

 Transmetalation-initiated three-component vicinal-diarylation of alkenes.
Transmetalation-initiated three-component vicinal-diarylation of alkenes.
Abstract
Herein, we report three-component vicinal-diarylation of non-conjugated alkenes initiated by transmetalation of arylboronic acids, which provides complementary access to β,γ-diaryl carbonyl compounds. We have also screened a large number of chiral ligands for developing an enantioselective version of this reaction and obtained the preliminary results (up to 79 : 21 e.r.). Notably, the methodology developed herein represents the first three component syn-vicinal-dicarbofunctionalization of non-conjugated alkenes involving palladium catalysis.
Introduction
Vicinal-diaryl structures are a common scaffold in various medicinally relevant molecules and can also be widely utilized as chemical feedstocks to access a number of natural products (Scheme 1, upper panel).1 Traditional reactions to access vicinal-diaryl structures generally suffer from disadvantages such as multi-step processes, limited substrate scope, and difficulties in accessing starting materials.1,2
Scheme 1. The usefulness of β,γ-diaryl carbonyl structures as well as the previous work and our design of three-component diarylation of olefins.

Alkenes are among the most abundant classes of organic molecules, available in bulk quantities from petrochemical feedstocks and renewable resources. Thus, the intermolecular diarylation of olefins represents one of the most powerful and straightforward methods to rapidly produce vicinal diaryl structures.3 For example, homodiarylation of alkenes with aryl metals or halides is a quick method to introduce two identical aryl groups across an olefin.4 On the other hand, two general approaches for the introduction of two different aryl moieties across an olefin have been developed: (1) a sequence involving the formation of an Ar–MX species via oxidative addition of an M0 catalyst (Ni0 or Pd0) to an aryl electrophile (El), alkene insertion, transmetalation with the aryl nucleophiles (Nu), and C–C reductive elimination (Scheme 1a, lower panel);5 (2) π-Lewis acid (PdII as the Lewis acid) activation of the alkene to enable attack of an aryl C–H nucleophile (limited to indoles and phenols) to form a carbopalladated Wacker-type intermediate, followed by the oxidative addition of aryl electrophiles (Ar–X) and C–C reductive elimination (Scheme 1b, lower panel).6
Transmetalation of aryl metal reagents with a palladium(ii) catalyst to generate the Ar–PdX intermediate has been well documented.7 In recent years, several groups have developed an array of methods for alkene functionalization triggered by PdII-catalyzed transmetalation of arylboronic acids such as the oxidative boron Heck reaction.8,9 We thus questioned whether three-component heterodiarylation of olefins with aryl halides and arylboronic acids could also be initiated by a transmetalation step (Scheme 1c, lower panel). Such a reaction would provide complementary access to the previous two approaches to vicinal-diaryl structures. Herein, we report the first transmetalation-initiated three-component vicinal-diarylation of γ-olefinic acids by using a directing-group strategy, which enables rapid construction of syn-β,γ-diaryl carbonyl compounds.
Results and discussion
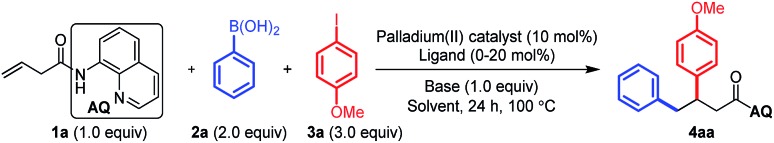
In a preliminary experiment, a 3-butenoic acid derivative 1a bearing an 8-aminoquinoline (AQ)-directing group (0.3 mmol) was treated with Pd(OAc)2 (10 mol%), K2CO3 (0.3 mmol), 2-phenylboronic acid 2a (0.6 mmol) and 4-methoxyphenyl iodide 3a (0.9 mmol) in hexafluoroisopropanol (HFIP, 2 mL) at 100 °C for 24 h, which was completely unreactive (Table 1, entry 1). Inspired by Toste's work,10 we changed the solvent to a mixture of DCM/H2O/CH3CN (2 mL : 0.4 mL : 0.2 mL) and the base to Na2CO3 (0.3 mmol). Then a variety of palladium(ii) catalysts were evaluated (Table 1, entries 2–8). (–)-SparteinePdCl2 was proven to be the best choice (entry 5). Subsequently, different bases were also screened (entries 9–11). All others gave inferior results. We further found that the addition of extra (–)-sparteine (20 mol%) would increase the yield to 83% (entry 12). The effect of each component of the mixed solvents was also investigated (entries 13–16). It has been proven that all the three solvents are necessary to ensure a satisfactory yield. Switching the solvent back to HFIP dramatically shut off the reaction (entry 17). Notably, we also checked the enantioselectivity of this reaction under the optimized conditions developed in this study. However, the product 4aa was always racemic at this stage. Different types of aryl boron compounds were also screened (entries 18–19).
