharlat A, Ducousso A, Charbit A, and Nassif X (2015). A new family of secreted toxins in pathogenic Neisseria species. PLoS Pathog 11, e1004592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janiak F, Dell VA, Abrahamson JK, Watson BS, Miller DL, and Johnson AE (1990). Fluorescence characterization of the interaction of various transfer RNA species with elongation factor Tu.GTP: evidence for a new functional role for elongation factor Tu in protein biosynthesis. Biochemistry 29, 4268–4277. [DOI] [PubMed] [Google Scholar]
- Janssen BD, and Hayes CS (2009). Kinetics of paused ribosome recycling in Escherichia coli. J Mol Biol 394, 251–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson PM, Beck CM, Morse RP, Garza-Sanchez F, Low DA, Hayes CS, and Goulding CW (2016a). Unraveling the essential role of CysK in CDI toxin activation. Proc Natl Acad Sci U S A 113, 9792–9797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson PM, Gucinski GC, Garza-Sanchez F, Wong T, Hung LW, Hayes CS, and Goulding CW (2016b). Functional Diversity of Cytotoxic tRNase/Immunity Protein Complexes from Burkholderia pseudomallei. J Biol Chem 291, 19387–19400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones AM, Garza-Sanchez F, So J, Hayes CS, and Low DA (2017). Activation of contact-dependent antibacterial tRNase toxins by translation elongation factors. Proc Natl Acad Sci U S A 114, E1951–E1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karplus PA, and Diederichs K (2012). Linking crystallographic model and data quality. Science 336, 1030–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koskiniemi S, Lamoureux JG, Nikolakakis KC, t'Kint de Roodenbeke C, Kaplan MD, Low DA, and Hayes CS (2013). Rhs proteins from diverse bacteria mediate intercellular competition. Proc Natl Acad Sci U S A 110, 7032–7037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levitz R, Chapman D, Amitsur M, Green R, Snyder L, and Kaufmann G (1990). The optional E. coli prr locus encodes a latent form of phage T4-induced anticodon nuclease. EMBO J 9, 1383–1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacIntyre DL, Miyata ST, Kitaoka M, and Pukatzki S (2010). The Vibrio cholerae type VI secretion system displays antimicrobial properties. Proc Natl Acad Sci U S A 107, 19520–19524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGinness KE, Baker TA, and Sauer RT (2006). Engineering controllable protein degradation. Mol. Cell 22, 701–707. [DOI] [PubMed] [Google Scholar]
- Michalska K, Gucinski GC, Garza-Sanchez F, Johnson PM, Stols LM, Eschenfeldt WH, Babnigg G, Low DA, Goulding CW, Joachimiak A, et al. (2017). Structure of a novel antibacterial toxin that exploits elongation factor Tu to cleave specific transfer RNAs. Nucleic Acids Res 45, 10306–10320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michalska K, Quan Nhan D, Willett JLE, Stols LM, Eschenfeldt WH, Jones AM, Nguyen JY, Koskiniemi S, Low DA, Goulding CW, et al. (2018). Functional plasticity of antibacterial EndoU toxins. Mol Microbiol 109, 509–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minor W, Cymborowski M, Otwinowski Z, and Chruszcz M (2006). HKL-3000: the integration of data reduction and structure solution--from diffraction images to an initial model in minutes. Acta Crystallogr D Biol Crystallogr 62, 859–866. [DOI] [PubMed] [Google Scholar]
- Morse RP, Nikolakakis KC, Willett JL, Gerrick E, Low DA, Hayes CS, and Goulding CW (2012). Structural basis of toxicity and immunity in contact-dependent growth inhibition (CDI) systems. Proc Natl Acad Sci U S A 109, 21480–21485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morse RP, Willett JL, Johnson PM, Zheng J, Credali A, Iniguez A, Nowick JS, Hayes CS, and Goulding CW (2015). Diversification of beta-Augmentation Interactions between CDI Toxin/Immunity Proteins. J Mol Biol 427, 3766–3784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murshudov GN, Vagin AA, and Dodson EJ (1997). Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr 53, 240–255. [DOI] [PubMed] [Google Scholar]
- Neubauer C, Gao YG, Andersen KR, Dunham CM, Kelley AC, Hentschel J, Gerdes K, Ramakrishnan V, and Brodersen DE (2009). The structural basis for mRNA recognition and cleavage by the ribosome-dependent endonuclease RelE. Cell 139, 1084–1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikolakakis K, Amber S, Wilbur JS, Diner EJ, Aoki SK, Poole SJ, Tuanyok A, Keim PS, Peacock S, Hayes CS, et al. (2012). The toxin/immunity network of Burkholderia pseudomallei contact-dependent growth inhibition (CDI) systems. Mol Microbiol 84, 516–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nissen P, Thirup S, Kjeldgaard M, and Nyborg J (1999). The crystal structure of Cys-tRNACys-EF-Tu-GDPNP reveals general and specific features in the ternary complex and in tRNA. Structure 7, 143–156. [DOI] [PubMed] [Google Scholar]
