

Abstract
Psoriasis (Ps) and psoriatic arthritis (PsA) represent a clinical and immunopathogenic continuum, called psoriatic disease, cumulatively affecting approximately 3% of the general population. Psoriatic disease is a chronic inflammatory disorder affecting the skin and musculoskeletal system. The immuno-pathogenesis is characterized by an activation of the TNF/IL-23/IL-17 cytokine axis, leading to an immunologic imbalance of T-cells resident in all affected tissues, mainly entheses. In the majority of cases, skin Ps predates rheumatological manifestations. Secondary to the higher incidence and the availability of mouse models, there is stronger data available on skin Ps, and data are, in most cases, relevant also to PsA. In a widely accepted model, environmental trigger factors like infections or trauma are capable of initiating an inflammatory cascade, ultimately creating a sustained state of chronic inflammation in genetically susceptible individuals. Besides well-known genetic susceptibility loci, epigenetic DNA modifications, which are associated with Ps development have been characterized recently and will be discussed in this article. The current evidence is promising in the possibility to provide new therapeutic avenues and fill the unmet need of patients, for whom current treatments either do not allow the disease to be controlled or must be continued for life.
Keywords: chronic inflammation, DNA methylation, histone modification, IL17, IL23, psoriasis, psoriatic arthritis, Th17
Introduction
Psoriasis (Ps) is a chronic, inflammatory predominantly skin-tropic disease that affects up to 3% of the general population with a geographical gradient.1–3 The most common form is plaques Ps or psoriasis vulgaris, which is characterized by red, well-demarcated plaques and silvery dry scale located predominantly on elbows, knees, and scalp, as well as in the umbilical and the lumbosacral area. Psoriatic skin lesions are histologically characterized by a hyperproliferation of premature keratinocytes and an incomplete cornification, leading to a thickened epidermis with elongated rete ridges.3,4 The dermis is infiltrated by an abnormal number of dendritic cells, macrophages, and T-cells.2,3
With a concordance rate of 20–70 % in monozygotic and 10–20 % in dizygotic twins, there is a strong genetic predisposition for the development of Ps,5–8 among the highest estimated for autoimmune or chronic inflammatory diseases.9 However, the mode of inheritance is complex and cannot fully explain Ps development. Several chromosomal loci and single nucleotide polymorphisms (SNP) with a significant association with susceptibility to Ps have been identified. The highest odds ratio for the heritability of Ps is associated with the PSORS1 locus on chromosome 6p spanning a segment in the class I region of the MHC (major histocompatibility complex), particularly the HLA-B and -C loci.2 Serological data suggest that the HLA-Cw6 antigen is responsible for Ps susceptibility within the PSORS1 locus; however, no specific variant has been identified so far.2,3 Furthermore, SNPs involved in the activation of interleukin (IL)17-producing cells (IL23R and IL12B gene) are associated with Ps development. Currently, it is widely accepted that, in such genetically susceptible individuals, environmental triggers such as streptococcal infection/superantigens, biomechanical stress (known as Koebner phenomenon in the skin, but also central to enthesitis development), stress, and smoking will initiate the disease.2,3 In as many as 30% of cases, Ps is accompanied by psoriatic arthritis (PsA), which may also be diagnosed in the absence of skin manifestations.3,10 PsA is characterized by a widespread musculoskeletal inflammation, which may affect the joints (arthritis), insertion sites of tendons and ligaments into bone (enthesitis), soft tissue of digits (dactylitis), and bone (osteitis) of the peripheral and axial skeleton.11 Family studies in PsA have demonstrated an increased risk of disease among first-degree relatives than among unrelated controls.12 As with Ps, PsA is associated with class I MHC alleles, but the reported HLA antigens and allelic variants differ from those in Ps. While being consistently associated with Ps, the association of HLA-C*06 with PsA is controversial, as most data show no, or only a weak, association with PsA.13–16 The HLA antigens B7 and B27 instead show an increased frequency in PsA.17 Even though HLA-B27 is clearly associated with PsA, particularly in the forms affecting the axial skeleton, the allele is not as frequent in PsA as it is in ankylosing spondylitis or reactive arthritis.18 In addition, the HLA-B*27:05:02, the HLA-B*08:01:01, and the HLA-C*07:01:01 haplotypes have been associated with different clinical subtypes of PsA and polymorphisms in the IL-23 receptor (IL23R),19 and the TNF-induced protein 3 (TNFAIP3) showed a stronger association with PsA than with Ps.20 In the present article, we will discuss the current immune-pathogenesis model of Ps leading to chronic skin inflammation and the progression to a systemic inflammatory disease primarily affecting the musculoskeletal system. In the second part of this article, we will emphasize the central role of epigenetics in the interplay between genetic susceptibility and environmental risk factors in Ps pathogenesis and discuss recent literature about epigenetics in Ps.