Table 1. Optimization of the directed three-component syn-vicinal-diarylation of alkene 1a with phenylboronic acids 2a and 4-methoxyphenyl iodide 3a a .
| ||||
| Entry | PdII cat. | Base | Solvent | Yield b [%] |
| 1 | Pd(OAc)2 | K2CO3 | HFIP (2 mL) | NR |
| 2 | Pd(OAc)2 | Na2CO3 | DCM/H2O/MeCN (1 : 0.2 : 0.1) | 54 |
| 3 | PdCl2 | Na2CO3 | DCM/H2O/MeCN (1 : 0.2 : 0.1) | 44 |
| 4 | Pd(PhCN)2Cl2 | Na2CO3 | DCM/H2O/MeCN (1 : 0.2 : 0.1) | 69 |
| 5 | (–)-SparteinePdCl2 | Na2CO3 | DCM/H2O/MeCN (1 : 0.2 : 0.1) | 71 |
| 6 | (–)-SparteinePd(OAc)2 | Na2CO3 | DCM/H2O/MeCN (1 : 0.2 : 0.1) | 6 |
| 7 | (Bipy)PdCl2 | Na2CO3 | DCM/H2O/MeCN (1 : 0.2 : 0.1) | 60 |
| 8 | PddppfCl2 | Na2CO3 | DCM/H2O/MeCN (1 : 0.2 : 0.1) | 11 |
| 9 | (–)-SparteinePdCl2 | K2CO3 | DCM/H2O/MeCN (1 : 0.2 : 0.1) | 66 |
| 10 | (–)-SparteinePdCl2 | Cs2CO3 | DCM/H2O/MeCN (1 : 0.2 : 0.1) | 44 |
| 11 | (–)-SparteinePdCl2 | K3PO4 | DCM/H2O/MeCN (1 : 0.2 : 0.1) | 53 |
| 12 c | (–)-SparteinePdCl 2 | Na 2 CO 3 | DCM/H 2 O/MeCN (1 : 0.2 : 0.1) | 83 |
| 13 c | (–)-SparteinePdCl2 | Na2CO3 | DCM/H2O/MeCN (1 : 0.1 : 0.1) | 72 |
| 14 c | (–)-SparteinePdCl2 | Na2CO3 | DCM/H2O (1 : 0.2) | 77 |
| 15 c | (–)-SparteinePdCl2 | Na2CO3 | DCM/CH3CN (1 : 0.1) | 14 |
| 16 c | (–)-SparteinePdCl2 | Na2CO3 | DCM (2 mL) | 18 |
| 17 c | (–)-SparteinePdCl2 | Na2CO3 | HFIP (2 mL) | NR |
| 18 c d | (–)-SparteinePdCl2 | Na2CO3 | DCM/H2O/MeCN (1 : 0.2 : 0.1) | 81 |
| 19 c e | (–)-SparteinePdCl2 | Na2CO3 | DCM/H2O/MeCN (1 : 0.2 : 0.1) | 35 |
aReactions were carried out by using PdII catalyst (10 mol%), ligand (0–20 mol%), base (0.3 mmol, 1.0 equiv.), 1a (0.3 mmol, 1.0 equiv.), phenylboronic acid 2a (2.0 equiv.), and 4-methoxyphenyl iodide 3a (3.0 equiv.) in the mixture of DCM/H2O/CH3CN (2 mL : 0.4 mL : 0.2 mL) for 24 h at 100 °C under a N2 atmosphere.
bIsolated yields.
c(–)-Sparteine (20 mol%) was added.
d(PhBO)3 instead of 2a.
ePhBNep instead of 2a.
