

Summary
One of the most important goals in the breeding of oilseed crops, including Brassica napus, is to improve the quality of edible vegetable oil, which is mainly determined by the seed fatty acid composition, particularly the C18:1 content. Previous studies have indicated that the C18:1 content is a polygenic trait, and no stable quantitative trait loci (QTLs) except for FAD2 have been reported. By performing a GWAS using 375 low erucic acid B. napus accessions genotyped with the Brassica 60K SNP array and constructing a high‐density SNP‐based genetic map of a 150 DH population, we identified a novel QTL on the A9 chromosome. The novel locus could explain 11.25%, 5.72% and 6.29% of phenotypic variation during three consecutive seasons and increased the C18:1 content by approximately 3%–5%. By fine mapping and gene expression analysis, we found three potential candidate genes and verified the fatty acids in a homologous gene mutant of Arabidopsis. A metal ion‐binding protein was found to be the most likely candidate gene in the region. Thus, the C18:1 content can be further increased to about 80% with this novel locus together with FAD2 mutant allele without compromise of agronomic performance. A closely linked marker, BnA129, for this novel QTL ( OLEA9) was developed so that we can effectively identify materials with high C18:1 content at an early growth stage by marker‐assisted selection. Our results may also provide new insight for understanding the complex genetic mechanism of fatty acid metabolism.
Keywords: rapeseed, oleic acid, quantitative trait loci
Introduction
Oilseed rape (Brassica napus L.) is one of the most important oil crops worldwide and provides high‐quality edible vegetable oil for human consumption. The edibility and processing quality of rapeseed oil are mainly determined by the fatty acid composition of the seeds, particularly the proportions of the three major unsaturated fatty acids: oleic acid (C18:1), linoleic acid (C18:2) and linolenic acid (C18:3) (Gillingham et al., 2011; Micha and Mozaffarian, 2009). Rapeseed oil with a high C18:1 content and a low C18:3 content is less susceptible to oxidation during storage, frying and food processing and is thus desirable for its thermal stability and long shelf life (Matthäus, 2006; Merrill et al., 2008). Currently, most of the oilseed rape cultivars in the world are of canola quality, that is with low erucic acid and glucosinolate contents and containing approximately 55%–65% oleic acid. One of the major goals for quality breeding of oilseed rape (after the removal of erucic acid) is to further increase the C18:1 content in the seeds. Therefore, identifying high oleic acid germplasm and understanding the genetic control of the trait are the prerequisites for high oleic acid breeding.
A number of quantitative trait loci (QTLs) for C18:1 content have been identified based on linkage (Bao et al., 2018; Burns et al., 2003; Chen et al., 2018; Hu et al., 2006; Smooker et al., 2011; Wen et al., 2015; Yan et al., 2011; Yang et al., 2012b; Zhao et al., 2008) and association (Bao et al., 2018; Körber et al., 2016; Qu et al., 2017) mapping studies. One major QTL located on A5 was detected (Hu et al., 2006; Yang et al., 2012b), and a homologous gene of Arabidopsis FAD2, BnaA.FAD2.a, was identified as the A5 major QTL (Hu et al., 2006; Yang et al., 2012b). Further analysis indicated that mutated alleles of BnaA.FAD2.a on A5 that contained a single‐nucleotide substitution (Hu et al., 2006) or a 4‐bp insertion (Yang et al., 2012b) in their coding region resulted in an increase in C18:1 content (from 64.1% to 75%). A new mutant of BnFAD2 with two single‐nucleotide polymorphisms (SNPs) in BnFAD2‐1 and BnFAD2‐2 again confirmed the importance of FAD2 (Long et al., 2018).
Four orthologues of FAD2 were identified in B. napus in our previous study (Yang et al., 2012b). In contrast to BnaA.FAD2.b, three other copies (BnaA.FAD2.a, BnaC.FAD2.a and BnaC.FAD2.b) appear intact and are likely to be functional. Most of the high oleic acid rapeseed germplasm that is currently available results from the mutation of BnaA.FAD2.a on A5. Silencing of the BnFAD2 gene by RNA interference (RNAi) (Peng et al., 2010) or CRISPR/Cas9‐mediated genome editing (Okuzaki et al., 2018) or knockout of three functional copies of FAD2 in B. napus (Wells et al., 2014) could result in a C18:1 content of up to 84%–85%.
Although great success has been achieved in the development of high oleic acid oilseed rape by manipulating the FAD2 gene family, the effects of enhanced oleic acid content in seeds on plant growth and development are less clear. Early studies in Arabidopsis showed that the loss‐of‐function mutant of FAD2 was hypersensitive to salt stress and low temperature (Miquel, 1994; Miquel et al., 1993; Zhang et al., 2012). The agronomic traits of some oilseed rape plants with an extremely high oleic acid content were also poor when the plants were grown at lower temperatures (Kinney, 1994). Most recently, a field experiment showed that mutant lines with more than 80% oleate in the seed oil had significantly lower seedling establishment and vigour, delayed flowering and reduced plant height at maturity, and 7%–11% reductions in seed oil content under field conditions (Bai et al., 2018).
It is thus highly desirable to develop novel genetic resources of B. napus with both increased C18:1 content and good agronomic performance. In this study, QTL mapping by both linkage and association analyses was performed in B. napus, and a novel QTL for C18:1 content was found on chromosome A9, which functions to increase the C18:1 content independently of the FAD2 gene. By means of fine mapping and gene expression analysis, together with mutant analysis in Arabidopsis, we identified a candidate gene for the locus. Our results thus provide novel information for a better understanding of the genetic architecture of the oleic acid content and fatty acid composition of B. napus seeds.
Results
A GWAS identified a novel locus for oleic acid content on chromosome A9
To search for novel loci for fatty acid variant in B. napus, we performed a genome‐wide association study (GWAS) using a panel of 375 accessions, which were genotyped in our previous study (Liu et al., 2016). All the accessions, which are selected from low erucic acid inbred lines (C22:1 < 3%, Figure 1a), were quantitated by fatty acid profiling. Extensive phenotypic variations in three major unsaturated fatty acids (C18:1, C18:2 and C18:3) were observed among the accessions (Figure 1a). The C18:1 content in the panel exhibited continuous variation, ranging from 48.8% to 76.2%, with an average of 64.7% (Figure 1b). The C18:1 content was significantly associated with the content of most other fatty acids in this low erucic acid association panel, except for the C20:1 content, with a notable high correlation coefficient between the C18:1 and C18:2 contents (−0.92) and between the C18:1 and C18:3 contents (−0.70) (Figure 1c).
Figure 1.
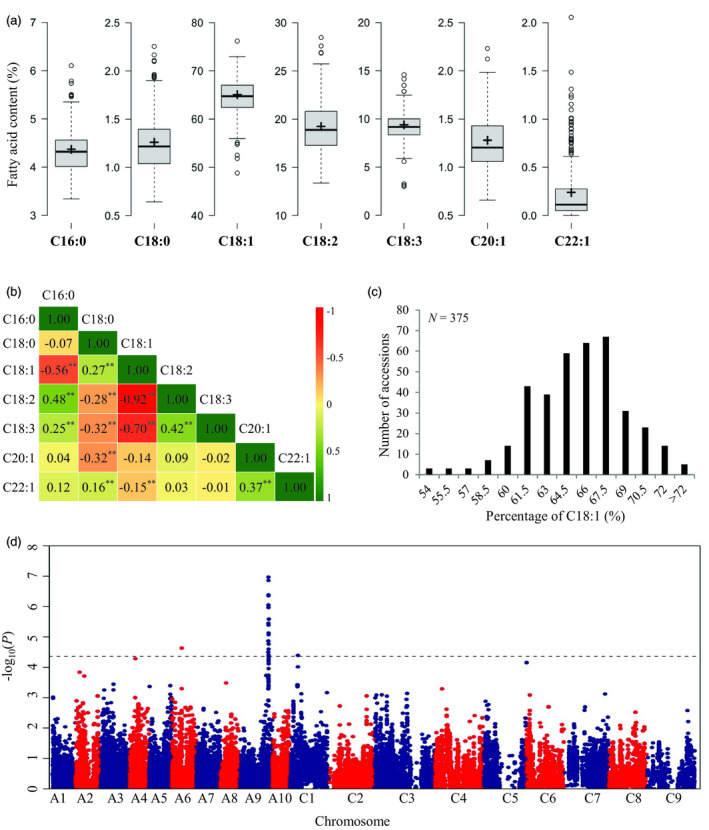
Genome‐wide association mapping for oleic acid (C18:1) content using an association panel of 375 Brassica napus accessions. (a) Box plot for fatty acid composition of the association panel. The middle line indicates the median; the plus sign indicates the mean; the box indicates the range of the 25–75th percentiles of the total data; the whiskers extend 1.5 times the interquartile range from the 25th and 75th percentiles; and the outer dots are outliers. (b) Distribution patterns of the C18:1 content in the 375 B. napus accessions. (c) Pairwise correlations for fatty acid composition. ** indicates correlations passed significance tests with P‐values < 0.01. (d) Manhattan plots for the GWAS of C18:1 content. The dashed horizontal line depicts the uniform significance threshold (−log101/23 168 = 4.36).