Immunological imbalance in Ps and PsA
It has been demonstrated in several models that immune imbalance of T-cells plays a key role in Ps and PsA pathogenesis.21–23 Experimental evidence suggests that an environmental trigger (particularly traumas as in the Koebner phenomenon) causes skin damage and the production of antimicrobial peptides by keratinocytes, particularly LL37. These peptides form complexes with DNA or RNA molecules, which can, in turn, activate plasmacytoid dentritic cells (pDC) via toll-like receptor (TLR) 7 and TLR9 signaling. pDCs produce type I interferons (IFNs), attracting myeloid dendritic cells and T-cells. The cytokines produced by myeloid DCs include IL-12 and IL-23. They activate and induce helper T (TH) cells to differentiate towards a TH1 and TH17 phenotype, respectively. The activated TH1 cells secrete IFN-γ and tumor necrosis factor α (TNF-α), whereas the TH17 cells produce IL-17 and IL-22. These proinflammatory cytokines induce the proliferation of keratinocytes and further sustain skin inflammation leading to psoriatic plaque formation (Figure 1).2,24–26 This pathogenetic model is supported by the high efficacy of novel biologic therapies, such as monoclonal antibodies against TNF-α, the p40 subunit shared by IL-12 and IL-23 (i.e. ustekinumab) and IL-17/IL-17-receptor (i.e. secukinumab, ixekizumab). These recently approved therapies, together with the small molecule inhibitor of phosphodiesterase 4 (PDE-4) apremilast, have become the new benchmarks in the therapy of moderate to severe Ps and PsA27–34 Apremilast inhibits the intracellular signal transduction involved in the secretion of several cytokines, mainly IL-17F, thus it acts directly on the immunologic imbalance observed in Ps.34
Figure 1.
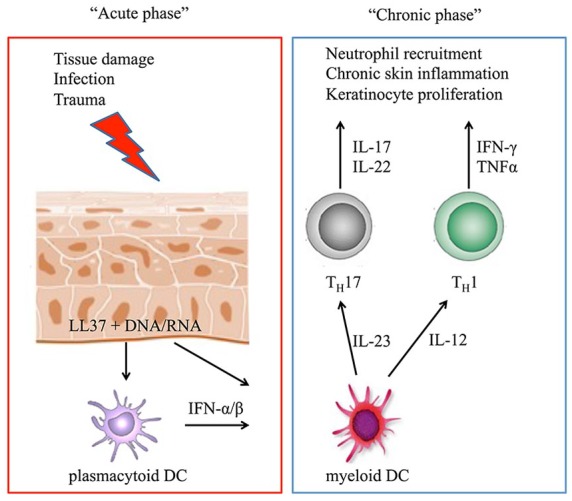
The proposed mechanisms of the immunological imbalance observed in psoriasis are summarized in the acute and chronic settings. In the acute phase of the disease, tissue damage induced, for example, by trauma or infection leads to the production of antimicrobial peptides by keratinocytes, particularly LL37. These peptides can form complexes with DNA or RNA molecules and, via toll-like receptor signaling, activate plasmacytoid dentritic cells (pDC), which produce type I interferons (IFN-α/β). Myeloid DCs are attracted and activated by IFN-α/β as well as from LL37/RNA complexes and secrete IL-12 and IL-23. They activate and induce helper T (TH) cells to develop a TH1 and TH17 phenotype, respectively, and initiate the chronic phase of the disease characterized by sustained production of the indicated proinflammatory cytokines, leading to neutrophil recruitment, chronic skin inflammation, and the formation of psoriatic plaques.