Having optimized the reaction conditions, we first investigated the substrate scope of aryl boronic acid nucleophiles by using the 3-butenoic acid derivative 1a as the alkene and 4-iodoanisole 3a as the electrophile (Scheme 2). To our delight, the reaction was found to tolerate an array of functional groups on the aromatic ring of boronic acid, including electron-neutral, electron-donating and electron-withdrawing substituents, at both the para- and meta-positions (4ba–oa). Substituents on the ortho-position led to a decreased yield, presumably due to steric effects (4pa). Multi-substituted aryl boronic acids (4ra–sa) and heteroaryl boronic acids were also reactive (4ta–ua). It is important to stress that these reaction conditions were compatible with a variety of functional groups such as halogens (F, Cl, and Br) and acetyl, ester, aldehyde, cyano, and methoxy groups on the aryl boronic acids, which could be subjected to further synthetic transformations.
Scheme 2. The substrate scope of aryl boronic acid nucleophiles.a aReactions were carried out by using (–)-sparteinePdCl2 (0.03 mmol, 10 mol%), (–)-sparteine (0.06 mmol, 20 mol%), Na2CO3 (0.3 mmol, 1 equiv.), 1a (0.3 mmol, 1.0 equiv.), arylboronic acid 2 (0.6 mmol, 2.0 equiv.), and 4-methoxyphenyl iodide 3a (0.9 mmol, 3.0 equiv.) in the mixture of DCM/H2O/CH3CN (2 mL : 0.4 mL : 0.2 mL) for 24 h at 100 °C under a N2 atmosphere. bAlkene 1a (0.2 mmol) was used in the reaction. cIodobenzene was used as the electrophile; dthe reaction was conducted for 48 h and 4.0 equiv. of 3a were used.

Subsequently, a variety of substituted aryl and heteroaryl iodide electrophiles were tested under the optimized conditions, and the results are summarized in Scheme 3. It was gratifying to find whether aryl iodides are electron-rich, electron-poor, or sterically hindered, and all of them afforded moderate to high yields (4ac–an). Heteroaryl iodides were also competent coupling partners (5n). Notably, a lot of important functional groups such as halogens (F, Br, and I), esters, and acetyl groups on the aryl iodides were tolerated, presenting the opportunity for subsequent diversification. Besides diverse (hetero)aryl iodides, the employment of methyl iodide as the electrophile also smoothly delivered the desired products in moderated yields in this reaction (Scheme 3, 4aq). X-ray analysis of a single crystal 4ah clearly confirmed that the diaryl groups of the structural connection order in the product is opposite to those vicinal-diaryl products produced by M0 (Ni0 or Pd0)-catalyzed diarylation of alkenes with aryl electrophiles and nucleophiles.5
Scheme 3. The scope of aryl iodide electrophiles.a aReactions were carried out by using (–)-sparteinePdCl2 (0.03 mmol, 10 mol%), (–)-sparteine (0.06 mmol, 20 mol%), Na2CO3 (0.3 mmol, 1.0 equiv.), 1a (0.3 mmol, 1.0 equiv.), phenylboronic acid 2a (0.6 mmol, 2 equiv.), and aryl iodide 3 (3–10 equiv.) in the mixture of DCM/H2O/CH3CN (2 mL : 0.4 mL : 0.2 mL) for 24 h at 100 °C under a N2 atmosphere. bA by-product 4af′ with 8% yield was observed in this reaction from the activation of the second C–I bond in the aromatic ring. cThe reaction was conducted for 36 h and 4.0 equiv. of aryl iodides were used. dThe reaction was conducted for 48 h and 10.0 equivalent of methyl iodide were used.

