

Summary
Years of selection for desirable fruit quality traits in dessert watermelon (Citrullus lanatus) has resulted in a narrow genetic base in modern cultivars. Development of novel genomic and genetic resources offers great potential to expand genetic diversity and improve important traits in watermelon. Here, we report a high‐quality genome sequence of watermelon cultivar ‘Charleston Gray’, a principal American dessert watermelon, to complement the existing reference genome from ‘97103’, an East Asian cultivar. Comparative analyses between genomes of ‘Charleston Gray’ and ‘97103’ revealed genomic variants that may underlie phenotypic differences between the two cultivars. We then genotyped 1365 watermelon plant introduction (PI) lines maintained at the U.S. National Plant Germplasm System using genotyping‐by‐sequencing (GBS). These PI lines were collected throughout the world and belong to three Citrullus species, C. lanatus, C. mucosospermus and C. amarus. Approximately 25 000 high‐quality single nucleotide polymorphisms (SNPs) were derived from the GBS data using the ‘Charleston Gray’ genome as the reference. Population genomic analyses using these SNPs discovered a close relationship between C. lanatus and C. mucosospermus and identified four major groups in these two species correlated to their geographic locations. Citrullus amarus was found to have a distinct genetic makeup compared to C. lanatus and C. mucosospermus. The SNPs also enabled identification of genomic regions associated with important fruit quality and disease resistance traits through genome‐wide association studies. The high‐quality ‘Charleston Gray’ genome and the genotyping data of this large collection of watermelon accessions provide valuable resources for facilitating watermelon research, breeding and improvement.
Keywords: watermelon, ‘Charleston Gray’, genome sequence, genetic diversity, Citrullus germplasm, genotyping‐by‐sequencing, genome‐wide association study, disease resistance
Introduction
Watermelon (Citrullus lanatus) is an important crop consumed throughout the world and is a rich source of lycopene, citrulline and other human health‐promoting compounds (Perkins‐Veazie et al., 2007). Nearly six per cent of all land used for growing vegetables is planted with watermelon, which had an annual global production of 117 million tonnes in 2016 (FAOSTAT, http://www.fao.org/faostat/en/). China is the largest producer and consumer of watermelon with 70 million tonnes produced per year. China and the United States both have intensive watermelon breeding programmes to meet varied consumer preferences. For example, cultivars of East Asian ancestry mainly produce small to mid‐sized globular fruits with a thin rind, while American cultivars tend to produce large, oblong fruits with a thick rind. Differential selection pressures in the two regions have led to distinct East Asian and American watermelon ecotypes (Guo et al., 2013; Sheng et al., 2012). Currently, the only published watermelon reference genome was constructed using the Chinese elite line ‘97103’, representing the East Asian type (Guo et al., 2013). Recent comparative genomic and pan‐genome studies have highlighted the importance of structural variations (SVs), particularly presence–absence variations (PAVs) and copy number variations (CNVs) (Gao et al., 2019; Zhang et al., 2015). Quantitative trait locus (QTL) and association mapping studies with American germplasm using the ‘97103’ genome as the reference would be limited to the genetic makeup of a single East Asian cultivar, potentially missing causal variants.
A large number of watermelon cultivars have been developed in the United States since the mid‐19th century (Levi et al., 2001), including ‘Charleston Gray’ which is considered the principal American dessert watermelon cultivar. ‘Charleston Gray’ was released in 1954 and was the most popular commercially grown watermelon in the United States for more than a decade. It produces large oblong fruits with a light green exterior, pink‐red flesh, and a thick and tough rind, as required for long‐distance shipping. It also harbours resistance to the soil‐borne disease Fusarium wilt and the foliar disease anthracnose. ‘Charleston Gray’ has been used in numerous watermelon breeding programmes (Wehner, 1999) and in the development of many improved cultivars. Therefore, unlocking the genome of ‘Charleston Gray’ would further facilitate watermelon breeding.
Many years of cultivation and selection for desirable fruit qualities have resulted in an overall narrowing of the genetic base among sweet dessert watermelon cultivars, and consequently contributed to their susceptibility to a large number of diseases and pests (Levi et al., 2001, 2017). As a result, there is a continual need to expand the genetic base of watermelon cultivars and enrich them with alleles conferring resistance to biotic stresses such as diseases, insects and nematodes in addition to alleles associated with desirable fruit quality. The Citrullus genus, whose centre of origin is Africa, includes seven recognized species (Chomicki and Renner, 2015). Two of these species, C. amarus (also known as citron) and C. mucosospermus (also known as egusi melon), were until recently considered to be subspecies of C. lanatus (Chomicki and Renner, 2015), and they can cross readily with cultivated watermelon. Citrullus lanatus is thought to be native to northern Africa, C. mucosospermus to sub‐Saharan western Africa, and C. amarus to southern Africa (Jarret et al., 1997; Levi et al., 2017; Paris, 2015). Unlike C. lanatus, C. mucosospermus is primarily cultivated for oil‐rich seeds. Citrullus mucosospermus and C. amarus are valuable sources of resistance to many diseases including Phytophthora fruit rot, powdery mildew, Fusarium wilt, gummy stem blight, anthracnose and various viruses (Levi et al., 2017). Therefore, these highly interfertile species have the potential to provide valuable alleles to broaden the genetic base of watermelon and increase disease and pest resistance in elite cultivars.
The U.S. National Plant Germplasm System (NPGS; https://www.ars-grin.gov/npgs/index.html) maintains a large collection of Citrullus accessions as Plant Introductions (PIs) collected/acquired from different geographic regions in Africa and throughout the world. The PIs within this Citrullus collection exhibit great phenotypic and genetic diversity (Levi et al., 2013; Nimmakayala et al., 2014) and thus represent a valuable resource for discovery of unique qualities useful for improving fruit nutritional contents and resistance to biotic and abiotic stresses in watermelon cultivars. Elucidating the genetic diversity and relationships among these PIs is an important step towards the identification of QTLs associated with beneficial attributes.
In this study, we sequenced and de novo assembled a high‐quality genome of the principal American watermelon cultivar ‘Charleston Gray’, as a complement to the genome of ‘97103’, an East Asian cultivar. Comparative genomic analyses of ‘Charleston Gray’ and ‘97103’ identified a number of genome variations overlapping with QTLs of important traits such as fruit weight and shape. We then genotyped 1365 Citrullus accessions from the NPGS (including C. lanatus, C. amarus and C. mucosospermus) using genotyping‐by‐sequencing (GBS; Elshire et al., 2011). Single nucleotide polymorphisms (SNPs) were called from the GBS data using the ‘Charleston Gray’ genome as the reference and were subsequently used to assess genetic diversity, phylogenetic relationships and population structure among these Citrullus accessions. The SNP set was also used to perform genome‐wide association studies (GWAS) for the identification of genomic regions associated with several important fruit quality and disease resistance traits.
Results
Genome sequencing, assembly, anchoring and quality evaluation
The ‘Charleston Gray’ genome was sequenced using the Illumina technology, which produced a total of 95.5 Gb of high‐quality cleaned sequences from paired‐end and mate‐pair libraries with insert sizes ranging from 400 bp to 20 kb (Table S1). These sequences represented approximately a 228× coverage of the ‘Charleston Gray’ genome with an estimated size of 419.2 Mb based on the k‐mer depth distribution analysis of the sequencing reads (Figure S1). De novo assembly yielded a draft genome of 396.4 Mb, representing 94.6% of the ‘Charleston Gray’ genome. The assembly consisted of 21 498 contigs and 2034 scaffolds (>500 bp), with N50 sizes of 36.7 kb and 7.47 Mb, respectively, and 90% of the genome draft was assembled into only 60 scaffolds (Table 1). The ‘Charleston Gray’ assembly had higher contiguity compared with that of the Chinese elite watermelon line ‘97103’ (Guo et al., 2013), as indicated by increased N50 and N90 scaffold lengths (Table 1).
Table 1.
Assembly statistics of watermelon ‘Charleston Gray’ and ‘97103’ genomes
| Charleston Gray | 97103 | |||||||
|---|---|---|---|---|---|---|---|---|
| Scaffold* | Contig | Scaffold* | Contig | |||||
| Size (bp) | Number | Size (bp) | Number | Size (bp) | Number | Size (bp) | Number | |
| Longest | 23 422 029 | 1 | 235 198 | 1 | 8 716 783 | 1 | 227 474 | 1 |
| N50 | 7 471 260 | 17 | 36 674 | 3073 | 2 378 183 | 42 | 26 377 | 3316 |
| N90 | 1 615 926 | 60 | 9605 | 10 630 | 374 692 | 184 | 3971 | 15 057 |
| Total | 396 351 412 | 2034 | 375 815 318 | 21 498 | 353 466 419 | 1793 | 321 373 230 | 43 342 |
Scaffolds longer than 500 bp in size were included in the assembly.