- Ogawa T, Tomita K, Ueda T, Watanabe K, Uozumi T, and Masaki H (1999). A cytotoxic ribonuclease targeting specific transfer RNA anticodons. Science 283, 2097–2100. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z (1991). Maximum likelihood refinement of heavy atom parameters. In Proceedings of the CCP4 Study Weekend 25–26 January 1991 (Daresbury Laboratory, Warrington, U.K.), pp. 80–85. [Google Scholar]
- Padilla JE, and Yeates TO (2003). A statistic for local intensity differences: robustness to anisotropy and pseudo-centering and utility for detecting twinning. Acta Crystallogr D Biol Crystallogr 59, 1124–1130. [DOI] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, and Ferrin TE (2004). UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem 25, 1605–1612. [DOI] [PubMed] [Google Scholar]
- Poole SJ, Diner EJ, Aoki SK, Braaten BA, T'Kint de Roodenbeke C, Low DA, and Hayes CS (2011). Identification of functional toxin/immunity genes linked to contact-dependent growth inhibition (CDI) and rearrangement hotspot (Rhs) systems. PLoS Genet. 7, e1002217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riley MA (1993a). Molecular mechanisms of colicin evolution. Mol Biol Evol 10, 1380–1395. [DOI] [PubMed] [Google Scholar]
- Riley MA (1993b). Positive selection for colicin diversity in bacteria. Mol Biol Evol 10, 1048–1059. [DOI] [PubMed] [Google Scholar]
- Ruhe ZC, Nguyen JY, Chen AJ, Leung NY, Hayes CS, and Low DA (2016). CDI Systems Are Stably Maintained by a Cell-Contact Mediated Surveillance Mechanism. PLoS Genet 12, e1006145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruhe ZC, Nguyen JY, Xiong J, Koskiniemi S, Beck CM, Perkins BR, Low DA, and Hayes CS (2017). CdiA Effectors Use Modular Receptor-Binding Domains To Recognize Target Bacteria. MBio 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruhe ZC, Subramanian P, Song K, Nguyen JY, Stevens TA, Low DA, Jensen GJ, and Hayes CS (2018). Programmed Secretion Arrest and Receptor-Triggered Toxin Export during Antibacterial Contact-Dependent Growth Inhibition. Cell 175, 921–933 e914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruhe ZC, Townsley L, Wallace AB, King A, Van der Woude MW, Low DA, Yildiz FH, and Hayes CS (2015). CdiA promotes receptor-independent intercellular adhesion. Mol Microbiol 98, 175–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schifano JM, Cruz JW, Vvedenskaya IO, Edifor R, Ouyang M, Husson RN, Nickels BE, and Woychik NA (2016). tRNA is a new target for cleavage by a MazF toxin. Nucleic Acids Res 44, 1256–1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheldrick GM (2008). A short history of SHELX. Acta Crystallogr A 64, 112–122. [DOI] [PubMed] [Google Scholar]
- Shneider MM, Buth SA, Ho BT, Basler M, Mekalanos JJ, and Leiman PG (2013). PAAR-repeat proteins sharpen and diversify the type VI secretion system spike. Nature 500, 350–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Souza DP, Oka GU, Alvarez-Martinez CE, Bisson-Filho AW, Dunger G, Hobeika L, Cavalcante NS, Alegria MC, Barbosa LR, Salinas RK, et al. (2015). Bacterial killing via a type IV secretion system. Nat Commun 6, 6453. [DOI] [PubMed] [Google Scholar]
- Steczkiewicz K, Muszewska A, Knizewski L, Rychlewski L, and Ginalski K (2012). Sequence, structure and functional diversity of PD-(D/E)XK phosphodiesterase superfamily. Nucleic Acids Res 40, 7016–7045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomason L, Court DL, Bubunenko M, Costantino N, Wilson H, Datta S, and Oppenheim A (2007). Recombineering: genetic engineering in bacteria using homologous recombination Current protocols in molecular biology / edited by Ausubel Frederick M. ... [et al. Chapter 1, Unit 1 16. [DOI] [PubMed] [Google Scholar]
- Tomita K, Ogawa T, Uozumi T, Watanabe K, and Masaki H (2000). A cytotoxic ribonuclease which specifically cleaves four isoaccepting arginine tRNAs at their anticodon loops. Proc Natl Acad Sci U S A 97, 8278–8283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Melderen L (2010). Toxin-antitoxin systems: why so many, what for? Curr Opin Microbiol 13, 781–785. [DOI] [PubMed] [Google Scholar]