We next evaluated the utility of this method for various non-conjugated alkenes by using phenylboronic acid 2a as the nucleophile and 4-methoxyphenyl iodide 3a as the electrophile (Scheme 4). The terminal alkene bearing mono-substitution at the α-position proceeded smoothly to afford the desired product in moderate yield; nevertheless harsher conditions are needed (Scheme 4, 5a). Furthermore, we proved that a variety of internal alkenes could also be efficiently converted into the syn-diastereomers with nearly negligible electronic effects of substituents (Scheme 4, 5b–m). The syn-selectivity of the reaction was confirmed by X-ray analysis of the crystal structure of 5m,11 which was in accordance with our proposed mechanism. Notably, both diastereomers could be accessed based on the stereochemistry of the alkene substrate (i.e., E-alkene to 5c and Z-alkene to 5d), suggesting that the alkene does not undergo isomerization in the Pd-catalyzed process used in this study. In addition, a pendant phthalimide-protected amine, benzyl-protected alcohol and ester were also well tolerated in the reaction (5g–i).
Scheme 4. The scope of various non-conjugated alkenes.a aReactions were carried out by using (–)-sparteinePdCl2 (0.03 mmol, 10 mol%), (–)-sparteine (0.06 mmol, 20 mol%), Na2CO3 (0.3 mmol, 1.0 equiv.), 1 (0.3 mmol, 1.0 equiv.), phenylboronic acid 2a (0.6 mmol, 2 equiv.), and 4-methoxyphenyl iodide 3a (0.9 mmol, 3 equiv.) in the mixture of DCM/H2O/CH3CN (2 mL : 0.4 mL : 0.2 mL) for 24 h at 100 °C under a N2 atmosphere. bThe reaction was conducted for 36 h and 4.0 equiv. of 3a were used. cAn alkene substrate (0.2 mmol) was used in the reaction.

In light of our success in the transmetalation-initiated three-component vicinal-diarylation of alkenes to synthesize racemic syn-β,γ-diaryl carbonyl compounds, we have tried to develop the enantioselective version of this reaction. After much work on ligand evaluation and optimization of the reaction conditions (see Fig. S2 and S3 in the ESI† for details of the optimization studies), we identified a pyridyl-oxazolidine ligand L41 as an effective ligand, producing a chiral product with moderate enantioselectivity (79 : 21 e.r.) and 60% yield (eqn (1)).
 |
1 |
To demonstrate the practicality of this method, we first scaled up the reaction to obtain ∼1 g of 4aa (Scheme 5, upper panel). Then, the AQ-directing group was directly removed by treatment with NaOH to yield the free carboxylic acid 6via two steps with an overall yield of 81%. Furthermore, the carboxylic acid 8, which has been employed as the intermediate to obtain the natural product pallidol, has been efficiently synthesized with 92% yield by using the Pd(ii)-catalyzed method (Scheme 5, lower panel). In contrast, the methods in the literature involved a non-catalytic procedure with three steps (86% yield), which shows the advantage and usefulness of our method in the modular synthesis of β,γ-diaryl carbonyl compounds.12
Scheme 5. The synthetic usefulness of our method.

On the basis of literature reports,7–9 we propose a PdII/PdIV catalytic cycle that starts with the transmetalation (TM) of arylboronic acids to form Ar–PdX species A (Scheme 6). Then the Ar–PdX species would coordinate with the directing group and alkene to form the intermediate B, which would involve the carbopalladation step resulting in the intermediate C. Because of the exclusive syn-addition during the carbopalladation step, the stereoselectivity of the as-synthesized diarylated products is opposite to the alkenes produced by the diarylation method involving a Wacker-type anti-nucleopalladation step.6 Moreover, the intermediate C would be intercepted by the oxidative addition of aryl iodides to form PdIV species D. Finally, reductive elimination from the high-valent palladium center and subsequent ligand exchange produce the product and regenerate the PdII catalyst.13 Alternatively, alkene coordination with the PdII catalyst could also happen first to form the intermediate A′, and then the transmetalation of aryl boronic acids to form the intermediate B.
Scheme 6. Proposed catalytic cycle of this three-component vicinal-diarylation of alkenes.