How chronic skin inflammation expands systemically to musculoskeletal tissues, and how PsA initiates in the absence of visible psoriatic skin lesions, is still a matter of debate. Besides the already described genetic risk factors for the development of PsA, environmental factors like mechanical stress have been associated with PsA development. In genetically susceptible hosts, trauma, which stimulates the musculoskeletal inflammatory disease, could act as a biomechanical trigger factor.35 Furthermore, there is an association with certain Ps skin phenotypes like scalp, inverse, and nail Ps.20,36 A severe form of Ps is also considered a risk factor for the development of PsA,20,36,37 and it was hypothesized that a higher burden of skin inflammation may lead to an increased systemic inflammation and trigger the onset of PsA.11 This is in line with the assumption of Ps as a systemic inflammatory disease that is associated with several comorbidities, including uveitis, inflammatory bowel disease, arteriosclerosis, and metabolic syndrome.3,11 Besides being a risk factor for major cardiovascular events, Ps causes vascular dysfunction at the site of inflammation (skin and synovial tissue). Due to the increased metabolic activity in the inflamed tissue, creating a hypoxic microenvironment, there is increased expression of angiogenic growth factors, including vascular endothelial growth factor (VEGF), and, therefore, neo-angiogenesis of immature, leaky blood vessels.3,10,38 The leaky blood vessels facilitate the leukocyte invasion of the inflamed tissues, and the hypoxic environment changes the metabolism of the immune cells and further enhances production of cytokines and increases the inflammation.3,10
Epigenetics at the crossroad of genetic susceptibility and environmental exposures
In most complex polygenic diseases, which include Ps and other chronic inflammatory diseases, the current etiopathogenic models assume that, in genetically susceptible individuals, exposure to certain environmental risk factors elicits a disease phenotype. The inherited genetic and acquired environmental contribution to disease development are thereby tightly connected by epigenetics.39 The term epigenetics summarizes all heritable changes in gene expression without alterations of the nucleotide sequence of the DNA. Mechanisms that can induce such heritable changes, and persist also in the absence of the signal that initiated them, include DNA methylation and specific histone modification.40–42 Noncoding RNAs are generally considered as epigenetic initiators that can ultimately lead to permanent heritable changes in gene expression.40,43 In the following paragraphs, we will focus on DNA and chromatin modifications.
The basic concepts and molecular mechanisms behind epigenetics have been reviewed extensively and their detailed discussion is beyond the scope of this article.41,44–47 In brief, DNA methylation of CpG dinucleotides by the action of DNA methyltransferases (DNMT) generally correlates with transcriptional repression. Post-translational modifications of histone proteins (e.g. acetylation and methylation) can cause either transcriptional activation or repression based on the location and nature of the modification. Both DNA and histone modifications can be maintained across cell cycles.41,44
In contrast to alterations of the DNA sequence, epigenetic modifications are relatively susceptible to environmental exposures (e.g. hypoxia, metabolites, drugs, chemical substances etc.).39 This is particularly the case during embryogenesis, where the degree of pluripotency is high. During cell differentiation, the epigenome is stabilized and becomes less responsive to environmental conditions.48 Indeed, in adult cell types, regulators of DNA methylation and chromatin modifications are less active,48 while, as demonstrated in monogenic twins, environmental conditions can affect the epigenome also postnatally.49 The transgenerational inheritance of epigenetic modifications remains controversial.47 During early embryonic development, most of the gamete cells epigenome is erased in a process called ‘reprogramming’ to initiate the de novo DNA and histone modifications necessary for cellular differentiation. A very small percentage of genes, however, keep their epigenetic marks during the process of reprogramming, and pass unchanged to the progeny.50 The most extensively studied example of epigenetic inheritance is the Dutch Hunger Winter, where DNA methylation patterns of metabolic genes, most likely induced by environmental exposure, was passed to the next generations, thereby affecting their susceptibility to metabolic disorders.47,51–53 Epigenetic modifications associated with disease can thus be either inherited, and contribute to an individual’s innate disease susceptibility, or can be induced by environmental exposures to risk factors during an individual’s lifespan.
Role of epigenetics in Ps and PsA
Given the discordance of Ps, particularly for PsA, in monogenic twins,54 the study of the epigenetic influence on the heritability and pathogenesis of Ps has become of major interest in recent years.55 Furthermore, the study of epigenetics might help discover novel targets for therapies that may change the DNA methylation or function of specific genes.
Several genome-wide methylation studies have been performed in Ps (Table 1).55,56 Gervin and colleagues analyzed CD4-positive and CD8-positive T-cells of monozygotic twins discordant for Ps. The authors did not identify significant differentially methylated sites comparing discordant co-twins.57 However, their data showed genes in CD4-positive T-cells with a correlation to differences in methylation status and differences in gene expression comparing unaffected with affected twins. Among these genes, several were involved in immune response and cytokine signaling pathways.57 Other groups have studied DNA methylation in subgroups of T-cells. Brandt and colleagues reported the hypomethylation of the interferon gamma gene (IFNG) of double-negative T cells in Ps patients using DNA methylation qPCR.58 Further studies have compared the methylation status of Ps-affected skin with adjacent unaffected skin using biopsy samples describing several differentially methylated regions (e.g., in Ps susceptibility regions).59–61 However, as some of these studies did not separate different cell types, and did not include adequate control groups, the differences observed may be secondary to inflammation rather than reflecting real permanent epigenetic changes. This is further supported by the fact that, after successful TNF-α inhibitor or UV-B therapy, CpG methylation status was reversed.60,62 Verma and Colleagues61 separated epidermal cells from skin biopsies from 6 psoriasis cases and 6 healthy controls and thereby ruled out changes in the methylation pattern most likely associated with cell type and the inflammatory response rather than with a real predisposition to the development of psoriasis. The study identified substantial differences in the methylation of genes involved in the Wnt and cadherin pathways comparing nonaffected psoriatic epidermis with healthy epidermis. The gene expression changes between the differentially methylated genes were, however, subtle and their functional significance remains unknown.61
Table 1.