Using an integrated high‐density genetic map (Ren et al., 2014), 90.3% (378.7 Mb) of the ‘Charleston Gray’ assembly were anchored to 11 linkage groups, and 90.5% of the anchored scaffolds (342.8 Mb) were oriented. We assigned orientation to an additional of 9.9 Mb of scaffolds, according to two other recently developed genetic maps (Branham et al., 2017; Reddy et al., 2014). Furthermore, based on the genome synteny between ‘97103’ and ‘Charleston Gray’, an additional 26.6 Mb of the ‘Charleston Gray’ scaffolds were oriented, and 3.8 Mb of scaffolds were anchored. One misassembled scaffold was identified according to the genetic map and mate‐pair read alignments, and was broken into two scaffolds. Finally, we obtained a chromosome‐scale assembly of the ‘Charleston Gray’ genome, of which 382.5 Mb (96.2% of the total assembly) in 100 scaffolds were constructed into 11 pseudomolecules and 379.2 Mb (95.4% of the assembly) were oriented (Table S2 and Figure 1).
Figure 1.
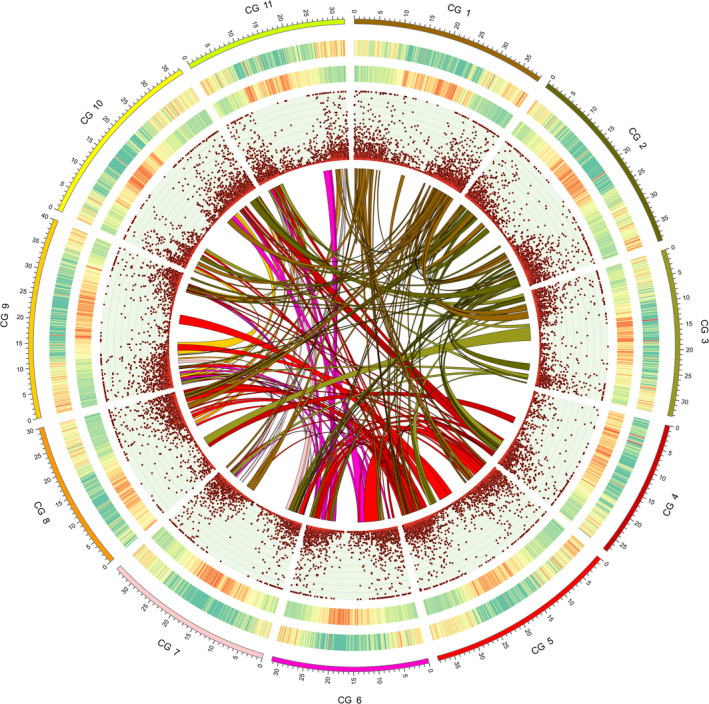
Genomic landscape of watermelon, ‘Charleston Gray’. The outermost circle is the ideogram of 11 chromosomes in Mb scale, followed by circles of gene density and TE density represented by percentage of genomic regions covered by genes and repeat sequences in 200‐kb windows, respectively (green to red, low to high), gene expression levels (RPKM; Maximum = 200) and syntenic blocks within the genome depicted by lines.
To evaluate the quality of the assembly, we first aligned RNA‐Seq reads and ESTs (expressed sequence tags; Levi et al., 2006) to the ‘Charleston Gray’ genome. Up to 94% of the RNA‐Seq reads were mapped to the genome, and more than 95% of the ESTs were covered by the genome (Table S3). The completeness of the ‘Charleston Gray’ assembly was further assessed with BUSCO (Simão et al., 2015), which showed that 91.8% of the core conserved plant genes were completely covered by the assembly, while another 1.7% were partially covered (Table S4). Together, these results confirmed the high quality of the ‘Charleston Gray’ assembly.
Comparative genomic analysis of ‘Charleston Gray’ and ‘97103’
We identified 194.9 Mb of repeat sequences in the ‘Charleston Gray’ genome, contributing to 51.7% of the genome (Table S5), which was higher than the 45.2% repetitive content in the ‘97103’ genome (Guo et al., 2013), mainly attributed to a better assembly of repeat regions in the ‘Charleston Gray’ genome. A total of 22 546 protein‐coding genes were predicted in ‘Charleston Gray’, among which 18 982 genes (84.2%) were assigned with biological functions. Comparison between orthologous genes in ‘Charleston Gray’ and ‘97103’ revealed a chromosome‐level syntenic relationship (Figure S2). Most differences between the two assemblies were observed around the centromeric regions (Figure S3). For example, the ‘big inversion’ on chromosome 1 might not be an actual SV between the two genomes but was likely caused by incorrect anchoring in the ‘97103’ genomes as a result of uncertainty in ordering short scaffolds within a close genetic distance (Figure S4a). In this case, the assembly of the ‘Charleston Gray’ scaffolds 15 and 19 spanning the ‘inversion’ breakpoints was well supported by the mate‐pair reads (Figure S4b), which suggested that, with a higher contiguity, the ‘Charleston Gray’ assembly provided more reliable pseudomolecule structures of the watermelon genome for facilitating QTL mapping and molecular genetic studies.
A total of 214 113 SNPs and 16 052 small indels were identified between the ‘Charleston Gray’ and ‘97103’ genomes, of which 184 567 SNPs and 13 664 small indels were located in the intergenic regions, and 29 546 SNPs and 2388 small indels were located in the introns of genes (23 947 SNPs and 2275 indels) or coding regions (5599 SNPs and 113 indels) (Table S6). Among genes with SNPs in the coding regions, 1810 had nonsynonymous mutations, 80 lost start or stop codons, and 1506 had synonymous mutations. We further identified a total of 3766 high‐confidence large SVs (≥50 bp) between the ‘Charleston Gray’ and ‘97103’ genomes, including 2913 indels and 853 tandem/repeat contractions/expansions (Table S7). The majority of these SVs (88.8%) were less than 1 kb in size (Figure S5). About 81.5% (3069) of the SVs were located in intergenic regions, 518 in introns of genes, and 179 affected the coding sequences (Table S7). A total of 815 SVs were found to overlap with 52 QTLs known to affect watermelon fruit traits (Table S8), and 400 were located within fruit shape and fruit weight QTLs (Table S8), which might be associated with the more elongated fruit shape and larger fruit size of ‘Charleston Gray’ as compared to ‘97103’, such as the 159‐bp deletion in the ClFS1 gene (ClCG03G016090) of ‘Charleston Gray’ known to be associated with elongated fruit shape (Dou et al., 2018).
Genotyping of the watermelon germplasm collection and variation identification
To facilitate the utilization of the Citrullus spp. germplasm stored in the NPGS for watermelon improvement, we performed GBS on 1365 Citrullus PIs collected throughout the world (Figure 2), including 1211 C. lanatus, 52 C. mucosospermus and 102 C. amarus PIs (Table S9). Most C. lanatus PIs are modern cultivars and are the result of many years of domestication and selection for desirable fruit qualities that do not exist in their closely related citron or egusi type watermelons. The C. lanatus and C. mucosospermus PIs in this study were collected in East Asia, North and South America, Europe, Central and West Asia, Oceania and different regions in Africa, while the majority of C. amarus PIs were collected in their natural surroundings in southern Africa (Figure 2 and Table S9).
Figure 2.

Geographical distribution of the 1365 Citrullus spp. accessions in the National Plant Germplasm System. The diameter of the circle is proportional to the number of accessions from each country.
A total of ~1.0 billion reads of 101 bp in length were generated using GBS, from which 388 293 unique tags with at least 10 read counts were obtained, corresponding to ~0.8 billion reads, and used for SNP calling. About 49.5% and 16.5% of these reads were aligned to unique and multiple positions in the ‘Charleston Gray’ genome, respectively, and the unaligned reads were mainly from the chloroplast and mitochondrion genomes. Approximately 2.4% of the ‘Charleston Gray’ genome was covered by GBS reads, which is typical for GBS data (Elshire et al., 2011). A total of 61 520 SNPs were identified, among which 25 308 were biallelic with a minor allele frequency (MAF) >0.01 and a missing data rate ≤50%, and were used in the subsequent phylogenetic and population structure analyses. These SNPs were distributed across the ‘Charleston Gray’ genome with an average of one SNP per 15.7 Kb (Figure S6).