To support our aforementioned continuous mechanism, several control experiments were performed. We found that no reaction occurred when N-(naphthalenyl)butenamide 9 was used as the alkene substrate (eqn (2)). This result indicates that the presence of the AQ directing group on the unconjugated alkene was indispensable. The Heck reaction between aryl iodides and alkene 1a did not happen under the optimized conditions and 80% starting material 1a was recovered (eqn (3)). It indicates that the Pd0 species might not be formed in this reaction system. Notably, the homodiarylated product with 8% yield was observed (eqn (3)). To obtain the intermediate C, the reaction of alkene 1a and phenylboronic acids 2a was run in the presence of a stoichiometric amount of the PdII catalyst (eqn (4)). The Heck product 1k (40% yield) and the starting material 1a (51% recovery) were isolated. It means that the transmetalation of aryl boronic acids and carbopalladation would preferentially occur in the presence of the PdII catalyst, which follows a β-H elimination step in the absence of an electrophile to form the Heck product. To rule out the possibility of the Heck product 11 as the intermediate for this three-component vicinal-diarylation of alkenes, we carried out the reaction of the Heck product 11 with aryl iodides with a stoichiometric amount of the Pd0 catalyst (eqn (5)). No desired product 4aa was observed. Finally, we exclude the possibility of the hydrocarbofunctionalized product 13 as the competent intermediate because only 7% diarylated product 4aa was yielded when the prepared hydrocarbofunctionalized intermediate 13 was exposed to the standard conditions in the absence of additional nucleophiles (eqn (6)).
 |
2 |
 |
3 |
 |
4 |
 |
5 |
 |
6 |
Conclusions
In conclusion, we have described the transmetalation-initiated three-component syn-β,γ-diarylation of olefins with aryl halides and arylboronic acids. The process is not only simple and convenient in terms of the reaction conditions, but also tolerates a variety of functional groups, thus constituting a practical route to β,γ-diaryl carbonyl compounds prevalent in bioactive molecules.
Conflicts of interest
There are no conflicts to declare.
Supplementary Material
Acknowledgments
We are grateful for the financial support from the National Natural Science Foundation of China (21602115), the 1000-Talent Youth Program (020/BF180181), the Natural Science Foundation of Tianjin (18JCYBJC20400), the Fundamental Research Funds for the Central Universities and Nankai University.
Footnotes
†Electronic supplementary information (ESI) available. CCDC 1852875 and 1852877. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc02182e
References