- Walker D, Lancaster L, James R, and Kleanthous C (2004). Identification of the catalytic motif of the microbial ribosome inactivating cytotoxin colicin E3. Protein Sci 13, 1603–1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webb JS, Nikolakakis KC, Willett JL, Aoki SK, Hayes CS, and Low DA (2013). Delivery of CdiA nuclease toxins into target cells during contact-dependent growth inhibition. PLoS ONE 8, e57609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitney JC, Beck CM, Goo YA, Russell AB, Harding BN, De Leon JA, Cunningham DA, Tran BQ, Low DA, Goodlett DR, et al. (2014). Genetically distinct pathways guide effector export through the type VI secretion system. Mol Microbiol 92, 529–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitney JC, Peterson SB, Kim J, Pazos M, Verster AJ, Radey MC, Kulasekara HD, Ching MQ, Bullen NP, Bryant D, et al. (2017). A broadly distributed toxin family mediates contact-dependent antagonism between gram-positive bacteria. Elife 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willett JL, Gucinski GC, Fatherree JP, Low DA, and Hayes CS (2015a). Contact-dependent growth inhibition toxins exploit multiple independent cell-entry pathways. Proc Natl Acad Sci U S A 112, 11341–11346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willett JL, Ruhe ZC, Goulding CW, Low DA, and Hayes CS (2015b). Contact-Dependent Growth Inhibition (CDI) and CdiB/CdiA Two-Partner Secretion Proteins. J Mol Biol 427, 3754–3765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AG, McCoy A, et al. (2011). Overview of the CCP4 suite and current developments. Acta Crystallogr D Biol Crystallogr 67, 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winther K, Tree JJ, Tollervey D, and Gerdes K (2016). VapCs of Mycobacterium tuberculosis cleave RNAs essential for translation. Nucleic Acids Res 44, 9860–9871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winther KS, and Gerdes K (2011). Enteric virulence associated protein VapC inhibits translation by cleavage of initiator tRNA. Proc Natl Acad Sci U S A 108, 7403–7407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yajima S, Inoue S, Ogawa T, Nonaka T, Ohsawa K, and Masaki H (2006). Structural basis for sequence-dependent recognition of colicin E5 tRNase by mimicking the mRNA-tRNA interaction. Nucleic Acids Res 34, 6074–6082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanagisawa T, Lee JT, Wu HC, and Kawakami M (1994). Relationship of protein structure of isoleucyl-tRNA synthetase with pseudomonic acid resistance of Escherichia coli. A proposed mode of action of pseudomonic acid as an inhibitor of isoleucyl-tRNA synthetase. J Biol Chem 269, 24304–24309. [PubMed] [Google Scholar]
- Zhang D, de Souza RF, Anantharaman V, Iyer LM, and Aravind L (2012). Polymorphic toxin systems: Comprehensive characterization of trafficking modes, processing, mechanisms of action, immunity and ecology using comparative genomics. Biol Direct 7, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang D, Iyer LM, and Aravind L (2011). A novel immunity system for bacterial nucleic acid degrading toxins and its recruitment in various eukaryotic and DNA viral systems. Nucleic Acids Res 39, 4532–4552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang D, Iyer LM, Burroughs AM, and Aravind L (2014). Resilience of biochemical activity in protein domains in the face of structural divergence. Curr Opin Struct Biol 26, 92–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Domain architectures of CdiAKp342 and CdiAEC3006, Related to Figures 1 & 2. The N-terminal signal sequence (ss) and TPS transport domain are required for secretion of CdiA across the cytoplasmic and outer membranes (respectively) of inhibitor cells. The filamentous hemagglutinin-1 (FHA-1) domain is composed of peptide repeats (Pfam: PF05594) that form a β-helix; and the FHA-2 repeat (PF13332) domain is predicted to translocate CdiA-CT toxins across the target-cell outer membrane. The receptor-binding domain (RBD) resides between the FHA repeat domains. The CdiAEC3006 RBD recognizes BamA on target cells (Ruhe et al., 2017), and the binding specificity of the RBD from CdiAKp342 has not been determined. The Tyr/Pro-enriched (YP) domain is required for stable presentation of CdiA on the surface of inhibitor cells (Ruhe et al., 2018). The pre-toxin (PT) domain is thought to act as an auto-protease, cleaving the C-terminal (CdiA-CT) region near the conserved VENN peptide motif to enable toxin delivery into target bacteria. The polymorphic CdiA-CT region is composed of two domains. The extreme C-terminal domains is typically a toxic nuclease, whereas the N-terminal "cytoplasm-entry" domain mediates toxin translocation into the target-cell cytosol. The entry domain from CdiAEC3006 hijacks the D-glucose transporter PtsG to translocate into target cells, whereas the entry domain of CdiAKp342 is uncharacterized. NCBI Refseq identifiers for each effector are given in parentheses.