Furthermore, a genome‐wide association analysis of C18:1 content was conducted with a mixed linear model (PCA + K model). A total of 19 SNPs were significantly associated with C18:1 content with a threshold of P < 4.32 × 10−5 (1/23 168; −log10 (1/23 168) > 4.36) (Figure 1d). Of these significant SNPs, two were detected on A6 and C1, explaining 6.48% and 6.16% of the phenotypic variation, respectively (Figure 1d). The 17 other SNPs were located on A9, and the most significant SNP was Bn‐A09‐p31549208 (−log10 (P) = 6.97), which explained 10.11% of the total phenotypic variation (Figure 1d).
OLEA9 increases oleic content additively with the BnaFad2.a locus
Two B. napus inbred lines with significant differences in C18:1 content were used as the parents of the DH population. One line was D126, with a C18:1 content of approximately 75.6%; this line was derived from SW Hickory, which harbours a fad2 mutation on chromosome A5 (Yang et al., 2012b). The other line was ZP1, with a C18:1 content of approximately 66.1% (Figure 2a). The C18:2 and C18:3 contents of D126 were significantly lower than those of ZP1 (Figure 2a). C18:1, C18:2 and C18:3 accounted for approximately 93% of the total fatty acids in both lines (Figure 2a). For other fatty acids (C16:0, C18:0, C20:1 and C22:1), no significant differences were detected between the two lines (Figure 2a).
Figure 2.
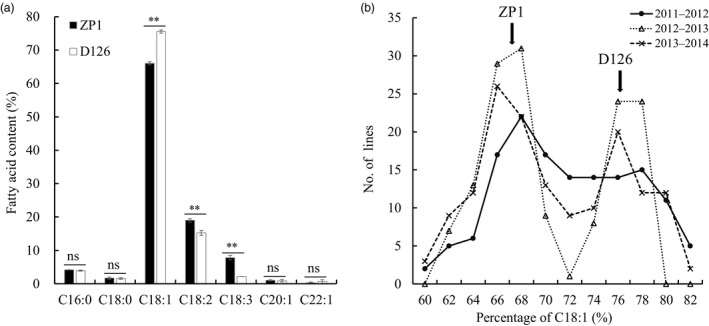
Fatty acid analysis of the two parental lines and the ZD‐DH population. (a) Fatty acid composition of the two parental lines ZP1 and D126, including palmitic acid (C16:0), stearic acid (C18:0), oleic acid (C18:1), linoleic acid (C18:2), linolenic acid (C18:3), eicosenoic acid (C20:1) and erucic acid (C22:1). ** indicates a significant difference at the level of P < 0.01 (Student's t‐test), and ns means the difference between the two lines is not statistically significant. (b) Distribution of the C18:1 content of the ZD‐DH population measured in three consecutive seasons from 2011 to 2014. Arrows indicate the fatty acid contents of the parental lines (average of three seasons).
In the ZD‐DH population, the distribution patterns of the C18:1 content of the three seasons were similar, with nearly a bimodal distribution and a cut‐off at 71% for the two groups (Figure 2b), indicating the involvement of a major gene for the trait. However, the two groups did not correspond to exactly a 1:1 ratio across the three seasons. A continuous distribution within the two groups suggested that some minor‐effect genes may be involved in the variation.
Since the segregation of oleic acid was not consistent with a single A5 mutation, we aimed to identify other possible loci contributing to the variation. We used a high‐density SNP‐based genetic map (Wu et al., 2019) for QTL mapping. In total, six QTLs were identified on six chromosomes (A2, A3, A4, A5, A9 and C1) by CIM, each of which explained 1.1%–86.3% of the phenotypic variation (Table 1). Among the QTLs, a major locus with the largest effect, OLEA5 on A5, was detected, which explained 67.18%, 86.30% and 69.26% of the phenotypic variation during the three seasons. The allele from D126 increased the C18:1 content by 4.88% (additive effect) on average (Table 1; Figure 3a). With the allele‐specific marker, OLEA5 was confirmed to be BnaA.FAD2.a (Yang et al., 2012b). In addition to OLEA5, another major QTL, OLEA9, was detected on A9, which explained 11.25%, 5.72% and 6.29% of the phenotypic variation during the three seasons, and the allele from ZP1 increased the C18:1 content by 2.14% (additive effect) on average (Table 1; Figure 3b). Based on the physical location of the SNP markers in the confidence intervals (bin1751‐bin1756) of OLEA9, we found that the QTL was located at 27.7–30.1 Mb on chromosome A9 (Figure 3c), which overlapped with the locus detected in GWAS analysis. The remaining four QTLs with minor effects were identified during only one season (Table 1).
Table 1.
QTLs for oleic acid (C18:1) content detected in the ZD‐DH population
| Season | QTL† | Peak‡ | LOD§ | A | R 2 (%)¶ | CI†† |
|---|---|---|---|---|---|---|
| 2011–2012 | OLEA5 | 142.51 | 45.17 | −4.54 | 67.18 | 141.7–143.1 |
| OLEA9 | 165.71 | 13.65 | 1.88 | 11.25 | 164.8–166.6 | |
| OLEC1 | 90.01 | 4.02 | 0.92 | 2.79 | 85.5–91.6 | |
| 2012–2013 | OLEA4 | 39.61 | 3.49 | 0.58 | 1.10 | 38.5–42.1 |
| OLEA5 | 142.11 | 75.08 | −5.03 | 86.30 | 141.6–142.9 | |
| OLEA9 | 166.51 | 12.10 | 3.07 | 5.72 | 166.3–167.2 | |
| 2013–2014 | OLEA2 | 93.81 | 3.82 | 0.96 | 1.86 | 92.4–95.3 |
| OLEA3 | 143.01 | 6.33 | −1.09 | 3.21 | 142.6–143.4 | |
| OLEA5 | 142.11 | 56.19 | −5.06 | 69.26 | 141.6–143 | |
| OLEA9 | 167.31 | 11.44 | 1.48 | 6.29 | 167–168.4 |
QTL is nomenclatured as trait name followed by chromosome number.
Peak map position (cM) of peak LOD scores.
A additive effect: positive additivity indicates that the allele from the female parent (ZP1) increases C18:1 content.
R 2 proportion for the phenotypic variation explained by the QTL.
CI Confidence intervals were obtained by marking positions ± 1 LOD from the peak.
Figure 3.
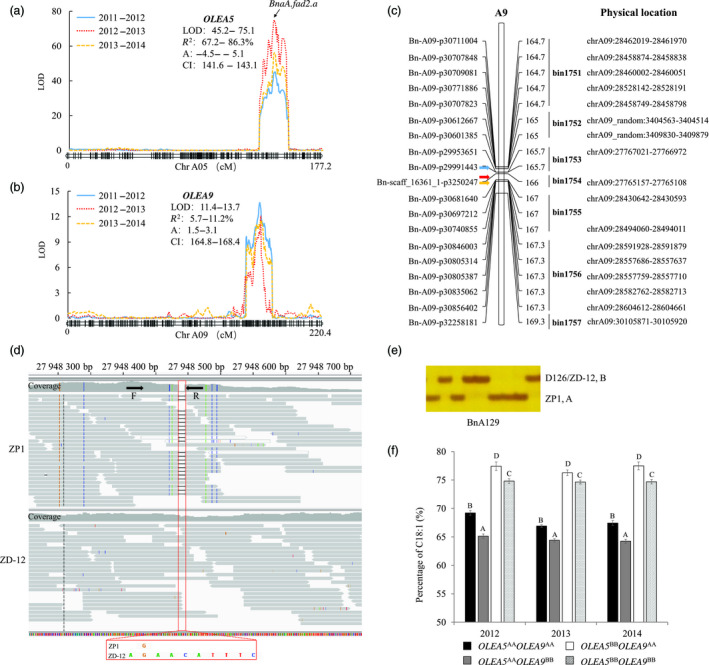
Identification of a novel QTL for oleic acid (C18:1) content on chromosome A9. (a and b) Two major QTLs of OLEA5 (a) and OLEA9 (b) were mapped separately. Curves of different colours represent QTL scanned from different seasons. The BnaA.fad2.a was responsible for OLEA5 demonstrated by our previous study (Yang et al., 2012b) and was located on the peak of OLEA5. (c) Physical localization of OLEA9 base on the physical location of the SNP markers in its confidence intervals (bin1751–bin1757). Arrows indicate the peak positions of OLEA9 detected in three seasons. (d) A closely linked InDel marker BnA129 (at 27.94 Mb) was developed for OLEA9 according to the resequencing data. The mapping result was displayed by Integrative Genomics Viewer. The red box indicates the positions of InDel. The arrows indicate the forward (F) and reverse (R) primer for BnA129. (e) The PCR‐amplified product of BnA129 was separated by 6% denaturing polyacrylamide gels and stained with silver. (f) Effects of individual or combined locus of OLEA5 and OLEA9 on C18:1 content in the ZD‐DH population as revealed by allelic genotype grouping. Allele‐specific marker YQ‐fad2a‐1 for OLEA5 (Yang et al., 2012b) and closely linked marker BnA129 for OLEA9 were used. AA and BB designate the allelic genotype same as parent ZP1 and D126 at a particular locus, respectively. Different uppercase letters indicate significant differences at P‐values < 0.01.