- (a) Ahmad I., Pathak V., Vasudev P. G., Maurya H. K., Gupta A. RSC Adv. 2014;4:24619. [Google Scholar]; (b) Miller W. H., Manley P. J., Cousins R. D., Erhard K. F., Heerding D. A., Kwon C., Ross S. T., Samanen J. M., Takata D. T., Uzinskas I. N., Yuan C. C. K., Haltiwanger R. C., Gress C. J., Lark M. W., Hwang S.-M., James I. E., Rieman D. J., Willette R. N., Yue T.-L., Azzarano L. M., Salyers K. L., Smith B. R., Ward K. W., Johanson K. O., Huffman W. F. Bioorg. Med. Chem. Lett. 2003;13:1483. doi: 10.1016/s0960-894x(03)00102-1. [DOI] [PubMed] [Google Scholar]; (c) Yous S., Durieux-Poissonnier S., Lipka-Belloli E., Guelzim H., Bochu C., Audinot V., Boutin J. A., Delagrange P., Bennejean C., Renard P., Lesieur D. Bioorg. Med. Chem. 2003;11:753. doi: 10.1016/s0968-0896(02)00473-x. [DOI] [PubMed] [Google Scholar]; (d) Watanabe T., Ohashi Y., Yoshino R., Komano N., Eguchi M., Maruyama S., Ishikawa T. Org. Biomol. Chem. 2003;1:3024. doi: 10.1039/b304216m. [DOI] [PubMed] [Google Scholar]; (e) Ishikawa T., Saito T., Ishii H. Tetrahedron. 1995;51:8447. [Google Scholar]; (f) Landvatter S. W., Katzenellenbogen J. A. J. Med. Chem. 1982;25:1300. doi: 10.1021/jm00353a006. [DOI] [PubMed] [Google Scholar]
- (a) Chen Z.-M., Hilton M. J., Sigman M. S. J. Am. Chem. Soc. 2016;138:11461. doi: 10.1021/jacs.6b06994. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Larionov E., Lin L., Guénée L., Mazet C. J. Am. Chem. Soc. 2014;136:16882. doi: 10.1021/ja508736u. [DOI] [PubMed] [Google Scholar]; (c) Zhang C., Yun J. Org. Lett. 2013;15:3416. doi: 10.1021/ol401468v. [DOI] [PubMed] [Google Scholar]; (d) Li T., Zhu J., Wu D., Li X., Wang S., Li H., Li J., Wang W. Chem.–Eur. J. 2013;19:9147. doi: 10.1002/chem.201300304. [DOI] [PubMed] [Google Scholar]; (e) Ziadi A., Martin R. Org. Lett. 2012;14:1266. doi: 10.1021/ol300119u. [DOI] [PubMed] [Google Scholar]; (f) Ding Z., Xue S., Wulff W. D. Chem.–Asian J. 2011;6:2130. doi: 10.1002/asia.201000804. [DOI] [PubMed] [Google Scholar]; (g) Durieux S., Chanu A., Bochu C., Audinot V., Coumailleau S., Boutin J. A., Delagrange P., Caignard D. H., Bennejean C., Renard P., Lesieur D., Berthelot P., Yous S. Bioorg. Med. Chem. 2009;17:2963. doi: 10.1016/j.bmc.2009.03.023. [DOI] [PubMed] [Google Scholar]; (h) Mandal S. K., Jana S., Roy S. C. Tetrahedron Lett. 2005;46:6115. [Google Scholar]; (i) Jung J. C., Lee J. H., Oh S., Lee J. G., Park O. S. Bioorg. Med. Chem. Lett. 2004;14:5527. doi: 10.1016/j.bmcl.2004.09.009. [DOI] [PubMed] [Google Scholar]; (j) Blaisdell T. P., Morken J. P. J. Am. Chem. Soc. 2015;137:8712. doi: 10.1021/jacs.5b05477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For some reviews on alkene difunctionalization, see: ; (a) Ping Y., Li Y., Zhu J., Kong W. Angew. Chem., Int. Ed. 2019;58:1562. doi: 10.1002/anie.201806088. [DOI] [PubMed] [Google Scholar]; (b) Giri R., KC S. J. Org. Chem. 2018;83:3013. doi: 10.1021/acs.joc.7b03128. [DOI] [PubMed] [Google Scholar]; (c) Xiong Y., Sun Y., Zhang G. Tetrahedron Lett. 2018;59:347. [Google Scholar]; (d) Yin G., Mu X., Liu G. Acc. Chem. Res. 2016;49:2413. doi: 10.1021/acs.accounts.6b00328. [DOI] [PubMed] [Google Scholar]; (e) McNeill E., Ritter T. Acc. Chem. Res. 2015;48:2330. doi: 10.1021/acs.accounts.5b00050. [DOI] [PubMed] [Google Scholar]; (f) DeLuca R. J., Stokes B. J., Sigman M. S. Pure Appl. Chem. 2014;86:395. [Google Scholar]; (g) McDonald R. I., Liu G., Stahl S. S. Chem. Rev. 2011;111:2981. doi: 10.1021/cr100371y. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Zeni G., Larock R. C. Chem. Rev. 2006;106:4644. doi: 10.1021/cr0683966. [DOI] [PubMed] [Google Scholar]