Figure S2. Homologs of CdiA-CTKp342 and CdiIKp342, Related to Figure 1C. A) Homologs of CdiA-CTKp342 were aligned using Clustal omega and rendered using the ESPript 3.0 server. Secondary structure elements for CdiA-CTKp342 are shown above the alignment. Residues that make direct contacts with CdiIKp342 are indicated by purple circles. Sequences are from the following bacteria with NCBI protein identifiers given in parentheses: K. pneumoniae 342 (WP_012542573.1), Dickeya zeae Ech586 (WP_012884592.1), Yersinia pseudotuberculosis IP 31758 (WP_012104437.1), Snodgrassella alvi wkB237 (WP_100141475.1), Burkholderia latens RF32-BP12 (WP_059545330.1), Achromobacter sp. 2789STDY5608608 (CUJ27995.1), Pseudomonas sp. BS3767 (SDI04727.1), Pseudomonas syringae Riq4 (WP_080370524.1), Pseudomonas sp. URIL14HWK12:I9 (WP_097084422.1). B) CdiIKp342 homologs were aligned as in panel A. Residues that make direct contacts with CdiA-CTKp342 are indicated by yellow circles. Sequences are from the following bacteria with NCBI protein identifiers given in parentheses: K. pneumoniae 342 (WP_041165391.1), D. zeae Ech586 (WP_012884593.1), Y. pseudotuberculosis IP 31758 (WP_011193122.1), S. alvi wkB237 (WP_100141474.1), B. latens RF32-BP12 (unannotated), Achromobacter sp. 2789STDY5608608 (unannotated), Pseudomonas sp. BS3767 (SDI04696.1), P. syringae Riq4 (WP_052966432.1), Pseudomonas sp. URIL14HWK12:I9 (WP_097084423.1).
Figure S3. Homologs of CdiA-CTEC3006 and CdiIEC3006, Related to Figure 2B. A) Homologs of CdiA-CTEC3006 were aligned using Clustal omega and rendered using the ESPript 3.0 server. Secondary structure elements for CdiA-CTEC3006 are shown above the alignment. Residues that make direct contacts with CdiIEC3006 are indicated by light blue circles. Sequences are from the following bacteria with NCBI protein identifiers given in parentheses: E. coli 3006 (EKI34460.1), Pectobacterium carotovorum F157 (PPE62299.1), Dickeya zeae MS2 (WP_102802545.1), Shigella sp. FC130 (WP_069368300.1), Paenibacillus sp. UNC451MF (WP_051620561.1), Kosakonia radicincitans YD4 (WP_052502032.1), Paenibacillaceae bacterium GAS479 (SDS42377.1), Cronobacter sakazakii cro795W1 (WP_105572611.1), Klebsiella michiganensis RC10 (WP_052698991.1). B) CdiIEC3006 homologs were aligned as in panel A. Residues that make direct contacts with CdiA-CTEC3006 are indicated by yellow circles. Sequences are from the following bacteria with NCBI protein identifiers given in parentheses: E. coli 3006 (EKI34459.1), P. carotovorum F157 (PPE62298.1), D. zeae MS2 (WP_102802544.1), Shigella sp. FC130 (WP_069368301.1), Paenibacillus sp. UNC451MF (WP_028549579.1), K. radicincitans YD4 (WP_043953995.1), Paenibacillaceae bacterium GAS479 (SDS42341.1), C. sakazakii cro795W1 (WP_087600961.1), K. michiganensis RC10 (WP_045783059.1).
Figure S4. Immunoblot analysis of chimeric CdiA proteins, Related to Figure 7. Chimeric CdiAEC93 proteins that carry the indicated CdiA-CTKp342 (panel A) or CdiA-CTEC3006 (panel B) sequences were analyzed by immunoblotting using polyclonal antibodies to the N-terminal TPS domain of CdiAEC93. Molecular mass standards (in kDa) appear in the red fluorescent channel.
Figure S5. Predicted inter-molecular clashes between EF-Tu and CdiA-CTKp342, Related to Table 2. CdiA-CTKp342 was superimposed onto the CdiA-CTNC101 toxin domain from PDB:5I4Q. EF-Tu secondary structure elements are labeled according to (Nissen et al., 1999).
Table S1. CDI toxin sequence types, Related to Figure 3A.
Table S2. PSI-BLAST search results, Related to Figure 3A.
Table S3. Oligonucleotides, Related to STAR Methods.
Data Availability Statement
Structure datasets generated during this study are available in the Protein Data Bank under accession numbers 6CP8 (http://www.rcsb.org/structure/6CP8) and 6CP9 (http://www.rcsb.org/structure/6CP9).