To examine the individual or combined effects of OLEA5 and OLEA9 on C18:1 content, a closely linked InDel marker, BnA129, located at 27.94 Mb, was developed based on the resequencing data for ZP1 and ZD‐12, a DH line from the ZD‐DH population with a null OLEA9 effect (Figure 3d, e). The DH population was then classified into four groups according to the genotypes of the allele‐specific marker for OLEA5 (YQ‐fad2a‐1) (Yang et al., 2012b) and OLEA9 (BnA129) (Figure 2d, e). For OLEA5, the average C18:1 content in the two groups with the alleles OLEA5 BB OLEA9 AA or OLEA5 BB OLEA9 BB was significantly higher than that in the groups with OLEA5 AA OLEA9 AA or OLEA5 AA OLEA9 BB, with the C18:1 content increased by 9.22%–10.13% (Figure 2f). For OLEA9, the average C18:1 content in the two groups with the alleles OLEA5 AA OLEA9 AA or OLEA5 BB OLEA9 AA was significantly higher than that in the groups with OLEA5 AA OLEA9 BB or OLEA5 BB OLEA9 BB, with the C18:1 content increased by 2.35%–3.26% (Figure 3f). The average C18:1 content in the group with both additive‐effect alleles (OLEA5 BB OLEA9 AA) was 77.07%, which was 12.48% higher than that in the group without additive‐effect alleles (OLEA5 AA OLEA9 BB) (Figure 3f). Our results thus showed that C18:1 can be further increased by integration of OLEA9 and OLEA5.
Fine mapping of OLEA9
For fine mapping of OLEA9, we aimed to construct a new bilateral mapping population using two parental lines without the interference of OLEA5. One of the two parental lines was ZP1, the original donor of the OLEA9 locus with a C18:1 content of 66.1%, and the other was ZD12, a DH line from the ZD‐DH population with a significantly lower C18:1 content (63.8%). ZD12 shared over 67% similarity with ZP1, as assayed with 15 622 SNP markers (Figure 4a). In addition to C18:1, the two parental lines also showed differences in C18:2 (Figure 4b). A segregating F2 population (ZD‐F2) and backcrossing (ZD‐BC) population were then generated for subsequent mapping analysis.
Figure 4.
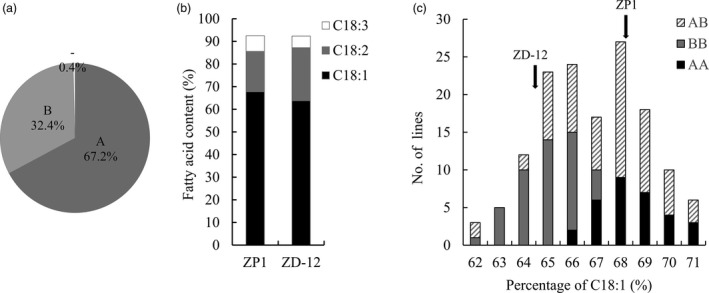
OLEA9 verification with an F2 population derived from the cross between ZP1 and ZD‐12 (a DH line from the ZD‐DH population). (a) Genetic background of ZD‐12 according to the genotypes of 15 622 SNP markers, ‘A’: the genotype same as parent line ZP1, ‘B’: the genotype same as parent line D126, and ‘−’: missing data. (b) The content of three major fatty acids, oleic acid (C18:1), linoleic acid (C18:2) and linolenic acid (C18:3) of the two parental lines ZP1 and ZD‐12 in 2015. (c) Distribution patterns of the C18:1 content in a random F2 subpopulation of 166 individuals. Three genotypes (AA, BB and AB) of BnA129 were classified with the marker of BnA129.
In an F2 subpopulation sampled from the ZD‐F2 population, 166 individual plants could be classified into three groups with the marker BnA129, that is homozygous with the parental genotype AA or BB and heterozygous (AB) (Figure 4c). AA and BB exhibited continuous distributions of C18:1 content that overlapped with each other, although the average content for AA (67.63%) was significantly higher than that for BB (64.68%, Figure 4c). The distribution for the AB group was even wider (Figure 4c). With InDel markers newly developed by resequencing the two parental lines, we were able to narrow down the region flanked by BnA144 and BnA153 in the F2 population (Figure S1). These data thus established that OLEA9 was responsible for the segregation of C18:1 content in the F2 population.
To further pinpoint the locus, we developed a BC3F2 NIL population. A local linkage map consisting of fifteen InDel and SNP markers was subsequently constructed. After three rounds of screening of 5184 BC3F2 individuals with flanking markers, 161 plants with recombination between the markers BnA144 and BnA153 were obtained to pinpoint the region where OLEA9 was located. SNP markers (Table S3) were developed within the interval of approximately 260 kb between markers BnA144 and BnA153 (Figure 5). Subsequently, these SNP markers, together with previously used markers (BnA129 and BnA153), were employed to genotype 161 BC3F2 individuals and some recombinant progeny of BC3F3 lines. The locus of OLEA9 was delimited to an interval of approximately 76 kb between SNP1 and SNP7 on the A9 chromosome (Figure 5). Based on the B. napus reference genome database (http://www.genoscope.cns.fr/brassicanapus/), twelve genes, designated BnaA09g39470D to BnaA09g39580D, were annotated within this region.
Figure 5.
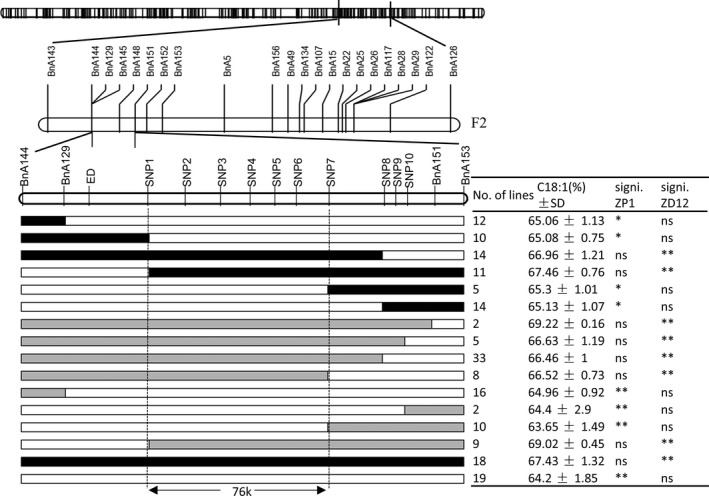
Fine mapping of the OLEA9 loci with a BC 3F2 segregating population. QTL region was narrowed to 76 kb containing 7 markers (SNP1 to SNP7) using BC3F2 and BC3F3 population. Black box indicates ZP1 genotype, white box indicates ZD12 genotype, and grey box indicates heterozygosis genotype. Statistical significance: *P < 0.05; **P < 0.01; ns, not significant.
Gene expression analysis of candidate genes
To further pinpoint the candidate genes in the QTL region, we compared the relative gene expression for each of the 12 genes between two NILs (designated H‐NIL NILAA and L‐NIL NILBB for the high and low oleic content lines, respectively) developed from the BC3F2 population. The expression levels of the genes at different stages during seed development were quantified by qPCR. Fold changes between NILAA and NILBB were calculated, and a heat map of gene expression was constructed (Figure 6) to illustrate the differences between the NILs for all the candidate genes.
Figure 6.