- (a) Anthony D., Lin Q., Baudet J., Diao T. Angew. Chem., Int. Ed. 2019;58:3198. doi: 10.1002/anie.201900228. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Kusunuru A. K., Jaladanki C. K., Tatina M. B., Bharatam P. V., Mukherjee D. Org. Lett. 2015;17:3742. doi: 10.1021/acs.orglett.5b01722. [DOI] [PubMed] [Google Scholar]; (c) Yahiaoui S., Fardost A., Trejos A., Larhed M. J. Org. Chem. 2011;76:2433. doi: 10.1021/jo1018188. [DOI] [PubMed] [Google Scholar]; (d) Trejos A., Fardost A., Yahiaoui S., Larhed M. Chem. Commun. 2009:7587. doi: 10.1039/b918358b. [DOI] [PubMed] [Google Scholar]; (e) Urkalan K. B., Sigman M. S. Angew. Chem., Int. Ed. 2009;48:3146. doi: 10.1002/anie.200900218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (a) Derosa J., Kleinmans R., Tran V. T., Karunananda M. K., Wisniewski S. R., Eastgate M. D., Engle K. M. J. Am. Chem. Soc. 2018;140:17878. doi: 10.1021/jacs.8b11942. [DOI] [PubMed] [Google Scholar]; (b) Basnet P., KC S., Dhungana R. K., Shrestha B., Boyle T. J., Giri R. J. Am. Chem. Soc. 2018;140:15586. doi: 10.1021/jacs.8b09401. [DOI] [PubMed] [Google Scholar]; (c) Gao P., Chen L.-A., Brown M. K. J. Am. Chem. Soc. 2018;140:10653. doi: 10.1021/jacs.8b05680. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Li W., Boon J. K., Zhao Y. Chem. Sci. 2018;9:600. doi: 10.1039/c7sc03149a. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Basnet P., Dhungana R. K., Thapa S., Shrestha B., KC S., Sears J. M., Giri R. J. Am. Chem. Soc. 2018;140:7782. doi: 10.1021/jacs.8b03163. [DOI] [PubMed] [Google Scholar]; (f) Thapa S., Dhungana R. K., Magar R. T., Shrestha B., Kc S., Giri R. Chem. Sci. 2018;9:904. doi: 10.1039/c7sc04351a. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Derosa J., Tran V. T., Boulous M. N., Chen J. S., Engle K. M. J. Am. Chem. Soc. 2017;139:10657. doi: 10.1021/jacs.7b06567. [DOI] [PubMed] [Google Scholar]; (h) Shrestha B., Basnet P., Dhungana R. K., Kc S., Thapa S., Sears J. M., Giri R. J. Am. Chem. Soc. 2017;139:10653. doi: 10.1021/jacs.7b06340. [DOI] [PubMed] [Google Scholar]; (i) Kuang Z., Yang K., Song Q. Org. Chem. Front. 2017;4:1224. [Google Scholar]; (j) Stokes B. J., Liao L., de Andrade A. M., Wang Q., Sigman M. S. Org. Lett. 2014;16:4666. doi: 10.1021/ol502279u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z., Zeng T., Yang K. S., Engle K. M. J. Am. Chem. Soc. 2016;138:15122. doi: 10.1021/jacs.6b09170. [DOI] [PubMed] [Google Scholar]
- (a) Dupont J. and Pfeffer M., Palladacycles, Wiley-VCH, Verlag GmbH & Co. KGaA, 2008. [Google Scholar]; (b) de Meijere A. and Diederich F., Metal-catalyzed Cross-coupling Reactions, Wiley-VCH, Weinheim, 2nd edn, 2004. [Google Scholar]; (c) Tsuji J., Palladium Reagents and Catalysts, Wiley, Hoboken, NJ, 2004. [Google Scholar]; (d) Negishi E. and de Meijere A., Handbook of Organopalladium Chemistry for Organic Synthesis, John Wiley & Sons, Inc., 2002. [Google Scholar]; (e) Tsuji J., Transition Metal Reagents and Catalysts, John Wiley & Sons, Ltd, 2000. [Google Scholar]