Genome-wide DNA methylation studies in psoriasis.
| Author (year) | Tissue/cells | N cases and controls | Major findings |
|---|---|---|---|
| Gervin (2013)57 | CD4-positive and CD8-positive T-cells | 54 (27 pairs of monozygotic twins discordant for Ps) | No significant difference in methylation status comparing discordant co-twins |
| Chandra (2018)59 | Skin (psoriatic and adjacent normal) | 39 Ps patients | Identification of differentially methylated CpG sites in several Ps susceptibility (PSORS) regions and inverse correlation between methylation and gene expression comparing psoriatic skin with adjacent normal skin |
| Roberson (2012)60 | Skin (psoriatic, uninvolved and normal) | 12 Ps patients and 10 controls | Identification of several differentially methylated CpG sites comparing psoriatic and control skin. With anti-TNF-α treatment, these methylation changes reverted back to baseline |
| Verma (2018)61 | Epidermis (psoriatic, uninvolved and normal) | 6 Ps patients and 6 controls | Identification of more than 2000 strongly differentially methylated sites with a striking overrepresentation of the Wnt and cadherin pathways |
| Gu (2015)62 | Epidermis | 12 patients undergoing narrow-band UVB phototherapy and 12 corresponding healthy controls | Identification of 3665 MVPs with an overall hypomethylation in Ps patient samples. DNA methylation pattern was reversed after successful phototherapy. |
| Zhou (2016)63 | Skin (psoriatic, uninvolved and normal), peripheral blood mononuclear cells | 114 Ps patients and 62 controls | Identification of several differentially methylated CpG sites comparing psoriatic skin to uninvolved skin and normal skin of healthy controls. |
MVP, methylation variable positions; Ps, psoriasis.
In addition to studies assessing epigenetic changes as risk factors for Ps development, and the contribution of epigenetics to the heritability of the disease, functional studies using Ps mouse models have demonstrated how epigenetic modifications affect Ps immune-pathogenesis.64,65 Li and colleagues demonstrated that a keratinocyte specific N-WASP (neural Wiskott-Aldrich syndrome protein) knock-out in mice provokes IL-23 expression of keratinocytes that leads to activation of IL-17 producing cells and to chronic skin inflammation.64 IL-23A expression was thereby regulated by histone 3 lysine 9 (H3K9) methylation of histones. In addition, they showed in human keratinocytes that TNF-α signaling via phosphorylation of N-WASP induces degradation of the two H3K9 methyltransferases G9a and GLP, leading to an increased expression of IL-23 thus ultimately suggesting a possible link between histone modifications and the TNF/IL-23/IL-17 cytokine cascade contributing to chronic skin inflammation (Figure 2).64
Figure 2.
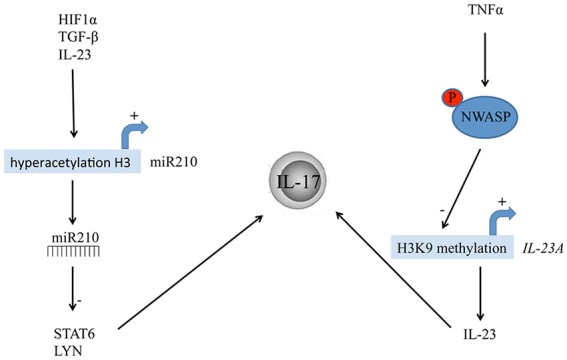
Epigenetic modifications involved in psoriasis immune-pathogenesis via activation of IL-17-producing cells. Hypoxia inducible factor 1α (HIF1α), induced by the hypoxic microenvironment, as well as IL-23 and TGF-β, cause hyperacetylation of histone H3 in the region of the miR-210 promoter. MiR-210 in turn targets the mRNA of STAT6 (Signal transducer and activator of transcription 6) and LYN (Lck/Yes novel tyrosine kinase), thereby inducing a TH17 phenotype and IL-17 production (left panel). TNF-α signaling via phosphorylation of N-WASP (neural Wiskott-Aldrich syndrome protein) activates IL-23A expression via decreased H3K9 methylation of its promotor. IL-23 is the main inducer of IL-17 producing cells (right panel).