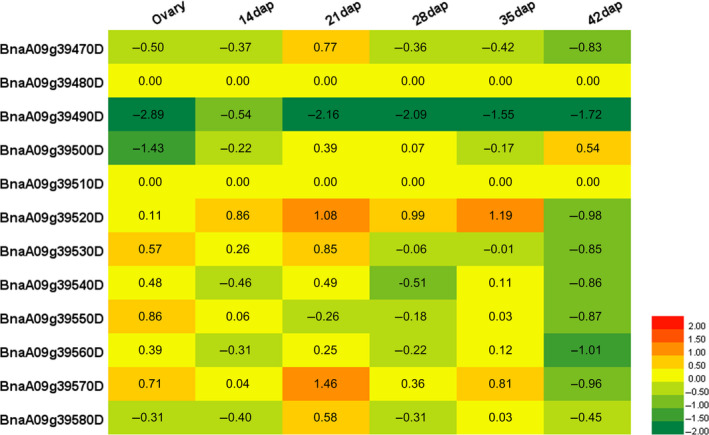
Expression levels of candidate genes in nearly isogenic lines at different stages quantitated by qRT PCR. dap, day after pollination. Gene expression level was calculated by log2 fold change (number shown) (NILAA /NILBB ). X‐axis represents different stages, and y‐axis represents different gene. Red colour indicates up‐regulated change, and green colour indicates down‐regulated change.
Since BnaA09g39480D and BnaA09g39510D had an average Cq value greater than 33 at all stages, they were regarded as having no expression. Among the remaining ten genes, the expression levels of BnaA09g39520D and BnaA09g39570D were dramatically higher in H‐NIL than in L‐NIL at almost all developing stages of seeds (except 42 dap), while BnaA09g39490D was dramatically lower in H‐NIL than in L‐NIL (Figure 6). Furthermore, BnaA09g39570D had a much greater fold change between the two NILs (log2 = 1.46) at 21 dap than at the other stages. The other 7 genes exhibited no significant changes in expression level (Figure 6).
The gene expression differences between H‐NIL and L‐NIL strongly suggest that oleic acid content is positively correlated with enhanced cand6 (BnaA09g39520D) or cand11 (BnaA09g39570D) or reduced cand3 (BnaA09g39490D) transcript levels. Thus, these three genes are the most likely candidate genes for OLEA9.
To confirm above results, we aligned the sequences with another reference genome of ‘ZS11’, a Chinese semi‐winter B. napus (Sun et al., 2017). We compared the genes and their locations in the target region in these two reference genomes against the orthologous region in Arabidopsis. It was found that three more genes are available in ZS11's genome compared with Darmor‐bzh. However, two of these three genes are aligned to their Arabidopsis orthologous located on different chromosomes. Another one, although corresponding to an orthologous at the same chromosome, is not on the same orthologous region. Apparently, the distribution of those three genes is not consistent to our QTL mapping results. Therefore, the three genes were ruled out from targeted candidate genes. We supplied the detailed mapping information for the target region in two reference genomes (Table S6).
Verification of the gene in the target region
To infer the potential roles of the candidate genes in determining C18:1 content, we annotated the candidate gene sequences based on the B. napus genome (http://www.genoscope.cns.fr/brassicanapus/) and Arabidopsis genome (http://www.arabidopsis.org/). Blast analyses showed that the 12 predicted genes from the B. napus reference genome were homologous to 12 genes on Arabidopsis chromosome 3 (Table 2). The gene sequence of BnaA09g39570D was highly similar to AT3G62010, which is described as a metal ion‐binding protein but without further information. BnaA09g39520D was similar to AT3G61950, which encodes a DNA‐binding protein (MYC67) in A. thaliana. BnaA09g39490D was annotated as UvrABC system protein, but no more information was given.
Table 2.
A list of candidate genes and gene annotations in Arabidopsis
| Gene alias | Arabidopsis | ChrA09:position (bp) | Description | |
|---|---|---|---|---|
| cand1 | BnaA09g39470D | AT3G61900.1 | 28000615–28001282 | SAUR‐like auxin‐responsive protein family |
| cand2 | BnaA09g39480D | AT3G61910.1 | 28004332–28005644 | NAC domain protein 66 |
| cand3 | BnaA09g39490D | AT3G61920.1 | 28012722–28013288 | – |
| cand4 | BnaA09g39500D | AT3G61930.1 | 28015922–28016255 | – |
| cand5 | BnaA09g39510D | AT3G61940.1 | 28018344–28019440 | Cation efflux family protein |
| cand6 | BnaA09g39520D | AT3G61950.1 | 28020457–28022135 | Basic helix–loop–helix (bHLH) DNA‐binding superfamily protein |
| cand7 | BnaA09g39530D | AT3G61960.1 | 28022399–28025991 | Protein kinase superfamily protein |
| cand8 | BnaA09g39540D | AT3G61970.1 | 28042200–28043079 | AP2/B3‐like transcriptional factor family protein |
| cand9 | BnaA09g39550D | AT3G61980.1 | 28046918–28047733 | Serine protease inhibitor, Kazal‐type family protein |
| cand10 | BnaA09g39560D | AT3G62000.1 | 28049650–28051588 | S‐adenosyl‐l‐methionine‐dependent methyltransferases superfamily protein |
| cand11 | BnaA09g39570D | AT3G62010.1 | 28058343–28064574 | – |
| cand12 | BnaA09g39580D | AT3G62020.1 | 28064637–28065509 | Germin‐like protein 10 |
Twelve genes in target region (namely cand1 to cand12 ordered by chromosome position) and their homologous gene in Arabidopsis.
Furthermore, Arabidopsis mutants of the three genes were collected (SALK_023939C for AT3G62010), and the fatty acid composition in mature seeds of the mutants was determined. In the A. thaliana mutant of AT3G62010 (SALK_023939C), a homolog for BnaA09g39570D, the oleic acid content was significantly higher than that in the other two mutants corresponding to cand6/cand3 and the wild type (Figure 7), suggesting that the BnaA09g39570D gene was most likely the candidate gene within OLEA9. The gene structure and sequence alignment for the two parental lines are displayed in Figure S2.
Figure 7.
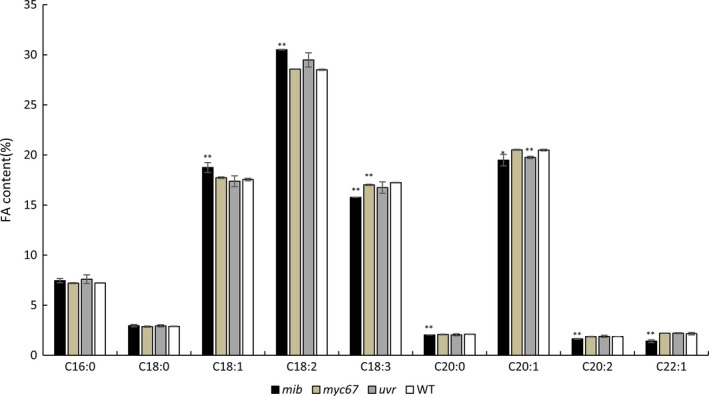
The fatty acid composition of three mutant lines and wild type. X‐axis shows different fatty acid. Y‐axis shows the content percentage of each composition. mib/myc67/uvr are three mutants for candidate genes in Arabidopsis. ** represents significance at P < 0.01 level and *P < 0.05.
Discussion
After the successful production of canola‐quality oil (low erucic acid and glucosinolate contents), selection for increased C18:1 content and reduced C18:3 content has become one of the most important goals for quality breeding in oilseed rape. To date, most high oleic acid germplasms reported are based on the mutation of FAD2 genes in Brassica species (Long et al., 2018; Peng et al., 2010; Wells et al., 2014). Such a narrow gene resource may lead to decreased genetic variation and thus compromise the improvement of yield and stress resistance, as illustrated in previous studies (Bai et al., 2018; Dar et al., 2017; Kinney, 1994; Miquel, 1994; Shi et al., 2017). The problem has also been noted in other oil crops, such as peanut and maize (Belo et al., 2008; Janila et al., 2016; Mikkilineni and Rocheford, 2003; Wang et al., 2015, 2018).
In this study, by both QTL mapping and GWAS, we identified a novel QTL (OLEA9) for C18:1 content in B. napus that further increased the C18:1 content by approximately 3% (Figures 1 and 3). The novel locus can function additively with fad2 alleles. With further increased oleic content, no negative effects on other traits were observed (Table S1). We had investigated the morphological performance in the NIL lines, and no obvious phenotypic variations other than fatty acid content were observed. For example, the initial flowering dates in the two subpopulations of the NIL were identical (156 days from sowing). It is unlikely that the change in fatty acid composition is due to the pleiotropic effects from other processes, such as flowering time or duration, besides fatty acid synthesis. Our finding thus provides a new genetic resource for the manipulation of the oleic acid content in seeds. We further checked the genotypes of the germplasms from China and other countries in GWAS population with the molecular marker ED‐1 which was developed in the study (Figure S3) and found that most materials had same genotype with low oleic (lower band in Figure S3). Furthermore, two groups of materials (from China and other countries) showed about similar frequencies of the mutated allele. It is likely that the mutated allele is rare. A further dissection of the molecular mechanism of the gene may uncover other possible pathways involved in fatty acid synthesis in Brassica oilseeds.