Phylogenetic relationships and population structure of Citrullus accessions
To infer phylogenetic relationships among the 1367 Citrullus accessions (including ‘Charleston Gray’ and ‘97103’ in addition to the 1365 accessions genotyped with GBS), we constructed a maximum‐likelihood tree, which revealed that C. lanatus and C. mucosospermus accessions exhibited close genetic relationships and were distant from C. amarus (Figure 3a). Within C. lanatus and C. mucosospermus, four major clades were identified (Figure 3a and Table S9). Group 1 included C. mucosospermus along with a subset of C. lanatus accessions collected in southern Africa, primarily Zimbabwe and Zambia. The remaining groups were all comprised of C. lanatus accessions primarily collected in Central and West Asia (Group 2), North America and East Asia (Group 3) and Europe (Group 4). For C. lanatus collected in northern Africa, four Sudanese accessions collected in Khartoum (PI 254622, PI 254623 and PI 260733; white flesh and beige seeds) and Darfur (PI 481871; white flesh and black/beige seeds) were closely related to C. mucosospermus in Group 1 (Table S9). Eight other accessions collected in northern Africa were in Group 2 (Central and West Asia) in one of the deepest branches of C. lanatus outside Group 1, including six collected in Egypt (PI 525090, PI 525086, PI 525087, PI 525084, PI 525091 and PI 525088), one in Sudan (PI 270545, red flesh and black seeds) and one in Algeria (PI 542617; white flesh and black seeds) (Table S9). For accessions collected in East Africa, five out of seven accessions collected in Ethiopia were closely related to accessions collected in Europe (Group 4), and five out of six accessions collected in Somalia were in Group 1 (Table S9). Similarly, accessions collected in Central and West Africa were placed in all four groups (Table S9), with more accessions (mostly collected in Central Africa) clustered with cultivars from North America and East Asia (Group 3). Despite the geographic distance, the North American C. lanatus accessions (including ‘Charleston Gray’) and the East Asian cultivars (including ‘97103’) were closely related, although they formed two distinguishable clades in Group 3, reflecting the two major cultivated ecotypes.
Figure 3.

Phylogenetic relationship and population structure of Citrullus spp. accessions. (a) Maximum‐likelihood tree of 1367 Citrullus spp. accessions. (b) Model‐based clustering analysis with K from 2 to 5. Each accession is represented by a vertical bar. Each colour represents one ancestral population, and the length of each coloured segment in each vertical bar represents the proportion contributed by ancestral populations. (c) Principal component analysis of 1367 watermelon accessions with PC1 and PC2 explaining 63.7% and 2.1% of variance, respectively. (d) Principal component analysis of C. lanatus and C. mucosospermus accessions with PC1 and PC2 explaining 4.6% and 2.3% of variance, respectively.
Population structure of the 1367 watermelon accessions was investigated. ΔK analysis showed that K = 2 ancestral types best explained the structure of this population and separated C. amarus from C. lanatus/C. mucosospermus (Figure S7). At K = 3 or 4, three primary clusters could be observed representing C. amarus, the C. mucosospermus/southern African C. lanatus group and the remaining C. lanatus accessions (Figure 3b). At K = 5, a new subgroup emerged, which included North American and East Asian cultivars, while most C. lanatus from Central and West Asia and Europe had similar backgrounds (Figure 3b). Principal component analysis (PCA) using all the accessions also revealed a clear separation of C. amarus from C. lanatus and C. mucosospermus (Figure 3c). According to the phylogenetic relationship and genetic background, a few accessions were likely misclassified or had ambiguous identity, including 26 accessions classified as C. lanatus, among which 11 might be C. amarus and 15 had mixed genetic background, and 14 accessions labelled as C. amarus but did not have a typical C. amarus ancestry pattern (Table S9). These accessions were excluded from the following genetic diversity and population divergence analyses. PCA using the remaining C. lanatus and C. mucosospermus accessions illustrated a pattern further separating C. mucosospermus, C. lanatus collected in southern Africa and the rest of the C. lanatus accessions, while the small portion of the total variance explained by the first two principal components further supported the close genetic relationship between C. lanatus and C. mucosospermus (Figure 3d). Together, the phylogeny, population structure and PCA results all indicated a distant relationship between C. amarus and C. lanatus/C. mucosospermus, and a similar genetic background in C. mucosospermus and a group of C. lanatus mostly from southern Africa.
Genetic diversity and population divergence in watermelons
The genetic diversity within C. amarus estimated by the average value of genome‐wide nucleotide diversity (π) was 3.375 × 10−4, higher than that within C. mucosospermus (π = 2.561 × 10−4) and those within different C. lanatus groups based on geographic locations, ranging from 2.405 × 10−4 (Europe) to 3.035 × 10−4 (Southern Africa) (Figure S8). Population divergence among different C. lanatus groups and C. mucosospermus was evaluated by pairwise fixation index (F ST). Multidimensional scaling visualization of pairwise F ST values clearly showed the distinction between C. mucosospermus and C. lanatus, and between C. lanatus collected in southern Africa and the rest of C. lanatus groups, and much less divergence among the C. lanatus groups outside southern Africa (Figure S8). The weighted pairwise F ST values between C. mucosospermus and different C. lanatus groups, and between the southern Africa C. lanatus group and the other groups ranged from 0.286 to 0.428, and from 0.238 to 0.385, respectively, while the values among the other C. lanatus groups were much lower, ranging from 0.020 to 0.149 (Table S10). The F ST values were largely consistent with relationships of different groups of Citrullus accessions inferred from phylogenetic and population structure analyses.
GWAS for fruit quality and disease resistance traits
We performed GWAS for several fruit quality and disease resistance traits to identify QTLs underlying these important traits. We collected historical phenotypic data from the NPGS for fruit flesh colour, fruit shape and rind stripe pattern for 788, 864 and 695 accessions, respectively, that were genotyped in this study. We also evaluated the resistance to bacterial fruit blotch (BFB), powdery mildew race 2W and Papaya ringspot virus‐watermelon strain (PRSV‐W) on 1125, 1147 and 908 PIs, respectively (Table S11). A group of SNPs on chromosome 4 (at ~15.3–15.8 Mb) were associated with red coloration of fruit flesh (Figure 4a and Table S12), and this genome region contained the LCYB gene (chromosome 4: 15 694 446–15 696 571) that converts lycopene to downstream carotenoids (Bang et al., 2010). In addition, 14 additional SNPs on other chromosomes were also associated with red flesh colour (Table S12). A peak associated with rind stripe pattern was identified on chromosome 6 (at ~30.2 Mb) (Figure 4a), overlapping with the ‘S’ locus that controls the foreground stripe pattern (Park et al., 2016). Nine additional SNPs were also significantly associated with rind stripe pattern. Five SNPs on chromosome 3 (at ~31.1 Mb) were associated with elongated fruit shape (Figure 4a and Table S12), which were located near the ClFS1 gene (~22.6 Kb distance; chromosome 3: 31 086 958–31 090 579) known to control watermelon fruit elongation (Dou et al., 2018). Two other SNPs associated with fruit shape were on chromosomes 2 and 6. Two peaks on chromosomes 10 (~32.6–32.9 Mb) and 6 (~10.9 Mb), respectively, were associated with resistance to BFB in fruit (Figure 4b and Table S12). A group of SNPs on chromosome 2 (~29.2–31.0 Mb) were associated with powdery mildew race 2W resistance in stems, and an additional 21 SNPs on other chromosomes were also associated (Figure 4b and Table S12). The same peak on chromosome 2 was also associated with powdery mildew race 2W resistance in leaves, and an overall similar association pattern was observed. For PRSV‐W resistance, 15 significantly associated SNPs were identified on multiple chromosomes (Figure 4b and Table S12).
Figure 4.

Genome‐wide association studies of fruit quality (a) and disease resistance (b) traits. BFBFruit, resistance to bacterial fruit blotch in fruits; PM2Stem, resistance to powdery mildew race 2W in stem; PM2Leaf: resistance to powdery mildew race 2W in leaf; PRSV: resistance to Papaya ringspot virus‐watermelon strain. Gray horizontal dashed lines on the Manhattan plots indicate the Bonferroni significance thresholds of GWAS (−log10(P) of 5.68 and 6.38, corresponding to α = 0.05 and α = 0.01, respectively).