Further evidence for the involvement of histone modifications in the immunological imbalance of Ps stems from a recent publication by Wu and colleagues.65 The authors used two mouse models of induced psoriatic skin lesions (subcutaneous injections of IL-23 and topical application of the TLR7 agonist imiquimod). The hypoxia-induced microRNA-210 (miR-210) was found to be upregulated in peripheral blood mononuclear cells (PBMCs) and in CD4-positive T-cells of both Ps patients and mouse models, miR210 knockdown or inhibition (by intradermal injection of antagomir-210) prevented Ps-like inflammation in both model systems. Mechanistically, miR-210 targeted the mRNA of STAT6 (signal transducer and activator of transcription 6) and LYN (Lck/Yes novel tyrosine kinase), thereby inducing the TH1 and TH17 phenotypes and inhibiting the TH2 phenotype. Furthermore, they demonstrated how the hypoxia inducible factor 1α (HIF1α), induced by the hypoxic microenvironment and further sustained by IL-23 and TGF-β, causes a hyperacetylation of histone H3 in the region of the miR-210 promoter, by recruiting the histone acetyltransferase p300 and thus promoting miR-210 expression (Figure 2).65 This study suggests a link between the psoriatic microenvironment and the maintenance of the immunological imbalance, highlighting a crucial role for miR-210, which might become a candidate as novel therapeutic target in Ps.
Conclusion
Despite remarkable progress in the understanding and treatment of Ps, several unanswered questions remain in the disease pathogenesis and preventive strategies. In recent years, an expanding number of susceptibility genes for Ps and PsA have been discovered.66–68 However, these susceptibility genes cannot fully explain the high degree of heritability of Ps suggested by twin studies.61,66 Epigenetic modifications inherited to the next generation might be, in part, responsible for this discrepancy. Furthermore, somatic epigenetic modifications might be triggered due to diverse environmental exposures and change the individual disease susceptibility. So far, only a few studies have addressed these questions in Ps and they are almost completely lacking for PsA. Genome-wide methylation studies performed on blood and skin samples have discovered several genes and pathways that are differentially methylated comparing Ps patients and healthy controls or psoriatic skin with adjacent normal skin.57,59–62,63 However, several limitation must be considered when interpreting these methylation studies, especially when they are performed on a complex tissue such as skin, which consists of different cell types. As every cell type has its unique epigenome, parts of the observed differences in DNA methylation might be explained by different cell- type composition between the samples. In addition, changes in DNA modifications caused by the microenvironment (e.g. inflammatory and hypoxic microenvironment of psoriatic skin or synovia) that are only transient in nature (i.e. not corresponding to the original definition of epigenetics) will also be identified. These DNA modifications do not help to identify Ps-prone individuals but are rather the result of the disease itself. Although their discovery will not help as biomarkers to identify individuals at risk for Ps or PsA development, the mechanistic understanding of the pathways involved in these ‘epigenetic’ modifications might contribute to a better understanding of Ps pathogenesis and can help to discover novel drug targets.
Footnotes
Funding: The author(s) received no financial support for the research, authorship, and publication of this article.
Conflict of interest statement: The authors declare that there is no conflict of interest.
ORCID iDs: Lukas Frischknecht  https://orcid.org/0000-0001-5666-5747
https://orcid.org/0000-0001-5666-5747
Carlo Selmi
https://orcid.org/0000-0002-0323-0376
Contributor Information
Lukas Frischknecht, Rheumatology and Clinical Immunology Unit, Humanitas Clinical and Research Center-IRCCS, Rozzano, Milan, Italy.
Matteo Vecellio, Rheumatology and Clinical Immunology Unit, Humanitas Clinical and Research Center-IRCCS, Rozzano, Milan, Italy; Nuffield Department of Orthopaedics, Rheumatology and Musculoskeletal Sciences, University of Oxford, Oxford, UK.
Carlo Selmi, Rheumatology and Clinical Immunology Unit, Humanitas Clinical and Research Center-IRCCS, via A. Manzoni 56, 20089 Rozzano, Milan, Italy; BIOMETRA Department, University of Milan, Milan, Italy.