Compared with the effect of mutations of BnFAD2 (on A5), that of OLEA9 is relatively small. However, the effect is stable, as demonstrated in multiple mapping populations in our study. For example, OLEA9 explained 5.72%–11.25% of the phenotypic variation in the ZD‐DH population during the three seasons (Figure 3 and Table 1). In the ZD‐DH population, the additive‐effect alleles of OLEA9 increased the C18:1 content from 64.59% (OLEA5 AA OLEA9 BB) to 67.85% (OLEA5 AA OLEA9 AA) or from 74.72% (OLEA5 BB OLEA9 BB) to 77.07% (OLEA5 BB OLEA9 AA) (Figure 4f). The effects of OLEA9 were further verified in an F2 population derived from a cross between ZP1 and ZD‐12, which had similar genetic backgrounds and a null OLEA5 effect (Figure 4). The results clearly showed that the locus can be effectively used to further modify the C18:1 content in B. napus.
There have been some gene function studies of major QTLs (Liu et al., 2015b; Shi et al., 2019; Xin et al., 2016) in rapeseed since the reference genome was released, but no minor QTLs or genes have been identified for the C18:1 content. Thus, it is challenging to fine‐map OLEA9 because of its small effect on the oleic content in B. napus, an allotetraploid species with a complex genome and multiple copies of a homologous gene in its two subgenomes (AA and CC). In other crops, such as rice, there have been some minor QTL studies (Deng et al., 2012; Zhang et al., 2016) despite the difficulties in separating minor‐effect QTLs. Because of the complexity of the rapeseed genome, gene cloning is difficult for minor QTLs. The detection of minor QTLs is more easily affected by the environment and other errors than that of major QTLs. The desired result can be obtained to some extent only by trying to decrease the effect of background differences in the mapping population or environment and phenotypic error.
Marker‐assisted selection (MAS) is an approach for precision plant breeding, and its effectiveness depends on reliable QTLs and closely linked molecular markers. The next‐generation sequencing technology offers an unprecedented powerful tool for developing large numbers of DNA markers, particularly SNP and InDel markers (Varshney et al., 2009; Yang et al., 2012a). InDel markers have become some of the most commonly used markers in plant genetic studies because they are relatively abundant, easy to use, PCR‐based and codominant (Păcurar et al., 2012). In this study, we performed resequencing analysis to develop a closely linked InDel marker (BnA129) for OLEA9 (Figure 3d, e). This marker is very user‐friendly because it can be easily distinguished based on the 9 bp differences between ZP1 and ZD‐12 (Figure 3d, e). This marker is a reliable, simple and accurate marker for high oleic acid content breeding via MAS in B. napus.
At present, it is not clear how the A9 gene increases the C18:1 content in rapeseed. The fatty acid desaturase genes FAD2 (Miquel, 1992) and FAD3 (Arondel et al., 1992) are two major genes controlling the C18:1 and C18:3 contents. Strong cosuppression was observed when FAD2 was overexpressed in the model plant Arabidopsis, and the cosuppression could be overcome in the Arabidopsis rdr6 mutant (Du et al., 2018). Of particular interest, 6% polyunsaturated fatty acids (C18:2 and C18:3) are still produced when the desaturation pathway is completely blocked by FAD2 in B. napus (Peng et al., 2010; Wells et al., 2014), suggesting that the remaining C18:2 and C18:3 likely originate from other fatty acid desaturases and/or other types of enzymes.
Based on our mapping results, the region does not harbour any known genes involved in fatty acid metabolism (Li‐Beisson et al., 2010). OLEA9 may participate in a new desaturation pathway that plays a role in subtly fine‐tuning fatty acids. Although the candidate gene was annotated as encoding a metal ion‐binding protein, little is known about the relationship between fatty acid synthesis and the function of metal ion‐binding proteins. Calcium ions play a central role in regulating plant growth, development and adaptation to environmental stresses. Calcium signals are primary regulators of plant growth, development and stress response, and calcium‐binding proteins have also been identified as some of the most important components of calcium signal transduction pathways in plants. A pathway (Cav1.1 → CaMKII → NOS) of normal skeletal muscle production in humans regulates the intracellular distribution of the fatty acid transport protein CD36, thus altering fatty acid metabolism. Blocking this pathway results in decreased mitochondrial oxidation and decreased energy expenditure (Georgiou et al., 2015). In plants, Ca2+ concentrations, as important classic secondary messengers, can regulate some lipid‐related compounds. For example, the activity of plant phospholipase C (PLC), a major membrane phospholipid‐hydrolysing enzyme, is regulated by various factors, including Ca2+ concentration. The activity of PI‐PLC depends on Ca2+, which regulates not only catalytic activity but also the subcellular localization and substrate preference of the enzyme. The PI‐PLCs can be translocated between the membrane and cytoplasm, depending on the Ca2+ concentrations in cells (Hong et al., 2016; Singh et al., 2015). Moreover, phospholipase Dα1 is the most abundant form of phospholipase D (PLD) in A. thaliana, and its activity is characterized by an in vitro requirement for millimolar concentrations of Ca2+ (Devaiah et al., 2007). The candidate gene might be involved in regulating fatty acid composition by combining Ca2+ and thus influencing the relative PLD/PLC participating in the pathway, ultimately changing the fatty acid component. Further gene cloning and functional analysis of OLEA9 in rapeseed will offer novel information for a more complete understanding of the fatty acid biosynthesis mechanism and provide valuable resources for high oleic acid content breeding in B. napus.
Materials and methods
Plant materials
An association panel with 375 rapeseed inbred lines was employed for association analysis of C18:1 content. The accessions were collected from the major breeding institutes across China, and detailed information is provided in Table S4. The association panel was grown in a randomized complete block design with two replicates. Self‐pollinated seeds were collected from each replicate for fatty acid profiling.
Two biparental segregating populations were used for primary mapping in this study. The first population was a doubled haploid (DH) population consisting of 150 individual DH lines, referred to as the ZD‐DH population. The population was developed from microspore culture of F1 buds of a cross between two B. napus inbred lines (ZP1 × D126). The second population was an F2 population derived from a cross between ZP1 and a DH line from the ZD‐DH population (ZD‐12) with a C18:1 content of approximately 62.71%. The F2 population was designated the ZD‐F2 population.
The DH population, along with its parental lines, was grown during three consecutive growing seasons in the years 2011–2012, 2012–2013 and 2013–2014. Self‐pollinated seeds were collected from individual DH plants during the season of 2011–2012 or from three plants (one plant from each of three replicates) of each DH line during the seasons of 2012–2013 and 2013–2014 for fatty acid profiling. The field experiments for the DH population followed a complete randomized design with one replicate (2011–2012) or a randomized complete block design with three replicates (2012–2013 and 2013–2014).
The ZD‐F2 population, together with its two parental lines, was grown during the 2014–2015 season. One hundred and sixty‐six F2 plants were randomly sampled from the F2 population for fatty acid profiling and genotyping.
A BC3F2 population and its derived BC3F3 progenies were constructed (designated ZD‐BC) by successive backcrossing using ZD12 (low oleic acid content) as a donor and ZP1 (high oleic acid content) as a recurrent parent followed by self‐pollination of F2 individuals, which were grown during the seasons of 2016–2017 and 2017–2018, respectively. Near‐isogenic lines (NILs) with high and low oleic acid contents (H‐NIL and L‐NIL, respectively) were developed. H‐NIL accumulated significantly more (~3%–5%) oleic acid than L‐NIL (Table S1).
All materials were grown during the winter (the oilseed rape growing season) on the experimental farm of Huazhong Agriculture University, Wuhan, China, and the field management generally followed regular breeding practices.
Wild‐type and mutant Arabidopsis plants (Columbia ecotype) were grown in composite soil at 21 °C under a 16‐h/8‐h day/night photoperiod in a plant growth room. All the T‐DNA insertion mutants were verified with LBb, LP and RP primers synthesized with T‐DNA Primer Design (http://signal.salk.edu/tdnaprimers.2.html). Table S5 lists the information for the mutants and PCR primers used for mutation verifications.
Phenotypic analysis
Fatty acid profiling of self‐pollinated seeds was conducted using gas–liquid chromatography (GC) with a Model 6890 GC analyser (Agilent Technologies, Inc., Wilmington, DE), following the protocol described by Thies (1971). The individual fatty acid contents were expressed as the percentage of total fatty acids in mature seeds.