Discussion
Watermelon is among the first cucurbit crops to have its genome sequenced, assembled and annotated (Zheng et al., 2019). Genome sequence of the East Asian watermelon cultivar, ‘97103’, was released in 2013 (Guo et al., 2013). To complement the ‘97103’ reference genome, and to capture genome information specifically present in the American watermelon ecotype, we assembled and annotated the genome of the principal American cultivar ‘Charleston Gray’, performed detailed characterization of sequence variations between ‘Charleston Gray’ and ‘97103’, and identified those overlapping with known watermelon fruit trait QTLs, which may underlie the phenotypic differences between the two cultivated ecotypes. Comparative analyses suggested that the assembled ‘Charleston Gray’ genome had higher continuity and quality than that of ‘97103’. The high‐quality ‘Charleston Gray’ assembly will facilitate its use as a reference for integrative genetic‐genomic studies aiming to identify QTLs associated with important agronomical traits.
We characterized the genetic composition of 1365 watermelon PIs maintained in the NPGS, belonging to three Citrullus species, C. lanatus, C. mucosospermus and C. amarus. The latter two are of particular interest because of the presence of potential genes/alleles associated with fruit quality or resistance to major cultivated watermelon diseases (Levi et al., 2013). In agreement with the known narrow genetic background in dessert watermelon, all C. lanatus groups from different geographic regions displayed low genetic diversity, with that of the southern Africa group being slightly higher, consistent with southern Africa being the likely centre of origin of Citrullus (Chomicki and Renner, 2015; Paris, 2015).
Our sampling included a large number of accessions collected in major dessert watermelon production areas and geographic regions where wild and primitive watermelons exist. This enabled us to reveal phylogenetic relationships among watermelon accessions collected in different geographic regions that may shed light on the domestication and dispersal of dessert watermelons. The distant relationship between C. amarus and C. lanatus/C. mucosospermus and their distinct genetic backgrounds revealed in the current study are consistent with previous genetic characterization of a few resequenced accessions (Guo et al., 2013). This result indicates that the dessert watermelon is unlikely to be descended from C. amarus in southern Africa, which is further supported by the evidence of genome organization differences based on rDNA chromosome landmarks (Guo et al., 2013; Reddy et al., 2013) and the finding that the emergence of dessert watermelon in Egypt predated the time when farming began in southern Africa (Paris, 2015). It has been hypothesized that the dessert watermelon was domesticated in northeastern Africa and a wild population of ‘cordophanus’ watermelons in Sudan with non‐bitter fruits and white pulp may be the living representatives of the progenitor of the cultivated dessert watermelon (Paris, 2015; Renner et al., 2017). Four Sudanese C. lanatus accessions bearing such fruit phenotypes, including two (PI 481871 and PI 254622) considered as dessert watermelon (Paris, 2015), are closely related to C. mucosospermus collected from Liberia, Ghana, Nigeria and Zaire, suggesting that C. lanatus accessions from Sudan and C. mucosospermus from central/West Africa could be derived from the same ancestral population; one selected for seed traits, and the other domesticated for fruit flesh characteristics. The placement of eight northern African C. lanatus accessions (six from Egypt) in one of the deepest branches of cultivated dessert watermelon groups on the phylogenetic tree (Figure 3a) further supports the hypothesis that modern dessert watermelons originated in northeastern Africa. Accessions from Central/West Asia are also located in relatively deep branches on the phylogenetic tree, suggesting that dessert watermelons might have been introduced from Africa to Central/West Asia, and then dispersed to the rest of the world. The accessions from East Asia and North America are closely related and yet clearly distinguishable, reflecting the differences in recent breeding efforts in the two areas. East Asian and North American accessions share a unique genetic background different from that of most current accessions in Central/West Asia and Europe, and a small group of European accessions (mostly from Hungary) were found in the deepest branch in the East Asia/North America clade, suggesting that watermelons from East Asia and North America might be derived from a single ancestral group and could be distributed through Europe. By including a large collection of C. lanatus accessions collected in southern Africa, our analyses showed a close relationship between C. lanatus collected in southern Africa and C. mucosospermus in Mali and Senegal. The C. lanatus accessions collected in southern Africa display various flesh colours and sweetness levels (https://npgsweb.ars-grin.gov/gringlobal/descriptors.aspx) and have a unique genetic background different from the other C. lanatus accessions collected in regions other than southern Africa, suggesting parallel evolution for increased flesh sugar content and coloration in different C. lanatus populations, or introduction of alleles controlling these trait from more improved cultivars to the southern African accessions. Knowledge of causal genes underlying domestication traits and distribution of functionally diverse alleles in different watermelon populations is needed to better demonstrate the evolutionary route of dessert watermelons.
The enhancement of disease resistance has been a major focus of current watermelon improvement programmes. Using the GBS SNP makers combined with GWAS, potential QTLs for resistance to BFB, powdery mildew and PRSV‐W were identified. BFB, caused by Acidovorax citrulli, is a devastating disease of most cucurbit crops including watermelon. Numerous BFB outbreaks have occurred in watermelon growing regions in the United States and in countries throughout the world. Recently, using a recombinant inbred line population, Branham et al. (2019) identified six QTLs significantly associated with foliar BFB resistance. Three of the QTLs (qAc‐1.1, qAc‐2.1 and qAc‐8.1, on chromosomes 1, 2 and 8, respectively) were consistent in multiple tests and explained the highest proportion of variation. Given the low heritability and large environmental component of disease response to A. citrulli, the three QTLs (qAc‐1.1, qAc‐2.1 and qAc‐8.1) identified in C. amarus and the two additional QTLs identified in this study on chromosomes 10 (~32.6–32.9 Mb) and 6 (~10.9 Mb) in C. lanatus/C. mucosospermus can be further explored for marker development and used to pyramid genes/loci for enhanced BFB resistance in elite watermelon cultivars.
Powdery mildew is a major disease of cucurbit crops, caused by the fungus Podosphaera xanthii. In the United States, two distinct powdery mildew races, 1W and 2W, are known for watermelon, and several cultivars and wild species highly resistant to these races have been identified (Davis et al., 2007; Tetteh et al., 2010). Recently, Kim et al. (2015) identified a major QTL (pmr2.1) for resistance to powdery mildew race 1W on chromosome 2 that explained 80.0% of the phenotypic variation. In the present study, GWAS analysis identified a major peak associated with powdery mildew race 2W resistance also on chromosome 2 (~29.2–31.0 Mb) overlapping with QTL pmr2.1, and additional SNPs on chromosomes 3, 6, 8, 9 and 10 associated with race 2W resistance (Figure 4b and Table S12). These results indicate the possibility that the QTL on chromosome 2 might be associated with resistance to both powdery mildew races 1W and 2W.
PRSV‐W is an important potyvirus causing serious economic damage to the watermelon crop. Resistance to PRSV‐W has been identified, and genetic inheritance mode of this important potyvirus in three C. amarus accessions (PI 244017, PI 244019 and PI 485583) has been determined, suggesting that the resistance is controlled by a single recessive gene (Guner et al., 2018). In contrast, a genetic inheritance study of PRSV‐W resistance in C. lanatus PI 595201 indicated that the inheritance of PRSV‐resistance is complex with additive genetic effects, and is controlled by multiple genes (de Azevedo et al., 2012). The GWAS analysis here, using the C. lanatus/C. mucosospermus PIs, identified SNPs on multiple chromosomes with significant associations with PRSV‐W resistance (Figure 4b and Table S12). These results suggest that resistance to PRSV‐W in C. lanatus might be controlled by several genes with additive effects while in C. amarus the resistance is controlled by a single gene.
GWAS analyses in the present study also successfully identified genomic regions known to underlie fruit quality traits such as flesh colour, fruit shape and rind pattern, and additional novel SNPs highly associated with these traits. The QTLs identified in this study together with QTLs identified in recent genetic inheritance studies provide a useful platform for the development of molecular markers for use in breeding programmes aiming to enhance disease resistance and fruit quality in elite watermelon cultivars.
Experimental procedures
Plant materials
For genome and transcriptome sequencing of ‘Charleston Gray’, seeds were obtained from a line descended from the original ‘Charleston Gray’ through self‐pollination over several generations at the U.S. Vegetable Laboratory (USVL) and germinated in a greenhouse at the USVL. For GBS, a total of 1365 PIs, including 1211 C. lanatus, 52 C. mucosospermus and 102 C. amarus, were obtained from self‐pollinated plants (one generation) and grown in the greenhouse at North Carolina State University.