References
- 1. Becher B, Pantelyushin S. Hiding under the skin: interleukin-17-producing γδ T cells go under the skin? Nat Med 2012; 18: 1748–1750. [DOI] [PubMed] [Google Scholar]
- 2. Lowes MA, Suárez-Fariñas M, Krueger JG. Immunology of psoriasis. Annu Rev Immunol 2014; 32: 227–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Nestle FO, Kaplan DH, Barker J. Psoriasis. N Engl J Med 2009; 361: 496–509. [DOI] [PubMed] [Google Scholar]
- 4. Perera GK, Di Meglio P, Nestle FO. Psoriasis. Annu Rev Pathol 2012; 7: 385–422. [DOI] [PubMed] [Google Scholar]
- 5. Duffy DL, Spelman LS, Martin NG. Psoriasis in Australian twins. J Am Acad Dermatol 1993; 29: 428–434. [DOI] [PubMed] [Google Scholar]
- 6. Farber EM, Nall ML, Watson W. Natural history of psoriasis in 61 twin pairs. Arch Dermatol 1974; 109: 207–211. [PubMed] [Google Scholar]
- 7. Bowcock AM. The genetics of psoriasis and autoimmunity. Annu Rev Genomics Hum Genet 2005; 6: 93–122. [DOI] [PubMed] [Google Scholar]
- 8. Lonnberg AS, Skov L, Skytthe A, et al. Heritability of psoriasis in a large twin sample. Br J Dermatol 2013; 169: 412–416. [DOI] [PubMed] [Google Scholar]
- 9. Generali E, Ceribelli A, Stazi MA, et al. Lessons learned from twins in autoimmune and chronic inflammatory diseases. J Autoimmun 2017; 83: 51–61. [DOI] [PubMed] [Google Scholar]
- 10. Veale DJ, Fearon U. The pathogenesis of psoriatic arthritis. Lancet 2018; 391: 2273–2284. [DOI] [PubMed] [Google Scholar]
- 11. Chimenti MS, Caso F, Alivernini S, et al. Amplifying the concept of psoriatic arthritis: the role of autoimmunity in systemic psoriatic disease. Autoimmun Rev 2019; 18: 565–575. [DOI] [PubMed] [Google Scholar]
- 12. Myers A, Kay LJ, Lynch SA, et al. Recurrence risk for psoriasis and psoriatic arthritis within sibships. Rheumatology (Oxford) 2005; 44: 773–776. [DOI] [PubMed] [Google Scholar]
- 13. Eder L, Chandran V, Pellet F, et al. Human leucocyte antigen risk alleles for psoriatic arthritis among patients with psoriasis. Ann Rheum Dis 2012; 71: 50–55. [DOI] [PubMed] [Google Scholar]
- 14. Ho PY, Barton A, Worthington J, et al. Investigating the role of the HLA-Cw*06 and HLA-DRB1 genes in susceptibility to psoriatic arthritis: comparison with psoriasis and undifferentiated inflammatory arthritis. Ann Rheum Dis 2008; 67: 677–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Winchester R, Minevich G, Steshenko V, et al. HLA associations reveal genetic heterogeneity in psoriatic arthritis and in the psoriasis phenotype. Arthritis Rheum 2012; 64: 1134–1144. [DOI] [PubMed] [Google Scholar]
- 16. Bowes J, Ashcroft J, Dand N, et al. Cross-phenotype association mapping of the MHC identifies genetic variants that differentiate psoriatic arthritis from psoriasis. Ann Rheum Dis 2017; 76: 1774–1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gladman DD, Anhorn KA, Schachter RK, et al. HLA antigens in psoriatic arthritis. J Rheumatol 1986; 13: 586–592. [PubMed] [Google Scholar]
- 18. Brandrup F, Hauge M, Henningsen K, et al. Psoriasis in an unselected series of twins. Arch Dermatol 1978; 114: 874–878. [PubMed] [Google Scholar]
- 19. Haroon M, Winchester R, Giles JT, et al. Certain class I HLA alleles and haplotypes implicated in susceptibility play a role in determining specific features of the psoriatic arthritis phenotype. Ann Rheum Dis 2016; 75: 155–162. [DOI] [PubMed] [Google Scholar]
- 20. Scher JU, Ogdie A, Merola JF, et al. Preventing psoriatic arthritis: focusing on patients with psoriasis at increased risk of transition. Nat Rev Rheumatol 2019; 15: 153–166. [DOI] [PubMed] [Google Scholar]
- 21. Boyman O, Hefti HP, Conrad C, et al. Spontaneous development of psoriasis in a new animal model shows an essential role for resident T cells and tumor necrosis factor-alpha. J Exp Med 2004; 199: 731–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gilliet M, Conrad C, Geiges M, et al. Psoriasis triggered by toll-like receptor 7 agonist imiquimod in the presence of dermal plasmacytoid dendritic cell precursors. Arch Dermatol 2004; 140: 1490–1495. [DOI] [PubMed] [Google Scholar]
- 23. van der Fits L, Mourits S, Voerman JS, et al. Imiquimod-induced psoriasis-like skin inflammation in mice is mediated via the IL-23/IL-17 axis. J Immunol 2009; 182: 5836–5845. [DOI] [PubMed] [Google Scholar]