Genome‑wide association analysis of the oleic acid content in B. napus
All the accessions in the association panel were genotyped using 60K SNP array chips as described in Liu et al. (2016). For mapping the physical localization of SNP markers, only the top blast hits were considered to be located at a chromosome using an e‐value threshold of e −10, while blast matches to multiple loci with the same top e‐value were not considered to be mapped. SNPs that could not be assigned to a B. napus chromosome were excluded for further analysis. A total of 23 168 SNPs were retained for further analysis. Principal component analysis (PCA) based on all 23 168 SNPs was performed using the GCTA tool (Yang et al., 2011). A relative kinship matrix of 375 B. napus lines was generated using SPAGeDi software (Hardy and Vekemans, 2002). Negative values between two accessions were set to 0. Trait–SNP association was performed using the PCA + K model implemented in TASSEL 4.0 (Bradbury et al., 2007). The significance of the associations between SNPs and the trait was based on a single threshold of P < 4.32 × 10−5 (P = 1/n, where n = the number of markers used; −log10 (1/23 168) = 4.36).
Primary QTL mapping of oleic acid content in the ZD‐DH population
The ZD‐DH population was genotyped with a 60k SNP array, and a high‐density SNP‐based genetic map was constructed as previously described (Wu et al., 2019). With the phenotypic data of fatty acids from the DH population, QTL mapping was performed by composite interval mapping (CIM) in QTL Cartographer 2.5 software (http://statgen.ncsu.edu/qtlcart/WQTLCart.htm). A significance threshold for QTLs at the level P = 0.05 was determined through permutation analysis using 1000 repetitions. The confidence interval of QTLs was determined by 1‐logarithm of the odds (LOD) intervals surrounding the QTL peak. QTLs detected during different seasons were considered the same QTL if they had overlapping confidence intervals. To determine the physical position of SNP markers, SNP probe sequences were used to perform a BlastN (Altschul et al., 1997) search against the B. napus ‘Darmor‐bzh’ reference genome (version 4.1) (Chalhoub et al., 2014) with an E‐value threshold of 1E‐20. Only the top BLAST hits were considered.
Development of InDel and SNP markers through resequencing
High‐quality genomic DNA from the parental lines (ZP1 and ZD‐12) was subjected to library construction using an Illumina® TruSeq™ DNA Sample Preparation Kit following the manufacturer's instructions. Resequencing was performed with an Illumina HiSeq X Ten sequencer at the Beijing Genomics Institute company to produce paired‐end reads that were 2 × 151 bp in length. After acquisition of the raw sequencing data, quality control for raw reads was conducted using the NGS QC Toolkit (Patel and Jain, 2012), including (i) removal of the reads containing primer/adaptor sequences and low‐quality reads (the number of bases whose PHRED‐like score (Q‐score) was <20 exceeded 30%), (ii) trimming the first ten base pairs of the reads that showed an unstable base composition, as determined by the percentages of four different nucleotides (A, T, C and G), and the low‐quality bases (Q‐score < 20) from the 3′ end of the reads, and (iii) removal of the reads <50 bp in length. High‐quality reads of ZP1 and ZD‐12 were then mapped to the B. napus ‘Darmor‐bzh’ reference genome (version 4.1) (Chalhoub et al., 2014) by Bowtie 2 v2.2.5 (Langmead and Salzberg, 2012) using the default parameters.
Insertion/deletion (InDel) polymorphism calling was carried out using SAMtools v 0.1.19 (Li et al., 2009) and VarScan v2.3 (Koboldt et al., 2012) with the default parameters. Primers for genome‐specific markers were designed using the software Primer 3. Subsequently, these markers were used to screen the parental lines and segregating population, and only the polymorphic markers located within the QTL interval were further employed in the segregating population.
Fine mapping with backcross populations
Genomic DNA was extracted from young leaves of the BC3F2‐segregating population consisting of 5184 plants (including the plants for RNA extraction) using the CTAB method. Selected SNPs identified in the target region were confirmed using the Kompetitive Allele‐Specific PCR (KASP) method following the instructions from the LGC company (Huang et al., 2017). The QTL position was analysed by examining the genotype of different recombination classes in combination with their phenotypic values. In brief, if the mean oleic acid content for several recombination groups was significantly higher than that of one parent, the QTL was located in the overlapping segment among these recombination groups. Phenotypic and genotypic data analyses were performed using Microsoft Excel.
RNA extraction and qRT‐PCR analysis
Individual flowers of the main raceme were tagged on the day of flowering, and developing seeds from two NILs at six developmental stages, namely the ovary and 14, 21, 28, 35 and 42 days after pollination (dap), were collected, immediately frozen in liquid nitrogen and stored at −80 °C for total RNA isolation. Total RNA was extracted using a TransZol Plant Total RNA Extraction Kit (TransGen, Beijing, China) following the manufacturer's instructions. RNA quality (purity and integrity) was monitored by running samples in a 1.0% agarose gel and with an Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA). RNA was quantified using a NanoDrop spectrophotometer (Thermo Fisher Scientific, Wilmington, DE, USA). Two genes [BnaC03g45080D (TIP41) and BnaA09g14410D (PP2A‐1)] were used as internal controls (Liu et al., 2015a; Wu et al., 2016). Primers were designed based on the reference genome sequences using Primer Premier 5.0 and are listed in Table S2. First‐strand cDNA was synthesized from 2 μg RNA per sample using TransScript One‐Step gDNA Remover and cDNA Synthesis Kit according to the manufacturer's instructions (TransGen). qRT‐PCR was performed using Hieff™ qPCR SYBR Green Master Mix (no ROX) (Yeasen, China) in a Bio‐Rad CFX‐384 Real‐Time PCR System (Bio‐Rad, Hercules, CA). Relative expression levels were calculated with LinReg (Ramakers et al., 2003). Data were collected from three biological and two technical replicates, and a heatmap of gene expression level was created with Heatmap Illustrator software (http://hemi.biocuckoo.org/).
Conflict of interest
The authors declare no conflict of interest.
Author contributions
YZ conceived the study. YZ, QZ and JW designed the experiment. QZ and MS performed the experiment. QZ, JW, GC and QY analysed the data. QZ, JW and YZ wrote the manuscript. All authors read and approved the final manuscript.
Supporting information
Figure S1 Genetic linkage map constructed by F2 population.
Figure S2 Gene structure and sequence alignment with IGV software.
Figure S3 Genotyping of partial germplasms from the GWAS population. (a) Germplasms from other countries than China. (b) Germplasms from China randomly sampled from the population.
Table S1 Seed fatty acid composition (%), oil content (%) and thousand seed weight (TSW) of NIL lines.
Table S2 Primer list for qRT PCR.
Table S3 InDel and KASP primer sequences in the OLEA9 target region.
Table S4 Detailed information for 375 rapeseed inbred lines employed for association analysis.
Table S5 Primer sequences for checking Arabidopsis mutant lines.
Table S6 Gene information in target region by using two reference genomes.
Acknowledgements
This work was supported by the National Key Research and Development Program of China (2016YFD0100506‐3) and International S&T Cooperation Program of China (2017YFE0104800), Ministry of Science and Technology of China.