Construction and sequencing of genomic and RNA‐Seq libraries
Genomic DNA was extracted from young fresh leaves of ‘Charleston Gray’ using the QIAGEN DNeasy Plant Mini Kit (QIAGEN, Valencia, CA) following the manufacturer's instructions. Two paired‐end genomic libraries with insert sizes of 400 bp and 1 kb, respectively, were prepared using the Genomic DNA Sample Prep kit (Illumina, San Diego, CA) according to the manufacturer's protocol, and sequenced on an Illumina GAIIx platform. The 400‐bp library was sequenced additionally on a HiSeq 2500 system. Another library with insert size of 400 bp was constructed with the DNA Library Preparation kit (Kapa Biosystems, Wilmington, MA) without PCR amplification, and sequenced on an Illumina MiSeq system. Four mate‐pair libraries with insert sizes of 3–5, 8–10 (2 libraries) and 15–20 kb were prepared with the Nextera Mate Pair Sample Preparation kit (Illumina), and sequenced on a HiSeq 2500 system. All libraries were sequenced with the paired‐end mode.
Total RNA was extracted from fruit flesh tissues of ‘Charleston Gray’ at 11, 20, 30 and 40 days after pollination, with two biological replicates for each stage, using the QIAGEN RNeasy Plant Mini Kit (QIAGEN). RNA‐Seq libraries were constructed using the NEB Next UltraTM RNA Library Prep Kit (NEB, Beverly, MA) and sequenced on the Illumina HiSeq 2500 system with the paired‐end mode.
De novo genome assembly
Duplicated read pairs, defined as having identical bases at positions of 14–90 in both left and right reads, were collapsed into unique read pairs. The non‐redundant reads were processed with Trimmomatic (Bolger et al., 2014) to remove adaptors and low‐quality sequences. The mate‐pair reads were processed with the ShortRead package (Morgan et al., 2009) to remove junction adaptors. The resulting reads were assembled into scaffolds using SOAPdenovo2 (Luo et al., 2012). Gaps in the scaffolds were filled with GapCloser (Luo et al., 2012). Pilon (Walker et al., 2014) was used to correct base errors, fix mis‐assemblies and fill additional gaps. The assembly was then aligned to NCBI non‐redundant nucleotide database using BLASTN with an E‐value cut‐off of 1e‐5. Scaffolds with more than 95% of their length similar to sequences of microorganisms, mitochondria or chloroplasts were considered contaminants and removed. Redundant scaffolds with sequence identity higher than 99% and more than 95% of their length covered by other scaffolds were removed.
Transposable element annotation and gene prediction
Long terminal repeat retrotransposon (LTR‐RT) and miniature inverted‐repeat transposable element (MITE) libraries were de novo constructed by screening the ‘Charleston Gray’ genome using LTRharvest (Ellinghaus et al., 2008) and MITE‐Hunter (Han and Wessler, 2010), respectively. After masking the assembly with these libraries using RepeatMasker (http://www.repeatmasker.org), we further searched for repeat elements in the unmasked sequences using RepeatModeler (http://www.repeatmasker.org/RepeatModeler.html). All the identified repetitive sequences were combined into a single repeat library and compared against the Swiss‐Prot database (Magrane and UniProt Consortium, 2011). Sequences that matched non‐TE proteins in the database were removed. TEs were classified using REPCLASS (Feschotte et al., 2009). The classified repeat library was then used to identify TEs in the ‘Charleston Gray’ genome with RepeatMasker.
The repeat‐masked ‘Charleston Gray’ assembly was used for gene prediction with MAKER (Cantarel et al., 2008) by integrating evidences from ab initio gene prediction, transcript mapping and protein homology to define confident gene models. SNAP (Korf, 2004) and AUGUSTUS (Stanke et al., 2006) were used for ab initio gene predictions. Two transcriptome assemblies from fruit RNA‐Seq data were obtained using Trinity (Grabherr et al., 2011) with the de novo mode and the genome‐guided mode, respectively, and aligned to the ‘Charleston Gray’ assembly using PASA2 (Haas, 2003). The resulting alignments were used as the transcript evidence. Protein sequences from Arabidopsis, watermelon, cucumber and melon, as well as the UniProt (Swiss‐Prot plant division) database, were aligned to the ‘Charleston Gray’ genome using Spaln (Iwata and Gotoh, 2012) to provide protein homology evidence. For gene annotation, protein sequences of the predicted ‘Charleston Gray’ genes were compared against the Arabidopsis protein and UniProt (Swiss‐Prot/TrEMBL) databases using BLAST, as well as the InterPro database using InterProScan (Jones et al., 2014). Blast2GO (Conesa et al., 2005) was used to obtain gene ontology (GO) annotations.
Synteny analysis
To identify syntenic regions between the ‘Charleston Gray’ and ‘97103’ genomes, protein sequences from the two cultivars were aligned using BLASTP, and high‐confidence collinear blocks were determined using MCScanX with an E‐value cut‐off of 1e‐10 (Wang et al., 2012). Ks values of orthologous gene pairs were calculated using the Yang–Nielsen algorithm implemented in PAML (Yang, 1997). LAST (v869; Kielbasa et al., 2011) was used to identify unique best alignments between the genomes of ‘Charleston Gray’ and ‘97103’.
Identification of SNPs and SVs
To detect SNPs and small indels between ‘97103’ and ‘Charleston Gray’, paired‐end genomic reads of ‘97103’ (Guo et al., 2013) were mapped to the ‘Charleston Gray’ genome using BWA (version 0.6.2) (Li and Durbin, 2009). Uniquely mapped reads were kept for variant detection. Genotypes were assigned to each genomic position based on the alignment mpileup files generated by SAMtools (Li et al., 2009). SNPs and small indels were then identified if they were supported by at least four mapped reads. To detect large SVs (≥50 bp), genome sequences of ‘97103’ and ‘Charleston Gray’ were aligned using Minimap2 (Li, 2018), and based on the alignment indels and tandem/repeat expansions/contractions were identified using Assemblytics (Nattestad and Schatz, 2016). To confirm the identified SVs, reads generated from one genome were aligned to the opposite genome using bowtie (Langmead et al., 2009) allowing one mismatch. SVs with junction sites at both ends having no more than five spanning reads were considered highly confident. Meantime, indels (≥50 bp) were also detected with SpeedSeq (Chiang et al., 2015) based on ‘97103’ genomic reads mapped to the ‘Charleston Gray’ genome and ‘Charleston Gray’ reads mapped to the ‘97103’ genome. Sequences flanking the identified indels (five kb on each side) from one genome were aligned to the other genome using BLAST with E‐value <1e‐10 and sequence identity >90%. Indels with flanking sequences aligned to the proper positions on the other genome were considered highly confident. The two high‐confidence sets of SVs by reference sequence comparison and read mapping, respectively, were integrated, and those spanning any gap regions in the assembled genomes were removed.
DNA extraction for GBS
About 100 mg fresh leaf tissue was collected from a young seedling representing each PI. The leaf tissue was freeze‐dried and then ground to a fine powder using 5/32” stainless steel balls (AbbottBall, West Hartford, CT) in a Retsch Mixer Mill (Retsch, Newtown, PA). DNA was isolated using the Plant DNA DS Kit (Omega Bio‐Tek, Norcross, GA). The DNA was quantified with the Quant‐iT PicoGreen dsDNA Kit (Invitrogen, Carlsbad, CA), and its quality was checked by electrophoresis of undigested and HindIII‐digested DNA on agarose gels.
GBS analysis and SNP calling
DNA samples of all PI accessions were subjected to GBS analysis following the protocol described in Elshire et al. (2011) using the ApeKI restriction enzyme (NEB). GBS libraries were sequenced on a HiSeq 2500 system to obtain reads with lengths of 101 bp. The TASSEL 5.0 GBS Discovery Pipeline (Glaubitz et al., 2014) was used for SNP identification using the ‘Charleston Gray’ genome as the reference. Tags were identified from raw reads possessing a barcode and a restriction enzyme cut site using GBSSeqToTagDBPlugin with parameters ‘‐kmerLength 90 ‐minKemrL 30 ‐mnQS 10 ‐c 10 ‐maKmerNum 200000000’. Tags supported by at least ten reads were retrieved and reformatted using TagExportToFastqPlugin, and mapped to the ‘Charleston Gray’ genome using BWA (v0.7.13; Li et al., 2009) with default parameters. Based on the alignments, positions of aligned tags were determined using SAMtoGBSdbPlugin, and SNPs were identified from the aligned tags using DiscoverySNPCallerPluginV2 with default parameters. SNPs were filtered using VCFtools (Danecek et al., 2011) to keep those that were biallelic and had missing rate ≤ 50% and MAF ≥1%.