- 24. Mak RK, Hundhausen C, Nestle FO. Progress in understanding the immunopathogenesis of psoriasis. Actas Dermosifiliogr 2009; 100(Suppl. 2): 2–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Fitzgerald O, Winchester R. Editorial: emerging evidence for critical involvement of the interleukin-17 pathway in both psoriasis and psoriatic arthritis. Arthritis Rheumatol 2014; 66: 1077–1080. [DOI] [PubMed] [Google Scholar]
- 26. Kim J, Krueger JG. The immunopathogenesis of psoriasis. Dermatol Clin 2015; 33: 13–23. [DOI] [PubMed] [Google Scholar]
- 27. Griffiths CE, Strober BE, van de Kerkhof P, et al. Comparison of ustekinumab and etanercept for moderate-to-severe psoriasis. N Engl J Med 2010; 362: 118–128. [DOI] [PubMed] [Google Scholar]
- 28. Gottlieb AB, Leonardi C, Kerdel F, et al. Efficacy and safety of briakinumab vs. etanercept and placebo in patients with moderate to severe chronic plaque psoriasis. Br J Dermatol 2011; 165: 652–660. [DOI] [PubMed] [Google Scholar]
- 29. Leonardi C, Matheson R, Zachariae C, et al. Anti-interleukin-17 monoclonal antibody ixekizumab in chronic plaque psoriasis. N Engl J Med 2012; 366: 1190–1199. [DOI] [PubMed] [Google Scholar]
- 30. Papp KA, Leonardi C, Menter A, et al. Brodalumab, an anti-interleukin-17-receptor antibody for psoriasis. N Engl J Med 2012; 366: 1181–1189. [DOI] [PubMed] [Google Scholar]
- 31. Mease PJ, Goffe BS, Metz J, et al. Etanercept in the treatment of psoriatic arthritis and psoriasis: a randomised trial. Lancet 2000; 356: 385–390. [DOI] [PubMed] [Google Scholar]
- 32. Bartlett BL, Tyring SK. Ustekinumab for chronic plaque psoriasis. Lancet 2008; 371: 1639–1640. [DOI] [PubMed] [Google Scholar]
- 33. Speeckaert R, van Geel N, Lambert J, et al. Secukinumab: IL-17A inhibition to treat psoriatic arthritis. Drugs Today (Barc) 2016; 52: 607–616. [DOI] [PubMed] [Google Scholar]
- 34. Pincelli C, Schafer PH, French LE, et al. Mechanisms underlying the clinical effects of apremilast for psoriasis. J Drugs Dermatol 2018; 17: 835–840. [PubMed] [Google Scholar]
- 35. Hsieh J, Kadavath S, Efthimiou P. Can traumatic injury trigger psoriatic arthritis? A review of the literature. Clin Rheumatol 2014; 33: 601–608. [DOI] [PubMed] [Google Scholar]
- 36. Wilson FC, Icen M, Crowson CS, et al. Incidence and clinical predictors of psoriatic arthritis in patients with psoriasis: a population-based study. Arthritis Rheum 2009; 61: 233–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Eder L, Haddad A, Rosen CF, et al. The incidence and risk factors for psoriatic arthritis in patients with psoriasis: a prospective cohort study. Arthritis Rheumatol 2016; 68: 915–923. [DOI] [PubMed] [Google Scholar]
- 38. Detmar M, Brown LF, Claffey KP, et al. Overexpression of vascular permeability factor/vascular endothelial growth factor and its receptors in psoriasis. J Exp Med 1994; 180: 1141–1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Feinberg AP, Fallin MD. Epigenetics at the crossroads of genes and the environment. JAMA 2015; 314: 1129–1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Berger SL, Kouzarides T, Shiekhattar R, et al. An operational definition of epigenetics. Genes Dev 2009; 23: 781–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Goldberg AD, Allis CD, Bernstein E. Epigenetics: a landscape takes shape. Cell 2007; 128: 635–638. [DOI] [PubMed] [Google Scholar]
- 42. Ptashne M. On the use of the word ‘epigenetic’. Curr Biol 2007; 17: R233–R236. [DOI] [PubMed] [Google Scholar]
- 43. Bernstein E, Allis CD. RNA meets chromatin. Genes Dev 2005; 19: 1635–1655. 2005/07/19. [DOI] [PubMed] [Google Scholar]
- 44. Allis CD, Jenuwein T. The molecular hallmarks of epigenetic control. Nat Rev Genet 2016; 17: 487–500. [DOI] [PubMed] [Google Scholar]
- 45. Berdasco M, Esteller M. Clinical epigenetics: seizing opportunities for translation. Nat Rev Genet 2019; 20: 109–127. [DOI] [PubMed] [Google Scholar]
- 46. Mazzio EA, Soliman KF. Basic concepts of epigenetics: impact of environmental signals on gene expression. Epigenetics 2012; 7: 119–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Skvortsova K, Iovino N, Bogdanović O. Functions and mechanisms of epigenetic inheritance in animals. Nat Rev Mol Cell Biol 2018; 19: 774–790. [DOI] [PubMed] [Google Scholar]