References
- Altschul, S.F. , Madden, T.L. , Schäffer, A.A. , Zhang, J. , Zhang, Z. , Miller, W. and Lipman, D.J. (1997) Gapped BLAST and PSI‐BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arondel, V. , Lemieux, B. , Hwang, I. , Gibson, S. , Goodman, H.M. and Somerville, C.R. (1992) Map‐based cloning of a gene controlling omega‐3 fatty acid desaturation in Arabidopsis. Science, 258, 1353–1355. [DOI] [PubMed] [Google Scholar]
- Bai, S. , Engelen, S. , Denolf, P. , Wallis, J.G. , Lynch, K. , Bengtsson, J.D. , Van Thournout, M. et al. (2018) Identification, characterization and field testing of Brassica napus mutants producing high‐oleic oils. Plant J. 98, 33–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao, B. , Chao, H. , Wang, H. , Zhao, W. , Zhang, L. , Raboanatahiry, N. , Wang, X. et al. (2018) Stable, environmental specific and novel QTL identification as well as genetic dissection of fatty acid metabolism in Brassica napus . Front. Plant Sci. 9, 1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belo, A. , Zheng, P. , Luck, S. , Shen, B. , Meyer, D.J. , Li, B. , Tingey, S. et al. (2008) Whole genome scan detects an allelic variant of fad2 associated with increased oleic acid levels in maize. Mol. Genet. Genomics, 279, 1–10. [DOI] [PubMed] [Google Scholar]
- Bradbury, P.J. , Zhang, Z. , Kroon, D.E. , Casstevens, T.M. , Ramdoss, Y. and Buckler, E.S. (2007) TASSEL: software for association mapping of complex traits in diverse samples. Bioinformatics, 23, 2633–2635. [DOI] [PubMed] [Google Scholar]
- Burns, M. , Barnes, S. , Bowman, J. and Clarke, M. (2003) QTL analysis of an intervarietal set of substitution lines in Brassica napus: (i) Seed oil content and fatty acid composition. Heredity, 90, 39. [DOI] [PubMed] [Google Scholar]
- Chalhoub, B. , Denoeud, F. , Liu, S. , Parkin, I.A. , Tang, H. , Wang, X. , Chiquet, J. et al. (2014) Early allopolyploid evolution in the post‐neolithic Brassica napus oilseed genome. Science, 345, 950–953. [DOI] [PubMed] [Google Scholar]
- Chen, F. , Zhang, W. , Yu, K. , Sun, L. , Gao, J. , Zhou, X. , Peng, Q. et al. (2018) Unconditional and conditional QTL analyses of seed fatty acid composition in Brassica napus L. BMC Plant Biol. 18, 49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dar, A.A. , Choudhury, A.R. , Kancharla, P.K. and Arumugam, N. (2017) The FAD2 gene in plants: occurrence, regulation, and role. Front. Plant Sci. 8, 1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng, H. , Liu, H. , Li, X. , Xiao, J. and Wang, S. (2012) A CCCH‐type zinc finger nucleic acid‐binding protein quantitatively confers resistance against rice bacterial blight disease. Plant Physiol. 158, 876–889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devaiah, S.P. , Pan, X. , Hong, Y. , Roth, M. , Welti, R. and Wang, X. (2007) Enhancing seed quality and viability by suppressing phospholipase D in Arabidopsis. Plant J. 50, 950–957. [DOI] [PubMed] [Google Scholar]
- Du, C. , Chen, Y. , Wang, K. , Yang, Z. , Zhao, C. , Jia, Q. , Taylor, D.C. et al. (2018) High‐frequency and strong FAD2 co‐suppression A bottleneck to increase polyunsaturated fatty acids in seeds. J. Exp. Bot. 70, 985–994. [DOI] [PubMed] [Google Scholar]
- Georgiou, D.K. , Dagnino‐Acosta, A. , Lee, C.S. , Griffin, D.M. , Wang, H. , Lagor, W.R. , Pautler, R.G. et al. (2015) Ca2+ binding/permeation via calcium channel, CaV1.1, regulates the intracellular distribution of the fatty acid transport protein, CD36, and fatty acid metabolism. J. Biol. Chem. 290, 23751–23765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillingham, L.G. , Harris‐Janz, S. and Jones, P.J. (2011) Dietary monounsaturated fatty acids are protective against metabolic syndrome and cardiovascular disease risk factors. Lipids, 46, 209–228. [DOI] [PubMed] [Google Scholar]
- Hardy, O.J. and Vekemans, X. (2002) SPAGeDi: a versatile computer program to analyse spatial genetic structure at the individual or population levels. Mol. Ecol. Notes, 2, 618–620. [Google Scholar]
- Hong, Y. , Zhao, J. , Guo, L. , Kim, S.C. , Deng, X. , Wang, G. , Zhang, G. et al. (2016) Plant phospholipases D and C and their diverse functions in stress responses. Prog. Lipid Res. 62, 55–74. [DOI] [PubMed] [Google Scholar]
- Hu, X. , Sullivan‐Gilbert, M. , Gupta, M. and Thompson, S.A. (2006) Mapping of the loci controlling oleic and linolenic acid contents and development of fad2 and fad3 allele‐specific markers in canola (Brassica napus L.). Theor. Appl. Genet. 113, 497–507. [DOI] [PubMed] [Google Scholar]
- Huang, Z. , Peng, G. , Liu, X. , Deora, A. , Falk, K.C. , Gossen, B.D. , McDonald, M.R. et al. (2017) Fine mapping of a clubroot resistance gene in Chinese cabbage using SNP markers identified from bulked segregant RNA sequencing. Front. Plant Sci. 8, 1448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janila, P. , Pandey, M.K. , Shasidhar, Y. , Variath, M.T. , Sriswathi, M. , Khera, P. , Manohar, S.S. et al. (2016) Molecular breeding for introgression of fatty acid desaturase mutant alleles (ahFAD2A and ahFAD2B) enhances oil quality in high and low oil containing peanut genotypes. Plant Sci. 242, 203–213. [DOI] [PubMed] [Google Scholar]
- Kinney, A.J. (1994) Genetic modification of the storage lipids of plants. Curr. Opin. Biotechnol. 5, 144–151. [Google Scholar]
- Koboldt, D.C. , Zhang, Q. , Larson, D.E. , Shen, D. , McLellan, M.D. , Lin, L. , Miller, C.A. et al. (2012) VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 22, 568–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Körber, N. , Bus, A. , Li, J. , Parkin, I.A. , Wittkop, B. , Snowdon, R.J. and Stich, B. (2016) Agronomic and seed quality traits dissected by genome‐wide association mapping in Brassica napus . Front. Plant Sci. 7, 386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead, B. and Salzberg, S.L. (2012) Fast gapped‐read alignment with Bowtie 2. Nat. Methods, 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, H. , Handsaker, B. , Wysoker, A. , Fennell, T. , Ruan, J. , Homer, N. , Marth, G. et al. (2009) The sequence alignment/map format and SAMtools. Bioinformatics, 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li‐Beisson, Y. , Shorrosh, B. , Beisson, F. , Andersson, M.X. , Arondel, V. , Bates, P.D. , Baud, S. et al. (2010) Acyl‐Lipid Metabolism. Arabidopsis Book, 8, e0133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, H. , Yang, Q. , Fan, C. , Zhao, X. , Wang, X. and Zhou, Y. (2015a) Transcriptomic basis of functional difference and coordination between seeds and the silique wall of Brassica napus during the seed‐filling stage. Plant Sci. 233, 186–199. [DOI] [PubMed] [Google Scholar]
- Liu, J. , Hua, W. , Hu, Z. , Yang, H. , Zhang, L. , Li, R. , Deng, L. et al. (2015b) Natural variation in ARF18 gene simultaneously affects seed weight and silique length in polyploid rapeseed. Proc. Natl Acad. Sci. USA, 112, E5123–E5132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, S. , Fan, C. , Li, J. , Cai, G. , Yang, Q. , Wu, J. , Yi, X. et al. (2016) A genome‐wide association study reveals novel elite allelic variations in seed oil content of Brassica napus . Theor. Appl. Genet. 129, 1203–1215. [DOI] [PubMed] [Google Scholar]
- Long, W. , Hu, M. , Gao, J. , Chen, S. , Zhang, J. , Cheng, L. and Pu, H. (2018) Identification and functional analysis of two new mutant BnFAD2 alleles that confer elevated oleic acid content in rapeseed. Front. Genet. 9, 399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthäus, B. (2006) Utilization of high‐oleic rapeseed oil for deep‐fat frying of French fries compared to other commonly used edible oils. Eur. J. Lipid Sci. Technol. 108, 200–211. [Google Scholar]
- Merrill, L.I. , Pike, O.A. , Ogden, L.V. and Dunn, M.L. (2008) Oxidative stability of conventional and high‐oleic vegetable oils with added antioxidants. J. Am. Oil Chem.’ Soc. 85, 771–776. [Google Scholar]
- Micha, R. and Mozaffarian, D. (2009) Trans fatty acids: effects on metabolic syndrome, heart disease and diabetes. Nat. Rev. Endocrinol. 5, 335–344. [DOI] [PubMed] [Google Scholar]
- Mikkilineni, V. and Rocheford, T.R. (2003) Sequence variation and genomic organization of fatty acid desaturase‐2 (fad2) and fatty acid desaturase‐6 (fad6) cDNAs in maize. Theor. Appl. Genet. 106, 1326–1332. [DOI] [PubMed] [Google Scholar]