- For some recent examples, see: ; (a) Xiao L.-J., Cheng L., Feng W.-M., Li M.-L., Xie J.-H., Zhou Q.-L. Angew. Chem., Int. Ed. 2018;57:461. doi: 10.1002/anie.201710735. [DOI] [PubMed] [Google Scholar]; (b) Matsuura R., Jankins T. C., Hill D. E., Yang K. S., Gallego G. M., Yang S., He M., Wang F., Marsters R. P., McAlpine I., Engle K. M. Chem. Sci. 2018;9:8363. doi: 10.1039/c8sc03081b. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Yuan Q., Sigman M. S. J. Am. Chem. Soc. 2018;140:6527. doi: 10.1021/jacs.8b02752. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Zhang C., Santiago C. B., Kou L., Sigman M. S. J. Am. Chem. Soc. 2015;137:7290. doi: 10.1021/jacs.5b04289. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Werner E. W., Sigman M. S. J. Am. Chem. Soc. 2010;132:13981. doi: 10.1021/ja1060998. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Delcamp J. H., Brucks A. P., White M. C. J. Am. Chem. Soc. 2008;130:11270. doi: 10.1021/ja804120r. [DOI] [PubMed] [Google Scholar]; (g) Delcamp J. H., White M. C. J. Am. Chem. Soc. 2006;128:15076. doi: 10.1021/ja066563d. [DOI] [PubMed] [Google Scholar]; (h) Satterfield A. D., Kubota A., Sanford M. S. Org. Lett. 2011;13:1076. doi: 10.1021/ol103121r. [DOI] [PMC free article] [PubMed] [Google Scholar]; (i) Kalyani D., Satterfield A. D., Sanford M. S. J. Am. Chem. Soc. 2010;132:8419. doi: 10.1021/ja101851v. [DOI] [PMC free article] [PubMed] [Google Scholar]; (j) Kalyani D., Sanford M. S. J. Am. Chem. Soc. 2008;130:2150. doi: 10.1021/ja0782798. [DOI] [PubMed] [Google Scholar]
- (a) KC S., Basnet P., Thapa S., Shrestha B., Giri R. J. Org. Chem. 2018;83:2920. doi: 10.1021/acs.joc.8b00184. [DOI] [PubMed] [Google Scholar]; (b) Thapa S., Basnet P., Giri R. J. Am. Chem. Soc. 2017;139:5700. doi: 10.1021/jacs.7b01922. [DOI] [PubMed] [Google Scholar]; (c) You W., Brown M. K. J. Am. Chem. Soc. 2015;137:14578. doi: 10.1021/jacs.5b10176. [DOI] [PubMed] [Google Scholar]; (d) Cong H., Fu G. C. J. Am. Chem. Soc. 2014;136:3788. doi: 10.1021/ja500706v. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) You W., Brown M. K. J. Am. Chem. Soc. 2014;136:14730. doi: 10.1021/ja509056j. [DOI] [PubMed] [Google Scholar]
- (a) He Y., Yang Z., Thornbury R. T., Toste F. D. J. Am. Chem. Soc. 2015;137:12207. doi: 10.1021/jacs.5b07795. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Talbot E. P., Fernandes Tde A., McKenna J. M., Toste F. D. J. Am. Chem. Soc. 2014;136:4101. doi: 10.1021/ja412881j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CCDC 1852875 (5f) and CCDC ; 1852877 (6k) contain the supplementary crystallographic data
- Klotter F., Studer A. Angew. Chem., Int. Ed. 2014;53:2473. doi: 10.1002/anie.201310676. [DOI] [PubMed] [Google Scholar]
- For reviews on high-valent Pd in catalysis, see: ; (a) Topczewski J. J., Sanford M. S. Chem. Sci. 2015;6:70. doi: 10.1039/c4sc02591a. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Hickman A. J., Sanford M. S. Nature. 2012;484:177. doi: 10.1038/nature11008. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Xu L. M., Li B. J., Yang Z., Shi Z. J. Chem. Soc. Rev. 2010;39:712. doi: 10.1039/b809912j. [DOI] [PubMed] [Google Scholar]; (d) Muniz K. Angew. Chem., Int. Ed. 2009;48:9412. doi: 10.1002/anie.200903671. [DOI] [PubMed] [Google Scholar]; (e) Canty A. J. Dalton Trans. 2009:10409. doi: 10.1039/b914080h. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.