- 48. Feil R, Fraga MF. Epigenetics and the environment: emerging patterns and implications. Nat Rev Genet 2012; 13: 97–109. [DOI] [PubMed] [Google Scholar]
- 49. Fraga MF, Ballestar E, Paz MF, et al. Epigenetic differences arise during the lifetime of monozygotic twins. Proc Natl Acad Sci U S A 2005; 102: 10604–10609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Norouzitallab P, Baruah K, Vanrompay D, et al. Can epigenetics translate environmental cues into phenotypes? Sci Total Environ 2019; 647: 1281–1293. [DOI] [PubMed] [Google Scholar]
- 51. Heijmans BT, Tobi EW, Stein AD, et al. Persistent epigenetic differences associated with prenatal exposure to famine in humans. Proc Natl Acad Sci U S A 2008; 105: 17046–17049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Painter RC, Osmond C, Gluckman P, et al. Transgenerational effects of prenatal exposure to the Dutch famine on neonatal adiposity and health in later life. BJOG 2008; 115: 1243–1249. [DOI] [PubMed] [Google Scholar]
- 53. Roseboom T, de Rooij S, Painter R. The Dutch famine and its long-term consequences for adult health. Early Hum Dev 2006; 82: 485–491. [DOI] [PubMed] [Google Scholar]
- 54. Pedersen OB, Svendsen AJ, Ejstrup L, et al. On the heritability of psoriatic arthritis. Disease concordance among monozygotic and dizygotic twins. Ann Rheum Dis 2008; 67: 1417–1421. [DOI] [PubMed] [Google Scholar]
- 55. O’Rielly DD, Rahman P. Genetic, epigenetic and pharmacogenetic aspects of psoriasis and psoriatic arthritis. Rheum Dis Clin North Am 2015; 41: 623–642. [DOI] [PubMed] [Google Scholar]
- 56. Pollock RA, Abji F, Gladman DD. Epigenetics of psoriatic disease: a systematic review and critical appraisal. J Autoimmun 2017; 78: 29–38. [DOI] [PubMed] [Google Scholar]
- 57. Gervin K, Vigeland MD, Mattingsdal M, et al. DNA methylation and gene expression changes in monozygotic twins discordant for psoriasis: identification of epigenetically dysregulated genes. PLoS Genet 2012; 8: e1002454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Brandt D, Sergon M, Abraham S, et al. TCR(+)CD3(+)CD4(-)CD8(-) effector T cells in psoriasis. Clin Immunol 2017; 181: 51–59. [DOI] [PubMed] [Google Scholar]
- 59. Chandra A, Senapati S, Roy S, et al. Epigenome-wide DNA methylation regulates cardinal pathological features of psoriasis. Clin Epigenetics 2018; 10: 108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Roberson ED, Liu Y, Ryan C, et al. A subset of methylated CpG sites differentiate psoriatic from normal skin. J Invest Dermatol 2012; 132: 583–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Verma D, Ekman AK, Bivik Eding C, et al. Genome-wide DNA methylation profiling identifies differential methylation in uninvolved psoriatic epidermis. J Invest Dermatol 2018; 138: 1088–1093. [DOI] [PubMed] [Google Scholar]
- 62. Gu X, Nylander E, Coates PJ, et al. Correlation between reversal of DNA methylation and clinical symptoms in psoriatic epidermis following narrow-B and UVB phototherapy. J Invest Dermatol 2015; 135: 2077–2083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Zhou F, Wang W, Shen C, et al. Epigenome-wide association analysis identified nine skin DNA methylation loci for psoriasis. J Invest Dermatol 2016; 136: 779–787. [DOI] [PubMed] [Google Scholar]
- 64. Li H, Yao Q, Mariscal AG, et al. Epigenetic control of IL-23 expression in keratinocytes is important for chronic skin inflammation. Nat Commun 2018; 9: 1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Wu R, Zeng J, Yuan J, et al. MicroRNA-210 overexpression promotes psoriasis-like inflammation by inducing Th1 and Th17 cell differentiation. J Clin Invest 2018; 128: 2551–2568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Tsoi LC, Spain SL, Ellinghaus E, et al. Enhanced meta-analysis and replication studies identify five new psoriasis susceptibility loci. Nat Commun 2015; 6: 7001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Tsoi LC, Spain SL, Knight J, et al. Identification of 15 new psoriasis susceptibility loci highlights the role of innate immunity. Nat Genet 2012; 44: 1341–1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Patrick MT, Stuart PE, Raja K, et al. Genetic signature to provide robust risk assessment of psoriatic arthritis development in psoriasis patients. Nat Commun 2018; 9: 4178. [DOI] [PMC free article] [PubMed] [Google Scholar]