- Miquel, M. (1992) Arabidopsis mutants deficient in polyunsaturated fatty acid synthesis. Biochemical and genetic characterization of a plant oleoyl‐phosphatidylcholine desaturase. J. Biol. Chem. 267, 1502–1509. [PubMed] [Google Scholar]
- Miquel, M.F. (1994) High‐oleate oilseeds fail to develop at low temperature. Plant Physiol. 106, 421–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miquel, M. , James, D. and Dooner, H. (1993) Arabidopsis requires polyunsaturated lipids for low‐temperature survival. Proc. Natl Acad. Sci. 90, 6208–6212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okuzaki, A. , Ogawa, T. , Koizuka, C. , Kaneko, K. , Inaba, M. , Imamura, J. and Koizuka, N. (2018) CRISPR/Cas9‐mediated genome editing of the fatty acid desaturase 2 gene in Brassica napus . Plant Physiol. Biochem. 131, 63–69. [DOI] [PubMed] [Google Scholar]
- Păcurar, D.I. , Păcurar, M.L. , Street, N. , Bussell, J.D. , Pop, T.I. , Gutierrez, L. and Bellini, C. (2012) A collection of INDEL markers for map‐based cloning in seven Arabidopsis accessions. J. Exp. Bot. 63, 2491–2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel, R.K. and Jain, M. (2012) NGS QC toolkit: a toolkit for quality control of next generation sequencing data. PLoS ONE, 7, e30619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng, Q. , Hu, Y. , Wei, R. , Zhang, Y. , Guan, C. , Ruan, Y. and Liu, C. (2010) Simultaneous silencing of FAD2 and FAE1 genes affects both oleic acid and erucic acid contents in Brassica napus seeds. Plant Cell Rep. 29, 317–325. [DOI] [PubMed] [Google Scholar]
- Qu, C. , Jia, L. , Fu, F. , Zhao, H. , Lu, K. , Wei, L. , Xu, X. et al. (2017) Genome‐wide association mapping and Identification of candidate genes for fatty acid composition in Brassica napus L. using SNP markers. BMC Genom. 18, 232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramakers, C. , Ruijter, J.M. , Deprez, R.H.L. and Moorman, A.F.M. (2003) Assumption‐free analysis of quantitative real‐time polymerase chain reaction (PCR) data. Neurosci. Lett. 339, 62–66. [DOI] [PubMed] [Google Scholar]
- Shi, J. , Lang, C. , Wang, F. , Wu, X. , Liu, R. , Zheng, T. , Zhang, D. et al. (2017) Depressed expression of FAE1 and FAD2 genes modifies fatty acid profiles and storage compounds accumulation in Brassica napus seeds. Plant Sci. 263, 177–182. [DOI] [PubMed] [Google Scholar]
- Shi, L. , Song, J. , Guo, C. , Wang, B. , Guan, Z. , Yang, P. , Chen, X. et al. (2019) A CACTA‐like transposable element in the upstream region of BnaA9.CYP78A9 acts as an enhancer to increase silique length and seed weight in rapeseed. Plant J. 98, 524–539. [DOI] [PubMed] [Google Scholar]
- Singh, A. , Bhatnagar, N. , Pandey, A. and Pandey, G.K. (2015) Plant phospholipase C family: regulation and functional role in lipid signaling. Cell Calcium, 58, 139–146. [DOI] [PubMed] [Google Scholar]
- Smooker, A.M. , Wells, R. , Morgan, C. , Beaudoin, F. , Cho, K. , Fraser, F. and Bancroft, I. (2011) The identification and mapping of candidate genes and QTL involved in the fatty acid desaturation pathway in Brassica napus . Theor. Appl. Genet. 122, 1075–1090. [DOI] [PubMed] [Google Scholar]
- Sun, F. , Fan, G. , Hu, Q. , Zhou, Y. , Guan, M. , Tong, C. , Li, J. et al. (2017) The high‐quality genome of Brassica napus cultivar ‘ZS11’ reveals the introgression history in semi‐winter morphotype. Plant J. 92, 452–468. [DOI] [PubMed] [Google Scholar]
- Thies, W. (1971) Schnelle und einfache Analysen der Fettsaurezusammensetzung in einzelnen Raps‐Kotyledonen. I. Gaschromatographische und papierchromatographische Methoden. Z Pflanzenzucht 65, 181–202. [Google Scholar]
- Varshney, R.K. , Nayak, S.N. , May, G.D. and Jackson, S.A. (2009) Next‐generation sequencing technologies and their implications for crop genetics and breeding. Trends Biotechnol. 27, 522–530. [DOI] [PubMed] [Google Scholar]
- Wang, M.L. , Khera, P. , Pandey, M.K. , Wang, H. , Qiao, L. , Feng, S. , Tonnis, B. et al. (2015) Genetic mapping of QTLs controlling fatty acids provided insights into the genetic control of fatty acid synthesis pathway in peanut (Arachis hypogaea L.). PLoS ONE, 10, e0119454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, M.L. , Chen, C.Y. , Tonnis, B. , Pinnow, D. , Davis, J. , An, Y.C. and Dang, P. (2018) Changes of seed weight, fatty acid composition, and oil and protein contents from different peanut FAD2 genotypes at different seed developmental and maturation stages. J. Agric. Food Chem. 66, 3658–3665. [DOI] [PubMed] [Google Scholar]
- Wells, R. , Trick, M. , Soumpourou, E. , Clissold, L. , Morgan, C. , Werner, P. , Gibbard, C. et al. (2014) The control of seed oil polyunsaturate content in the polyploid crop species Brassica napus . Mol. Breed. 33, 349–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen, J. , Xu, J. , Long, Y. , Xu, H. , Wu, J. , Meng, J. and Shi, C. (2015) Mapping QTLs controlling beneficial fatty acids based on the embryo and maternal plant genomes in Brassica napus L. J. Am. Oil Chem.’ Soc. 92, 541–552. [Google Scholar]
- Wu, J. , Zhao, Q. , Yang, Q. , Liu, H. , Li, Q. , Yi, X. , Cheng, Y. et al. (2016) Comparative transcriptomic analysis uncovers the complex genetic network for resistance to Sclerotinia sclerotiorum in Brassica napus . Sci. Rep. 6, 19007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, J. , Chen, P. , Zhao, Q. , Cai, G. , Hu, Y. , Xiang, Y. , Yang, Q. et al. (2019) Co‐location of QTL for Sclerotinia stem rot resistance and flowering time in Brassica napus . Crop J. 7, 227–237. [Google Scholar]
- Xin, Q. , Shen, Y. , Li, X. , Lu, W. , Wang, X. , Han, X. , Dong, F. et al. (2016) MS5 mediates early meiotic progression and its natural variants may have applications for hybrid production in Brassica napus . Plant Cell, 28, 1263–1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan, X.Y. , Li, J.N. , Wang, R. , Jin, M.Y. , Chen, L. , Qian, W. , Wang, X.N. et al. (2011) Mapping of QTLs controlling content of fatty acid composition in rapeseed (Brassica napus). Genes Genom. 33, 365–371. [Google Scholar]
- Yang, J. , Lee, S.H. , Goddard, M.E. and Visscher, P.M. (2011) GCTA: a tool for genome‐wide complex trait analysis. Am. J. Hum. Genet. 88, 76–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, H. , Tao, Y. , Zheng, Z. , Li, C. , Sweetingham, M.W. and Howieson, J.G. (2012a) Application of next‐generation sequencing for rapid marker development in molecular plant breeding: a case study on anthracnose disease resistance in Lupinus angustifolius L. BMC Genom. 13, 318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, Q. , Fan, C. , Guo, Z. , Qin, J. , Wu, J. , Li, Q. , Fu, T. et al. (2012b) Identification of FAD2 and FAD3 genes in Brassica napus genome and development of allele‐specific markers for high oleic and low linolenic acid contents. Theor. Appl. Genet. 125, 715–729. [DOI] [PubMed] [Google Scholar]
- Zhang, J. , Liu, H. , Sun, J. , Li, B. , Zhu, Q. , Chen, S. and Zhang, H. (2012) Arabidopsis fatty acid desaturase FAD2 is required for salt tolerance during seed germination and early seedling growth. PLoS ONE, 7, e30355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, H.W. , Fan, Y.Y. , Zhu, Y.J. , Chen, J.Y. , Yu, S.B. and Zhuang, J.Y. (2016) Dissection of the qTGW1.1 region into two tightly‐linked minor QTLs having stable effects for grain weight in rice. BMC Genet. 17, 98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao, J. , Dimov, Z. , Becker, H.C. , Ecke, W. and Möllers, C. (2008) Mapping QTL controlling fatty acid composition in a doubled haploid rapeseed population segregating for oil content. Mol. Breed. 21, 115–125. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Genetic linkage map constructed by F2 population.
Figure S2 Gene structure and sequence alignment with IGV software.
Figure S3 Genotyping of partial germplasms from the GWAS population. (a) Germplasms from other countries than China. (b) Germplasms from China randomly sampled from the population.
Table S1 Seed fatty acid composition (%), oil content (%) and thousand seed weight (TSW) of NIL lines.
Table S2 Primer list for qRT PCR.
Table S3 InDel and KASP primer sequences in the OLEA9 target region.
Table S4 Detailed information for 375 rapeseed inbred lines employed for association analysis.
Table S5 Primer sequences for checking Arabidopsis mutant lines.
Table S6 Gene information in target region by using two reference genomes.