Phylogenetic and population genomic analyses
Biallelic SNPs with MAF ≥1% and missing rate ≤50% were further filtered to remove those in high linkage disequilibrium (LD) blocks using SNPhylo (v20140701; Lee et al., 2014) with the parameter ‘‐l 0.1’, and the remaining SNPs were used in phylogenetic and population structure analyses. A maximum‐likelihood tree was constructed using IQTREE with 1000 bootstraps (v1.6.8; Nguyen et al., 2015), using C. amarus accessions as the outgroup. ggtree (v1.10.5; Yu et al., 2017) was used to visualize the phylogenetic tree.
PCA was performed using Plink (v1.9; Purcell et al., 2007). Population structure analysis was performed using STRUCTURE (v2.3.4; Falush et al., 2003). To determine the most likely cluster number, STRUCTURE analyses were run 20 times for each K value ranging from 2 to 20, using 3715 SNPs filtered by LD information using SNPhylo described above, with an admixture model. After the best K (K = 2) was determined, population structure of the watermelon accessions was inferred using fastStructure (v1.0; Raj et al., 2014) with all SNPs for each K (K = 2‐5).
Nucleotide diversity (π) and population fixation index (F ST) were calculated using vcftools (v 0.1.15; Danecek et al., 2011) using unfiltered raw SNPs. The average nucleotide diversity was calculated as the sum of nucleotide diversity at each site divided by the total bases covered by GBS reads in the genome.
Phenotypic data collection and disease symptom rating
Historical phenotypic data for watermelon fruit quality traits were downloaded from the GRIN database (https://npgsweb.ars-grin.gov/gringlobal/descriptors.aspx). Fruit flesh colour was rated as red (red and pink) and non‐red (green, white, yellow and orange). Fruit shape was rated as round and elongated (oblate, oblong and elongated). Rind stripe pattern was rated as striped and solid. For PRSV‐W disease screening, young watermelon seedlings were rated at 14, 21 and 28 days post‐inoculation (DPI), and the average of the three ratings was used. BFB disease data were collected from young fruits at 21 DPI. Powdery mildew race 2W disease data were collected from young seedlings at 14 and 28 DPI, and the average of the two ratings was used. All three diseases were rated using a visual scale of 0‐9, where 0 = no symptoms and 9 = dead plant. Phenotypic data for accessions genotyped in the present study were used for GWAS.
Genome‐wide association studies
Citrullus lanatus and C. mucosospermus accessions were used in GWAS. A Balding–Nichols kinship matrix (Balding and Nichols, 1995) constructed using the 57 294 unfiltered biallelic SNPs was used to correct population structure. Missing genotypes were imputed using the k‐nearest neighbour algorithm implemented in fillGenotype (Huang et al., 2010) with an optimal combination of parameters (w = 30, k = 9, p = −9, r = 0.8) determined based on filling rate and imputation accuracy (Table S13). Each combination of the following parameters: w (20, 30, 50, 65, 80), p (−3, −5, −7, −9), k (3, 5, 7, 9), and r (0.65, 0.7, 0.75, 0.8), was tested with accessions having lowest missing rates in each of the two Citrullus species (C. mucosospermus PI 306780, and C. lanatus PI 635626), and 10%, 20% and 30% SNP sites were randomly masked as missing genotypes for imputing. Only biallelic imputed SNPs within C. lanatus and C. mucosospermus accessions (a total of 24 065 SNPs) were used for GWAS. GWAS were performed using the linear mixed model implemented in EMMAX (Kang et al., 2010). Genome‐wide significance thresholds of GWAS were determined using the Bonferroni correction at α = 0.05 and α = 0.01 for significant and extremely significant associations, respectively, as described in Li et al. (2012).
Supporting information
Figure S1 K‐mer distribution of Illumina genomic sequencing reads of ‘Charleston Gray’.
Figure S2 Syntenic orthologous gene blocks between ‘Charleston Gray’ and ‘97103’.
Figure S3 Collinearity between the ‘Charleston Gray’ and ‘97103’ genomes.
Figure S4 Collinearity between ‘Charleston Gray’ and ‘97103’ chromosome 1.
Figure S5 Size distribution of indels between genomes of ‘Charleston Gray’ and ‘97103’.
Figure S6 SNP density across the 11 watermelon ‘Charleston Gray’ chromosomes.
Figure S7 Plot of ΔK values with K from 2 to 20 in the STRUCTURE analysis for the 1367 watermelon accessions using GBS SNPs.
Figure S8 Multidimensional scaling of pairwise F ST between C. mucosospermus (CM) and C. lanatus accessions from different geographic regions.
Table S1 Summary of ‘Charleston Gray’ genome sequencing data.
Table S2 Statistics of ‘Charleston Gray’ pseudomolecules.
Table S3 Mapping of RNA‐Seq reads and ESTs to the ‘Charleston Gray’ genome.
Table S4 Assessment of ‘Charleston Gray’ genome assembly completeness by BUSCO.
Table S5 Summary of repeat sequences in the ‘Charleston Gray’ genome.
Table S6 Summary of SNPs and small indels between ‘Charleston Gray’ and ‘97103’.
Table S7 Structural variations between ‘Charleston Gray’ and ‘97103’.
Table S8 Structural variations between ‘Charleston Gray’ and ‘97103’ overlapping with known QTLs.
Table S9 List of the 1365 watermelon accessions from the NPGS collection.
Table S10 Weighted pairwise F ST values between C. mucosospermus and C. lanatus accessions from different geographic regions.
Table S11 Phenotypic data for GWAS.
Table S12 SNPs significantly associated with agronomic traits identified through GWAS.
Table S13 Evaluation of the SNP imputation using KNN under the optimal parameters ‘w = 50, k = 7, p = ‐9, r = 0.8’.
Acknowledgements
This research was supported by grants from USDA National Institute of Food and Agriculture Specialty Crop Research Initiative (2015‐51181‐24285), the National Watermelon Promotion Board, National Watermelon Association, and USDA Agricultural Research Service. The authors declare no conflict of interest.
Contributor Information
Amnon Levi, Email: Amnon.Levi@ars.usda.gov.
Zhangjun Fei, Email: zf25@cornell.edu.
Data availability
The genome sequence of ‘Charleston Gray’ has been deposited at DDBJ/ENA/GenBank under the accessions CP026477‐CP026488. Raw genome and transcriptome sequencing reads have been deposited into the NCBI sequence read archive (SRA) under accessions SRP183199 and SRP183523, respectively. The genome sequence of ‘Charleston Gray’ and the GBS SNPs are also available at the Cucurbit Genomics Database (http://cucurbitgenomics.org; Zheng et al., 2019).
References
- de Azevedo, S.M. , Maluf, W.R. , Faria, M.V. , de Resende, J.T.V. , de Menezes, C.B. and do Nascimento, I.R. (2012) Inheritance of resistance to the papaya ringspot virus‐watermelon strain (PRSV‐W) from watermelon accession ‘PI 595201’. Crop Breed. Appl. Biotechnol., 12, 67–75. [Google Scholar]
- Balding, D.J. and Nichols, R.A. (1995) A method for quantifying differentiation between populations at multi‐allelic loci and its implications for investigating identity and paternity. Genetica 96, 3–12. [DOI] [PubMed] [Google Scholar]
- Bang, H. , Davis, A.R. , Kim, S. , Leskovar, D.I. and King, S.R. (2010) Flesh color inheritance and gene interactions among canary yellow, pale yellow, and red watermelon. J. Am. Soc. Hortic. Sci. 135, 362–368. [Google Scholar]
- Bolger, A.M. , Lohse, M. and Usadel, B. (2014) Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Branham, S.E. , Levi, A. , Farnham, M.W. and Wechter, W.P. (2017) A GBS‐SNP‐based linkage map and quantitative trait loci (QTL) associated with resistance to Fusarium oxysporum f. sp. niveum race 2 identified in Citrullus lanatus var. citroides . Theor. Appl. Genet. 130, 319–330. [DOI] [PubMed] [Google Scholar]
- Branham, S.E. , Levi, A. , Katawczik, M.L. and Wechter, W.P. (2019) QTL mapping of resistance to bacterial fruit blotch in Citrullus amarus . Theor. Appl. Genet. 132, 1463–1471. [DOI] [PubMed] [Google Scholar]
- Cantarel, B.L. , Korf, I. , Robb, S.M.C. , Parra, G. , Ross, E. , Moore, B. , Holt, C. et al. (2008) MAKER: an easy‐to‐use annotation pipeline designed for emerging model organism genomes. Genome Res. 18, 188–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang, C. , Layer, R.M. , Faust, G.G. , Lindberg, M.R. , Rose, D.B. , Garrison, E.P. , Marth, G.T. et al. (2015) SpeedSeq: ultra‐fast personal genome analysis and interpretation. Nat. Methods 12, 966–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chomicki, G. and Renner, S.S. (2015) Watermelon origin solved with molecular phylogenetics including Linnaean material: another example of museomics. New Phytol. 205, 526–532. [DOI] [PubMed] [Google Scholar]
- Conesa, A. , Gotz, S. , Garcia‐Gomez, J.M. , Terol, J. , Talon, M. and Robles, M. (2005) Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21, 3674–3676. [DOI] [PubMed] [Google Scholar]
- Danecek, P. , Auton, A. , Abecasis, G. , Albers, C.A. , Banks, E. , DePristo, M.A. , Handsaker, R.E. et al. (2011) The variant call format and VCFtools. Bioinformatics 27, 2156–2158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis, A.R. , Levi, A. , Tetteh, A. , Wehner, T. , Russo, V. and Pitrat, M. (2007) Evaluation of watermelon and related species for resistance to race 1W powdery mildew. J. Am. Soc. Hort. Sci. 132, 790–795. [Google Scholar]
- Dou, J. , Zhao, S. , Lu, X. , He, N. , Zhang, L. , Ali, A. , Kuang, H. et al. (2018) Genetic mapping reveals a candidate gene (ClFS1) for fruit shape in watermelon (Citrullus lanatus L.). Theor. Appl. Genet. 131, 947–958. [DOI] [PubMed] [Google Scholar]
- Ellinghaus, D. , Kurtz, S. and Willhoeft, U. (2008) LTRharvest, an efficient and flexible software for de novo detection of LTR retrotransposons. BMC Bioinformatics 9, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elshire, R.J. , Glaubitz, J.C. , Sun, Q. , Poland, J.A. , Kawamoto, K. , Buckler, E.S. and Mitchell, S.E. (2011) A robust, simple genotyping‐by‐sequencing (GBS) approach for high diversity species. PLoS ONE 6, e19379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falush, D. , Stephens, M. and Pritchard, J.K. (2003) Inference of population structure using multilocus genotype data: linked loci and correlated allele frequencies. Genetics 164, 1567–1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feschotte, C. , Keswani, U. , Ranganathan, N. , Guibotsy, M.L. and Levine, D. (2009) Exploring repetitive DNA landscapes using REPCLASS, a tool that automates the classification of transposable elements in eukaryotic genomes. Genome Biol. Evol. 1, 205–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao, L. , Gonda, I. , Sun, H. , Ma, Q. , Bao, K. , Tieman, D.M. , Burzynski‐Chang, E.A. et al. (2019) The tomato pan‐genome uncovers new genes and a rare allele regulating fruit flavor. Nat. Genet. (in press). [DOI] [PubMed] [Google Scholar]
- Glaubitz, J.C. , Casstevens, T.M. , Lu, F. , Harriman, J. , Elshire, R.J. , Sun, Q. and Buckler, E.S. (2014) TASSEL‐GBS: a high capacity genotyping by sequencing analysis pipeline. PLoS ONE 9, e90346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grabherr, M.G. , Haas, B.J. , Yassour, M. , Levin, J.Z. , Thompson, D.A. , Amit, I. et al. (2011) Full‐length transcriptome assembly from RNA‐Seq data without a reference genome. Nat. Biotechnol. 29, 644–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guner, N. , Pesic‐VanEsbroeck, Z. , Rivera‐Burgos, L.A. and Wehner, T.C. (2018) Inheritance of resistance to Papaya ringspot virus‐watermelon strain in watermelon. HortScience 53, 624–627. [Google Scholar]
- Guo, S. , Zhang, J. , Sun, H. , Salse, J. , Lucas, W.J. , Zhang, H. et al. (2013) The draft genome of watermelon (Citrullus lanatus) and resequencing of 20 diverse accessions. Nat. Genet. 45, 51–58. [DOI] [PubMed] [Google Scholar]
- Haas, B.J. (2003) Improving the Arabidopsis genome annotation using maximal transcript alignment assemblies. Nucleic Acids Res. 31, 5654–5666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han, Y. and Wessler, S.R. (2010) MITE‐Hunter: a program for discovering miniature inverted‐repeat transposable elements from genomic sequences. Nucleic Acids Res. 38, e199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, X. , Wei, X. , Sang, T. , Zhao, Q. , Feng, Q. , Zhao, Y. et al. (2010) Genome‐wide association studies of 14 agronomic traits in rice landraces. Nat. Genet. 42, 961–967. [DOI] [PubMed] [Google Scholar]
- Iwata, H. and Gotoh, O. (2012) Benchmarking spliced alignment programs including Spaln2, an extended version of Spaln that incorporates additional species‐specific features. Nucleic Acids Res. 40, e161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarret, R.L. , Merrick, L.C. , Holms, T. , Evans, J. and Aradhya, M.K. (1997) Simple sequence repeats in watermelon [Citrullus lanatus (Thunb.) Matsum. & Nakai]. Genome 40, 433–441. [DOI] [PubMed] [Google Scholar]
- Jones, P. , Binns, D. , Chang, H.‐Y. , Fraser, M. , Li, W. , McAnulla, C. et al. (2014) InterProScan 5: genome‐scale protein function classification. Bioinformatics 30, 1236–1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang, H.M. , Sul, J.H. , Service, S.K. , Zaitlen, N.A. , Kong, S. , Freimer, N.B. , Sabatti, C. et al. (2010) Variance component model to account for sample structure in genome‐wide association studies. Nat. Genet., 42, 348–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kielbasa, S.M. , Wan, R. , Sato, K. , Horton, P. and Frith, M.C. (2011) Adaptive seeds tame genomic sequence comparison. Genome Res. 21, 487–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, K.H. , Hwang, J.H. , Han, D.Y. , Park, M. , Kim, S. , Choi, D. et al. (2015) Major quantitative trait loci and putative candidate genes for powdery mildew resistance and fruit‐related traits revealed by an intraspecific genetic map for watermelon (Citrullus lanatus var. lanatus). PLoS ONE 10, e0145665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korf, I. (2004) Gene finding in novel genomes. BMC Bioinformatics 5, 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead, B. , Trapnell, C. , Pop, M. and Salzberg, S.L. (2009) Ultrafast and memory‐efficient alignment of short DNA sequences to the human genome. Genome Biol. 10, R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, T.‐H. , Guo, H. , Wang, X. , Kim, C. and Paterson, A.H. (2014) SNPhylo: a pipeline to construct a phylogenetic tree from huge SNP data. BMC Genom. 15, 162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levi, A. , Thomas, C.E. , Wehner, T.C. and Zhang, X. (2001) Low genetic diversity indicates the need to broaden the genetic base of cultivated watermelon. HortScience 36, 1096–1101. [Google Scholar]
- Levi, A. , Davis, A. , Hernandez, A. , Wechter, P. , Thimmapuram, J. , Trebitsh, T. et al. (2006) Genes expressed during the development and ripening of watermelon fruit. Plant Cell Rep. 25, 1233–1245. [DOI] [PubMed] [Google Scholar]
- Levi, A. , Thies, J.A. , Wechter, W.P. , Harrison, H.F. , Simmons, A.M. , Reddy, U.K. et al. (2013) High frequency oligonucleotides: targeting active gene (HFO‐TAG) markers revealed wide genetic diversity among Citrullus spp. accessions useful for enhancing disease or pest resistance in watermelon cultivars. Genet. Resour. Crop Evol. 60, 427–440. [Google Scholar]
- Levi, A. , Jarret, R. , Kousik, S. , Patrick Wechter, W. , Nimmakayala, P. and Reddy, U.K. (2017) Genetic Resources of Watermelon. In Genetics and Genomics of Cucurbitaceae( Grumet, R. , Katzir, N. and Garcia‐Mas, J. , eds), pp. 87–110. Cham: Springer. [Google Scholar]
- Li, H. (2018) Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, H. and Durbin, R. (2009) Fast and accurate short read alignment with Burrows‐Wheeler transform. Bioinformatics 25, 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, H. , Handsaker, B. , Wysoker, A. , Fennell, T. , Ruan, J. , Homer, N. et al. (2009) The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, M.‐X. , Yeung, J.M.Y. , Cherny, S.S. and Sham, P.C. (2012) Evaluating the effective numbers of independent tests and significant p‐value thresholds in commercial genotyping arrays and public imputation reference datasets. Hum. Genet. 131, 747–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo, R. , Liu, B. , Xie, Y. , Li, Z. , Huang, W. , Yuan, J. et al. (2012) SOAPdenovo2: an empirically improved memory‐efficient short‐read de novo assembler. Gigascience 1, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magrane, M. and UniProt Consortium (2011) UniProt Knowledgebase: a hub of integrated protein data. Database 2011, bar009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan, M. , Anders, S. , Lawrence, M. , Aboyoun, P. , Pagès, H. and Gentleman, R. (2009) ShortRead: a bioconductor package for input, quality assessment and exploration of high‐throughput sequence data. Bioinformatics 25, 2607–2608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nattestad, M. and Schatz, M.C. (2016) Assemblytics: a web analytics tool for the detection of variants from an assembly. Bioinformatics 32, 3021–3023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen, L.‐T. , Schmidt, H.A. , von Haeseler, A. and Minh, B.Q. (2015) IQ‐TREE: a fast and effective stochastic algorithm for estimating maximum‐likelihood phylogenies. Mol. Biol. Evol. 32, 268–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nimmakayala, P. , Levi, A. , Abburi, L. , Abburi, V. , Tomason, Y.R. , Saminathan, T. et al. (2014) Single nucleotide polymorphisms generated by genotyping by sequencing to characterize genome‐wide diversity, linkage disequilibrium, and selective sweeps in cultivated watermelon. BMC Genom. 15, 767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paris, H.S. (2015) Origin and emergence of the sweet dessert watermelon, Citrullus lanatus . Ann. Bot. 116, 133–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park, S. , Kim, K.‐T. , Kang, S.‐C. and Yang, H.‐B. (2016) Rapid and practical molecular marker development for rind traits in watermelon. Hortic. Environ. Biotechnol. 57, 385–391. [Google Scholar]
- Perkins‐Veazie, P. , Collins, J.K. and Clevidence, B. (2007) Watermelon and health. Acta Hortic. 731, 121–128. [Google Scholar]
- Purcell, S. , Neale, B. , Todd‐Brown, K. , Thomas, L. , Ferreira, M.A.R. , Bender, D. et al. (2007) PLINK: a tool set for whole‐genome association and population‐based linkage analyses. Am. J. Hum. Genet. 81, 559–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raj, A. , Stephens, M. and Pritchard, J.K. (2014) fastSTRUCTURE: variational inference of population structure in large SNP data sets. Genetics 197, 573–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy, U.K. , Aryal, N. , Islam‐Faridi, N. , Tomason, Y.R. , Levi, A. and Nimmakayala, P. (2013) Cytomolecular characterization of rDNA distribution in various Citrullus species using fluorescent in situ hybridization. Genet. Resour. Crop Evol. 60, 2091–2100. [Google Scholar]
- Reddy, U.K. , Nimmakayala, P. , Levi, A. , Abburi, V.L. , Saminathan, T. , Tomason, Y.R. et al. (2014) High‐resolution genetic map for understanding the effect of genome‐wide recombination rate on nucleotide diversity in watermelon. G3 (Bethesda) 4, 2219–2230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren, Y. , McGregor, C. , Zhang, Y. , Gong, G. , Zhang, H. , Guo, S. et al. (2014) An integrated genetic map based on four mapping populations and quantitative trait loci associated with economically important traits in watermelon (Citrullus lanatus). BMC Plant Biol. 14, 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renner, S.S. , Sousa, A. and Chomicki, G. (2017) Chromosome numbers, Sudanese wild forms, and classification of the watermelon genus Citrullus, with 50 names allocated to seven biological species. Taxon 66, 1393–1405. [Google Scholar]
- Sheng, Y. , Luan, F. , Zhang, F. and Davis, A.R. (2012) Genetic diversity within Chinese watermelon ecotypes compared with germplasm from other countries. J. Am. Soc. Hortic. Sci. 137, 144–152. [Google Scholar]
- Simão, F.A. , Waterhouse, R.M. , Ioannidis, P. , Kriventseva, E.V. and Zdobnov, E.M. (2015) BUSCO: assessing genome assembly and annotation completeness with single‐copy orthologs. Bioinformatics 31, 3210–3212. [DOI] [PubMed] [Google Scholar]
- Stanke, M. , Tzvetkova, A. and Morgenstern, B. (2006) AUGUSTUS at EGASP: using EST, protein and genomic alignments for improved gene prediction in the human genome. Genome Biol. 7, S11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tetteh, A.Y. , Wehner, T.C. and Davis, A.R. (2010) Identifying resistance to powdery mildew race 2W in the USDA‐ARS watermelon germplasm collection. Crop Sci. 50, 933–939. [Google Scholar]
- Walker, B.J. , Abeel, T. , Shea, T. , Priest, M. , Abouelliel, A. , Sakthikumar, S. et al. (2014) Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE 9, e112963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Y. , Tang, H. , Debarry, J.D. , Tan, X. , Li, J. , Wang, X. et al. (2012) MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 40, e49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wehner, T.C. (1999) Vegetable cultivar descriptions for North America, List 25. HortScience 34, 957–1012. [Google Scholar]
- Yang, Z. (1997) PAML: a program package for phylogenetic analysis by maximum likelihood. Bioinformatics 13, 555–556. [DOI] [PubMed] [Google Scholar]
- Yu, G. , Smith, D.K. , Zhu, H. , Guan, Y. and Lam, T.T.‐Y. (2017) ggtree: an r package for visualization and annotation of phylogenetic trees with their covariates and other associated data. Methods Ecol. Evol. 8, 28–36. [Google Scholar]
- Zhang, Z. , Mao, L. , Chen, H. , Bu, F. , Li, G. , Sun, J. et al. (2015) Genome‐wide mapping of structural variations reveals a copy number variant that determines reproductive morphology in cucumber. Plant Cell 27, 1595–1604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng, Y. , Wu, S. , Bai, Y. , Sun, H. , Jiao, C. , Guo, S. et al. (2019) Cucurbit Genomics Database (CuGenDB): a central portal for comparative and functional genomics of cucurbit crops. Nucleic Acids Res. 47, D1128–D1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 K‐mer distribution of Illumina genomic sequencing reads of ‘Charleston Gray’.
Figure S2 Syntenic orthologous gene blocks between ‘Charleston Gray’ and ‘97103’.
Figure S3 Collinearity between the ‘Charleston Gray’ and ‘97103’ genomes.
Figure S4 Collinearity between ‘Charleston Gray’ and ‘97103’ chromosome 1.
Figure S5 Size distribution of indels between genomes of ‘Charleston Gray’ and ‘97103’.
Figure S6 SNP density across the 11 watermelon ‘Charleston Gray’ chromosomes.
Figure S7 Plot of ΔK values with K from 2 to 20 in the STRUCTURE analysis for the 1367 watermelon accessions using GBS SNPs.
Figure S8 Multidimensional scaling of pairwise F ST between C. mucosospermus (CM) and C. lanatus accessions from different geographic regions.
Table S1 Summary of ‘Charleston Gray’ genome sequencing data.
Table S2 Statistics of ‘Charleston Gray’ pseudomolecules.
Table S3 Mapping of RNA‐Seq reads and ESTs to the ‘Charleston Gray’ genome.
Table S4 Assessment of ‘Charleston Gray’ genome assembly completeness by BUSCO.
Table S5 Summary of repeat sequences in the ‘Charleston Gray’ genome.
Table S6 Summary of SNPs and small indels between ‘Charleston Gray’ and ‘97103’.
Table S7 Structural variations between ‘Charleston Gray’ and ‘97103’.
Table S8 Structural variations between ‘Charleston Gray’ and ‘97103’ overlapping with known QTLs.
Table S9 List of the 1365 watermelon accessions from the NPGS collection.
Table S10 Weighted pairwise F ST values between C. mucosospermus and C. lanatus accessions from different geographic regions.
Table S11 Phenotypic data for GWAS.
Table S12 SNPs significantly associated with agronomic traits identified through GWAS.
Table S13 Evaluation of the SNP imputation using KNN under the optimal parameters ‘w = 50, k = 7, p = ‐9, r = 0.8’.
Data Availability Statement
The genome sequence of ‘Charleston Gray’ has been deposited at DDBJ/ENA/GenBank under the accessions CP026477‐CP026488. Raw genome and transcriptome sequencing reads have been deposited into the NCBI sequence read archive (SRA) under accessions SRP183199 and SRP183523, respectively. The genome sequence of ‘Charleston Gray’ and the GBS SNPs are also available at the Cucurbit Genomics Database (http://cucurbitgenomics.org; Zheng et al., 2019).